
Current Targeted Therapy for Metastatic Colorectal Cancer
by
 , and
, and
Tomokazu Ohishi
1,2,*,
Mika K. Kaneko
3,
Yukihiro Yoshida
4,
Atsuo Takashima
5,
Yukinari Kato
3,6,* and
and
Manabu Kawada
2 1
Institute of Microbial Chemistry (BIKAKEN), Numazu, Microbial Chemistry Research Foundation, 18-24 Miyamoto, Numazu-shi 410-0301, Shizuoka, Japan
2
Institute of Microbial Chemistry (BIKAKEN), Laboratory of Oncology, Microbial Chemistry Research Foundation, 3-14-23 Kamiosaki, Shinagawa-ku 141-0021, Tokyo, Japan
3
Department of Antibody Drug Development, Tohoku University Graduate School of Medicine, 2-1 Seiryo-machi, Aoba-ku, Sendai 980-8575, Miyagi, Japan
4
Department of Thoracic Surgery, National Cancer Center Hospital, 5-1-1 Tsukiji, Chuo-ku 104-0045, Tokyo, Japan
5
Department of Gastrointestinal Medical Oncology, National Cancer Center Hospital, Tsukiji, Chuo-ku 104-0045, Tokyo, Japan
6
Department of Molecular Pharmacology, Tohoku University Graduate School of Medicine, 2-1 Seiryo-machi, Aoba-ku, Sendai 980-8575, Miyagi, Japan
*
Authors to whom correspondence should be addressed.
Int. J. Mol. Sci. 2023, 24(2), 1702; https://doi.org/10.3390/ijms24021702
Submission received: 2 November 2022
/
Revised: 7 January 2023
/
Accepted: 13 January 2023
/
Published: 15 January 2023
(This article belongs to the Special Issue Molecular Mechanisms of Drug Resistance in Cancer: From Chemotherapy to Immunotherapy)
Abstract
:Colorectal cancer (CRC) is the third most common type of cancer and the second leading cause of cancer deaths worldwide. Surgery or surgery plus radiotherapy and/or chemotherapy for patients with metastatic CRC (mCRC) were accepted as the main therapeutic strategies until the early 2000s, when targeted drugs, like cetuximab and bevacizumab, were developed. The use of targeted drugs in clinical practice has significantly increased patients’ overall survival. To date, the emergence of several types of targeted drugs has opened new possibilities and revealed new prospects for mCRC treatment. Therapeutic strategies are continually being updated to select the most suitable targeted drugs based on the results of clinical trials that are currently underway. This review discusses the up-to date molecular evidence of targeted therapy for mCRC and summarizes the Food and Drug Administration-approved targeted drugs including the results of clinical trials. We also explain their mechanisms of action and how these affect the choice of a suitable targeted therapy.
1. Introduction
Colorectal cancer (CRC) arises from the epithelial cells lining the colon or rectum of the gastrointestinal tract and is the third most common cancer type among men and women in the United States [1]. CRC is also the third most commonly diagnosed cancer and was the second leading cause of cancer deaths in 2020 worldwide [2]. Surgery or surgery plus radiotherapy and chemotherapy in the adjuvant setting has improved the survival of patients with CRC, and the 5-year survival rate for CRC is 65%. However, because this falls to 15% for metastatic CRC (mCRC), the development of new therapeutic approaches to mCRC are critical [1,3].
Although complete surgical resection of the tumor and its metastatic sites improves overall survival (OS) in patients with CRC, approximately 25% of CRCs are diagnosed at an advanced stage with metastases in distant organs, which is difficult to manage surgically [4]. Unresectable advanced or recurrent CRC is treated with chemotherapy along with targeted therapy and/or radiotherapy to reduce the tumor size and prolong patient survival [5]. Regarding chemotherapy and targeted therapy, there are several first-line therapeutic options, and understanding the gene mutation status in CRC and resistance mechanisms is crucial to choose the best therapeutic option [6]. Notably, on rare occasions, the treatment may facilitate tumor downstaging, thereby improving the opportunity for resection.
Cytotoxic chemotherapy remains the standard treatment strategy for mCRC. Fluoropyrimidines play an important role as the backbone of combination regimens. Chemotherapy, such as FOLFOX (fluorouracil, leucovorin, and oxaliplatin), FOLFIRI (fluorouracil, leucovorin, and irinotecan), or FOLFOXIRI (fluorouracil, leucovorin, oxaliplatin, and irinotecan), combined with or without targeted drugs (anti-epidermal growth factor receptor [EGFR] antibody or anti-vascular endothelial growth factor [VEGF] antibody) is considered the first-line treatment for mCRC [7]. When chemotherapy is used, the doctor should give the patient as much information as possible about side effects because their severity depends on various factors, such as the type of cytotoxic drugs used and the duration of treatment [8,9]. Therefore, the active management of side effects is crucial so that the patient can continue chemotherapeutic treatment.
Several targeted drugs have been developed and studied. These drugs target the molecules involved in tumorigenesis and their related signaling pathways in cancer cells that make them different from normal cells [10]. Additionally, the tumor microenvironment, including blood vessels in the tissue surrounding the tumor and immune cells, are also affected by these targeted drugs to impede tumor growth and improve antitumor immune surveillance and attack [11]. The major types of targeted drugs are monoclonal antibodies and small molecule inhibitors. Such drugs are advantageous because, unlike chemotherapy, they can be chosen based on the molecular characteristics of tumor types [12]. However, a large number of patients experience disease progression even after receiving standard regimens. In the future, personalized medicine, in which drugs and drug combinations are optimized based on each patient’s available data including their genetic and epigenetic features and alterations, will improve the efficacy of treatments, reduce side effects, and benefit cancer patients [13,14].
This review summarizes the up-to-date evidence of clinical successes using targeted therapies to treat patients with mCRC. The molecular mechanisms of action and how these affect the choice of a suitable targeted therapy are also discussed.
2. mCRC Treatment Strategies
In the 1990s, fluorouracil-based chemotherapy improved the OS of patients with mCRC to 14 months. Later, the additional combination of leucovorin and oxaliplatin (FOLFOX) prolonged the OS to 19.5 months [15,16]. In 2004, the first Food and Drug Administration (FDA)-approved targeted drug was the anti-EGFR antibody cetuximab [17]. Since then, many targeted drugs for mCRC have been approved by FDA (Table 1).
The progression and spread of mCRC involves mediation with receptors in several signaling pathways. These include EGFRs, fibroblast growth factor receptors (FGFRs), vascular endothelia growth factor receptors (VEGFRs), and tropomyosin receptor kinases (TRKs) [29,30]. Furthermore, tumor cells express B7-1 (CD80)/B7-2 (CD80) and programmed cell death ligand 1 (PD-L1), which bind to cytotoxic T-lymphocyte-associated protein-4 (CTLA-4) and programmed cell death 1 (PD-1), respectively, on T cells to escape immune surveillance [30]. Therefore, these molecules and their related pathways must be inhibited by targeted drugs (Figure 1).
3. EGFR-Targeting Strategy
3.1. Molecular Mechanism of EGFR Signaling
EGFR is a member of the ErbB family of receptors, a subfamily of four receptor tyrosine kinases, including EGFR (ErbB-1), HER2 (ErbB-2), HER3 (ErbB-3), and HER4 (ErbB-4). It consists of extracellular, transmembrane, and intracellular domains and regulates cell proliferation, survival, differentiation, and migration [31]. The binding of ligands to the extracellular domain of EGFR promotes receptor dimerization, activating downstream signaling pathways, such as RAS/Raf/mitogen-activated protein kinase (MEK)/extracellular signal-regulated kinase (ERK), phosphoinositide 3-kinase (PI3K)/Akt, Janus kinase (JAK)/signal transducer and activator of transcription (STAT), and phospholipase C (PLC)-γ/protein kinase C (PKC), leading to the activation of gene transcription and playing key roles in cancer initiation and progression [32,33,34,35]. Several tumors, including mCRCs, overexpress EGFR, and the aberrant EGFR signaling is associated with poor prognosis. Thus, this receptor is a promising target for mCRC treatment [36].
3.2. Cetuximab and Panitumumab
Cetuximab and panitumumab are FDA-approved agents targeting EGFR (Table 1 and Figure 1). They are both distinct monoclonal antibodies and are used in monotherapy or in combination therapy with chemotherapy to treat patients with RAS wild-type mCRC [37]. Multiple reports have demonstrated that responses to either cetuximab or panitumumab occur exclusively in patients with mCRC without mutations in KRAS and NRAS codons 12 and 13 of exon 2, codons 59 and 61 of exon 3, and codons 117 and 146 of exon 4 [38].
Cetuximab is a chimeric mouse-human monoclonal antibody of the IgG1 subclass. It had the highest capacity to stimulate antibody-dependent cell-mediated cytotoxicity (ADCC) compared with other isotypes (such as IgG2, IgG3, and IgG4) [39,40]. It is thought that ADCC is mainly mediated by natural killer cells or macrophages, and it is one of the important modes of action of therapeutic antibodies [41]. Cetuximab showed great potential in an initial phase II clinical trial [42]. This was also confirmed by a randomized phase II trial (BOND trial) of cetuximab plus irinotecan or single irinotecan, which reported an OS of 22.9 months (218 subjects) vs. 10.8 months (111 subjects), respectively, in irinotecan-refractory patients [43] (Table 2).
Panitumumab is a fully human monoclonal IgG2 antibody. Whereas cetuximab, a chimeric mouse-human monoclonal antibody, might induce immunogenic reactions, there is less fear of this happening with panitumumab [46]. In fact, panitumumab was reported to show a lower risk of hypersensitivity reactions than cetuximab [47]. On the other hand, unlike cetuximab, panitumumab does not induce ADCC [48]. In a randomized phase III trial (PRIME trial), panitumumab plus FOLFOX4 showed improved progression-free survival (PFS) and OS of patients with mCRC compared with FOLFOX4 alone [44] (Table 2).
Importantly, cetuximab and panitumumab recognize and bind to domain III of EGFR and are effective in patients with wild-type RAS mCRC; both agents showed similar OS in a phase III trial (ASPECCT trial) [49,50]. Panitumumab shows effectiveness following cetuximab failure, and cetuximab is effective following panitumumab failure, indicating that the mechanisms of action of these two agents differ [51,52].
Although cetuximab and panitumumab have been used to treat mCRC, their use is limited to patients with wild-type RAS because patients with RAS mutations do not benefit from anti-EGFR treatment [53,54,55,56,57,58,59,60,61,62,63,64,65]. Notably, although approximately 50% of patients with CRC harbor RAS mutations (KRAS: 36% and NRAS: 3%) [66], not all patients with KRAS mutations are resistant to EGFR-targeted therapy [67]. There are conflicting reports with respect to the KRAS codon G13D mutation. Several retrospective studies demonstrated that cetuximab confers clinical benefits to patients with KRAS codon G13D-mutated mCRC compared with patients harboring other KRAS mutations [68,69]. A cell line from a tumor with a KRAS codon G12V mutation was unresponsive to cetuximab and panitumumab, whereas cell lines from a tumor with a KRAS codon G13D mutation showed an intermediate responsive to cetuximab and panitumumab in comparison with resistant KRAS codon mutation G12V and wild-type cells [70]. De Roock et al. investigated the association of response and survival between patients with KRAS codon G13D and other mutations (data set of 579 patients with chemotherapy-refractory CRC treated with cetuximab). They found that cetuximab treatment was associated with longer PFS and OS [71]. Although this finding was obtained from a retrospective study, in a randomized phase II study, Nakamura et al. showed that cetuximab-based treatment benefited patients with chemotherapy-resistant, refractory KRAS codon G13D-mutated mCRC [72]. Because of the limitations of the low number of patients with KRAS mutations in the datasets, further clinical studies with larger sample sizes are necessary to evaluate the differences in the efficacy of the EGFR-targeting strategy for the patients with the KRAS codon G13D mutation and those with KRAS mutations other than codon G13D.
It is important to note that tumor-sidedness has been identified as an important prognostic factor found in several clinical studies [73,74]. It has been reported that patients with wild-type KRAS/RAS having right-sided tumors are associated with worse prognosis to anti-EGFR treatment than left-sided tumors regardless of the first-line treatment given [74,75]. In addition, retrospective analyses have revealed that wild-type RAS with right-sided mCRC also leads to poor prognosis in second and later line anti-EGFR treatments than left-sided tumors [76]. Since the left and right colon have different embryologic origins, the left colon is from the foregut and right from midgut, this may explain the difference in treatment effects. Based on the results and randomized clinical trial results above, patients with right-sided mCRC with wild-type KRAS/NRAS/BRAF mutations should use anti-EGFR containing chemotherapy regimens [77,78].
Rechallenge with anti-EGFR treatment in patients who have responded to first-line regimens containing an EGFR targeting drug is emerging as a valid therapeutic strategy in patients with mCRC. In a phase II single-arm clinical trial (CRICKET trial), Cremolini et al. demonstrated that cetuximab rechallenge strategy is beneficial in patients with RAS and BRAF wild-type mCRC with acquired resistance to first-line cetuximab-based treatment (median PFS, 4.0 vs. 1.9 months, p < 0.001) [79]. In this trial, liquid biopsies are used to assess the circulating tumor DNA (ctDNA), and it is found that only patients with no RAS and BRAF mutations detected in their ctDNA may have a clinical benefit by the rechallenge with anti-EGFR treatment. In a single arm, open-label phase II study (CHRONOS trial), compared with standard third-line treatments, liquid biopsy-driven anti-EGFR rechallenge with panitumumab maximizes the therapeutic effects and avoids adverse effects [80].
4. VEGF/VEGFR Targeting Strategy
Angiogenesis is the process whereby new vessels are formed or reformed from existing vessels, and tumor angiogenesis plays an important role in tumor growth. The VEGF/VEGFR signaling pathway is recognized as one of the most predominant factors contributing to tumor angiogenesis, which participates in the multiple processes of tumor progression by activating host vascular endothelial cells [81].
Although, a VEGF/VEGFR-targeted strategy has been used in the patients with CRC with or without RAS mutations, patients with left-sided mCRC and right-sided mCRC with KRAS/NRAS/BRAF mutations should be considered for a VEGF/VEGFR-targeted drug containing chemotherapy regimens.
4.1. Molecular Mechanism of VEGF/VEGFR Signaling
VEGF family proteins and VEGFRs are key factors of tumor growth and metastasis that regulate normal and pathological tumor angiogenesis, leading to the activation of several signaling pathways [81]. The VEGF family consists of five members (VEGF-A, B, C, D, and placenta growth factor [PlGF]) that bind to endothelial cells through VEGFRs, including VEGFR-1, VEGFR-2, and VEGFR-3 [82]. While VEGF-A, VEGF-B, and PlGF mainly induce angiogenesis, VEGF-C and VEGF-D tend to regulate lymphangiogenesis [83]. VEGF-A, VEGF-B, and PlGF bind to VEGFR-1; VEGF-A, VEGF-C, and VEGF-D bind to VEGFR-2; and VEGF-C and VEGF-D bind to VEGFR-3; respectively, leading to various biological responses [84]. VEGFs cause VEGFR dimerization, which activates intrinsic tyrosine kinase, leading to the activation of signaling pathways, such as RAS/Raf/MEK/ERK, PI3K/Akt, and PLC-γ/PKC, to enhance tumor angiogenesis and proliferation. Among them, VEGFR-1 and VEGFR-2, which are common receptors for VEGF-A, are considered promising targets against cancer in clinical settings [85,86].
4.2. Bevacizumab
Bevacizumab is a humanized anti-VEGF-A monoclonal IgG1 antibody that inhibits VEGF-A binding to VEGFR-1 and VEGFR-2, and it is approved by the FDA to treat mCRC [87] (Table 1 and Figure 1). Hurwitz et al. demonstrated that relative to placebo plus IFL (irinotecan, fluorouracil, and leucovorin), the addition of bevacizumab to IFL when treating patients with mCRC significantly improved the 1-year survival rate (74.3% in 402 subjects vs. 63.4% in 411 subjects), OS (20.3 vs. 15.6 months), PFS (10.6 vs. 6.2 months), and response rate (RR) (44.8% vs. 34.8%) [88] (Table 3). In a randomized phase III trial (AVEX study), the addition of bevacizumab to capecitabine showed a tolerable safety profile and efficient administration and significantly improved OS (20.7 months in 140 subjects vs. 16.8 months in 140 subjects) and PFS (9.1 vs. 5.1 months) relative to single capecitabine in elderly patients (aged > 70 years) [89] (Table 3). Furthermore, in a randomized phase III trial (TRIBE trial), Cremolini et al. showed that a combination regimen of bevacizumab with the FOLFOXIRI showed better efficacy than that with FOLFIRI (OS: 31.0 vs. 25.8 months, p = 0.125; PFS: 12.1 vs. 9.7 months, p = 0.006; RR: 65% vs. 53%, p = 0.006; respectively) [90].
Although some clinical trials using bevacizumab with chemotherapy showed partial improvement in OS or PFS, bevacizumab in combination with chemotherapy is similarly effective in patients with KRAS wild-type and KRAS mutant mCRC [95]. In addition, Yoshimatsu et al. showed that there was no difference in therapeutic effect of bevacizumab for mCRC patient with RAS wild-type and RAS mutant [96]. Although both the EGFR and VEGF/VEGFR signaling pathways have been identified as possible therapeutic targets to treat patients with KRAS wild-type mCRC, data from several clinical trials (FIRE-3 trial and PEAK trial) shows that anti-EGFR treatment (cetuximab or panitumumab) appears superior to anti-VGFR treatment (bevacizumab) [77,78].
Importantly, Grothey et al. performed two large observational cohort studies and showed that after failure of bevacizumab combination chemotherapy for first-line treatment, continued bevacizumab beyond initial progression (BBP) improves OS and post progression survival [97,98]. In addition, during a randomized phase III trial (ML18147), relative to chemotherapy alone, second-line changing chemotherapy with bevacizumab at the same dose as used in the first-line treatment beyond disease progression improved median OS (11.2 months in 409 subjects vs. 9.8 months in 411 subjects) and PFS (6.9 vs. 4.67 months) [99]. From several other clinical studies including ARIES [100], RAISE [94], and VELOUR [101], BBP might have clinical benefits in patients who have experienced disease progression during a first-line bevacizumab containing chemotherapy.
4.3. Aflibercept
Aflibercept (also known as ziv-aflibercept) is a soluble molecule composed of the critical ligand-binding domains of human VEGFR-1 and VEGFR-2 fused with the Fc fragment of human IgG1. It functions as a decoy receptor by binding to VEGF-A, VEGF-B, and PlGF [102,103] (Table 1 and Figure 1). Aflibercept binds to VEGF-A with higher affinity and a faster association rate than bevacizumab [103]. In a randomized phase III trial (VELOUR trial), relative to placebo plus FOXFIRI, aflibercept plus FOXFIRI regimen improved OS (13.5 months in 612 subjects vs. 12.06 months in 614 subjects), PFS (6.9 vs. 4.67 months), and RR (19.8% vs. 11.1%) in patients with mCRC who progressed after receiving an oxaliplatin-based regimen [91] (Table 3). From these results, the FDA-approved aflibercept for the treatment of mCRC when given in combination with the FOLFIRI in 2012. However, in a randomized phase II trial (AFFIRM study), adding aflibercept to first-line modified FOLFOX6 (mFOLFOX6) did not show significant efficacy (aflibercept plus mFOLFOX6 in 119 subjects vs. mFOLFOX6 in 116 subjects, PFS: 8.48 vs. 8.77 months, RR: 49.1% vs. 45.9%). Use as a second-line treatment after progression following first-line treatment, aflibercept in combination with FOLFIRI showed efficacy following bevacizumab plus FOLFOXIRI for unresectable or mCRC in single arm phase II trials [104]. Therefore, the use of aflibercept-based regimens is recommended in second-line settings.
4.4. Ramucirumab
Ramucirumab is a fully human anti-VEGF-A monoclonal IgG1 antibody that inhibits VEGFR-2 and its downstream angiogenesis pathways. It was approved by the FDA for second-line use in combination with FOLFIRI for patients with mCRC who progressed during or after treatment with bevacizumab, oxaliplatin, and fluoropyrimidine [105]. In a second-line randomized phase III study (RAISE), relative to the placebo, ramucirumab in combination with FOLFIRI significantly improved OS (13.3 months in 536 subjects) vs. (11.7 months in 536 subjects) and PFS (5.7 vs. 4.5 months) with FOLFIRI [94] (Table 3). This was also confirmed by a retrospective study of ramucirumab plus FOLFIRI as a second- or later line therapy, which reported a positive impact on survival outcomes, with the PFS on second-line ramucirumab of 5.4 months (26 subjects) being equivalent to that observed in the RAISE trial [106]. Additionally, another retrospective study of ramucirumab plus FOLFIRI as second-line therapy reported an OS of 17.0 months (74 subjects) and PFS of 6.2 months (74 subjects), which was also equivalent to the RAISE trial [107].
4.5. Regorafenib
Regorafenib is a multikinase inhibitor that inhibits multiple intracellular and membrane-bound receptor tyrosine kinases, including VEGFR and FGFR involved in the regulation of tumor angiogenesis. It was approved by the FDA to treat previously treated patients with mCRC [108] (Table 1 and Figure 1). In a clinical phase III trial (CORRECT trial), Grothey et al. demonstrated that compared with the placebo, regorafenib significantly improved the OS (6.4 months in 505 subjects vs. 5.0 months in 255 subjects) and PFS (1.9 vs. 1.7 months) in treatment-refractory mCRC [92] (Table 3). This was also confirmed by a randomized phase III trial (CONCUR trial) of regorafenib plus best supportive care (BSC) or placebo plus BSC, which reported an OS of 8.8 months (138 subjects) vs. 6.3 months (68 subjects) and PFS of 3.2 vs. 1.7 months [93] (Table 3). Tyrosine kinase inhibitors (TKIs) have become one of the standard treatment regimens for patients with non-small-cell lung cancers [109]. Several TKIs have been tested for the patients with mCRC in clinical settings over recent years.
4.6. Fruquintinib
Fruquintinib is a potent and selective small molecule tyrosine kinase inhibitor of VEGFR 1, 2, and 3 [110]. It was approved in China for use in the treatment of mCRC. The FDA granted a fast track designation for the development of fruquintinib for the treatment of patients with mCRC who have received prior fluoropyrimidine-, oxaliplatin-, and irinotecan-based chemotherapy; EGFR-targeted therapy; and VEGF-targeted therapy. In a randomized, multicenter, phase III FRESCO trial conducted in China (NCT02314819), fruquintinib treatment in patients with mCRC who progressed after standard second-line therapy showed significant improvement in the PFS and OS [111]. Furthermore, in a randomized phase III FRESCO-2 trial (NCT04322539), with a total of 687 patients with refractory mCRC for more than third-line treatment, fruquintinib treatment showed significant PFS and OS [112]. These results indicate the clinical benefit of Fruquintinib for the patients with mCRC beyond third-line therapy.
5. Immune Checkpoint Targeting Immunotherapies
Immunotherapy is a novel treatment option against several types of tumors. Tumor immunotherapy induces an immune cell-mediated immune response through the neoantigen expressed on a broad range of tumors [113]. The success of tumor immunotherapy in achieving long-lasting antitumor responses has demonstrated that immune cells, mainly T cells, could be utilized to eliminate tumor cells [114].
Microsatellite instability (MSI) is an indicator of defective DNA mismatch repair (dMMR) and an MSI/dMMR status is observed in approximately 5% of mCRC cases [115]. Because MMR pathways are responsible for correcting DNA replication errors, MSI-high tumors carry various somatic mutations, leading to a high neoantigen exposure that favors the initiation of an antitumor immune response [116,117]. Therefore, MSI-high tumors respond well to immunotherapy.
5.1. Molecular Mechanism of Immune Checkpoints
Programmed cell death-1 (PD-1) and CTLA-4 are expressed on the surface of activated immune cells, including T cells, and are key immune checkpoint molecules that inactivate immune cells through distinct mechanisms [118]. PD-1 inhibits T cell response via interaction with its two ligands, PD-L1 and PD-L2, to mediate an inhibitory signal in T cells, which suppresses cellular and humoral immune responses [119]. Blockage of PD-1 or PD-L1 can inhibit the checkpoint and induce T cell activation to drive immunity [120,121].
CTLA-4 is expressed on activated T cells and regulatory T cells (Tregs). It inhibits T cell response by interacting with B7-1 and B7-2 ligands to provide an inhibitory signal in T cells [122]. Inhibition of CTLA-4 binding to these ligands results in the reactivation and proliferation of T cells and decreases immunosuppressive Tregs, leading to increased activation of the immune system in the tumor microenvironment [123].
5.2. Pembrolizumab, Nivolumab, and Ipilimumab
Pembrolizumab is a humanized IgG4 anti-PD-1 monoclonal antibody and nivolumab is a human IgG4 anti-PD-1 monoclonal antibody. They have been approved by the FDA for patients with multiple tumors, including MSI-high/dMMR mCRC [124] (Table 1 and Figure 1). In a phase II study (NCT01876511), pembrolizumab treatment in patients with MSI-high/dMMR mCRC in second-line settings showed an objective RR (ORR) of 52% (40 subjects), disease control rate of 82%, 2-year OS of 72%, and 2-year PFS of 59% [125] (Table 4). Furthermore, in an open-label phase III trial (KEYNOTE-177), pembrolizumab as first-line treatment for patients with MSI-high/dMMR mCRC was superior to chemotherapy with respect to PFS (16.5 months in 153 subjects vs. 8.2 months in 154 subjects) and ORR (43.8% vs. 33.1%). Importantly, treatment-related adverse effects of grade 3 or higher occurred in 22% of the patients in the pembrolizumab-treated group compared with 66% in the chemotherapy-treated group [126] (Table 4). In addition to pembrolizumab, in an open-label phase II trial (CheckMate 142), second-line treatment with nivolumab in patients with MSI-high/dMMR mCRC showed an ORR of 31% (74 subjects) and disease control for 12 weeks or longer of 69% [127] (Table 4). From these results, pembrolizumab and nivolumab are thought to be effective and show a manageable safety profile in patients with MSI-high/dMMR mCRC.
Ipilimumab is a fully human IgG1 anti-CTLA-4 monoclonal antibody. It has been approved by the FDA for combination therapy with nivolumab in patients with MSI-high/dMMR mCRC after progression following chemotherapy [25] (Table 1 and Figure 1). In an open-label phase II trial (CheckMate-142), relative to nivolumab, nivolumab in combination with ipilimumab in patients with MSI-high/dMMR mCRC improved 1-year OS (85% in 119 subjects vs. 73% in 74 subjects), ORR (55% vs. 31%), and disease control for 12 weeks or longer (80% vs. 69%) [128] (Table 4). These results indicated that the combination of nivolumab and ipilimumab showed a favorable impact on the quality of life for patients with MSI-high/dMMR mCRC.
6. NTRK Signaling Pathway and Targeting Strategy
The neurotrophic tropomyosin receptor kinases (NTRK) family consists of three members (NTRK1, NTRK2, and NTRK3) that code for TRKA, TRKB, and TRKC, respectively, and they are mainly expressed in neural and neuronal tissues [129]. They perform biological functions by homodimerization, which activates their downstream pathways, including RAS/Raf/MEK/ERK, PI3K/Akt, and PLC-γ/PKC, leading to the activation of gene transcription, cell survival, and progression [130,131]. Multiple NTRK fusions are one of the most common oncogenic events, leading to constitutive activation of their downstream pathways [132]. Besides NTRK, anaplastic lymphoma kinase (ALK) and c-ros oncogene 1 (ROS1) fusions also occur in CRC. ALK and ROS1 also encode tyrosine kinases that are constitutively activated by fusion, leading to the activation downstream signaling pathways for tumor cell growth and progression [133,134]. Their gene rearrangements are thought to be mutually exclusive in lung cancers despite ALK and ROS1 tyrosine kinase domains sharing high homology [135]. Although these fusions occur in less than 2.5% of CRC cases, they are considered oncogenic drivers and a potential target for mCRC treatment [136,137].
Larotrectinib and Entrectinib
Larotrectinib and entrectinib are first-generation TRK inhibitors. They were approved by the FDA for patients with solid tumors that harbor NTRK gene fusions [138] (Table 1). Larotrectinib is a selective inhibitor of TRK that was tested in three phase I/II clinical trials of 153 cancer patients carrying NTRK fusions (age: 48–67 years) and showed an impressive ORR of 79% and good tolerability [139] (Figure 1).
7. BRAF Signaling Pathway and Targeting Strategy
BRAF is a downstream effector of RAS in the RAS/Raf/MEK/ERK signaling pathway and is a well-known oncogenic driver [140]. Mutations in BRAF V600E are present in approximately 10% of mCRCs and associated with chemotherapy resistance and worse prognosis [141,142]. Interestingly, non-V600 BRAF mutation was independently and significantly associated with improved OS [142]. Given that monotherapy with a BRAF-targeting tyrosine kinase inhibitor for patients with mCRC has failed, some combination regimens have been tested in multiple clinical trials [143].
Encorafenib
Encorafenib is a kinase inhibitor that targets BRAF V600E as well as wild-type BRAF and shows more prolonged pharmacodynamic activity than other BRAF inhibitors [144] (Table 1 and Figure 1). In an open-label phase III trial (BEACON), compared with the control (cetuximab plus chemotherapy), encorafenib in combination with cetuximab showed a significantly improved median OS (8.4 months in 220 subjects vs. 5.4 months in 221 subjects) and confirmed RR (20% vs. 2%) [45] (Table 2). Additionally, in BEACON trial, compared with the control, a triple regimen containing encorafenib, cetuximab, and a MEK inhibitor, binimetinib also showed a significantly improved median OS (9.0 months in 224 subjects vs. 5.4 months in 221 subjects) and confirmed RR (24% vs. 2%) [45] (Table 2). From these results, in 2020, the FDA-approved encorafenib for the treatment of mCRC with a BRAF V600E mutation after prior therapy.
8. Concluding Remarks and Future Perspectives
This review focused on FDA-approved targeted drugs that are currently available to treat patients with mCRC. Our objective was to address the efficacy of these treatments. We included most clinical trials that were associated with FDA approvals, reviewed trial-associated publications, and explored other relevant clinical trials.
Recent advancements in sequencing technologies have led to a better understanding of comprehensive genomic and proteomic alterations in mCRC, which helps to choose the correct treatment strategy. Although the latest therapeutic strategies have achieved substantial progress in patients with mCRC, emerging resistance and lack of predictive biomarkers to current targeted therapies remains a major problem in clinical settings. Further understanding of resistance mechanisms and exploration of biomarkers that can predict the treatment sensitivity and efficacy/toxicity are required to test new evidence-based therapeutic strategies, expand treatment options, and realize personalized medicine [145,146,147,148].
To further improve the efficacy of treatments, innovations, such as CRC multicellular 3D models, patient-derived xenograft models, and single-cell sequencing strategy, and applying new antibody-based therapies strategies, including chimeric antigen receptor T cell therapy [149], antibody drug conjugates [150], radioimmunotherapy [151], and photoimmunotherapy [152], may provide a survival benefit for patients with mCRC. To date, we have developed several cancer-specific monoclonal antibodies, including anti-EGFR antibodies using the CasMab method [153,154,155,156,157,158,159,160,161,162,163]. Currently, we are trying to further develop these antibodies for the above-mentioned antibody-based therapy.
Several promising targets including HER2, KRAS codon G12C mutation, and RET have been revealed and their targeted therapies are under development and/or clinical evaluation [164,165,166]. Continued comprehensive understanding of mCRC pathology and molecular drivers, and developing novel therapeutic approaches are necessary to further refine the treatment of mCRC. While validated targets, including RAS, BRAF, TRK, and MSI/dMMR, are important to choose the correct therapeutic strategy for patients with mCRC [167]. This is because disclosure of additional targets may lead to the discovery of potential predictive biomarkers of treatment response and the correct selection of patients. Further studies, including clinical trials, hold promise for improved outcomes and progress toward personalized medicine for mCRC.
Author Contributions
Project design, Outline of the whole manuscript, T.O., M.K.K., Y.Y., A.T., Y.K. and M.K.; Writing, T.O. Funding acquisition, Y.K., T.O. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported in part by Japan Agency for Medical Research and Development (AMED) under grant numbers JP21bm1004001 (to Y.K.), JP21am0401013 (to Y.K., T.O.), JP22ama121008 (to Y.K.) and JP21am0101078 (to Y.K.) and by the Japan Society for the Promotion of Science (JSPS) Grants-in-Aid for Scientific Research (KAKENHI) under grant numbers 18K08693 (T.O.), 22K06783 (T.O.).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We would like to thank Akiko Harakawa, Shun-ichi Ohba, and Hiroyuki Inoue (Institute of Microbial Chemistry (BIKAKEN), Numazu, Microbial Chemistry Research Foundation) for their helpful suggestions. We also appreciate laboratory members for their contributing discussion.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Kuipers, E.J.; Grady, W.M.; Lieberman, D.; Seufferlein, T.; Sung, J.J.; Boelens, P.G.; van de Velde, C.J.; Watanabe, T. Colorectal cancer. Nat. Rev. Dis. Prim. 2015, 1, 15065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Stok, E.P.; Spaander, M.C.W.; Grunhagen, D.J.; Verhoef, C.; Kuipers, E.J. Surveillance after curative treatment for colorectal cancer. Nat. Rev. Clin. Oncol. 2017, 14, 297–315. [Google Scholar] [CrossRef] [PubMed]
- Graham, D.M.; Coyle, V.M.; Kennedy, R.D.; Wilson, R.H. Molecular Subtypes and Personalized Therapy in Metastatic Colorectal Cancer. Curr. Color. Cancer Rep. 2016, 12, 141–150. [Google Scholar] [CrossRef] [Green Version]
- Motta, R.; Cabezas-Camarero, S.; Torres-Mattos, C.; Riquelme, A.; Calle, A.; Montenegro, P.; Sotelo, M.J. Personalizing first-line treatment in advanced colorectal cancer: Present status and future perspectives. J. Clin. Transl. Res. 2021, 7, 771–785. [Google Scholar]
- Marques, R.P.; Duarte, G.S.; Sterrantino, C.; Pais, H.L.; Quintela, A.; Martins, A.P.; Costa, J. Triplet (FOLFOXIRI) versus doublet (FOLFOX or FOLFIRI) backbone chemotherapy as first-line treatment of metastatic colorectal cancer: A systematic review and meta-analysis. Crit. Rev. Oncol. Hematol. 2017, 118, 54–62. [Google Scholar] [CrossRef]
- Sunakawa, Y.; Bekaii-Saab, T.; Stintzing, S. Reconsidering the benefit of intermittent versus continuous treatment in the maintenance treatment setting of metastatic colorectal cancer. Cancer Treat. Rev. 2016, 45, 97–104. [Google Scholar] [CrossRef]
- Miyo, M.; Kato, T.; Yoshino, T.; Yamanaka, T.; Bando, H.; Satake, H.; Yamazaki, K.; Taniguchi, H.; Oki, E.; Kotaka, M.; et al. Protocol of the QUATTRO-II study: A multicenter randomized phase II study comparing CAPOXIRI plus bevacizumab with FOLFOXIRI plus bevacizumab as a first-line treatment in patients with metastatic colorectal cancer. BMC Cancer 2020, 20, 687. [Google Scholar] [CrossRef]
- DeStefanis, R.A.; Kratz, J.D.; Emmerich, P.B.; Deming, D.A. Targeted Therapy in Metastatic Colorectal Cancer: Current Standards and Novel Agents in Review. Curr. Color. Cancer Rep. 2019, 15, 61–69. [Google Scholar] [CrossRef]
- Nappi, A.; Berretta, M.; Romano, C.; Tafuto, S.; Cassata, A.; Casaretti, R.; Silvestro, L.; Divitiis, C.; Alessandrini, L.; Fiorica, F.; et al. Metastatic Colorectal Cancer: Role of Target Therapies and Future Perspectives. Curr. Cancer Drug Targets 2018, 18, 421–429. [Google Scholar] [CrossRef]
- Baraibar, I.; Ros, J.; Mulet, N.; Salva, F.; Argiles, G.; Martini, G.; Cuadra, J.L.; Sardo, E.; Ciardiello, D.; Tabernero, J.; et al. Incorporating traditional and emerging biomarkers in the clinical management of metastatic colorectal cancer: An update. Expert Rev. Mol. Diagn. 2020, 20, 653–664. [Google Scholar] [CrossRef]
- Singh, M.P.; Rai, S.; Pandey, A.; Singh, N.K.; Srivastava, S. Molecular subtypes of colorectal cancer: An emerging therapeutic opportunity for personalized medicine. Genes Dis. 2021, 8, 133–145. [Google Scholar] [CrossRef]
- Wu, C.W.K.; Reid, M.; Leedham, S.; Lui, R.N. The emerging era of personalized medicine in advanced colorectal cancer. J. Gastroenterol. Hepatol. 2022, 37, 1411–1425. [Google Scholar] [CrossRef] [PubMed]
- Douillard, J.Y.; Cunningham, D.; Roth, A.D.; Navarro, M.; James, R.D.; Karasek, P.; Jandik, P.; Iveson, T.; Carmichael, J.; Alakl, M.; et al. Irinotecan combined with fluorouracil compared with fluorouracil alone as first-line treatment for metastatic colorectal cancer: A multicentre randomised trial. Lancet 2000, 355, 1041–1047. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, R.M.; Sargent, D.J.; Morton, R.F.; Fuchs, C.S.; Ramanathan, R.K.; Williamson, S.K.; Findlay, B.P.; Pitot, H.C.; Alberts, S.R. A randomized controlled trial of fluorouracil plus leucovorin, irinotecan, and oxaliplatin combinations in patients with previously untreated metastatic colorectal cancer. J. Clin. Oncol. 2004, 22, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Caballero Servin, I.A. Evaluation and application of microbiological methods to the sanitary control of restaurants. Salud Publica Mex. 1967, 9, 221–235. [Google Scholar] [PubMed]
- US Food and Drug Administration. New treatments for colorectal cancer. FDA Consum 2004, 38, 17. [Google Scholar]
- US Food and Drug Administration. The FDA approves drugs for colorectal cancer, lung cancer. FDA Consum 2007, 41, 5. [Google Scholar]
- FDA approves aflibercept (Zaltrap) for metastatic colorectal cancer. Oncology 2012, 26, 842–873.
- FDA approves regorafenib (Stivarga) for metastatic colorectal cancer. Oncology 2012, 26, 896.
- Venook, A.P. The value and effectiveness of angiogenesis inhibitors for colorectal cancer. Clin. Adv. Hematol. Oncol. 2015, 13, 561–563. [Google Scholar] [PubMed]
- Geanta, M.; Cioroboiu, C. The FDA Changed Everything. Biomed. Hub 2017, 2, 52–54. [Google Scholar] [CrossRef] [PubMed]
- Ciombor, K.K.; Goldberg, R.M. Hypermutated Tumors and Immune Checkpoint Inhibition. Drugs 2018, 78, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Garcia, C.R.; Jayswal, R.; Adams, V.; Anthony, L.B.; Villano, J.L. Multiple sclerosis outcomes after cancer immunotherapy. Clin. Transl. Oncol. 2019, 21, 1336–1342. [Google Scholar] [CrossRef] [PubMed]
- Valeri, N. Streamlining Detection of Fusion Genes in Colorectal Cancer: Having “Faith” in Precision Oncology in the (Tissue) “Agnostic” Era. Cancer Res. 2019, 79, 1041–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullard, A. FDA notches up third tissue-agnostic cancer approval. Nat. Rev. Drug Discov. 2019, 18, 737. [Google Scholar] [CrossRef]
- Koumaki, K.; Kontogianni, G.; Kosmidou, V.; Pahitsa, F.; Kritsi, E.; Zervou, M.; Chatziioannou, A.; Souliotis, V.L.; Papadodima, O.; Pintzas, A. BRAF paradox breakers PLX8394, PLX7904 are more effective against BRAFV600Epsilon CRC cells compared with the BRAF inhibitor PLX4720 and shown by detailed pathway analysis. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166061. [Google Scholar] [CrossRef]
- Kheder, E.S.; Hong, D.S. Emerging Targeted Therapy for Tumors with NTRK Fusion Proteins. Clin. Cancer Res. 2018, 24, 5807–5814. [Google Scholar] [CrossRef] [Green Version]
- Guler, I.; Askan, G.; Klostergaard, J.; Sahin, I.H. Precision medicine for metastatic colorectal cancer: An evolving era. Expert Rev. Gastroenterol. Hepatol. 2019, 13, 919–931. [Google Scholar] [CrossRef]
- Wieduwilt, M.J.; Moasser, M.M. The epidermal growth factor receptor family: Biology driving targeted therapeutics. Cell Mol. Life Sci. 2008, 65, 1566–1584. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.C.; Hung, M.C. Nuclear translocation of the epidermal growth factor receptor family membrane tyrosine kinase receptors. Clin. Cancer Res. 2009, 15, 6484–6489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinemann, V.; Stintzing, S.; Kirchner, T.; Boeck, S.; Jung, A. Clinical relevance of EGFR- and KRAS-status in colorectal cancer patients treated with monoclonal antibodies directed against the EGFR. Cancer Treat. Rev. 2009, 35, 262–271. [Google Scholar] [CrossRef]
- Yarden, Y.; Pines, G. The ERBB network: At last, cancer therapy meets systems biology. Nat. Rev. Cancer 2012, 12, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Bertotti, A.; Papp, E.; Jones, S.; Adleff, V.; Anagnostou, V.; Lupo, B.; Sausen, M.; Phallen, J.; Hruban, C.A.; Tokheim, C.; et al. The genomic landscape of response to EGFR blockade in colorectal cancer. Nature 2015, 526, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Jeong, W.J.; Cha, P.H.; Choi, K.Y. Strategies to overcome resistance to epidermal growth factor receptor monoclonal antibody therapy in metastatic colorectal cancer. World J. Gastroenterol. 2014, 20, 9862–9871. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Cervantes, A.; Adam, R.; Sobrero, A.; Van Krieken, J.H.; Aderka, D.; Aranda Aguilar, E.; Bardelli, A.; Benson, A.; Bodoky, G.; et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann. Oncol. 2016, 27, 1386–1422. [Google Scholar] [CrossRef]
- Allegra, C.J.; Rumble, R.B.; Hamilton, S.R.; Mangu, P.B.; Roach, N.; Hantel, A.; Schilsky, R.L. Extended RAS Gene Mutation Testing in Metastatic Colorectal Carcinoma to Predict Response to Anti-Epidermal Growth Factor Receptor Monoclonal Antibody Therapy: American Society of Clinical Oncology Provisional Clinical Opinion Update 2015. J. Clin. Oncol. 2016, 34, 179–185. [Google Scholar] [CrossRef]
- Goldstein, N.I.; Prewett, M.; Zuklys, K.; Rockwell, P.; Mendelsohn, J. Biological efficacy of a chimeric antibody to the epidermal growth factor receptor in a human tumor xenograft model. Clin. Cancer Res. 1995, 1, 1311–1318. [Google Scholar]
- Trotta, A.M.; Ottaiano, A.; Romano, C.; Nasti, G.; Nappi, A.; De Divitiis, C.; Napolitano, M.; Zanotta, S.; Casaretti, R.; D’Alterio, C.; et al. Prospective Evaluation of Cetuximab-Mediated Antibody-Dependent Cell Cytotoxicity in Metastatic Colorectal Cancer Patients Predicts Treatment Efficacy. Cancer Immunol. Res. 2016, 4, 366–374. [Google Scholar] [CrossRef] [Green Version]
- Lo Nigro, C.; Macagno, M.; Sangiolo, D.; Bertolaccini, L.; Aglietta, M.; Merlano, M.C. NK-mediated antibody-dependent cell-mediated cytotoxicity in solid tumors: Biological evidence and clinical perspectives. Ann. Transl. Med 2019, 7, 105. [Google Scholar] [CrossRef] [Green Version]
- Saltz, L.B.; Meropol, N.J.; Loehrer, P.J., Sr.; Needle, M.N.; Kopit, J.; Mayer, R.J. Phase II trial of cetuximab in patients with refractory colorectal cancer that expresses the epidermal growth factor receptor. J. Clin. Oncol. 2004, 22, 1201–1208. [Google Scholar] [CrossRef]
- Cunningham, D.; Humblet, Y.; Siena, S.; Khayat, D.; Bleiberg, H.; Santoro, A.; Bets, D.; Mueser, M.; Harstrick, A.; Verslype, C.; et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N. Engl. J. Med. 2004, 351, 337–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Douillard, J.Y.; Siena, S.; Cassidy, J.; Tabernero, J.; Burkes, R.; Barugel, M.; Humblet, Y.; Bodoky, G.; Cunningham, D.; Jassem, J.; et al. Final results from PRIME: Randomized phase III study of panitumumab with FOLFOX4 for first-line treatment of metastatic colorectal cancer. Ann. Oncol. 2014, 25, 1346–1355. [Google Scholar] [CrossRef]
- Kopetz, S.; Grothey, A.; Yaeger, R.; Van Cutsem, E.; Desai, J.; Yoshino, T.; Wasan, H.; Ciardiello, F.; Loupakis, F.; Hong, Y.S.; et al. Encorafenib, Binimetinib, and Cetuximab in BRAF V600E-Mutated Colorectal Cancer. N. Engl. J. Med. 2019, 381, 1632–1643. [Google Scholar] [CrossRef] [Green Version]
- Keating, G.M. Panitumumab: A review of its use in metastatic colorectal cancer. Drugs 2010, 70, 1059–1078. [Google Scholar] [CrossRef] [PubMed]
- Fakih, M.; Vincent, M. Adverse events associated with anti-EGFR therapies for the treatment of metastatic colorectal cancer. Curr. Oncol. 2010, 17 (Suppl. 1), S18–S30. [Google Scholar] [CrossRef] [Green Version]
- Yarom, N.; Jonker, D.J. The role of the epidermal growth factor receptor in the mechanism and treatment of colorectal cancer. Discov. Med. 2011, 11, 95–105. [Google Scholar] [PubMed]
- Koefoed, K.; Steinaa, L.; Soderberg, J.N.; Kjaer, I.; Jacobsen, H.J.; Meijer, P.J.; Haurum, J.S.; Jensen, A.; Kragh, M.; Andersen, P.S.; et al. Rational identification of an optimal antibody mixture for targeting the epidermal growth factor receptor. MAbs 2011, 3, 584–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, T.J.; Peeters, M.; Kim, T.W.; Li, J.; Cascinu, S.; Ruff, P.; Suresh, A.S.; Thomas, A.; Tjulandin, S.; Zhang, K.; et al. Panitumumab versus cetuximab in patients with chemotherapy-refractory wild-type KRAS exon 2 metastatic colorectal cancer (ASPECCT): A randomised, multicentre, open-label, non-inferiority phase 3 study. Lancet Oncol. 2014, 15, 569–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saif, M.W.; Kaley, K.; Chu, E.; Copur, M.S. Safety and efficacy of panitumumab therapy after progression with cetuximab: Experience at two institutions. Clin. Color. Cancer 2010, 9, 315–318. [Google Scholar] [CrossRef] [PubMed]
- Hata, A.; Katakami, N.; Kitajima, N. Successful cetuximab therapy after failure of panitumumab rechallenge in a patient with metastatic colorectal cancer: Restoration of drug sensitivity after anti-EGFR monoclonal antibody-free interval. J. Gastrointest. Cancer 2014, 45, 506–507. [Google Scholar] [CrossRef] [PubMed]
- Di Fiore, F.; Blanchard, F.; Charbonnier, F.; Le Pessot, F.; Lamy, A.; Galais, M.P.; Bastit, L.; Killian, A.; Sesboue, R.; Tuech, J.J.; et al. Clinical relevance of KRAS mutation detection in metastatic colorectal cancer treated by Cetuximab plus chemotherapy. Br. J. Cancer 2007, 96, 1166–1169. [Google Scholar] [CrossRef] [Green Version]
- Benvenuti, S.; Sartore-Bianchi, A.; Di Nicolantonio, F.; Zanon, C.; Moroni, M.; Veronese, S.; Siena, S.; Bardelli, A. Oncogenic activation of the RAS/RAF signaling pathway impairs the response of metastatic colorectal cancers to anti-epidermal growth factor receptor antibody therapies. Cancer Res. 2007, 67, 2643–2648. [Google Scholar] [CrossRef] [Green Version]
- De Roock, W.; Piessevaux, H.; De Schutter, J.; Janssens, M.; De Hertogh, G.; Personeni, N.; Biesmans, B.; Van Laethem, J.L.; Peeters, M.; Humblet, Y.; et al. KRAS wild-type state predicts survival and is associated to early radiological response in metastatic colorectal cancer treated with cetuximab. Ann. Oncol. 2008, 19, 508–515. [Google Scholar] [CrossRef]
- Karapetis, C.S.; Khambata-Ford, S.; Jonker, D.J.; O’Callaghan, C.J.; Tu, D.; Tebbutt, N.C.; Simes, R.J.; Chalchal, H.; Shapiro, J.D.; Robitaille, S.; et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N. Engl. J. Med. 2008, 359, 1757–1765. [Google Scholar] [CrossRef] [PubMed]
- Lievre, A.; Bachet, J.B.; Boige, V.; Cayre, A.; Le Corre, D.; Buc, E.; Ychou, M.; Bouche, O.; Landi, B.; Louvet, C.; et al. KRAS mutations as an independent prognostic factor in patients with advanced colorectal cancer treated with cetuximab. J. Clin. Oncol. 2008, 26, 374–379. [Google Scholar] [CrossRef]
- Amado, R.G.; Wolf, M.; Peeters, M.; Van Cutsem, E.; Siena, S.; Freeman, D.J.; Juan, T.; Sikorski, R.; Suggs, S.; Radinsky, R.; et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J. Clin. Oncol. 2008, 26, 1626–1634. [Google Scholar] [CrossRef]
- Bokemeyer, C.; Bondarenko, I.; Hartmann, J.T.; de Braud, F.; Schuch, G.; Zubel, A.; Celik, I.; Schlichting, M.; Koralewski, P. Efficacy according to biomarker status of cetuximab plus FOLFOX-4 as first-line treatment for metastatic colorectal cancer: The OPUS study. Ann. Oncol. 2011, 22, 1535–1546. [Google Scholar] [CrossRef]
- Allegra, C.J.; Jessup, J.M.; Somerfield, M.R.; Hamilton, S.R.; Hammond, E.H.; Hayes, D.F.; McAllister, P.K.; Morton, R.F.; Schilsky, R.L. American Society of Clinical Oncology provisional clinical opinion: Testing for KRAS gene mutations in patients with metastatic colorectal carcinoma to predict response to anti-epidermal growth factor receptor monoclonal antibody therapy. J. Clin. Oncol. 2009, 27, 2091–2096. [Google Scholar] [CrossRef] [Green Version]
- Van Cutsem, E.; Kohne, C.H.; Hitre, E.; Zaluski, J.; Chang Chien, C.R.; Makhson, A.; D’Haens, G.; Pinter, T.; Lim, R.; Bodoky, G.; et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N. Engl. J. Med. 2009, 360, 1408–1417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bokemeyer, C.; Kohne, C.H.; Ciardiello, F.; Lenz, H.J.; Heinemann, V.; Klinkhardt, U.; Beier, F.; Duecker, K.; van Krieken, J.H.; Tejpar, S. FOLFOX4 plus cetuximab treatment and RAS mutations in colorectal cancer. Eur. J. Cancer 2015, 51, 1243–1252. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Lenz, H.J.; Kohne, C.H.; Heinemann, V.; Tejpar, S.; Melezinek, I.; Beier, F.; Stroh, C.; Rougier, P.; van Krieken, J.H.; et al. Fluorouracil, leucovorin, and irinotecan plus cetuximab treatment and RAS mutations in colorectal cancer. J. Clin. Oncol. 2015, 33, 692–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peeters, M.; Oliner, K.S.; Price, T.J.; Cervantes, A.; Sobrero, A.F.; Ducreux, M.; Hotko, Y.; Andre, T.; Chan, E.; Lordick, F.; et al. Analysis of KRAS/NRAS Mutations in a Phase III Study of Panitumumab with FOLFIRI Compared with FOLFIRI Alone as Second-line Treatment for Metastatic Colorectal Cancer. Clin. Cancer Res. 2015, 21, 5469–5479. [Google Scholar] [CrossRef] [Green Version]
- Douillard, J.Y.; Oliner, K.S.; Siena, S.; Tabernero, J.; Burkes, R.; Barugel, M.; Humblet, Y.; Bodoky, G.; Cunningham, D.; Jassem, J.; et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N. Engl. J. Med. 2013, 369, 1023–1034. [Google Scholar] [CrossRef] [Green Version]
- Wilson, C.Y.; Tolias, P. Recent advances in cancer drug discovery targeting RAS. Drug Discov. Today 2016, 21, 1915–1919. [Google Scholar] [CrossRef] [PubMed]
- Misale, S.; Di Nicolantonio, F.; Sartore-Bianchi, A.; Siena, S.; Bardelli, A. Resistance to anti-EGFR therapy in colorectal cancer: From heterogeneity to convergent evolution. Cancer Discov. 2014, 4, 1269–1280. [Google Scholar] [CrossRef] [Green Version]
- Tejpar, S.; Celik, I.; Schlichting, M.; Sartorius, U.; Bokemeyer, C.; Van Cutsem, E. Association of KRAS G13D tumor mutations with outcome in patients with metastatic colorectal cancer treated with first-line chemotherapy with or without cetuximab. J. Clin. Oncol. 2012, 30, 3570–3577. [Google Scholar] [CrossRef]
- Mao, C.; Huang, Y.F.; Yang, Z.Y.; Zheng, D.Y.; Chen, J.Z.; Tang, J.L. KRAS p.G13D mutation and codon 12 mutations are not created equal in predicting clinical outcomes of cetuximab in metastatic colorectal cancer: A systematic review and meta-analysis. Cancer 2013, 119, 714–721. [Google Scholar] [CrossRef]
- Kumar, S.S.; Price, T.J.; Mohyieldin, O.; Borg, M.; Townsend, A.; Hardingham, J.E. KRAS G13D Mutation and Sensitivity to Cetuximab or Panitumumab in a Colorectal Cancer Cell Line Model. Gastrointest. Cancer Res. 2014, 7, 23–26. [Google Scholar]
- De Roock, W.; Jonker, D.J.; Di Nicolantonio, F.; Sartore-Bianchi, A.; Tu, D.; Siena, S.; Lamba, S.; Arena, S.; Frattini, M.; Piessevaux, H.; et al. Association of KRAS p.G13D mutation with outcome in patients with chemotherapy-refractory metastatic colorectal cancer treated with cetuximab. JAMA 2010, 304, 1812–1820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, M.; Aoyama, T.; Ishibashi, K.; Tsuji, A.; Takinishi, Y.; Shindo, Y.; Sakamoto, J.; Oba, K.; Mishima, H. Randomized phase II study of cetuximab versus irinotecan and cetuximab in patients with chemo-refractory KRAS codon G13D metastatic colorectal cancer (G13D-study). Cancer Chemother. Pharmacol. 2017, 79, 29–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Missiaglia, E.; Jacobs, B.; D’Ario, G.; Di Narzo, A.F.; Soneson, C.; Budinska, E.; Popovici, V.; Vecchione, L.; Gerster, S.; Yan, P.; et al. Distal and proximal colon cancers differ in terms of molecular, pathological, and clinical features. Ann. Oncol. 2014, 25, 1995–2001. [Google Scholar] [CrossRef]
- Loupakis, F.; Yang, D.; Yau, L.; Feng, S.; Cremolini, C.; Zhang, W.; Maus, M.K.; Antoniotti, C.; Langer, C.; Scherer, S.J.; et al. Primary tumor location as a prognostic factor in metastatic colorectal cancer. J. Natl. Cancer Inst. 2015, 107, dju427. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Yu, Q.; Ning, R.; Zhao, W.; Wei, C. Efficacy of bevacizumab versus epidermal growth factor receptor inhibitors for wild-type RAS metastatic colorectal cancer: A meta-analysis. OncoTargets Ther. 2018, 11, 4271–4281. [Google Scholar] [CrossRef] [Green Version]
- Boeckx, N.; Koukakis, R.; Op de Beeck, K.; Rolfo, C.; Van Camp, G.; Siena, S.; Tabernero, J.; Douillard, J.Y.; Andre, T.; Peeters, M. Effect of Primary Tumor Location on Second- or Later-line Treatment Outcomes in Patients With RAS Wild-type Metastatic Colorectal Cancer and All Treatment Lines in Patients With RAS Mutations in Four Randomized Panitumumab Studies. Clin. Color. Cancer 2018, 17, 170–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinemann, V.; von Weikersthal, L.F.; Decker, T.; Kiani, A.; Vehling-Kaiser, U.; Al-Batran, S.E.; Heintges, T.; Lerchenmuller, C.; Kahl, C.; Seipelt, G.; et al. FOLFIRI plus cetuximab versus FOLFIRI plus bevacizumab as first-line treatment for patients with metastatic colorectal cancer (FIRE-3): A randomised, open-label, phase 3 trial. Lancet Oncol. 2014, 15, 1065–1075. [Google Scholar] [CrossRef]
- Schwartzberg, L.S.; Rivera, F.; Karthaus, M.; Fasola, G.; Canon, J.L.; Hecht, J.R.; Yu, H.; Oliner, K.S.; Go, W.Y. PEAK: A randomized, multicenter phase II study of panitumumab plus modified fluorouracil, leucovorin, and oxaliplatin (mFOLFOX6) or bevacizumab plus mFOLFOX6 in patients with previously untreated, unresectable, wild-type KRAS exon 2 metastatic colorectal cancer. J. Clin. Oncol. 2014, 32, 2240–2247. [Google Scholar] [CrossRef]
- Cremolini, C.; Rossini, D.; Dell’Aquila, E.; Lonardi, S.; Conca, E.; Del Re, M.; Busico, A.; Pietrantonio, F.; Danesi, R.; Aprile, G.; et al. Rechallenge for Patients With RAS and BRAF Wild-Type Metastatic Colorectal Cancer With Acquired Resistance to First-line Cetuximab and Irinotecan: A Phase 2 Single-Arm Clinical Trial. JAMA Oncol. 2019, 5, 343–350. [Google Scholar] [CrossRef] [Green Version]
- Sartore-Bianchi, A.; Pietrantonio, F.; Lonardi, S.; Mussolin, B.; Rua, F.; Crisafulli, G.; Bartolini, A.; Fenocchio, E.; Amatu, A.; Manca, P.; et al. Circulating tumor DNA to guide rechallenge with panitumumab in metastatic colorectal cancer: The phase 2 CHRONOS trial. Nat. Med. 2022, 28, 1612–1618. [Google Scholar] [CrossRef]
- Carmeliet, P.; Jain, R.K. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat. Rev. Drug Discov. 2011, 10, 417–427. [Google Scholar] [CrossRef]
- Mariotti, V.; Fiorotto, R.; Cadamuro, M.; Fabris, L.; Strazzabosco, M. New insights on the role of vascular endothelial growth factor in biliary pathophysiology. JHEP Rep. 2021, 3, 100251. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Shibuya, M. The vascular endothelial growth factor (VEGF)/VEGF receptor system and its role under physiological and pathological conditions. Clin. Sci. 2005, 109, 227–241. [Google Scholar] [CrossRef] [Green Version]
- Byrne, A.M.; Bouchier-Hayes, D.J.; Harmey, J.H. Angiogenic and cell survival functions of vascular endothelial growth factor (VEGF). J. Cell Mol. Med. 2005, 9, 777–794. [Google Scholar] [CrossRef]
- Wada, S.; Tsunoda, T.; Baba, T.; Primus, F.J.; Kuwano, H.; Shibuya, M.; Tahara, H. Rationale for antiangiogenic cancer therapy with vaccination using epitope peptides derived from human vascular endothelial growth factor receptor 2. Cancer Res. 2005, 65, 4939–4946. [Google Scholar] [CrossRef] [Green Version]
- Ishizaki, H.; Tsunoda, T.; Wada, S.; Yamauchi, M.; Shibuya, M.; Tahara, H. Inhibition of tumor growth with antiangiogenic cancer vaccine using epitope peptides derived from human vascular endothelial growth factor receptor 1. Clin. Cancer Res. 2006, 12, 5841–5849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Fei, D.; Vanderlaan, M.; Song, A. Biological activity of bevacizumab, a humanized anti-VEGF antibody in vitro. Angiogenesis 2004, 7, 335–345. [Google Scholar] [CrossRef]
- Hurwitz, H.; Fehrenbacher, L.; Novotny, W.; Cartwright, T.; Hainsworth, J.; Heim, W.; Berlin, J.; Baron, A.; Griffing, S.; Holmgren, E.; et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N. Engl. J. Med. 2004, 350, 2335–2342. [Google Scholar] [CrossRef] [Green Version]
- Cunningham, D.; Lang, I.; Marcuello, E.; Lorusso, V.; Ocvirk, J.; Shin, D.B.; Jonker, D.; Osborne, S.; Andre, N.; Waterkamp, D.; et al. Bevacizumab plus capecitabine versus capecitabine alone in elderly patients with previously untreated metastatic colorectal cancer (AVEX): An open-label, randomised phase 3 trial. Lancet Oncol. 2013, 14, 1077–1085. [Google Scholar] [CrossRef]
- Cremolini, C.; Loupakis, F.; Antoniotti, C.; Lupi, C.; Sensi, E.; Lonardi, S.; Mezi, S.; Tomasello, G.; Ronzoni, M.; Zaniboni, A.; et al. FOLFOXIRI plus bevacizumab versus FOLFIRI plus bevacizumab as first-line treatment of patients with metastatic colorectal cancer: Updated overall survival and molecular subgroup analyses of the open-label, phase 3 TRIBE study. Lancet Oncol. 2015, 16, 1306–1315. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Tabernero, J.; Lakomy, R.; Prenen, H.; Prausova, J.; Macarulla, T.; Ruff, P.; van Hazel, G.A.; Moiseyenko, V.; Ferry, D.; et al. Addition of aflibercept to fluorouracil, leucovorin, and irinotecan improves survival in a phase III randomized trial in patients with metastatic colorectal cancer previously treated with an oxaliplatin-based regimen. J. Clin. Oncol. 2012, 30, 3499–3506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grothey, A.; Van Cutsem, E.; Sobrero, A.; Siena, S.; Falcone, A.; Ychou, M.; Humblet, Y.; Bouche, O.; Mineur, L.; Barone, C.; et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): An international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013, 381, 303–312. [Google Scholar] [CrossRef]
- Li, J.; Qin, S.; Xu, R.; Yau, T.C.; Ma, B.; Pan, H.; Xu, J.; Bai, Y.; Chi, Y.; Wang, L.; et al. Regorafenib plus best supportive care versus placebo plus best supportive care in Asian patients with previously treated metastatic colorectal cancer (CONCUR): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2015, 16, 619–629. [Google Scholar] [CrossRef]
- Tabernero, J.; Yoshino, T.; Cohn, A.L.; Obermannova, R.; Bodoky, G.; Garcia-Carbonero, R.; Ciuleanu, T.E.; Portnoy, D.C.; Van Cutsem, E.; Grothey, A.; et al. Ramucirumab versus placebo in combination with second-line FOLFIRI in patients with metastatic colorectal carcinoma that progressed during or after first-line therapy with bevacizumab, oxaliplatin, and a fluoropyrimidine (RAISE): A randomised, double-blind, multicentre, phase 3 study. Lancet Oncol. 2015, 16, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.C.; Shi, Y.; Wang, Y.R.; Lv, Y.; Yan, H.; Mao, H.; Wang, Z.K.; Wu, Z.Y.; Shi, W.W.; Dai, G.H. KRAS mutation and primary tumor location do not affect efficacy of bevacizumab-containing chemotherapy in stagae IV colorectal cancer patients. Sci. Rep. 2017, 7, 14368. [Google Scholar] [CrossRef] [Green Version]
- Yoshimatsu, K.; Satake, M.; Sano, M.; Asaka, S.; Yamada, Y.; Okayama, S.; Yano, Y.; Yokomizo, H.; Usui, T.; Yamaguchi, K.; et al. Standard Chemotherapy with Bevacizumab as First-Line Therapy for Metastatic Colorectal Cancer with RAS Mutation. Gan Kagaku Ryoho 2017, 44, 918–920. [Google Scholar]
- Grothey, A.; Sugrue, M.M.; Purdie, D.M.; Dong, W.; Sargent, D.; Hedrick, E.; Kozloff, M. Bevacizumab beyond first progression is associated with prolonged overall survival in metastatic colorectal cancer: Results from a large observational cohort study (BRiTE). J. Clin. Oncol. 2008, 26, 5326–5334. [Google Scholar] [CrossRef]
- Grothey, A.; Flick, E.D.; Cohn, A.L.; Bekaii-Saab, T.S.; Bendell, J.C.; Kozloff, M.; Roach, N.; Mun, Y.; Fish, S.; Hurwitz, H.I. Bevacizumab exposure beyond first disease progression in patients with metastatic colorectal cancer: Analyses of the ARIES observational cohort study. Pharmacoepidemiol. Drug Saf. 2014, 23, 726–734. [Google Scholar] [CrossRef]
- Bennouna, J.; Sastre, J.; Arnold, D.; Osterlund, P.; Greil, R.; Van Cutsem, E.; von Moos, R.; Vieitez, J.M.; Bouche, O.; Borg, C.; et al. Continuation of bevacizumab after first progression in metastatic colorectal cancer (ML18147): A randomised phase 3 trial. Lancet Oncol. 2013, 14, 29–37. [Google Scholar] [CrossRef]
- Bendell, J.C.; Bekaii-Saab, T.S.; Cohn, A.L.; Hurwitz, H.I.; Kozloff, M.; Tezcan, H.; Roach, N.; Mun, Y.; Fish, S.; Flick, E.D.; et al. Treatment patterns and clinical outcomes in patients with metastatic colorectal cancer initially treated with FOLFOX-bevacizumab or FOLFIRI-bevacizumab: Results from ARIES, a bevacizumab observational cohort study. Oncologist 2012, 17, 1486–1495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Cutsem, E.; Joulain, F.; Hoff, P.M.; Mitchell, E.; Ruff, P.; Lakomy, R.; Prausova, J.; Moiseyenko, V.M.; van Hazel, G.; Cunningham, D.; et al. Aflibercept Plus FOLFIRI vs. Placebo Plus FOLFIRI in Second-Line Metastatic Colorectal Cancer: A Post Hoc Analysis of Survival from the Phase III VELOUR Study Subsequent to Exclusion of Patients who had Recurrence During or Within 6 Months of Completing Adjuvant Oxaliplatin-Based Therapy. Target. Oncol. 2016, 11, 383–400. [Google Scholar] [CrossRef] [PubMed]
- Chu, Q.S. Aflibercept (AVE0005): An alternative strategy for inhibiting tumour angiogenesis by vascular endothelial growth factors. Expert Opin. Biol. Ther. 2009, 9, 263–271. [Google Scholar] [CrossRef]
- Papadopoulos, N.; Martin, J.; Ruan, Q.; Rafique, A.; Rosconi, M.P.; Shi, E.; Pyles, E.A.; Yancopoulos, G.D.; Stahl, N.; Wiegand, S.J. Binding and neutralization of vascular endothelial growth factor (VEGF) and related ligands by VEGF Trap, ranibizumab and bevacizumab. Angiogenesis 2012, 15, 171–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satake, H.; Ando, K.; Oki, E.; Shimokawa, M.; Makiyama, A.; Saeki, H.; Tsuji, A.; Mori, M. Protocol of the EFFORT study: A prospective study of FOLFIRI plus aflibercept as second-line treatment after progression on FOLFOXIRI plus bevacizumab or during maintenance treatment in patients with unresectable/metastatic colorectal cancer. BMC Cancer 2020, 20, 1116. [Google Scholar] [CrossRef]
- Debeuckelaere, C.; Murgioni, S.; Lonardi, S.; Girardi, N.; Alberti, G.; Fano, C.; Gallimberti, S.; Magro, C.; Ahcene-Djaballah, S.; Daniel, F.; et al. Ramucirumab: The long and winding road toward being an option for mCRC treatment. Expert Opin. Biol. Ther. 2019, 19, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Yoshihiro, T.; Kusaba, H.; Makiyama, A.; Kobayashi, K.; Uenomachi, M.; Ito, M.; Doi, Y.; Mitsugi, K.; Aikawa, T.; Takayoshi, K.; et al. Efficacy and safety of ramucirumab plus modified FOLFIRI for metastatic colorectal cancer. Int. J. Clin. Oncol. 2019, 24, 508–515. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Shinozaki, E.; Osumi, H.; Nakayama, I.; Ota, Y.; Ichimura, T.; Ogura, M.; Wakatsuki, T.; Ooki, A.; Takahari, D.; et al. Second-line FOLFIRI plus ramucirumab with or without prior bevacizumab for patients with metastatic colorectal cancer. Cancer Chemother. Pharmacol. 2019, 84, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Carter, N.J. Regorafenib: A review of its use in previously treated patients with progressive metastatic colorectal cancer. Drugs Aging 2014, 31, 67–78. [Google Scholar] [CrossRef]
- Dong, J.; Li, B.; Lin, D.; Zhou, Q.; Huang, D. Advances in Targeted Therapy and Immunotherapy for Non-small Cell Lung Cancer Based on Accurate Molecular Typing. Front. Pharmacol. 2019, 10, 230. [Google Scholar] [CrossRef]
- Sun, Q.; Zhou, J.; Zhang, Z.; Guo, M.; Liang, J.; Zhou, F.; Long, J.; Zhang, W.; Yin, F.; Cai, H.; et al. Discovery of fruquintinib, a potent and highly selective small molecule inhibitor of VEGFR 1, 2, 3 tyrosine kinases for cancer therapy. Cancer Biol. Ther. 2014, 15, 1635–1645. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Qu, T.; Zhang, H.; Sun, Y.; Cui, C.; Chi, Y.; Zhang, W.; Wang, X.; Yang, L. The Real-World Practice of Fruquintinib for Chinese Patients with Metastatic Colorectal Cancer. Cancer Manag. Res. 2021, 13, 6199–6205. [Google Scholar] [CrossRef]
- Dasari, A.; Sobrero, A.; Yao, J.; Yoshino, T.; Schelman, W.; Yang, Z.; Chien, C.; Kania, M.; Tabernero, J.; Eng, C. FRESCO-2: A global Phase III study investigating the efficacy and safety of fruquintinib in metastatic colorectal cancer. Future Oncol. 2021, 17, 3151–3162. [Google Scholar] [CrossRef]
- Hu, L.F.; Lan, H.R.; Huang, D.; Li, X.M.; Jin, K.T. Personalized Immunotherapy in Colorectal Cancers: Where Do We Stand? Front. Oncol. 2021, 11, 769305. [Google Scholar] [CrossRef]
- Darvin, P.; Toor, S.M.; Sasidharan Nair, V.; Elkord, E. Immune checkpoint inhibitors: Recent progress and potential biomarkers. Exp. Mol. Med. 2018, 50, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colle, R.; Cohen, R.; Cochereau, D.; Duval, A.; Lascols, O.; Lopez-Trabada, D.; Afchain, P.; Trouilloud, I.; Parc, Y.; Lefevre, J.H.; et al. Immunotherapy and patients treated for cancer with microsatellite instability. Bull Cancer 2017, 104, 42–51. [Google Scholar] [CrossRef] [Green Version]
- Schrock, A.B.; Ouyang, C.; Sandhu, J.; Sokol, E.; Jin, D.; Ross, J.S.; Miller, V.A.; Lim, D.; Amanam, I.; Chao, J.; et al. Tumor mutational burden is predictive of response to immune checkpoint inhibitors in MSI-high metastatic colorectal cancer. Ann. Oncol. 2019, 30, 1096–1103. [Google Scholar] [CrossRef]
- Li, K.; Luo, H.; Huang, L.; Luo, H.; Zhu, X. Microsatellite instability: A review of what the oncologist should know. Cancer Cell Int. 2020, 20, 16. [Google Scholar] [CrossRef] [Green Version]
- Myint, Z.W.; Goel, G. Role of modern immunotherapy in gastrointestinal malignancies: A review of current clinical progress. J. Hematol. Oncol. 2017, 10, 86. [Google Scholar] [CrossRef]
- Wang, J.; Yuan, R.; Song, W.; Sun, J.; Liu, D.; Li, Z. PD-1, PD-L1 (B7-H1) and Tumor-Site Immune Modulation Therapy: The Historical Perspective. J. Hematol. Oncol. 2017, 10, 34. [Google Scholar] [CrossRef] [Green Version]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef] [Green Version]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef] [Green Version]
- Peggs, K.S.; Quezada, S.A.; Allison, J.P. Cell intrinsic mechanisms of T-cell inhibition and application to cancer therapy. Immunol. Rev. 2008, 224, 141–165. [Google Scholar] [CrossRef]
- Simpson, T.R.; Li, F.; Montalvo-Ortiz, W.; Sepulveda, M.A.; Bergerhoff, K.; Arce, F.; Roddie, C.; Henry, J.Y.; Yagita, H.; Wolchok, J.D.; et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J. Exp Med. 2013, 210, 1695–1710. [Google Scholar] [CrossRef]
- Sun, J.; Zheng, Y.; Mamun, M.; Li, X.; Chen, X.; Gao, Y. Research progress of PD-1/PD-L1 immunotherapy in gastrointestinal tumors. Biomed. Pharmacother. 2020, 129, 110504. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [Green Version]
- Andre, T.; Shiu, K.K.; Kim, T.W.; Jensen, B.V.; Jensen, L.H.; Punt, C.; Smith, D.; Garcia-Carbonero, R.; Benavides, M.; Gibbs, P.; et al. Pembrolizumab in Microsatellite-Instability-High Advanced Colorectal Cancer. N. Engl. J. Med. 2020, 383, 2207–2218. [Google Scholar] [CrossRef]
- Overman, M.J.; McDermott, R.; Leach, J.L.; Lonardi, S.; Lenz, H.J.; Morse, M.A.; Desai, J.; Hill, A.; Axelson, M.; Moss, R.A.; et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): An open-label, multicentre, phase 2 study. Lancet Oncol. 2017, 18, 1182–1191. [Google Scholar] [CrossRef]
- Overman, M.J.; Lonardi, S.; Wong, K.Y.M.; Lenz, H.J.; Gelsomino, F.; Aglietta, M.; Morse, M.A.; Van Cutsem, E.; McDermott, R.; Hill, A.; et al. Durable Clinical Benefit With Nivolumab Plus Ipilimumab in DNA Mismatch Repair-Deficient/Microsatellite Instability-High Metastatic Colorectal Cancer. J. Clin. Oncol. 2018, 36, 773–779. [Google Scholar] [CrossRef]
- Huang, E.J.; Reichardt, L.F. Neurotrophins: Roles in neuronal development and function. Annu. Rev. Neurosci. 2001, 24, 677–736. [Google Scholar] [CrossRef] [Green Version]
- Amatu, A.; Sartore-Bianchi, A.; Bencardino, K.; Pizzutilo, E.G.; Tosi, F.; Siena, S. Tropomyosin receptor kinase (TRK) biology and the role of NTRK gene fusions in cancer. Ann. Oncol. 2019, 30 (Suppl. 8), viii5–viii15. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.; Kim, N.; Murakami, T.; Sim, T. Anti-Tumor Activity of AZD4547 Against NTRK1 Fusion Positive Cancer Cells Through Inhibition of NTRKs. Front. Oncol. 2021, 11, 757598. [Google Scholar] [CrossRef] [PubMed]
- Davare, M.A.; Tognon, C.E. Detecting and targetting oncogenic fusion proteins in the genomic era. Biol. Cell 2015, 107, 111–129. [Google Scholar] [CrossRef] [Green Version]
- Hallberg, B.; Palmer, R.H. The role of the ALK receptor in cancer biology. Ann. Oncol. 2016, 27 (Suppl. 3), iii4–iii15. [Google Scholar] [CrossRef] [PubMed]
- Akhoundova, D.; Hussung, S.; Sivakumar, S.; Topfer, A.; Rechsteiner, M.; Kahraman, A.; Arnold, F.; Angst, F.; Britschgi, C.; Zoche, M.; et al. ROS1 genomic rearrangements are rare actionable drivers in microsatellite stable colorectal cancer. Int. J. Cancer 2022, 151, 2161–2171. [Google Scholar] [CrossRef] [PubMed]
- Bergethon, K.; Shaw, A.T.; Ou, S.H.; Katayama, R.; Lovly, C.M.; McDonald, N.T.; Massion, P.P.; Siwak-Tapp, C.; Gonzalez, A.; Fang, R.; et al. ROS1 rearrangements define a unique molecular class of lung cancers. J. Clin. Oncol. 2012, 30, 863–870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietrantonio, F.; Di Nicolantonio, F.; Schrock, A.B.; Lee, J.; Tejpar, S.; Sartore-Bianchi, A.; Hechtman, J.F.; Christiansen, J.; Novara, L.; Tebbutt, N.; et al. ALK, ROS1, and NTRK Rearrangements in Metastatic Colorectal Cancer. J. Natl. Cancer Inst. 2017, 109, djx089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doebele, R.C.; Drilon, A.; Paz-Ares, L.; Siena, S.; Shaw, A.T.; Farago, A.F.; Blakely, C.M.; Seto, T.; Cho, B.C.; Tosi, D.; et al. Entrectinib in patients with advanced or metastatic NTRK fusion-positive solid tumours: Integrated analysis of three phase 1-2 trials. Lancet Oncol. 2020, 21, 271–282. [Google Scholar] [CrossRef]
- Ratti, M.; Grizzi, G.; Passalacqua, R.; Lampis, A.; Cereatti, F.; Grassia, R.; Hahne, J.C. NTRK fusions in colorectal cancer: Clinical meaning and future perspective. Expert Opin. Ther. Targets 2021, 25, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.S.; DuBois, S.G.; Kummar, S.; Farago, A.F.; Albert, C.M.; Rohrberg, K.S.; van Tilburg, C.M.; Nagasubramanian, R.; Berlin, J.D.; Federman, N.; et al. Larotrectinib in patients with TRK fusion-positive solid tumours: A pooled analysis of three phase 1/2 clinical trials. Lancet Oncol. 2020, 21, 531–540. [Google Scholar] [CrossRef]
- He, Z.; Thorrez, L.; Siegfried, G.; Meulemans, S.; Evrard, S.; Tejpar, S.; Khatib, A.M.; Creemers, J.W.M. The proprotein convertase furin is a pro-oncogenic driver in KRAS and BRAF driven colorectal cancer. Oncogene 2020, 39, 3571–3587. [Google Scholar] [CrossRef]
- Jones, J.C.; Renfro, L.A.; Al-Shamsi, H.O.; Schrock, A.B.; Rankin, A.; Zhang, B.Y.; Kasi, P.M.; Voss, J.S.; Leal, A.D.; Sun, J.; et al. (Non-V600) BRAF Mutations Define a Clinically Distinct Molecular Subtype of Metastatic Colorectal Cancer. J. Clin. Oncol. 2017, 35, 2624–2630. [Google Scholar] [CrossRef]
- Chu, J.E.; Johnson, B.; Kugathasan, L.; Morris, V.K.; Raghav, K.; Swanson, L.; Lim, H.J.; Renouf, D.J.; Gill, S.; Wolber, R.; et al. Population-based Screening for BRAF (V600E) in Metastatic Colorectal Cancer Reveals Increased Prevalence and Poor Prognosis. Clin. Cancer Res. 2020, 26, 4599–4605. [Google Scholar] [CrossRef]
- Takeda, H.; Sunakawa, Y. Management of BRAF Gene Alterations in Metastatic Colorectal Cancer: From Current Therapeutic Strategies to Future Perspectives. Front. Oncol. 2021, 11, 602194. [Google Scholar] [CrossRef] [PubMed]
- Delord, J.P.; Robert, C.; Nyakas, M.; McArthur, G.A.; Kudchakar, R.; Mahipal, A.; Yamada, Y.; Sullivan, R.; Arance, A.; Kefford, R.F.; et al. Phase I Dose-Escalation and -Expansion Study of the BRAF Inhibitor Encorafenib (LGX818) in Metastatic BRAF-Mutant Melanoma. Clin. Cancer Res. 2017, 23, 5339–5348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, V.; Kalita, J.; Pal, M. Predictive and prognostic biomarkers in colorectal cancer: A systematic review of recent advances and challenges. Biomed. Pharmacother. 2017, 87, 8–19. [Google Scholar] [CrossRef]
- Castro, F.; Leite Pereira, C.; Helena Macedo, M.; Almeida, A.; Jose Silveira, M.; Dias, S.; Patricia Cardoso, A.; Jose Oliveira, M.; Sarmento, B. Advances on colorectal cancer 3D models: The needed translational technology for nanomedicine screening. Adv. Drug Deliv. Rev. 2021, 175, 113824. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Chen, Y.; Lu, J.; He, K.; Chen, Y.; Ding, Y.; Jin, K.; Wang, H.; Zhang, H.; Wang, H.; et al. Patient-derived xenograft models for gastrointestinal tumors: A single-center retrospective study. Front. Oncol. 2022, 12, 985154. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Wei, Y.; Fu, Y.; Li, J.; Han, L. A novel enterocyte-related 4-gene signature for predicting prognosis in colon adenocarcinoma. Front. Immunol. 2022, 13, 1052182. [Google Scholar] [CrossRef]
- Petty, A.J.; Heyman, B.; Yang, Y. Chimeric Antigen Receptor Cell Therapy: Overcoming Obstacles to Battle Cancer. Cancers 2020, 12, 842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, M.; Spreafico, A.; Vashisht, K.; Hinrichs, M.J. Patient Selection Strategies to Maximize Therapeutic Index of Antibody-Drug Conjugates: Prior Approaches and Future Directions. Mol. Cancer Ther. 2020, 19, 1770–1783. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, H. Radioimmunotherapy: A specific treatment protocol for cancer by cytotoxic radioisotopes conjugated to antibodies. Sci. World J. 2014, 2014, 492061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishimura, T.; Mitsunaga, M.; Sawada, R.; Saruta, M.; Kobayashi, H.; Matsumoto, N.; Kanke, T.; Yanai, H.; Nakamura, K. Photoimmunotherapy targeting biliary-pancreatic cancer with humanized anti-TROP2 antibody. Cancer Med. 2019, 8, 7781–7792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, Y.; Kaneko, M.K. A cancer-specific monoclonal antibody recognizes the aberrantly glycosylated podoplanin. Sci. Rep. 2014, 4, 5924. [Google Scholar] [CrossRef] [Green Version]
- Itai, S.; Yamada, S.; Kaneko, M.K.; Chang, Y.W.; Harada, H.; Kato, Y. Establishment of EMab-134, a Sensitive and Specific Anti-Epidermal Growth Factor Receptor Monoclonal Antibody for Detecting Squamous Cell Carcinoma Cells of the Oral Cavity. Monoclon. Antibodies Immunodiagn. Immunother. 2017, 36, 272–281. [Google Scholar] [CrossRef]
- Kaneko, M.K.; Yamada, S.; Itai, S.; Chang, Y.W.; Nakamura, T.; Yanaka, M.; Kato, Y. Elucidation of the critical epitope of an anti-EGFR monoclonal antibody EMab-134. Biochem. Biophys. Rep. 2018, 14, 54–57. [Google Scholar] [CrossRef]
- Sano, M.; Kaneko, M.K.; Aasano, T.; Kato, Y. Epitope Mapping of an Antihuman EGFR Monoclonal Antibody (EMab-134) Using the REMAP Method. Monoclon. Antibodies Immunodiagn. Immunother. 2021, 40, 191–195. [Google Scholar] [CrossRef]
- Hosono, H.; Takei, J.; Ohishi, T.; Sano, M.; Asano, T.; Sayama, Y.; Nakamura, T.; Yanaka, M.; Kawada, M.; Harada, H.; et al. AntiEGFR monoclonal antibody 134mG2a exerts antitumor effects in mouse xenograft models of oral squamous cell carcinoma. Int. J. Mol. Med. 2020, 46, 1443–1452. [Google Scholar] [CrossRef]
- Tateyama, N.; Nanamiya, R.; Ohishi, T.; Takei, J.; Nakamura, T.; Yanaka, M.; Hosono, H.; Saito, M.; Asano, T.; Tanaka, T.; et al. Defucosylated Anti-Epidermal Growth Factor Receptor Monoclonal Antibody 134-mG2a-f Exerts Antitumor Activities in Mouse Xenograft Models of Dog Epidermal Growth Factor Receptor-Overexpressed Cells. Monoclon. Antibodies Immunodiagn. Immunother. 2021, 40, 177–183. [Google Scholar] [CrossRef]
- Li, G.; Ohishi, T.; Kaneko, M.K.; Takei, J.; Mizuno, T.; Kawada, M.; Saito, M.; Suzuki, H.; Kato, Y. Defucosylated Mouse-Dog Chimeric Anti-EGFR Antibody Exerts Antitumor Activities in Mouse Xenograft Models of Canine Tumors. Cells 2021, 10, 3599. [Google Scholar] [CrossRef] [PubMed]
- Itai, S.; Kaneko, M.K.; Fujii, Y.; Yamada, S.; Nakamura, T.; Yanaka, M.; Saidoh, N.; Handa, S.; Chang, Y.W.; Suzuki, H.; et al. Development of EMab-51, a Sensitive and Specific Anti-Epidermal Growth Factor Receptor Monoclonal Antibody in Flow Cytometry, Western Blot, and Immunohistochemistry. Monoclon. Antibodies Immunodiagn. Immunother. 2017, 36, 214–219. [Google Scholar] [CrossRef]
- Nanamiya, R.; Sano, M.; Asano, T.; Yanaka, M.; Nakamura, T.; Saito, M.; Tanaka, T.; Hosono, H.; Tateyama, N.; Kaneko, M.K.; et al. Epitope Mapping of an Anti-Human Epidermal Growth Factor Receptor Monoclonal Antibody (EMab-51) Using the RIEDL Insertion for Epitope Mapping Method. Monoclon. Antibodies Immunodiagn. Immunother. 2021, 40, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Takei, J.; Kaneko, M.K.; Ohishi, T.; Kawada, M.; Harada, H.; Kato, Y. A novel anti-EGFR monoclonal antibody (EMab-17) exerts antitumor activity against oral squamous cell carcinomas via antibody-dependent cellular cytotoxicity and complement-dependent cytotoxicity. Oncol. Lett 2020, 19, 2809–2816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohishi, T.; Kato, Y.; Kaneko, M.K.; Ohba, S.I.; Inoue, H.; Harakawa, A.; Kawada, M. Anti-Metastatic Activity of an Anti-EGFR Monoclonal Antibody against Metastatic Colorectal Cancer with KRAS p.G13D Mutation. Int. J. Mol. Sci. 2020, 21, 6037. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Wang, X.; Xin, Y.; Wang, Z.; Gong, J.; Zhang, X.; Li, Y.; Ji, C.; Sun, Y.; Zhao, F.; et al. Trastuzumab combined with irinotecan in patients with HER2-positive metastatic colorectal cancer: A phase II single-arm study and exploratory biomarker analysis. Cancer Res. Treat. 2022. [Google Scholar] [CrossRef]
- Patelli, G.; Tosi, F.; Amatu, A.; Mauri, G.; Curaba, A.; Patane, D.A.; Pani, A.; Scaglione, F.; Siena, S.; Sartore-Bianchi, A. Strategies to tackle RAS-mutated metastatic colorectal cancer. ESMO Open 2021, 6, 100156. [Google Scholar] [CrossRef] [PubMed]
- Kohno, T.; Tabata, J.; Nakaoku, T. REToma: A cancer subtype with a shared driver oncogene. Carcinogenesis 2020, 41, 123–129. [Google Scholar] [CrossRef]
- Crutcher, M.; Waldman, S. Biomarkers in the development of individualized treatment regimens for colorectal cancer. Front. Med. 2022, 9, 1062423. [Google Scholar] [CrossRef]
Figure 1.
Comprehensive overview of FDA-approved targeted drugs for mCRC. Abbreviations: EGFR: epidermal growth factor receptor; FGFR: fibroblast growth factor receptor; VEGFR: vascular endothelia growth factor receptor; TRK: tropomyosin receptor kinase; VEGF: vascular endothelial growth factor; PlGF: placenta growth factor; PD-1: programmed cell death 1; PD-L1: programmed cell death ligand 1; CTLA-4: cytotoxic T-lymphocyte-associated protein-4; B7: CD80/CD86; MEK: mitogen-activated protein kinase; ERK: extracellular signal-regulated kinase; PI3K: phosphoinositide 3-kinase; mTOR: mechanistic target of rapamycin; PLC-γ: phospholipase C-γ; PKC: protein kinase C; NF-kB: nuclear factor-kappa B; JAK: Janus kinase; STAT: transducer and activator of transcription.
Figure 1.
Comprehensive overview of FDA-approved targeted drugs for mCRC. Abbreviations: EGFR: epidermal growth factor receptor; FGFR: fibroblast growth factor receptor; VEGFR: vascular endothelia growth factor receptor; TRK: tropomyosin receptor kinase; VEGF: vascular endothelial growth factor; PlGF: placenta growth factor; PD-1: programmed cell death 1; PD-L1: programmed cell death ligand 1; CTLA-4: cytotoxic T-lymphocyte-associated protein-4; B7: CD80/CD86; MEK: mitogen-activated protein kinase; ERK: extracellular signal-regulated kinase; PI3K: phosphoinositide 3-kinase; mTOR: mechanistic target of rapamycin; PLC-γ: phospholipase C-γ; PKC: protein kinase C; NF-kB: nuclear factor-kappa B; JAK: Janus kinase; STAT: transducer and activator of transcription.


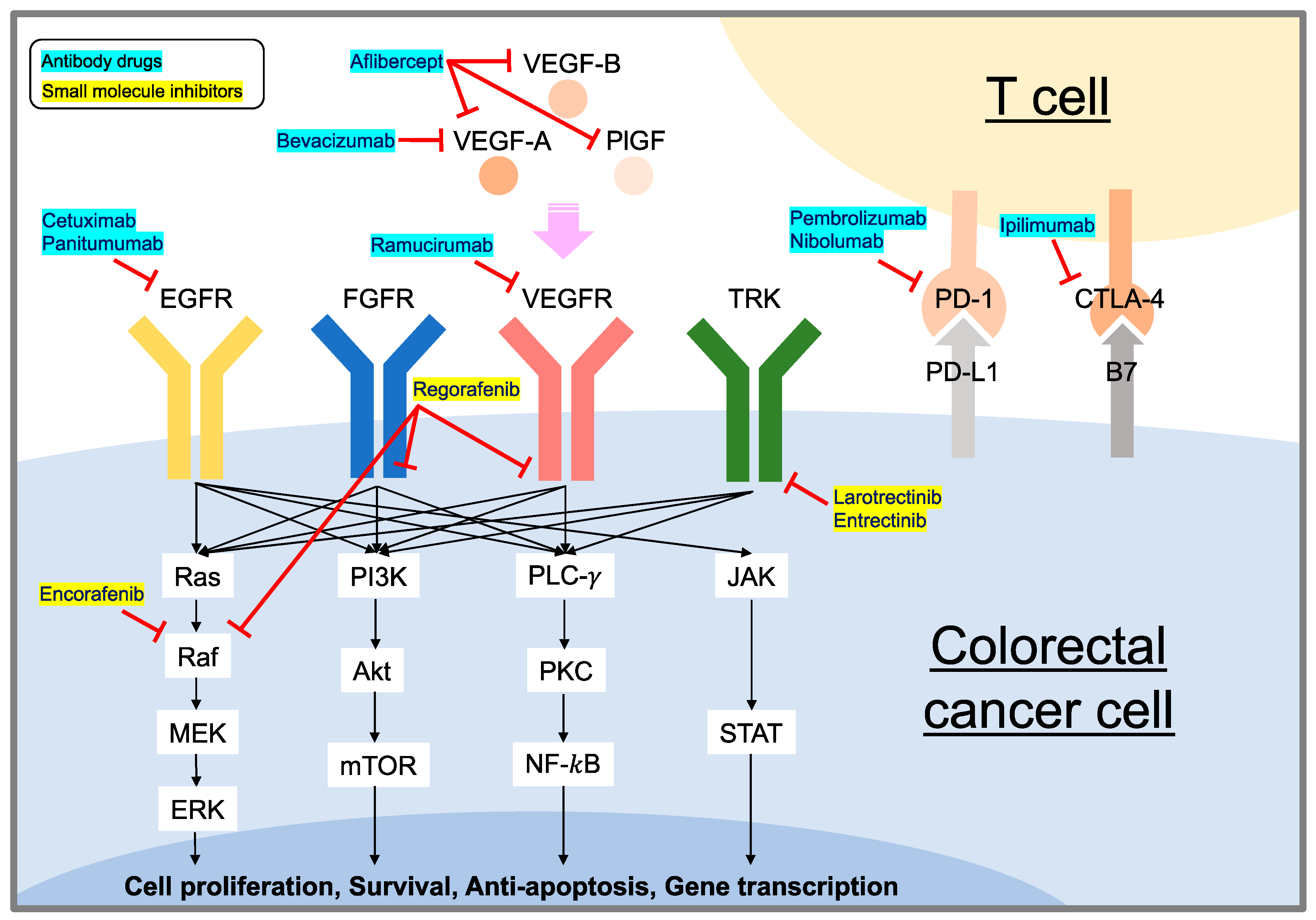{kind=link}
Table 1.
FDA-approved targeted drugs for mCRC.
| Year Approved by the FDA | Drugs | Targets | Drug Details | Ref |
|---|---|---|---|---|
| 2004 | Cetuximab | EGFR | Chimeric mouse/human mAb (IgG1) | [18] |
| Bevacizumab | VEGF-A | Humanized mAb (IgG1) | [18] | |
| 2006 | Panitumumab | EGFR | Fully human mAb (IgG1) | [19] |
| 2012 | Aflibercept | VEGF-A, VEGF-B, PlGF | Fusion protein which consists of the binding portions of VEGF from VEGF-1 and 2 fused to the Fc portion ofimmunoglobulin G1 (IgG1) | [20] |
| Regorafenib | VEGFR, FGFR, KIT, PDGFR, BRAF | Small molecule inhibitor of membrane-bound and intracellular receptor tyrosine kinases | [21] | |
| 2015 | Ramucirumab | VEGFR-2 | Fully human mAb (IgG1) | [22] |
| 2017 | Pembrolizumab | PD-1 | Humanized mAb (IgG4) | [23] |
| Nivolumab | PD-1 | Fully human mAb (IgG4) | [24] | |
| 2018 | Ipilimumab | CTLA-4 | Fully human mAb (IgG1) | [25] |
| Larotrectinib | TRK | Small molecule of tyrosine kinase inhibitor | [26] | |
| 2019 | Entrectinib | TRK, ALK, ROS1 | Small molecule of tyrosine kinase inhibitor | [27] |
| 2020 | Encorafenib | BRAF (V600E-mutant) | Small molecule kinase inhibitor | [28] |
Abbreviations: EGFR: epidermal growth factor receptor; VEGF: vascular endothelial growth factor; PlGF: placenta growth factor; VEGFR: vascular endothelia growth factor receptor; FGFR: fibroblast growth factor receptor; KIT: mast/stem cell growth factor receptor; PDGFR: platelet-derived growth factor receptor; BRAF: v-Raf murine sarcoma viral oncogene homolog B1; PD-1: programmed cell death 1; CTLA-4: cytotoxic T-lymphocyte-associated protein-4; TRK: tropomyosin receptor kinase; ALK: anaplastic lymphoma kinase; ROS1: c-ros oncogene 1; mAb: monoclonal antibody.
Table 2.
Selected clinical trials for EGFR (± BRAF)-targeting drugs.
| Targeted Drugs | Study | Phase | Study Regimen | Number | Results | Ref |
|---|---|---|---|---|---|---|
| Cetuximab | BOND | II | Cetuximab + Irinotecan /Irinotecan (Refractory to Irinotecan) (Third-line) | 218/111 | OS: 22.9/10.8 months (p = 0.007) Disease control: 55.5%/32.4% (p < 0.001) | Cunningham et al. [43] |
| Panitumumab | PRIME | III | Panitumumab + FOLFOX4 /FOLFOX4 (First-line) | 325/331 | OS: 23.8/19.4 months (p = 0.03) PFS: 10.0/8.6 months (p = 0.01) | Douillard et al. [44] |
| Cetuximab + Encorafenib | BEACON | III | Cetuximab + Encorafenib /Cetuximab + Chemotherapy 1 (Second- or later-line) | 220/221 | Median OS: 8.4/5.4 months (p < 0.001) Confirmed RR: 20%/2% (p < 0.001) | Kopetz et al. [45] |
| Cetuximab + Encorafenib + Binimetinib | BEACON | III | Cetuximab + Encorafenib + Binimetinib /Cetuximab + Chemotherapy 1 (Second- or later-line) | 224/221 | Median OS: 9.0/5.4 months (p < 0.001) Confirmed RR: 26%/2% (p < 0.001) | Kopetz et al. [45] |
1 The investigators’ choice of either cetuximab and irinotecan or cetuximab and FOLFIRI (folinic acid, fluorouracil, and irinotecan. Abbreviations: OS: overall survival; PFS: progression-free survival; RR: response rate.
Table 3.
Selected clinical trials for VEGF/VEGFR-targeting drugs.
| Targeted Drugs | Study | Phase | Study Regimen | Number | Results | Ref |
|---|---|---|---|---|---|---|
| Bevacizumab | Clinical study | III | Bevacizumab + IFL/Placebo + IFL (First-line) | 402/411 | OS: 20.3/15.6 months (p < 0.001) PFS: 10.6/6.2 months (p < 0.001) RR: 44.8%/34.8% (p = 0.004) 1-year survival rate: 74.3%/63.4% (p < 0.001) | Hurwitz et al. [88] |
| Bevacizumab | AVEX | III | Bevacizumab + Capecitabine/Capecitabine (First-line) | 140/140 | OS: 20.7/16.8 months (p = 0.18) PFS: 9.1/5.1 months (p < 0.001) | Cunningham et al. [89] |
| Aflibercept | VELOUR (NCT00561470) | III | Aflibercept + FOLFILI/Placebo + FOLFILI (Second-line) | 612/614 | OS: 13.5/12.06 months (p = 0.0032) PFS: 6.9/4.67 months (p < 0.0001) RR: 19.8%/11.1% (p = 0.0001) | Van Cutsem et al. [91] |
| Regorafenib | CORRECT (NCT01103323) | III | Regorafenib/Placebo (Third- or later-line) | 505/255 | OS: 6.4/5.0 months (p = 0.0052) PFS: 1.9/1.7 months (p < 0.0001) | Grothey et al. [92] |
| Regorafenib | CONCUR (NCT01103323) | III | Regorafenib/Placebo (Third- or later-line) | 138/68 | OS: 8.8/6.3 months (p = 0.00016) PFS: 3.2/1.7 months (p < 0.0001) | Li et al. [93] |
| Ramucirumab | RAISE (NCT01183780) | III | Ramucirumab + FOLFIRI/Placebo + FOLFIRI (Second-line) | 536/536 | OS: 13.3/11.7 months (p = 0.0219) PFS: 5.7/4.5 months (p < 0.0005) | Tabernero et al. [94] |
Abbreviations: OS: overall survival; PFS: progression-free survival; RR: response rate.
Table 4.
Selected clinical trials for immune checkpoint-targeting drugs.
| Targeted Drugs | Study | Phase | Study Regimen | Number | Results | Ref |
|---|---|---|---|---|---|---|
| Pembrolizumab | Clinical study (NCT01876511) | II | Pembrolizumab (Second-line) (Patients with MSI-high/dMMR mCRC) | 40 | ORR: 52% Disease control rate: 82% 2-year OS: 72% 2-year PFS: 59% | Le et al. [125] |
| Pembrolizumab | KEYNOTE-177 (NCT02563002) | II | Pembrolizumab/Chemotherapy (First-line) (Patients with MSI-high/dMMR mCRC) | 153/154 | PFS: 16.5/8.2 months ORR: 43.8%/33.1% | Andre et al. [126] |
| Nivolumab | CheckMate-142 (NCT02060188) | II | Nivolumab (Second-line) (Patients with MSI-high/dMMR mCRC) | 74 | ORR: 31% Disease control for 12 weeks or longer: 69% | Overman et al. [127] |
| Nivolumab +Ipilimumab | CheckMate-142 (NCT02060188) | II | Nivolumab + Ipilimumab/Nivolumab (Second-line) (Patients with MSI-high/dMMR mCRC) | 119/74 | 1-year OS: 85%/73% ORR: 55%/31% Disease control for 12 weeks or longer: 80%/69% | Overman et al. [128] |
Abbreviations: ORR: objective response rate; OS: overall survival; PFS: progression-free survival.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ohishi, T.; Kaneko, M.K.; Yoshida, Y.; Takashima, A.; Kato, Y.; Kawada, M. Current Targeted Therapy for Metastatic Colorectal Cancer. Int. J. Mol. Sci. 2023, 24, 1702. https://doi.org/10.3390/ijms24021702
AMA Style
Ohishi T, Kaneko MK, Yoshida Y, Takashima A, Kato Y, Kawada M. Current Targeted Therapy for Metastatic Colorectal Cancer. International Journal of Molecular Sciences. 2023; 24(2):1702. https://doi.org/10.3390/ijms24021702
Chicago/Turabian StyleOhishi, Tomokazu, Mika K. Kaneko, Yukihiro Yoshida, Atsuo Takashima, Yukinari Kato, and Manabu Kawada. 2023. "Current Targeted Therapy for Metastatic Colorectal Cancer" International Journal of Molecular Sciences 24, no. 2: 1702. https://doi.org/10.3390/ijms24021702
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.