
PEBP4 Directs the Malignant Behavior of Hepatocellular Carcinoma Cells via Regulating mTORC1 and mTORC2

 , , and
, , and
Abstract
:1. Introduction
2. Results
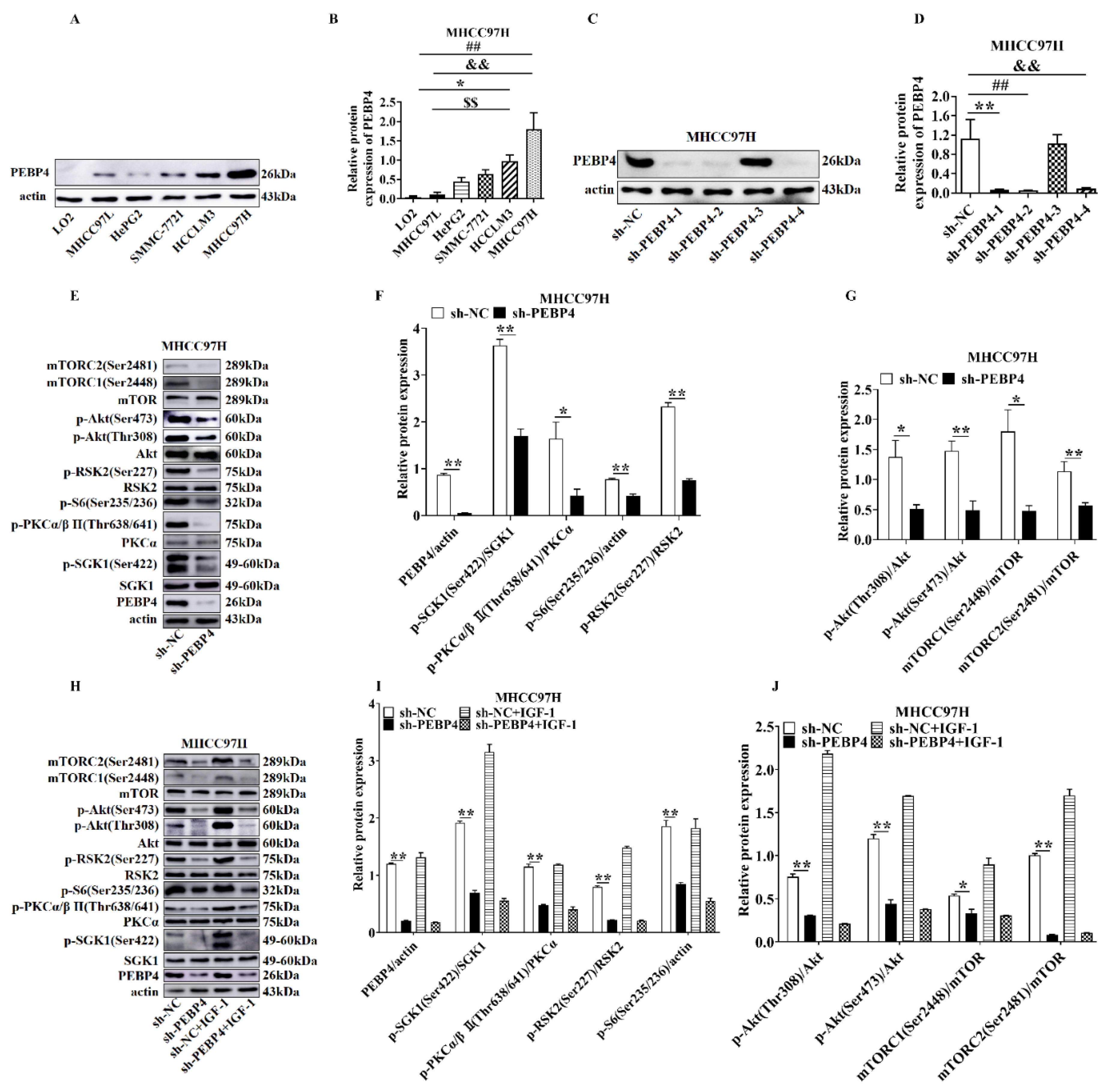
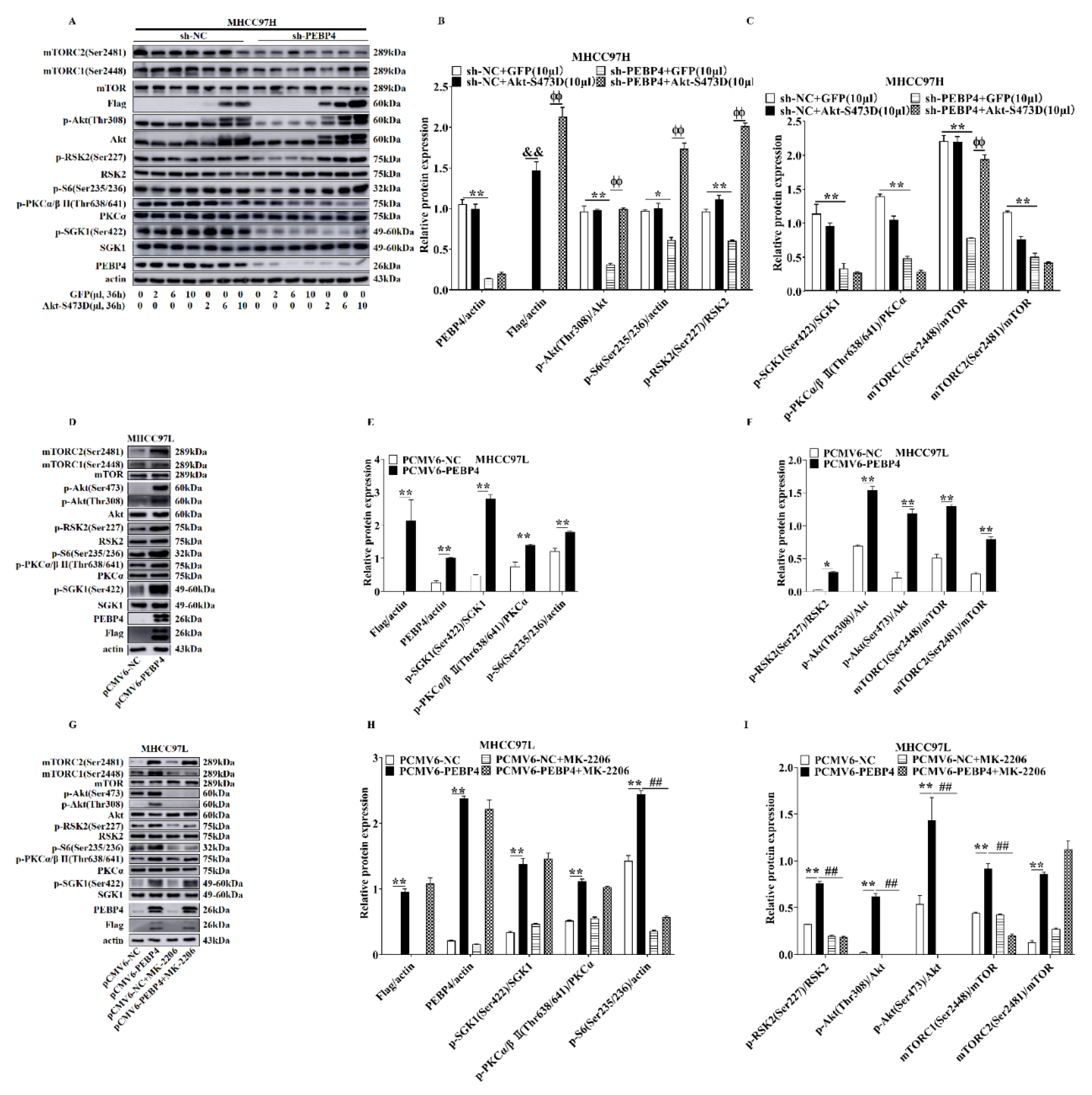
2.1. The Effects of PEBP4 Expression on the Signal Transduction Pathways Downstream of mTORCs
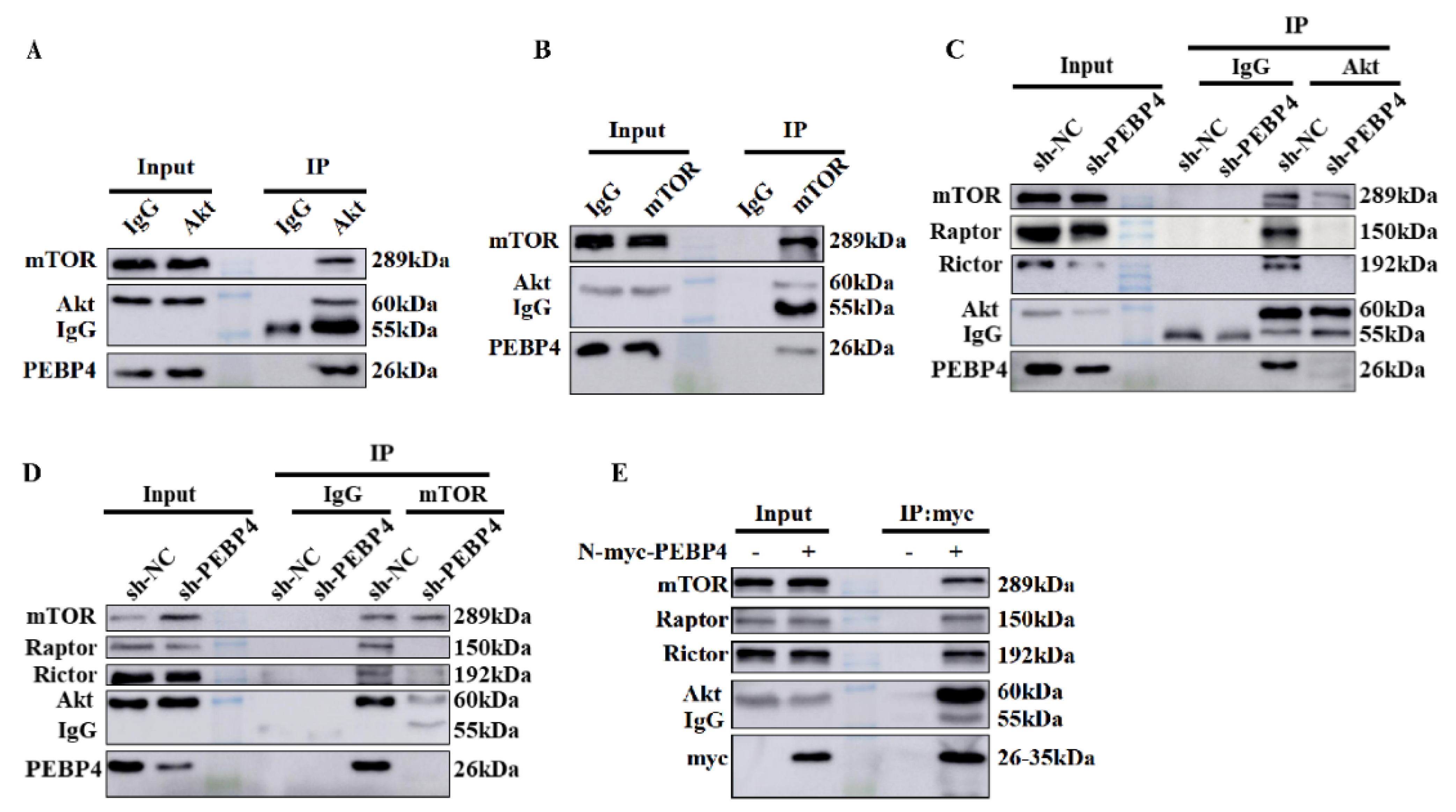
2.2. PEBP4 Associates with Akt and mTORCs
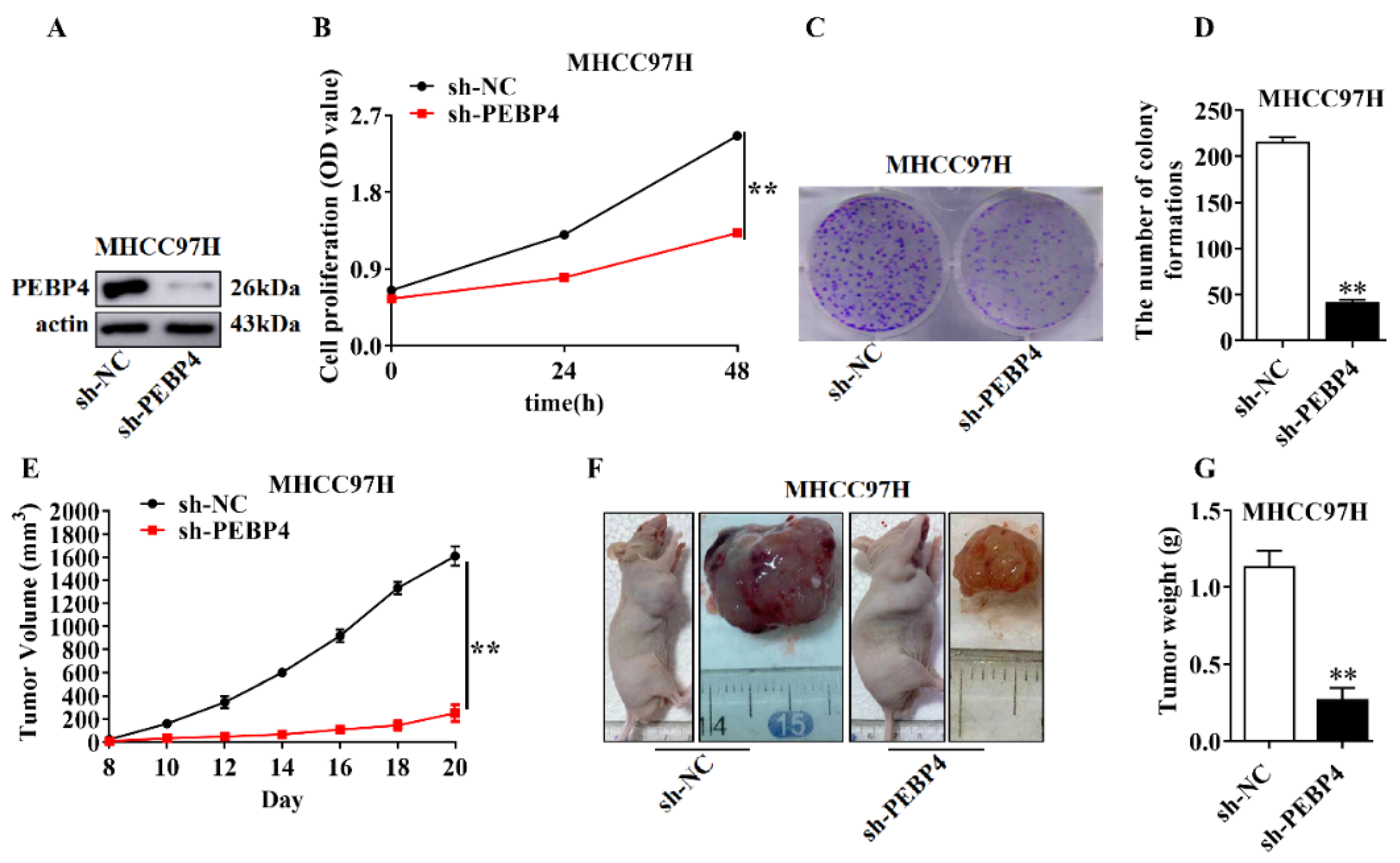
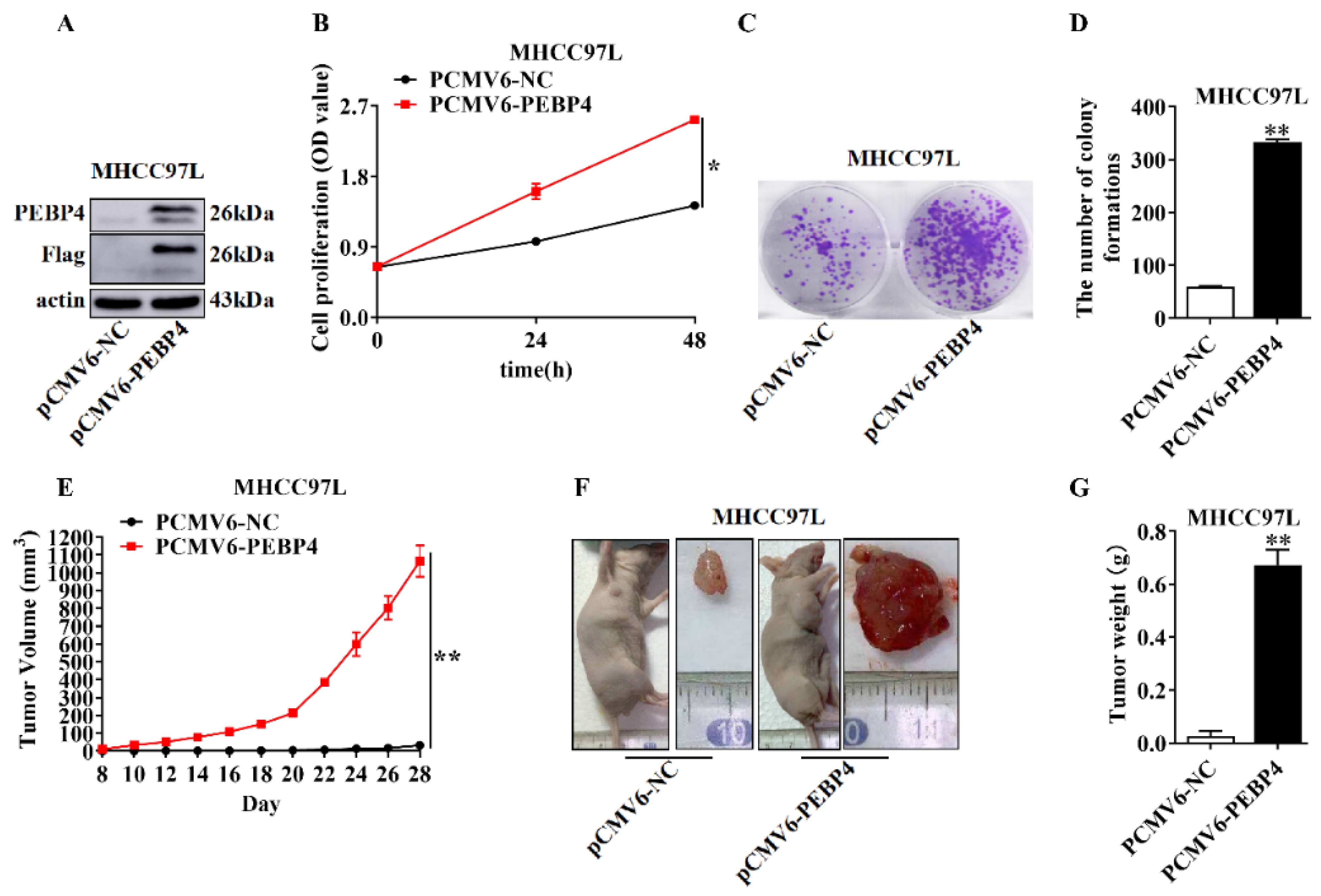
2.3. The Role of PEBP4 in Cell Proliferation and Tumor Growth
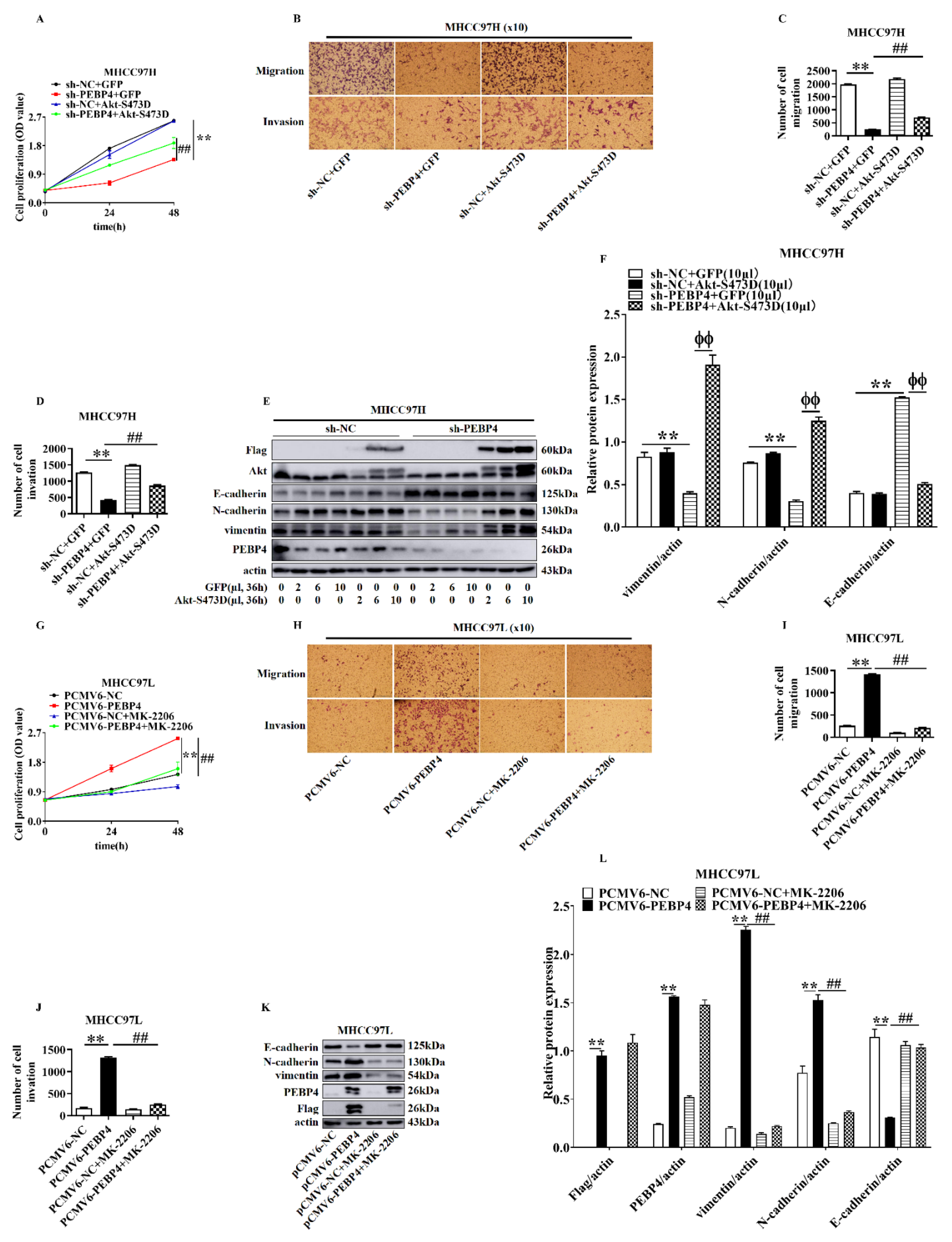
2.4. The Role of Akt in Mediating the Effect of PEBP4 on Proliferation, Migration, Invasion, and EMT of HCC
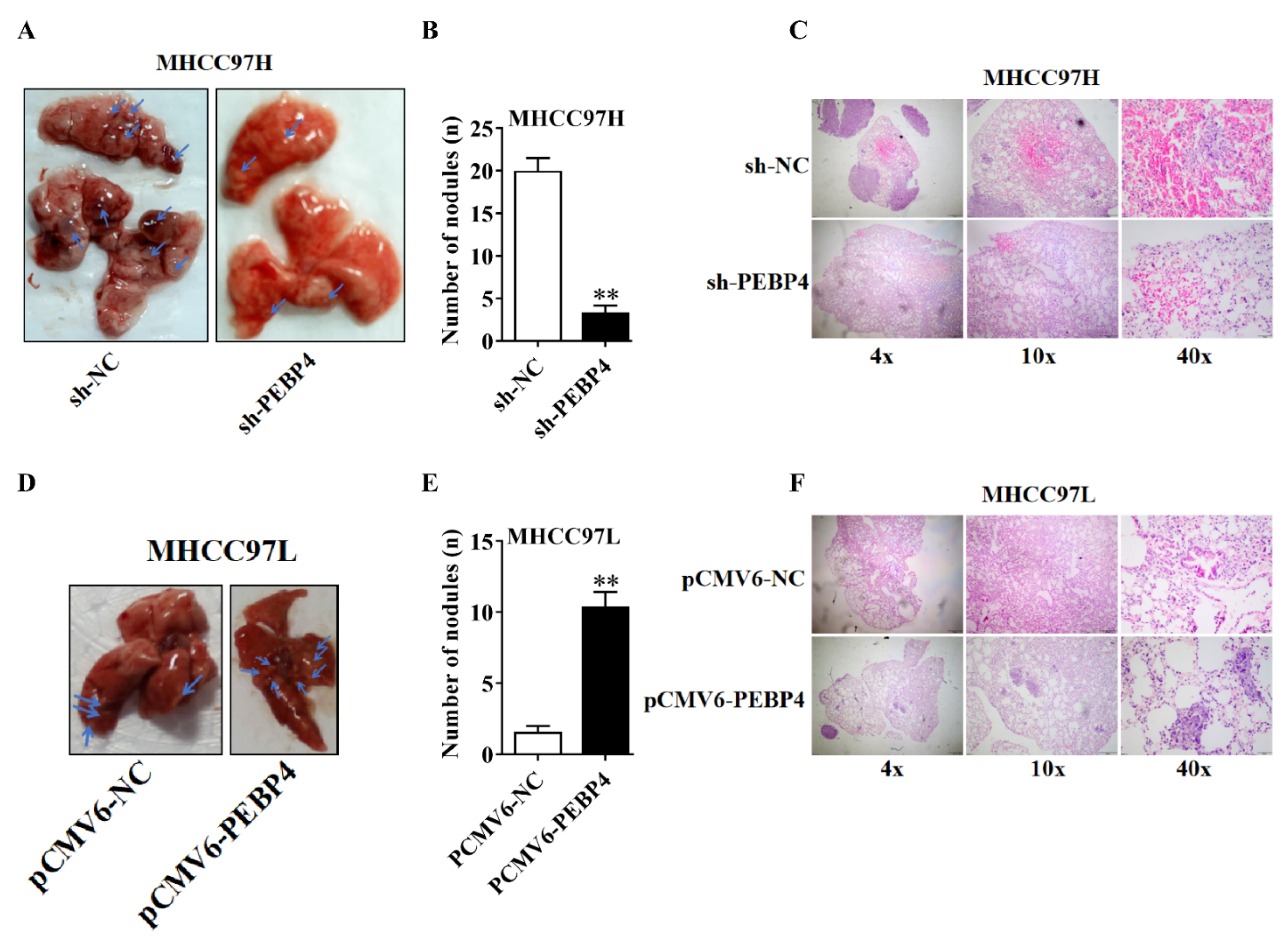
2.5. PEBP4 Stimulates Metastasis of HCC
3. Discussion
4. Materials and Methods
4.1. Materials and Reagents
4.2. Cell Culture
4.3. Virus Production and Transfection
4.4. Adenovirus Infection
4.5. Western Blot and Immunoprecipitation
4.6. Cell Proliferation Assay
4.7. Cell Migration and Invasion Assays
4.8. Colony Formation Assay
4.9. Hematoxylin and Eosin (H&E) Staining
4.10. Xenograft Tumor Model
4.11. Lung Metastasis Assay
4.12. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- He, H.; Liu, D.; Lin, H.; Jiang, S.; Ying, Y.; Chun, S.; Deng, H.; Zaia, J.; Wen, R.; Luo, Z. Phosphatidylethanolamine binding protein 4 (PEBP4) is a secreted protein and has multiple functions. Biochim. Biophys. Acta 2016, 1863 Pt A, 1682–1689. [Google Scholar] [CrossRef]
- Kim, I.J.; Quigley, D.; To, M.D.; Pham, P.; Lin, K.; Jo, B.; Jen, K.Y.; Raz, D.; Kim, J.; Mao, J.H.; et al. Rewiring of human lung cell lineage and mitotic networks in lung adenocarcinomas. Nat. Commun. 2013, 4, 1701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, S.; Pieri, K.; Nanni, P.; Tica, J.; Barratt, J.; Didangelos, A. Phosphatidylethanolamine binding protein-4 (PEBP4) is increased in IgA nephropathy and is associated with IgA-positive B-cells in affected kidneys. J. Autoimmun. 2019, 105, 102309. [Google Scholar] [CrossRef]
- Baka, R.; Eckersall, D.; Horvatic, A.; Gelemanovic, A.; Mrljak, V.; McLaughlin, M.; Athanasiou, L.V.; Papaioannou, N.; Stylianaki, I.; Hanh, H.Q.; et al. Quantitative proteomics of cerebrospinal fluid using tandem mass tags in dogs with recurrent epileptic seizures. J. Proteom. 2021, 231, 103997. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Guo, H.; Zhu-Salzman, K.; Ge, F.; Sun, Y. PEBP balances apoptosis and autophagy in whitefly upon arbovirus infection. Nat. Commun. 2022, 13, 846. [Google Scholar] [CrossRef]
- Wen, Y.; Dai, B.; Zhang, X.; Zhu, H.; Xie, C.; Xia, J.; Sun, Y.; Zhu, M.; Tong, J.; Shen, Y. Retinal Transcriptomics Analysis Reveals the Underlying Mechanism of Disturbed Emmetropization Induced by Wavelength Defocus. Curr. Eye Res. 2022, 47, 908–917. [Google Scholar] [CrossRef]
- An, L.P.; Maeda, T.; Sakaue, T.; Takeuchi, K.; Yamane, T.; Du, P.G.; Ohkubo, I.; Ogita, H. Purification, molecular cloning and functional characterization of swine phosphatidylethanolamine-binding protein 4 from seminal plasma. Biochem. Biophys. Res. Commun. 2012, 423, 690–696. [Google Scholar] [CrossRef] [PubMed]
- Whitwell, H.J.; Worthington, J.; Blyuss, O.; Gentry-Maharaj, A.; Ryan, A.; Gunu, R.; Kalsi, J.; Menon, U.; Jacobs, I.; Zaikin, A.; et al. Improved early detection of ovarian cancer using longitudinal multimarker models. Br. J. Cancer 2020, 122, 847–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, P.; Zhou, C.; Shen, H. Diagnostic value of phosphatidylethanolamine binding protein 4 levels in patients receiving nursing interventions for advanced chronic kidney disease. J. Int. Med. Res. 2021, 49, 300060521996179. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, X.; Xiang, Z.; Li, H.; Qiu, J.; Sun, Q.; Wan, T.; Li, N.; Cao, X.; Wang, J. Promotion of cellular migration and apoptosis resistance by a mouse eye-specific phosphatidylethanolamine-binding protein. Int. J. Mol. Med. 2007, 19, 55–63. [Google Scholar] [CrossRef]
- Garcia, R.; Grindlay, J.; Rath, O.; Fee, F.; Kolch, W. Regulation of human myoblast differentiation by PEBP4. EMBO Rep. 2009, 10, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Huang, B.; Chen, G.; Mi, Y. Phosphatidylethanolamine-binding protein 4 promotes lung cancer cells proliferation and invasion via PI3K/Akt/mTOR axis. J. Thorac. Dis. 2015, 7, 1806–1816. [Google Scholar] [PubMed]
- Li, W.; Dong, Y.; Zhang, B.; Kang, Y.; Yang, X.; Wang, H. PEBP4 silencing inhibits hypoxia-induced epithelial-to-mesenchymal transition in prostate cancer cells. Biomed. Pharm. 2016, 81, 1–6. [Google Scholar] [CrossRef]
- Zhang, D.; Dai, Y.; Cai, Y.; Suo, T.; Liu, H.; Wang, Y.; Cheng, Z.; Liu, H. PEBP4 promoted the growth and migration of cancer cells in pancreatic ductal adenocarcinoma. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2016, 37, 1699–1705. [Google Scholar] [CrossRef]
- Wang, S.C.; Zhou, F.; Zhou, Z.Y.; Hu, Z.; Chang, L.; Ma, M.D. Knockdown of PEBP4 suppresses proliferation, migration and invasion of human breast cancer cells. Biomed. Pharm. 2017, 90, 659–664. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.Q.; Wang, S.Q.; Zhu, Q.B.; Guo, S.C.; Shi, D.L.; Chen, F.; Fang, Y.C.; Chen, R.; Lu, Y.C. Knockdown of PEBP4 inhibits human glioma cell growth and invasive potential via ERK1/2 signaling pathway. Mol. Carcinog. 2019, 58, 135–143. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Kong, Q.; Li, B.; He, Y.; Li, P.; Jia, B. Expression of PEBP4 protein correlates with the invasion and metastasis of colorectal cancer. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2012, 33, 267–273. [Google Scholar] [CrossRef]
- Wu, Z.; Liu, B.; Zheng, X.; Hou, H.; Li, Y. Role of the PEBP4 protein in the development and metastasis of gastric cancer. Oncotarget 2017, 8, 18177–18184. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.P.; Chen, G.Q.; Wu, S.; Shen, K.; Ji, Y. The expression of PEBP4 protein in lung squamous cell carcinoma. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2011, 32, 1257–1263. [Google Scholar] [CrossRef]
- Huang, R.Q.; Shi, D.L.; Huang, W.; Chen, F.; Lu, Y.C. Increased expression of phosphatidylethanolamine-binding protein 4 (PEBP4) strongly associates with human gliomas grade. J. Neurooncol. 2016, 127, 235–242. [Google Scholar] [CrossRef]
- Huang, R.Q.; Chen, F.; Jiang, Y.B.; Jin, Y.M.; Wang, S.Q.; Liang, H.H.; Chen, Z.P.; Qian, J. Overexpression of phosphatidylethanolamine-binding protein 4 (PEBP4) associates with recurrence of meningiomas. Clin. Neurol. Neurosurg. 2022, 214, 107148. [Google Scholar] [CrossRef]
- Li, H.; Huang, F.; Fan, L.; Jiang, Y.; Wang, X.; Li, J.; Wang, Q.; Pan, H.; Sun, J.; Cao, X.; et al. Phosphatidylethanolamine-binding protein 4 is associated with breast cancer metastasis through Src-mediated Akt tyrosine phosphorylation. Oncogene 2014, 33, 4589–4598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, G.P.; Huang, B.; Chen, G.Q.; Wu, S.; Ji, Y.; Shen, Z.Y. PEBP4 gene expression and its significance in invasion and metastasis of non-small cell lung cancer. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2012, 33, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Shen, Z.; Chen, G.; Teng, X.; Hu, Y.; Huang, B. PEBP4 enhanced HCC827 cell proliferation and invasion ability and inhibited apoptosis. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2013, 34, 91–98. [Google Scholar] [CrossRef]
- Yu, G.; Zhong, N.; Chen, G.; Huang, B.; Wu, S. Downregulation of PEBP4, a target of miR-34a, sensitizes drug-resistant lung cancer cells. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2014, 35, 10341–10349. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Zhang, L.; Yao, Q.; Tao, Z. miR-15b regulates cisplatin resistance and metastasis by targeting PEBP4 in human lung adenocarcinoma cells. Cancer Gene 2015, 22, 108–114. [Google Scholar] [CrossRef]
- Jian, W.; Bai, Y.; Li, X.; Kang, J.; Lei, Y.; Xue, Y. Phosphatidylethanolamine-binding protein 4 promotes the epithelial-to-mesenchymal transition in non-small cell lung cancer cells by activating the sonic hedgehog signaling pathway. J. Cell. Biochem. 2019, 120, 5386–5395. [Google Scholar] [CrossRef]
- Liu, G.Y.; Sabatini, D.M. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef]
- Menon, S.; Manning, B.D. Common corruption of the mTOR signaling network in human tumors. Oncogene 2008, 27 (Suppl. S2), S43–S51. [Google Scholar] [CrossRef] [Green Version]
- Pearce, L.R.; Komander, D.; Alessi, D.R. The nuts and bolts of AGC protein kinases. Nat. Rev. Mol. Cell Biol. 2010, 11, 9–22. [Google Scholar] [CrossRef]
- Liu, P.; Gan, W.; Chin, Y.R.; Ogura, K.; Guo, J.; Zhang, J.; Wang, B.; Blenis, J.; Cantley, L.C.; Toker, A.; et al. PtdIns(3,4,5)P3-Dependent Activation of the mTORC2 Kinase Complex. Cancer Discov. 2015, 5, 1194–1209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebner, M.; Sinkovics, B.; Szczygieł, M.; Ribeiro, D.W.; Yudushkin, I. Localization of mTORC2 activity inside cells. J. Cell Biol. 2017, 216, 343–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humphrey, S.J.; Yang, G.; Yang, P.; Fazakerley, D.J.; Stöckli, J.; Yang, J.Y.; James, D.E. Dynamic adipocyte phosphoproteome reveals that Akt directly regulates mTORC2. Cell Metab. 2013, 17, 1009–1020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, G.; Murashige, D.S.; Humphrey, S.J.; James, D.E. A Positive Feedback Loop between Akt and mTORC2 via SIN1 Phosphorylation. Cell Rep. 2015, 12, 937–943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iida, M.; Brand, T.M.; Campbell, D.A.; Starr, M.M.; Luthar, N.; Traynor, A.M.; Wheeler, D.L. Targeting AKT with the allosteric AKT inhibitor MK-2206 in non-small cell lung cancer cells with acquired resistance to cetuximab. Cancer Biol. Ther. 2013, 14, 481–491. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Q.; Zhang, X.; Dai, X.; Han, S.; Wu, X.; Wang, L.; Wei, W.; Zhang, N.; Xie, W.; Guo, J. S6K1-mediated phosphorylation of PDK1 impairs AKT kinase activity and oncogenic functions. Nat. Commun. 2022, 13, 1548. [Google Scholar] [CrossRef]
- Kondo, N.; Ishii, Y.; Kwon, Y.W.; Tanito, M.; Horita, H.; Nishinaka, Y.; Nakamura, H.; Yodoi, J. Redox-sensing release of human thioredoxin from T lymphocytes with negative feedback loops. J. Immunol. 2004, 172, 442–448. [Google Scholar] [CrossRef] [Green Version]
- Gardella, S.; Andrei, C.; Ferrera, D.; Lotti, L.V.; Torrisi, M.R.; Bianchi, M.E.; Rubartelli, A. The nuclear protein HMGB1 is secreted by monocytes via a non-classical, vesicle-mediated secretory pathway. EMBO Rep. 2002, 3, 995–1001. [Google Scholar] [CrossRef] [Green Version]
- Jiang, S.; Wang, Y.; Luo, L.; Shi, F.; Zou, J.; Lin, H.; Ying, Y.; Luo, Y.; Zhan, Z.; Liu, P.; et al. AMP-activated protein kinase regulates cancer cell growth and metabolism via nuclear and mitochondria events. J. Cell. Mol. Med. 2019, 23, 3951–3961. [Google Scholar] [CrossRef] [Green Version]
- Zang, M.; Hayne, C.; Luo, Z. Interaction between active Pak1 and Raf-1 is necessary for phosphorylation and activation of Raf-1. J. Biol. Chem. 2002, 277, 4395–4405. [Google Scholar] [CrossRef] [Green Version]
- Jiang, S.; Shi, F.; Lin, H.; Ying, Y.; Luo, L.; Huang, D.; Luo, Z. Inonotus obliquus polysaccharides induces apoptosis of lung cancer cells and alters energy metabolism via the LKB1/AMPK axis. Int. J. Biol. Macromol. 2020, 151, 1277–1286. [Google Scholar] [CrossRef] [PubMed]
- Li, N.S.; Zou, J.R.; Lin, H.; Ke, R.; He, X.L.; Xiao, L.; Huang, D.; Luo, L.; Lv, N.; Luo, Z. LKB1/AMPK inhibits TGF-β1 production and the TGF-β signaling pathway in breast cancer cells. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2016, 37, 8249–8258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Hsu, S.H.; Majumder, S.; Kutay, H.; Huang, W.; Jacob, S.T.; Ghoshal, K. TGFbeta-mediated upregulation of hepatic miR-181b promotes hepatocarcinogenesis by targeting TIMP3. Oncogene 2010, 29, 1787–1797. [Google Scholar] [CrossRef] [Green Version]
- Hu, Q.; Hu, Z.; Chen, Q.; Huang, Y.; Mao, Z.; Xu, F.; Zhou, X. BML-111 equilibrated ACE-AngII-AT1R and ACE2-Ang-(1-7)-Mas axis to protect hepatic fibrosis in rats. Prostaglandins Other Lipid Mediat. 2017, 131, 75–82. [Google Scholar] [CrossRef]
- Zhangyuan, G.; Wang, F.; Zhang, H.; Jiang, R.; Tao, X.; Yu, D.; Jin, K.; Yu, W.; Liu, Y.; Yin, Y.; et al. VersicanV1 promotes proliferation and metastasis of hepatocellular carcinoma through the activation of EGFR-PI3K-AKT pathway. Oncogene 2020, 39, 1213–1230. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Catalog No. | Source |
|---|---|---|
| β-actin | TA-09 | Zhongshan Golden Bridge (Beijing, China) |
| PEBP4 | ab139074 | Abcam (Cambridge, UK) |
| p-SGK1 (Ser422) | ab55281 | Abcam |
| Akt | ab32505 | Abcam |
| Rictor | ab70374 | Abcam |
| SGK1 | 28454-1-AP | Proteintech (Wuhan, China) |
| PKCα | 21991-1-Ig | Proteintech |
| RSK2 | 23762-1-AP | Proteintech |
| mTOR (2448) | 67778-1-Ig | Proteintech |
| E-cadherin | 00077111 | Proteintech |
| N-cadherin | 00059200 | Proteintech |
| Vimentin | 00066574 | Proteintech |
| myc | 60003-2-Ig | Proteintech |
| p-PKCα/β (Thr638/641) | 9375 | Cell Signaling Tech. (Danvers, MA, USA) |
| p-S6 (Ser235/236) | 4858 | Cell Signaling Tech. |
| p-RSK2 (Ser227) | 3556 | Cell Signaling Tech. |
| p-Akt (Thr308) | 13038s | Cell Signaling Tech. |
| p-Akt (Ser473) | 4060s | Cell Signaling Tech. |
| mTOR | 2983 | Cell Signaling Tech. |
| integrinβ-1 | 34971T | Cell Signaling Tech. |
| Raptor | 2280 | Cell Signaling Tech. |
| mTOR (2481) | PA5104898 | Thermo scientific (Waltham, MA, USA) |
| Flag | T0053 | Affinity (Miami, FL, USA) |
| Rabbit IgG | A7016 | Beyotime (Shanghai, China) |
| Plasmid | Sequences |
|---|---|
| sh-PEBP4-1 | ACCTCCTGGATGGAGCCGATAGTCAAGTT |
| sh-PEBP4-2 | TGGCTTCCATCGCTACCAGTTCTTTGTCT |
| sh-PEBP4-3 | GTTGGACAATGAGGCTGGTTACAGCAGCA |
| sh-PEBP4-4 | GGACAGATTTCTGAACCGCTTCCACCTGG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Q.; Jin, J.; Guo, W.; Tang, Z.; Luo, Y.; Ying, Y.; Lin, H.; Luo, Z. PEBP4 Directs the Malignant Behavior of Hepatocellular Carcinoma Cells via Regulating mTORC1 and mTORC2. Int. J. Mol. Sci. 2022, 23, 8798. https://doi.org/10.3390/ijms23158798
Chen Q, Jin J, Guo W, Tang Z, Luo Y, Ying Y, Lin H, Luo Z. PEBP4 Directs the Malignant Behavior of Hepatocellular Carcinoma Cells via Regulating mTORC1 and mTORC2. International Journal of Molecular Sciences. 2022; 23(15):8798. https://doi.org/10.3390/ijms23158798
Chicago/Turabian StyleChen, Qiongfeng, Jingguang Jin, Wenhui Guo, Zhimin Tang, Yunfei Luo, Ying Ying, Hui Lin, and Zhijun Luo. 2022. "PEBP4 Directs the Malignant Behavior of Hepatocellular Carcinoma Cells via Regulating mTORC1 and mTORC2" International Journal of Molecular Sciences 23, no. 15: 8798. https://doi.org/10.3390/ijms23158798