
Targeted-Deletion of a Tiny Sequence via Prime Editing to Restore SMN Expression

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
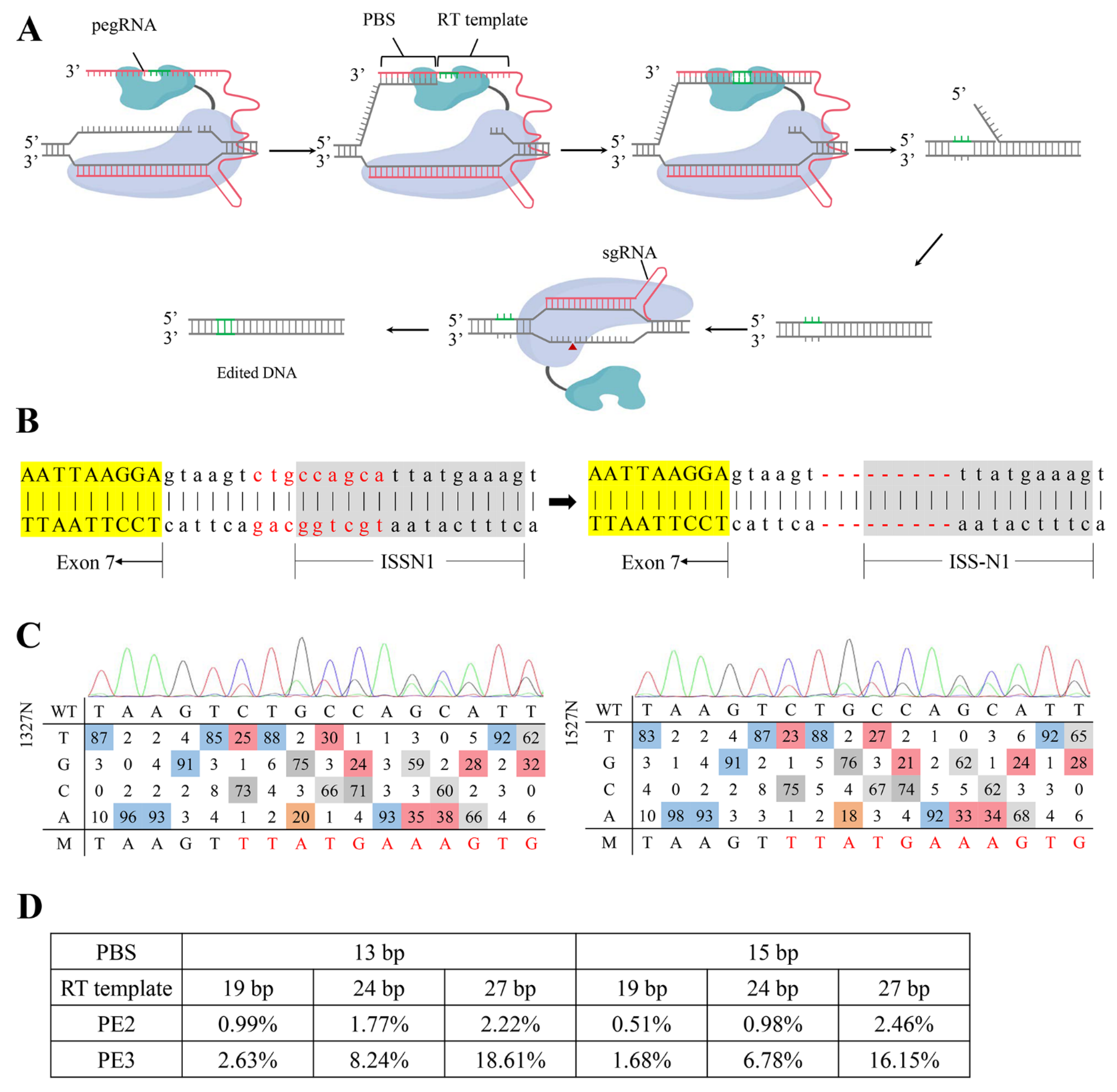
2.1. Designing and Screening of an Efficient ISS-N1-Targeted PE System
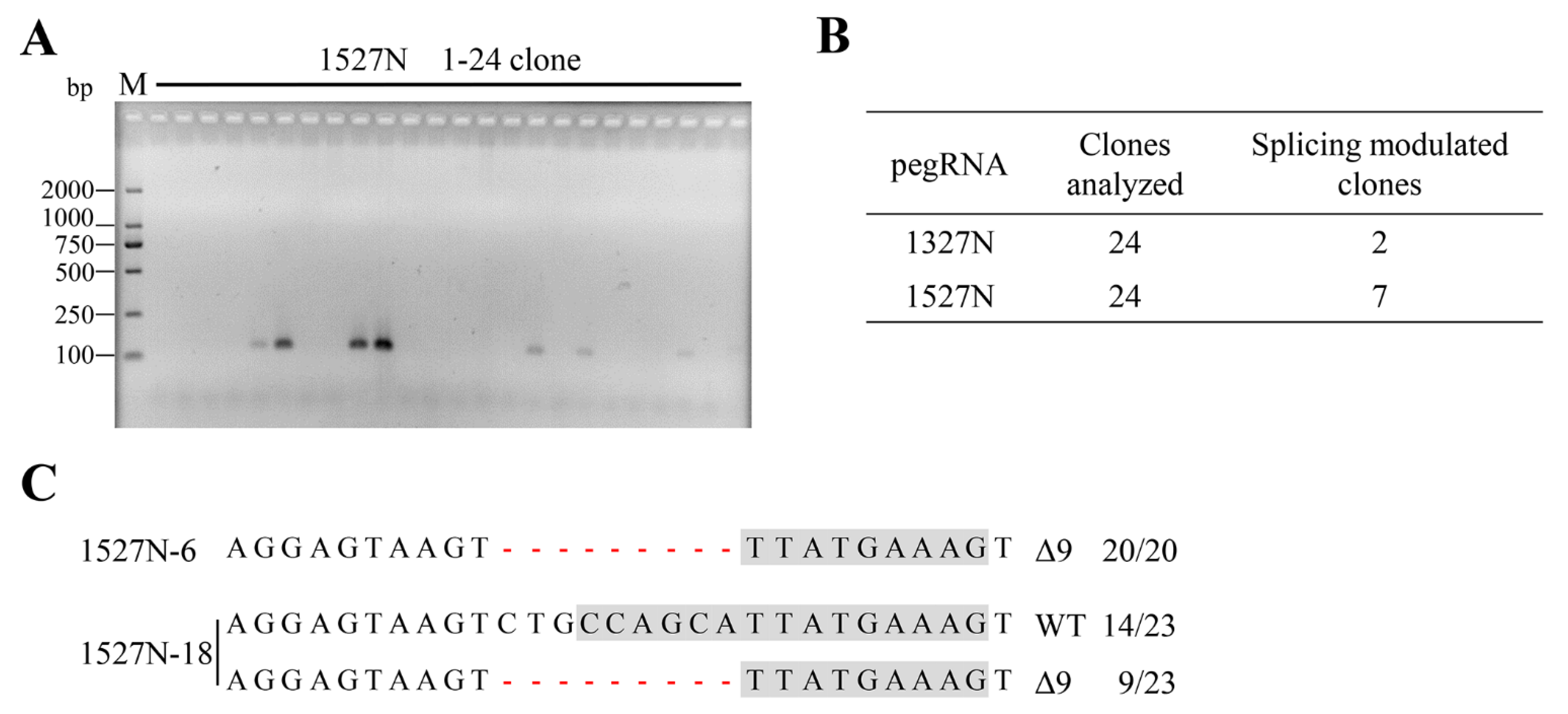
2.2. Targeted-Deletion ISS-N1 of SMN2 in SMA-iPSCs by PE
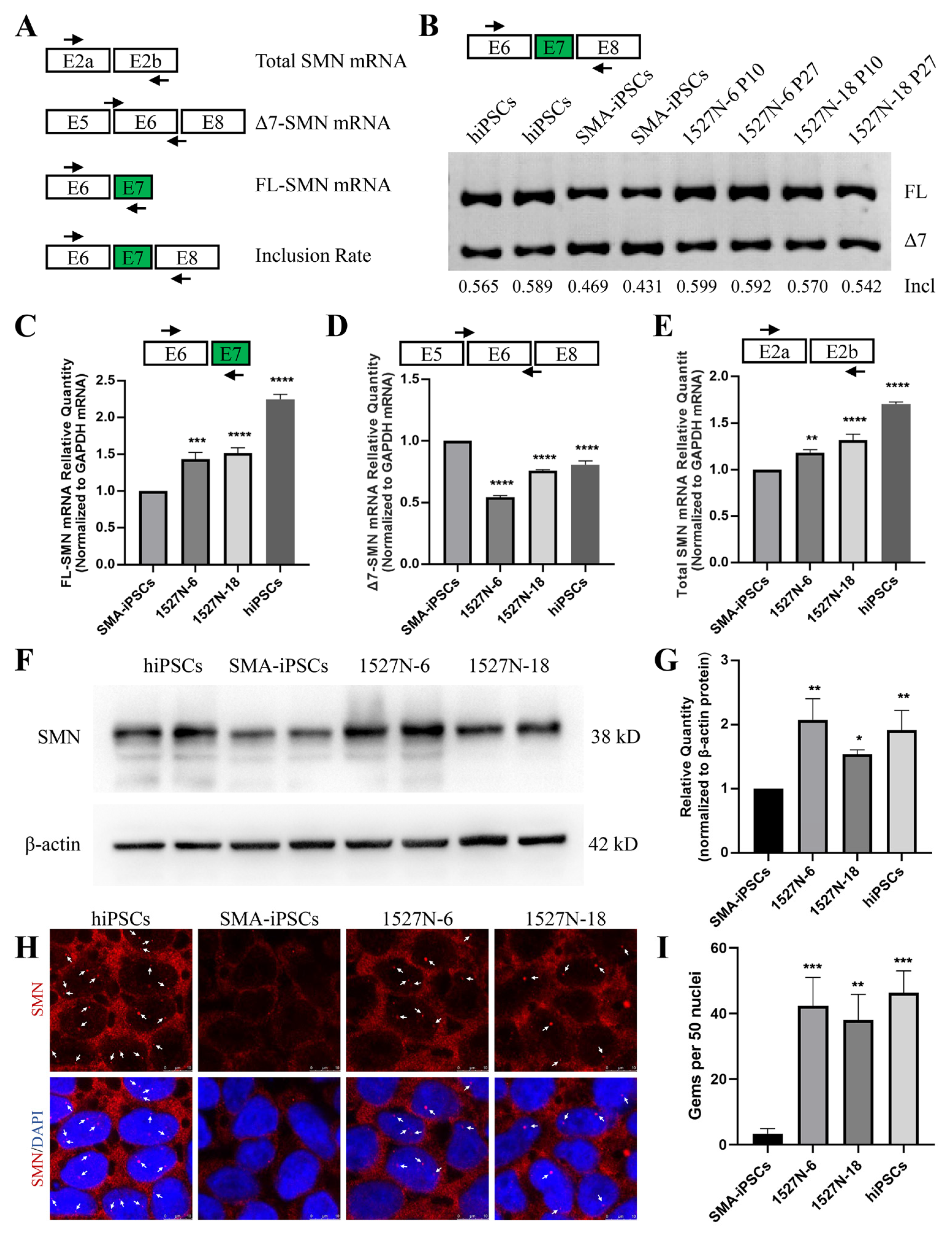
2.3. SMN Expression in Targeted-Deletion iPSCs Clones
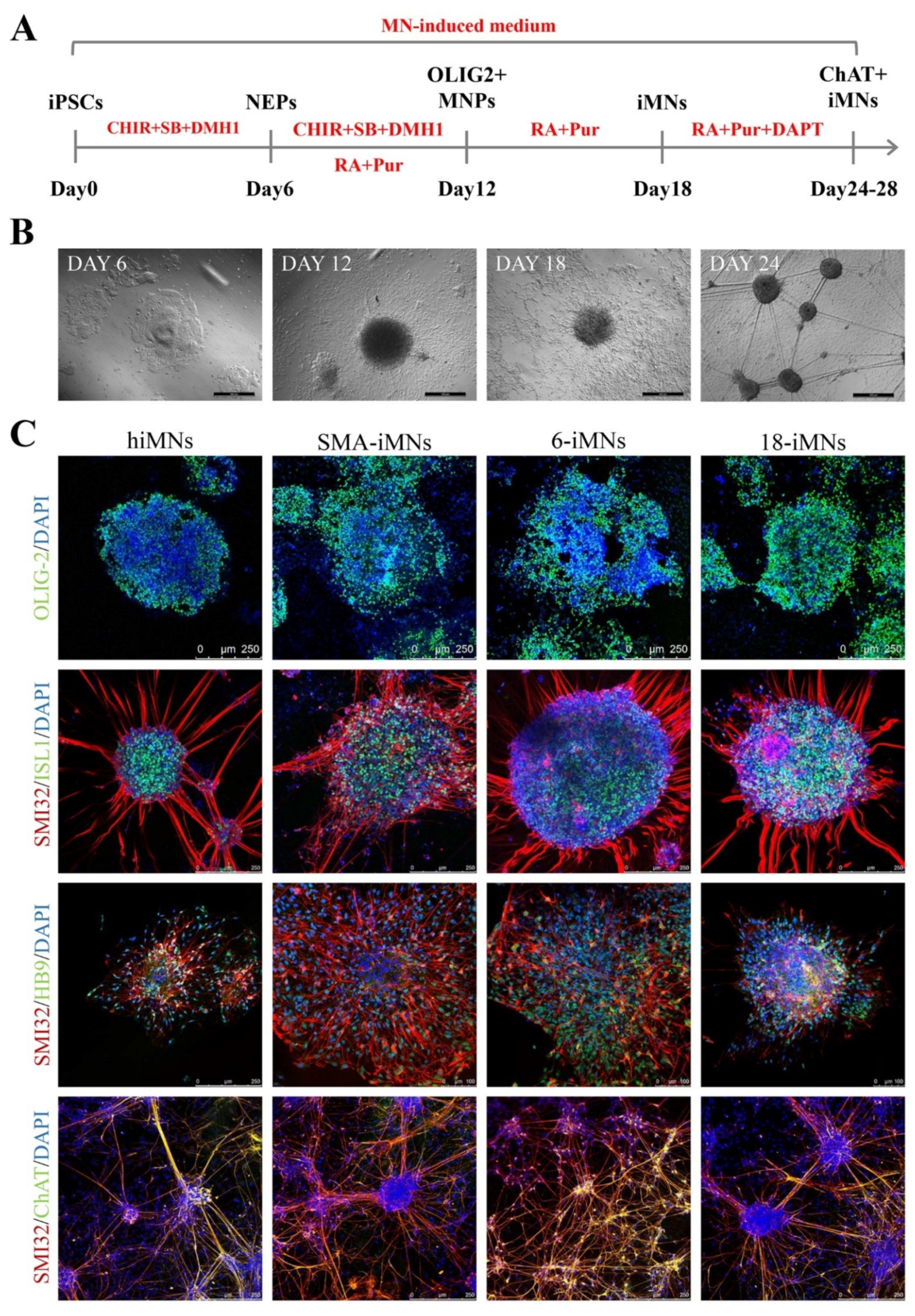
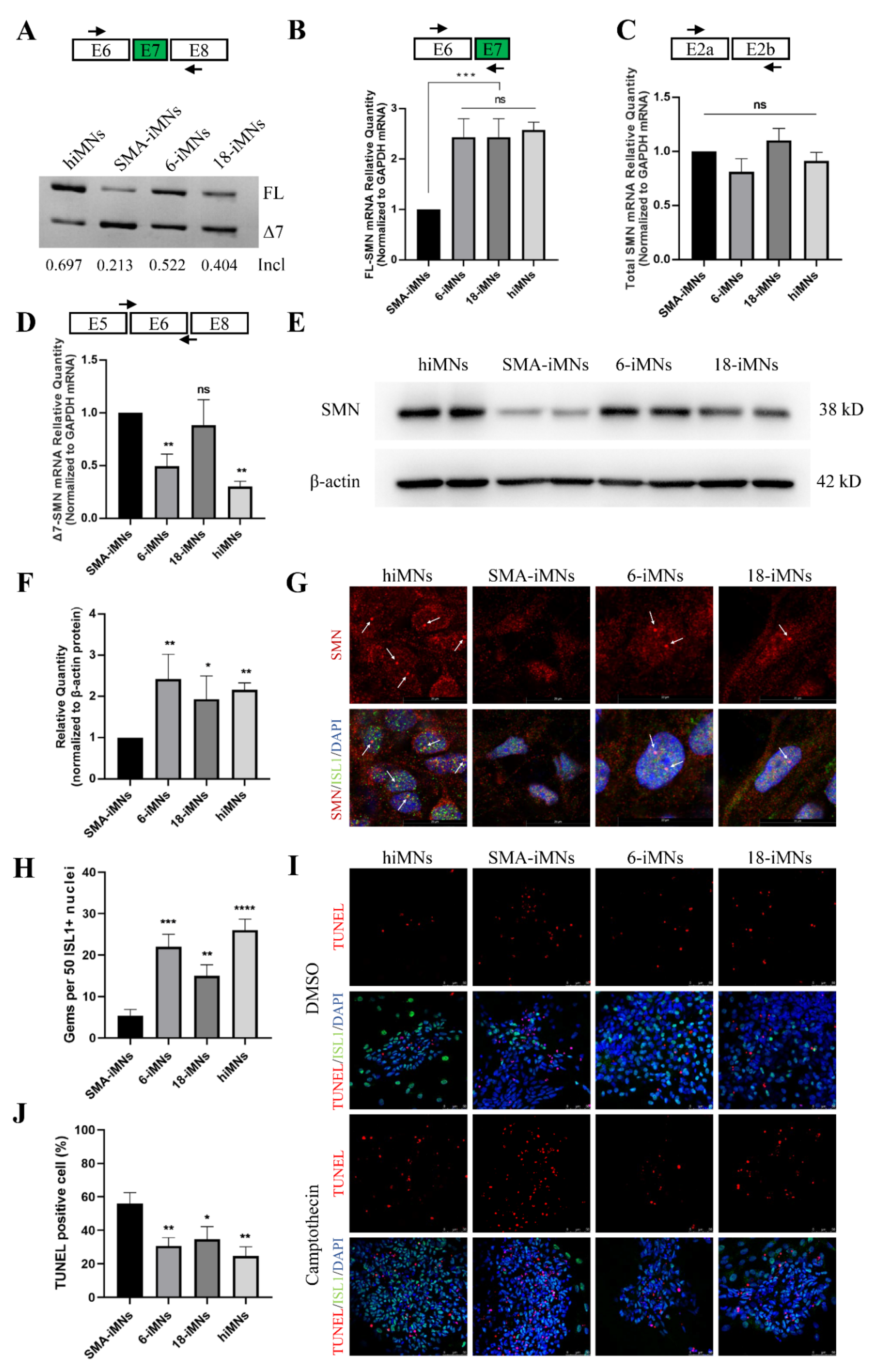
2.4. Restoration of SMN Expression in Targeted-Deletion iPSC-Derived MNs
3. Discussion
4. Materials and Methods
4.1. PE Design and Construction
4.2. PE Efficiency Evaluation
4.3. Gene Targeting and PCR Detection of Targeted-Deletion Clones
4.4. Detection of Potential Off-Target Sites
4.5. iPSCs Differentiation into MNs
4.6. Reverse Transcription PCR (RT-PCR) and Quantitative RT-PCR (qRT-PCR) Analysis
4.7. Western Blot
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pearn, J. Classification of spinal muscular atrophies. Lancet 1980, 1, 919–922. [Google Scholar] [CrossRef]
- Verhaart, I.E.C.; Robertson, A.; Wilson, I.J.; Aartsma-Rus, A.; Cameron, S.; Jones, C.C.; Cook, S.F.; Lochmuller, H. Prevalence, incidence and carrier frequency of 5q-linked spinal muscular atrophy—A literature review. Orphanet J. Rare Dis. 2017, 12, 124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Z.; Chen, M.; Zhang, C.; Li, Z.; Feng, M.; Wu, L.; Zhou, M.; Liang, D. Cas14a1-Mediated Nucleic Acid Diagnostics for Spinal Muscular Atrophy. Biosensors 2022, 12, 268. [Google Scholar] [CrossRef] [PubMed]
- Yuan, P.; Jiang, L. Clinical characteristics of three subtypes of spinal muscular atrophy in children. Brain Dev. 2015, 37, 537–541. [Google Scholar] [CrossRef]
- Mercuri, E.; Bertini, E.; Iannaccone, S.T. Childhood spinal muscular atrophy: Controversies and challenges. Lancet Neurol. 2012, 11, 443–452. [Google Scholar] [CrossRef]
- Ng, S.Y.; Soh, B.S.; Rodriguez-Muela, N.; Hendrickson, D.G.; Price, F.; Rinn, J.L.; Rubin, L.L. Genome-wide RNA-Seq of Human Motor Neurons Implicates Selective ER Stress Activation in Spinal Muscular Atrophy. Cell Stem Cell 2015, 17, 569–584. [Google Scholar] [CrossRef] [Green Version]
- Lefebvre, S.; Burglen, L.; Reboullet, S.; Clermont, O.; Burlet, P.; Viollet, L.; Benichou, B.; Cruaud, C.; Millasseau, P.; Zeviani, M.; et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995, 80, 155–165. [Google Scholar] [CrossRef] [Green Version]
- Alias, L.; Bernal, S.; Fuentes-Prior, P.; Barcelo, M.J.; Also, E.; Martinez-Hernandez, R.; Rodriguez-Alvarez, F.J.; Martin, Y.; Aller, E.; Grau, E.; et al. Mutation update of spinal muscular atrophy in Spain: Molecular characterization of 745 unrelated patients and identification of four novel mutations in the SMN1 gene. Hum. Genet. 2009, 125, 29–39. [Google Scholar] [CrossRef]
- Cartegni, L.; Hastings, M.L.; Calarco, J.A.; de Stanchina, E.; Krainer, A.R. Determinants of exon 7 splicing in the spinal muscular atrophy genes, SMN1 and SMN2. Am. J. Hum. Genet. 2006, 78, 63–77. [Google Scholar] [CrossRef] [Green Version]
- Elsheikh, B.; Prior, T.; Zhang, X.; Miller, R.; Kolb, S.J.; Moore, D.; Bradley, W.; Barohn, R.; Bryan, W.; Gelinas, D.; et al. An analysis of disease severity based on SMN2 copy number in adults with spinal muscular atrophy. Muscle Nerve 2009, 40, 652–656. [Google Scholar] [CrossRef]
- Lorson, C.L.; Hahnen, E.; Androphy, E.J.; Wirth, B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc. Natl. Acad. Sci. USA 1999, 96, 6307–6311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cartegni, L.; Krainer, A.R. Disruption of an SF2/ASF-dependent exonic splicing enhancer in SMN2 causes spinal muscular atrophy in the absence of SMN1. Nat. Genet. 2002, 30, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Deshaies, J.E.; Shkreta, L.; Moszczynski, A.J.; Sidibe, H.; Semmler, S.; Fouillen, A.; Bennett, E.R.; Bekenstein, U.; Destroismaisons, L.; Toutant, J.; et al. TDP-43 regulates the alternative splicing of hnRNP A1 to yield an aggregation-prone variant in amyotrophic lateral sclerosis. Brain 2018, 141, 1320–1333. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.L.; Lorson, C.L.; Androphy, E.J.; Zhou, J. An in vivo reporter system for measuring increased inclusion of exon 7 in SMN2 mRNA: Potential therapy of SMA. Gene Ther. 2001, 8, 1532–1538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osman, E.Y.; Yen, P.F.; Lorson, C.L. Bifunctional RNAs targeting the intronic splicing silencer N1 increase SMN levels and reduce disease severity in an animal model of spinal muscular atrophy. Mol. Ther. 2012, 20, 119–126. [Google Scholar] [CrossRef] [Green Version]
- Li, J.J.; Lin, X.; Tang, C.; Lu, Y.Q.; Hu, X.; Zuo, E.; Li, H.; Ying, W.; Sun, Y.; Lai, L.L.; et al. Disruption of splicing-regulatory elements using CRISPR/Cas9 to rescue spinal muscular atrophy in human iPSCs and mice. Natl. Sci. Rev. 2020, 7, 92–101. [Google Scholar] [CrossRef] [Green Version]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019, 576, 149–157. [Google Scholar] [CrossRef]
- Singh, N.N.; Shishimorova, M.; Cao, L.C.; Gangwani, L.; Singh, R.N. A short antisense oligonucleotide masking a unique intronic motif prevents skipping of a critical exon in spinal muscular atrophy. RNA Biol. 2009, 6, 341–350. [Google Scholar] [CrossRef] [Green Version]
- Kluesner, M.G.; Nedveck, D.A.; Lahr, W.S.; Garbe, J.R.; Abrahante, J.E.; Webber, B.R.; Moriarity, B.S. EditR: A Method to Quantify Base Editing from Sanger Sequencing. CRISPR J. 2018, 1, 239–250. [Google Scholar] [CrossRef]
- Zhou, M.; Hu, Z.; Qiu, L.; Zhou, T.; Feng, M.; Hu, Q.; Zeng, B.; Li, Z.; Sun, Q.; Wu, Y.; et al. Seamless Genetic Conversion of SMN2 to SMN1 via CRISPR/Cpf1 and Single-Stranded Oligodeoxynucleotides in Spinal Muscular Atrophy Patient-Specific Induced Pluripotent Stem Cells. Hum. Gene Ther. 2018, 29, 1252–1263. [Google Scholar] [CrossRef]
- Stemmer, M.; Thumberger, T.; Del Sol Keyer, M.; Wittbrodt, J.; Mateo, J.L. CCTop: An Intuitive, Flexible and Reliable CRISPR/Cas9 Target Prediction Tool. PLoS ONE 2015, 10, e0124633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pang, J.; Wu, Y.; Li, Z.; Hu, Z.; Wang, X.; Hu, X.; Wang, X.; Liu, X.; Zhou, M.; Liu, B.; et al. Targeting of the human F8 at the multicopy rDNA locus in Hemophilia A patient-derived iPSCs using TALENickases. Biochem. Biophys. Res. Commun. 2016, 472, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Zhou, M.; Wu, Y.; Li, Z.; Liu, X.; Wu, L.; Liang, D. ssODN-Mediated In-Frame Deletion with CRISPR/Cas9 Restores FVIII Function in Hemophilia A-Patient-Derived iPSCs and ECs. Mol. Ther. Nucleic Acids 2019, 17, 198–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Zhou, M.; Wang, Z.; Wu, L.; Hu, Z.; Liang, D. Ectopic Expression of FVIII in HPCs and MSCs Derived from hiPSCs with Site-Specific Integration of ITGA2B Promoter-Driven BDDF8 Gene in Hemophilia A. Int. J. Mol. Sci. 2022, 23, 623. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, T.; Dougan, S.K.; Truttmann, M.C.; Bilate, A.M.; Ingram, J.R.; Ploegh, H.L. Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nat. Biotechnol. 2015, 33, 538–542. [Google Scholar] [CrossRef]
- Yoshimi, K.; Oka, Y.; Miyasaka, Y.; Kotani, Y.; Yasumura, M.; Uno, Y.; Hattori, K.; Tanigawa, A.; Sato, M.; Oya, M.; et al. Combi-CRISPR: Combination of NHEJ and HDR provides efficient and precise plasmid-based knock-ins in mice and rats. Hum. Genet. 2021, 140, 277–287. [Google Scholar] [CrossRef]
- Lei, Z.N.; Teng, Q.X.; Wu, Z.X.; Ping, F.F.; Song, P.; Wurpel, J.N.D.; Chen, Z.S. Overcoming multidrug resistance by knockout of ABCB1 gene using CRISPR/Cas9 system in SW620/Ad300 colorectal cancer cells. MedComm 2021, 2, 765–777. [Google Scholar] [CrossRef]
- Wang, T.; Zhang, C.; Zhang, H.; Zhu, H. CRISPR/Cas9-Mediated Gene Editing Revolutionizes the Improvement of Horticulture Food Crops. J. Agric. Food Chem. 2021, 69, 13260–13269. [Google Scholar] [CrossRef]
- Lin, X.; Li, J.J.; Qian, W.J.; Zhang, Q.J.; Wang, Z.F.; Lu, Y.Q.; Dong, E.L.; He, J.; Wang, N.; Ma, L.X.; et al. Modeling the differential phenotypes of spinal muscular atrophy with high-yield generation of motor neurons from human induced pluripotent stem cells. Oncotarget 2017, 8, 42030–42042. [Google Scholar] [CrossRef] [Green Version]
- Chang, T.; Zheng, W.; Tsark, W.; Bates, S.; Huang, H.; Lin, R.J.; Yee, J.K. Brief report: Phenotypic rescue of induced pluripotent stem cell-derived motoneurons of a spinal muscular atrophy patient. Stem Cells 2011, 29, 2090–2093. [Google Scholar] [CrossRef]
- Cheung, A.K.; Hurley, B.; Kerrigan, R.; Shu, L.; Chin, D.N.; Shen, Y.; O’Brien, G.; Sung, M.J.; Hou, Y.; Axford, J.; et al. Discovery of Small Molecule Splicing Modulators of Survival Motor Neuron-2 (SMN2) for the Treatment of Spinal Muscular Atrophy (SMA). J. Med. Chem. 2018, 61, 11021–11036. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, S. Risdiplam: First Approval. Drugs 2020, 80, 1853–1858. [Google Scholar] [CrossRef] [PubMed]
- Keil, J.M.; Seo, J.; Howell, M.D.; Hsu, W.H.; Singh, R.N.; DiDonato, C.J. A short antisense oligonucleotide ameliorates symptoms of severe mouse models of spinal muscular atrophy. Mol. Ther. Nucleic Acids 2014, 3, e174. [Google Scholar] [CrossRef]
- Hammond, S.M.; Hazell, G.; Shabanpoor, F.; Saleh, A.F.; Bowerman, M.; Sleigh, J.N.; Meijboom, K.E.; Zhou, H.; Muntoni, F.; Talbot, K.; et al. Systemic peptide-mediated oligonucleotide therapy improves long-term survival in spinal muscular atrophy. Proc. Natl. Acad. Sci. USA 2016, 113, 10962–10967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Ydewalle, C.; Ramos, D.M.; Pyles, N.J.; Ng, S.Y.; Gorz, M.; Pilato, C.M.; Ling, K.; Kong, L.; Ward, A.J.; Rubin, L.L.; et al. The Antisense Transcript SMN-AS1 Regulates SMN Expression and Is a Novel Therapeutic Target for Spinal Muscular Atrophy. Neuron 2017, 93, 66–79. [Google Scholar] [CrossRef] [Green Version]
- Passini, M.A.; Bu, J.; Richards, A.M.; Treleaven, C.M.; Sullivan, J.A.; O’Riordan, C.R.; Scaria, A.; Kells, A.P.; Samaranch, L.; San Sebastian, W.; et al. Translational fidelity of intrathecal delivery of self-complementary AAV9-survival motor neuron 1 for spinal muscular atrophy. Hum. Gene Ther. 2014, 25, 619–630. [Google Scholar] [CrossRef] [PubMed]
- Rashnonejad, A.; Amini Chermahini, G.; Gunduz, C.; Onay, H.; Aykut, A.; Durmaz, B.; Baka, M.; Su, Q.; Gao, G.; Ozkinay, F. Fetal Gene Therapy Using a Single Injection of Recombinant AAV9 Rescued SMA Phenotype in Mice. Mol. Ther. 2019, 27, 2123–2133. [Google Scholar] [CrossRef]
- Mercuri, E.; Darras, B.T.; Chiriboga, C.A.; Day, J.W.; Campbell, C.; Connolly, A.M.; Iannaccone, S.T.; Kirschner, J.; Kuntz, N.L.; Saito, K.; et al. Nusinersen versus Sham Control in Later-Onset Spinal Muscular Atrophy. N. Engl. J. Med. 2018, 378, 625–635. [Google Scholar] [CrossRef]
- Hoy, S.M. Onasemnogene Abeparvovec: First Global Approval. Drugs 2019, 79, 1255–1262. [Google Scholar] [CrossRef]
- Silva, A.C.; Lobo, D.D.; Martins, I.M.; Lopes, S.M.; Henriques, C.; Duarte, S.P.; Dodart, J.C.; Nobre, R.J.; Pereira de Almeida, L. Antisense oligonucleotide therapeutics in neurodegenerative diseases: The case of polyglutamine disorders. Brain 2020, 143, 407–429. [Google Scholar] [CrossRef]
- Kelaini, S.; Chan, C.; Cornelius, V.A.; Margariti, A. RNA-Binding Proteins Hold Key Roles in Function, Dysfunction, and Disease. Biology 2021, 10, 366. [Google Scholar] [CrossRef]
- Aguti, S.; Malerba, A.; Zhou, H. The progress of AAV-mediated gene therapy in neuromuscular disorders. Expert Opin. Biol. Ther. 2018, 18, 681–693. [Google Scholar] [CrossRef]
- Hinderer, C.; Katz, N.; Buza, E.L.; Dyer, C.; Goode, T.; Bell, P.; Richman, L.K.; Wilson, J.M. Severe Toxicity in Nonhuman Primates and Piglets Following High-Dose Intravenous Administration of an Adeno-Associated Virus Vector Expressing Human SMN. Hum. Gene Ther. 2018, 29, 285–298. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, M.; Tang, S.; Duan, N.; Xie, M.; Li, Z.; Feng, M.; Wu, L.; Hu, Z.; Liang, D. Targeted-Deletion of a Tiny Sequence via Prime Editing to Restore SMN Expression. Int. J. Mol. Sci. 2022, 23, 7941. https://doi.org/10.3390/ijms23147941
Zhou M, Tang S, Duan N, Xie M, Li Z, Feng M, Wu L, Hu Z, Liang D. Targeted-Deletion of a Tiny Sequence via Prime Editing to Restore SMN Expression. International Journal of Molecular Sciences. 2022; 23(14):7941. https://doi.org/10.3390/ijms23147941
Chicago/Turabian StyleZhou, Miaojin, Shuqing Tang, Nannan Duan, Mi Xie, Zhuo Li, Mai Feng, Lingqian Wu, Zhiqing Hu, and Desheng Liang. 2022. "Targeted-Deletion of a Tiny Sequence via Prime Editing to Restore SMN Expression" International Journal of Molecular Sciences 23, no. 14: 7941. https://doi.org/10.3390/ijms23147941