
Leptin in Osteoarthritis and Rheumatoid Arthritis: Player or Bystander?

 , ,
, ,  , , , ,
, , , ,
Abstract
:1. Introduction
2. Leptin Physiology
3. Leptin Receptors
Leptin Receptor Signaling
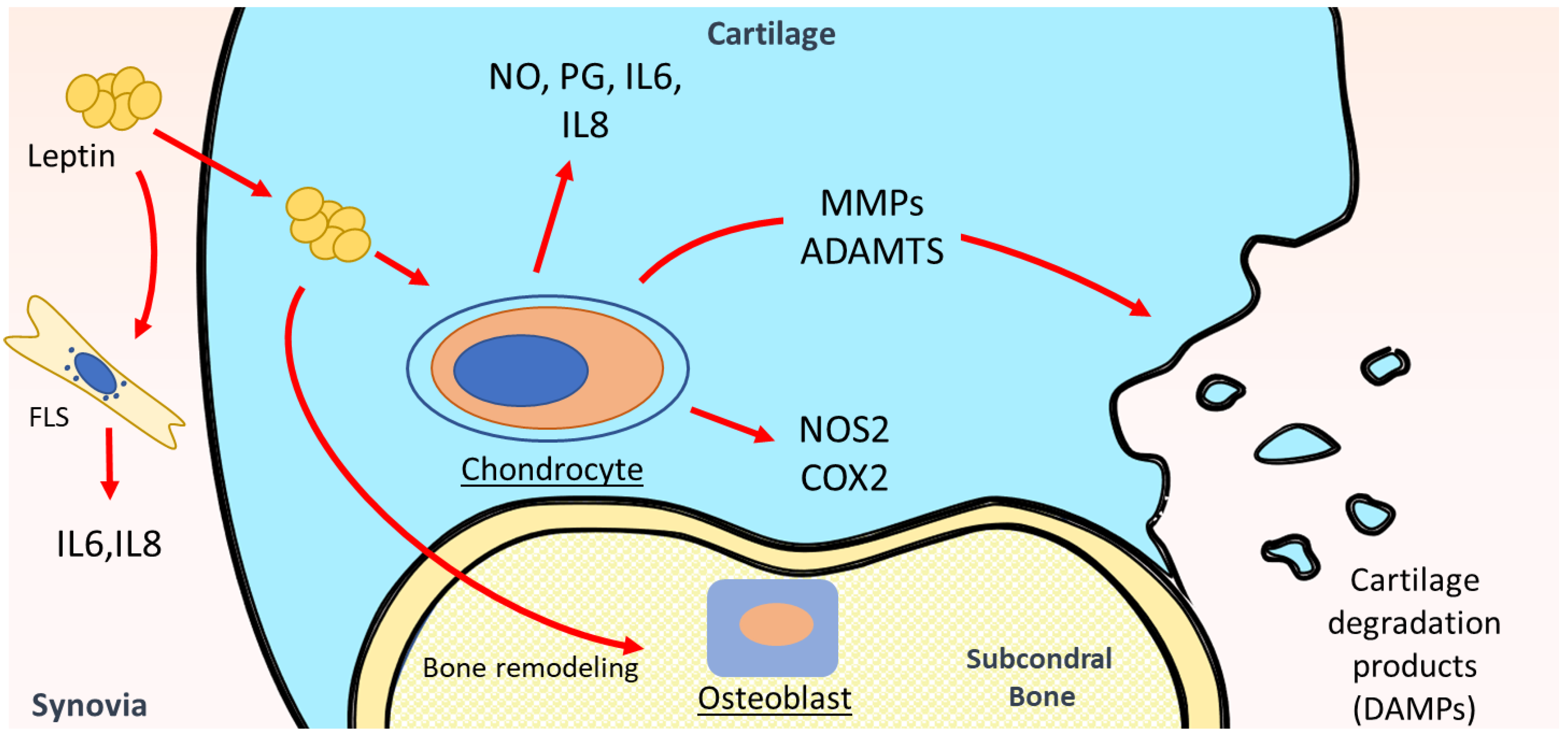
4. Leptin and Osteoarthritis
4.1. Leptin in Serum and SF of OA Patients

{kind=link}
{kind=link}
| Author | Sample | Patients | Leptin Levels | Relation with the Disease |
|---|---|---|---|---|
| Dumond et al. (2003) [51] | Synovial fluid | 20 OA, 2H | Detected in SF and correlated with BMI | Yes |
| Simopoulou et al. (2007) [46] | SF/Serum | 17 OA, 5H | Significantly much higher in OA patients | Yes |
| Ku et al. (2009) [49] | SF | 42 OA, 10H | Significantly higher in OA patients | Yes |
| Min et al. (2020) [45] | Serum | 148 OA, 101H | Significantly higher in OA patients | Yes |
| Lübekke et al. (2013) [48] | SF from hip and knee | 219 | High leptin levels in SF were correlated with joint pain | Yes |
| Xiong et al. (2018) [57] | SF | 13 OA, 7H | Significantly higher in OA patients than all the other groups | Yes |
| Kroon et al. (2019) [58] | Serum from hand and knee | 6408 | Leptin levels were positively associated with OA | Yes |
| Massengale et al. (2012) [55] | Serum from hand | 2477 | No significant difference between symptomatic, asymptomatic, and no hand OA | No |
| Yusuf et al. (2011) [54] | Serum from hand | 248 | Not associated with hand OA progression | No |
| De Boer et al. (2012) [14] | Serum from knee | 172 OA, 132H | Significant difference between OA patients and control | Yes |
| Morales Abaunza et al. (2020) [56] | Serum from hand | 44 OA, 30H | Significantly higher in patients with hand OA | Yes |
| Bas et al. (2014) [47] | Serum and SF from hip and knee | 112 hip OA, 92 knee OA | Higher in knee OA than in hip OA joints | Yes |
4.2. Leptin in Metabolism and Inflammation of Articular Cartilage
4.3. Leptin in Other Intraarticular Tissues of OA
4.4. Leptin and Obesity in OA
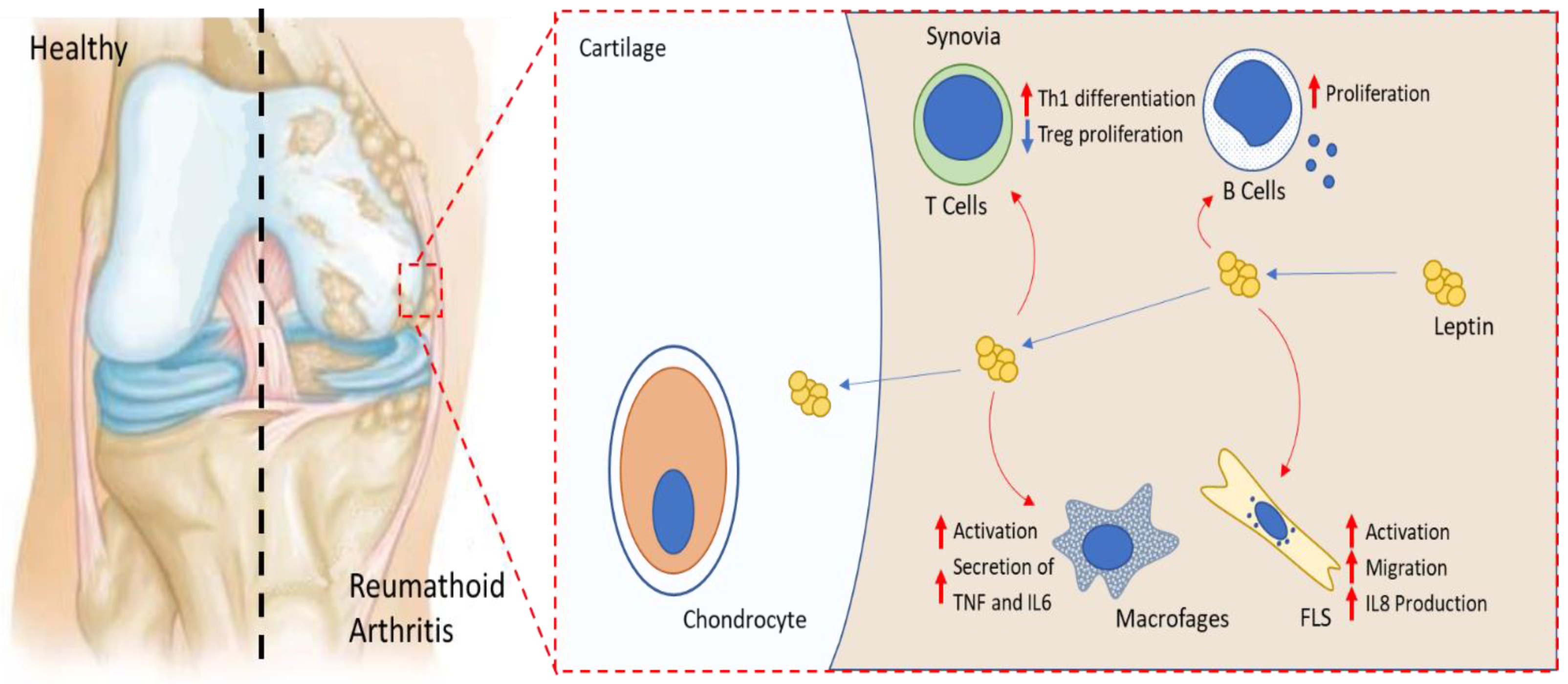
5. Leptin and Rheumatoid Arthritis
5.1. SF and Serum Leptin Levels in RA Patients
| Author | Sample | Patients | Leptin Levels | Evidence of Relation with the Disease |
|---|---|---|---|---|
| Petra et al. (2020) [99] | Serum | 84 RA, 44H | Significantly higher in RA patients than in controls | Yes |
| Lee et al. (2007) [86] | Serum | 50 RA | RA patients had higher mean leptin levels | Yes |
| Bokarewa et al. (2003) [84] | Serum and SF | 76 RA, 34H | Higher in RA patients | Yes |
| Rho et al. (2009) [93] | Serum | 167 RA, 91H | Significantly higher in RA patients | Yes |
| Abdalla et al. (2014) [87] | Serum | 60 RA, 30H | Significantly higher in RA patients | Yes |
| Olama et al. (2012) [92] | Serum and SF | 40 RA, 30H | Increased in RA patients | Yes |
| Seven et al. (2009) [85] | SF and serum | 20 RA, 25H | Significantly higher in RA patients | Yes |
| Hizmetli et al. (2005) [88] | SF and plasma | 41 RA, 25H | No significant difference between RA patients and healthy controls | No |
| Oner et al. (2015) [89] | Serum | 106 RA, 52H | No significant difference between RA patients and healthy controls | No |
| Otero et al. (2006) [100] | Plasma | 31 RA, 18H | Markedly increased in RA patients | Yes |
| Allam et al. (2012) [101] | Serum | 37 RA, 34H | Higher in RA patients | Yes |
| Anders et al. (1999) [91] | Serum | 58 RA, 16H | No significant difference between RA patients and healthy controls | No |
| Popa et al. (2005) [90] | Plasma | 31 RA, 18H | No significant difference between RA patients and healthy controls | No |
| Wislowska, M. et al. (2007) [97] | Serum | 30 RA, 30OA | No difference between OA and RA | No |
| Chihara et al. (2020) [12] | Serum | 136 RA, 78H | Higher in RA patients | No |
| Toussirot et al. (2013) [102] | Serum | 30 RA, 51H | No difference between RA patients and healthy controls | No |
5.2. Leptin in Animal Models of RA
5.3. Leptin and Major Effector Cells in RA
6. Leptin as a Potential Therapy in OA and RA
7. Conclusions
Funding
Conflicts of Interest
References
- Carrión, M.; Frommer, K.W.; Pérez-García, S.; Müller-Ladner, U.; Gomariz, R.P.; Neumann, E. The adipokine network in rheumatic joint diseases. Int. J. Mol. Sci. 2019, 20, 4091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martel-Pelletier, J.; Barr, A.J.; Cicuttini, F.M.; Conaghan, P.G.; Cooper, C.; Goldring, M.B.; Goldring, S.R.; Jones, G.; Teichtahl, A.J.; Pelletier, J.P. Osteoarthritis. Nat. Rev. Dis. Prim. 2016, 2, 16072 . [Google Scholar] [CrossRef] [Green Version]
- Hunter, D.J.; Bierma-Zeinstra, S. Osteoarthritis. Lancet 2019, 393, 1745–1759. [Google Scholar] [CrossRef]
- Smolen, J.S.; Aletaha, D.; Barton, A.; Burmester, G.R.; Emery, P.; Firestein, G.S.; Kavanaugh, A.; McInnes, I.B.; Solomon, D.H.; Strand, V.; et al. Rheumatoid arthritis. Nat. Rev. Dis. Prim. 2018, 4, 18001. [Google Scholar] [CrossRef]
- Firestein, G.S.; McInnes, I.B. Immunopathogenesis of Rheumatoid Arthritis. Immunity 2017, 46, 183–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldring, M.B.; Otero, M. Inflammation in osteoarthritis. Curr. Opin. Rheumatol. 2011, 23, 471–478. [Google Scholar] [CrossRef] [PubMed]
- King, L.K.; March, L.; Anandacoomarasamy, A. Obesity & osteoarthritis. Indian J. Med. Res. 2013, 138, 185–193. [Google Scholar] [PubMed]
- Abuhelwa, A.Y.; Hopkins, A.M.; Sorich, M.J.; Proudman, S.; Foster, D.J.R.; Wiese, M.D. Association between obesity and remission in rheumatoid arthritis patients treated with disease-modifying anti-rheumatic drugs. Sci. Rep. 2020, 10, 18634. [Google Scholar] [CrossRef]
- Francisco, V.; Pérez, T.; Pino, J.; López, V.; Franco, E.; Alonso, A.; Gonzalez-Gay, M.A.; Mera, A.; Lago, F.; Gómez, R.; et al. Biomechanics, obesity, and osteoarthritis. The role of adipokines: When the levee breaks. J. Orthop. Res. 2018, 36, 594–604. [Google Scholar] [CrossRef] [Green Version]
- Smekal, A.; Vaclavik, J. Adipokines and cardiovascular disease: A comprehensive review. Biomed. Pap. 2017, 161, 31–40. [Google Scholar] [CrossRef] [Green Version]
- Lago, F.; Dieguez, C.; Gómez-Reino, J.; Gualillo, O. The emerging role of adipokines as mediators of inflammation and immune responses. Cytokine Growth Factor Rev. 2007, 18, 313–325. [Google Scholar] [CrossRef]
- Chihara, K.; Hattori, N.; Ichikawa, N.; Matsuda, T.; Saito, T. Re-evaluation of serum leptin and adiponectin concentrations normalized by body fat mass in patients with rheumatoid arthritis. Sci. Rep. 2020, 10, 15932. [Google Scholar] [CrossRef]
- Neumann, E.; Frommer, K.W.; Vasile, M.; Müller-Ladner, U. Adipocytokines as driving forces in rheumatoid arthritis and related inflammatory diseases? Arthritis Rheum. 2011, 63, 1159–1169. [Google Scholar] [CrossRef]
- de Boer, T.N.; van Spil, W.E.; Huisman, A.M.; Polak, A.A.; Bijlsma, J.W.J.; Lafeber, F.P.J.G.; Mastbergen, S.C. Serum adipokines in osteoarthritis; comparison with controls and relationship with local parameters of synovial inflammation and cartilage damage. Osteoarthr. Cartil. 2012, 20, 846–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, P.F.; Bao, J.P.; Wu, L.D. The emerging role of adipokines in osteoarthritis: A narrative review. Mol. Biol. Rep. 2011, 38, 873–878. [Google Scholar] [CrossRef] [PubMed]
- Wada, N.; Hirako, S.; Takenoya, F.; Kageyama, H.; Okabe, M.; Shioda, S. Leptin and its receptors. J. Chem. Neuroanat. 2014, 61, 191–199. [Google Scholar] [CrossRef]
- Kelesidis, T.; Kelesidis, I.; Chou, S.; Mantzoros, C.S. Narrative review: The role of leptin in human physiology: Emerging clinical applications. Ann. Intern. Med. 2010, 152, 93–100. [Google Scholar] [CrossRef]
- Conde, J.; Scotece, M.; Gómez, R.; Gómez-Reino, J.J.; Lago, F.; Gualillo, O. At the crossroad between immunity and metabolism: Focus on leptin. Expert Rev. Clin. Immunol. 2010, 6, 801–808. [Google Scholar] [CrossRef]
- Zhang, F.; Chen, Y.; Heiman, M.; DiMarchi, R. Leptin: Structure, Function and Biology. Vitam. Horm. 2005, 71, 345–372. [Google Scholar] [CrossRef]
- Margetic, S.; Gazzola, C.; Pegg, G.G.; Hill, R.A. Leptin: A review of its peripheral actions and interactions. Int. J. Obes. 2002, 26, 1407–1433. [Google Scholar] [CrossRef] [Green Version]
- Hoogard, N.; Hunter, L.; Duncan, J.S.; Williams, L.M.; Trayhurn, P.; Mercer, J.G. Leptin and leptin receptor mRNA and protein expression in the murine fetus and placenta. Proc. Natl. Acad. Sci. USA 1997, 94, 11073–11078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bado, A.; Levasseur, S.; Attoub, S.; Kermorgant, S.; Laigneau, J.P.; Bortoluzzi, M.N.; Moizo, L.; Lehy, T.; Guerre-Millo, M.; Marchand-Brustel, L.; et al. The stomach is a source of leptin. Nature 1998, 394, 790–793. [Google Scholar] [CrossRef]
- Mantzoros, C.S.; Magkos, F.; Brinkoetter, M.; Sienkiewicz, E.; Dardeno, T.A.; Kim, S.Y.; Hamnvik, O.P.R.; Koniaris, A. Leptin in human physiology and pathophysiology. Am. J. Physiol.-Endocrinol. Metab. 2011, 301, E567–E584. [Google Scholar] [CrossRef] [PubMed]
- Masuzaki, H.; Ogawa, Y.; Hosoda, K.; Miyawaki, T.; Hanaoka, I.; Hiraoka, J.; Yasuno, A.; Nishimura, H.; Yoshimasa, Y.; Nishi, S.; et al. Glucocorticoid regulation of leptin synthesis and secretion in humans: Elevated plasma leptin levels in Cushing’s syndrome. J. Clin. Endocrinol. Metab. 1997, 82, 2542–2547. [Google Scholar] [CrossRef] [PubMed]
- Kolaczynski, J.W.; Nyce, M.R.; Considine, R.V.; Boden, G.; Nolan, J.J.; Henry, R.; Mudaliar, S.R.; Olefsky, J.; Caro, J.F. Acute and chronic effects of insulin on leptin production in humans: Studies in vivo and in vitro. Diabetes 1996, 45, 699–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wauters, M.; Considine, R.V.; Van Gaal, L.F. Human leptin: From an adipocyte hormone to an endocrine mediator. Eur. J. Endocrinol. 2000, 143, 293–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, H.; Shimomura, Y.; Nakanishi, Y.; Futawatari, T.; Ohtani, K.; Sato, N.; Mori, M. Estrogen increases in vivo leptin production in rats and human subjects. J. Endocrinol. 1997, 154, 285–292. [Google Scholar] [CrossRef]
- Fain, J.N.; Leffler, C.W.; Cowan, J.; Buffington, C.; Pouncey, L.; Bahouth, S.W. Stimulation of leptin release by arachidonic acid and prostaglandin E2 in adipose tissue from obese humans. Metabolism 2001, 50, 921–928. [Google Scholar] [CrossRef]
- Fain, J.N.; Bahouth, S.W. Regulation of leptin release by mammalian adipose tissue. Biochem. Biophys. Res. Commun. 2000, 274, 571–575. [Google Scholar] [CrossRef]
- Ntambi, M.J.; Kim, Y.-C. Symposium: Adipocyte Function, Differentiation and Metabolism Regulation of Leptin Production in Humans. J. Nutr. 2000, 130, 3127–3131. [Google Scholar]
- Bakshi, A.; Singh, R.; Rai, U. Trajectory of leptin and leptin receptor in vertebrates: Structure, function and their regulation. Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 2021, 257, 110652. [Google Scholar] [CrossRef]
- La Cava, A. Leptin in inflammation and autoimmunity. Cytokine 2017, 98, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Bruno, A.; Conus, S.; Schmid, I.; Simon, H.-U. Apoptotic Pathways Are Inhibited by Leptin Receptor Activation in Neutrophils. J. Immunol. 2005, 174, 8090–8096. [Google Scholar] [CrossRef] [Green Version]
- Hutcheson, J. Adipokines influence the inflammatory balance in autoimmunity. Cytokine 2015, 75, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Hausman, G.J.; Barb, C.R.; Lents, C.A. Leptin and reproductive function. Biochimie 2012, 94, 2075–2081. [Google Scholar] [CrossRef] [PubMed]
- Chehab, F.F.; Lim, M.E.; Lu, R. Correction of the sterility defect in homozygous obese female mice by treatment with the human recombinant leptin. Nat. Genet. 1996, 12, 318–320. [Google Scholar] [CrossRef]
- Tartaglia, L.A. The leptin receptor. J. Biol. Chem. 1997, 272, 6093–6096. [Google Scholar] [CrossRef] [Green Version]
- Gorska, E.; Popko, K.; Stelmaszczyk-Emmel, A.; Ciepiela, O.; Kucharska, A.; Wasik, M. Leptin receptors. Eur. J. Med. Res. 2010, 15, 50. [Google Scholar] [CrossRef] [Green Version]
- Gualillo, O.; Eiras, S.; White, D.W.; Diéguez, C.; Casanueva, F.F. Leptin promotes the tyrosine phosphorylation of SHC proteins and SHC association with GRB2. Mol. Cell. Endocrinol. 2002, 190, 83–89. [Google Scholar] [CrossRef]
- Zabeau, L.; Defeau, D.; Iserentant, H.; Vandekerckhove, J.; Peelman, F.; Tavernier, J. Leptin receptor activation depends on critical cysteine residues in its fibronectin type III subdomains. J. Biol. Chem. 2005, 280, 22632–22640. [Google Scholar] [CrossRef] [Green Version]
- Münzberg, H.; Morrison, C.D. Structure, production and signaling of leptin Heike. Metabolism 2014, 64, 13–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banks, A.S.; Davis, S.M.; Bates, S.H.; Myers, M.G. Activation of downstream signals by the long form of the leptin receptor. J. Biol. Chem. 2000, 275, 14563–14572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.K.; Ahima, R.S. Leptin signaling. F1000Prime Rep. 2014, 6, 73. [Google Scholar] [CrossRef] [PubMed]
- Scanzello, C.R.; Goldring, S.R. The role of synovitis in osteoarthritis pathogenesis. Bone 2012, 51, 249–257. [Google Scholar] [CrossRef] [Green Version]
- Min, S.; Shi, T.; Han, X.; Chen, D.; Xu, Z.; Shi, D.; Teng, H.; Jiang, Q. Serum levels of leptin, osteopontin, and sclerostin in patients with and without knee osteoarthritis. Clin. Rheumatol. 2021, 40, 287–294. [Google Scholar] [CrossRef]
- Simopoulou, T.; Malizos, K.N.; Iliopoulos, D.; Stefanou, N.; Papatheodorou, L.; Ioannou, M.; Tsezou, A. Differential expression of leptin and leptin’s receptor isoform (Ob-Rb) mRNA between advanced and minimally affected osteoarthritic cartilage; effect on cartilage metabolism. Osteoarthr. Cartil. 2007, 15, 872–883. [Google Scholar] [CrossRef] [Green Version]
- Bas, S.; Finckh, A.; Puskas, G.J.; Suva, D.; Hoffmeyer, P.; Gabay, C.; Lübbeke, A. Adipokines correlate with pain in lower limb osteoarthritis: Different associations in hip and knee. Int. Orthop. 2014, 38, 2577–2583. [Google Scholar] [CrossRef]
- Lübbeke, A.; Finckh, A.; Puskas, G.J.; Suva, D.; Lädermann, A.; Bas, S.; Fritschy, D.; Gabay, C.; Hoffmeyer, P. Do synovial leptin levels correlate with pain in end stage arthritis? Int. Orthop. 2013, 37, 2071–2079. [Google Scholar] [CrossRef] [Green Version]
- Ku, J.H.; Lee, C.K.; Joo, B.S.; An, B.M.; Choi, S.H.; Wang, T.H.; Cho, H.L. Correlation of synovial fluid leptin concentrations with the severity of osteoarthritis. Clin. Rheumatol. 2009, 28, 1431–1435. [Google Scholar] [CrossRef]
- Lambova, S.N.; Batsalova, T.; Moten, D.; Stoyanova, S.; Georgieva, E.; Belenska-Todorova, L.; Kolchakova, D.; Dzhambazov, B. Serum Leptin and Resistin Levels in Knee Osteoarthritis—Clinical and Radiologic Links: Towards Precise Definition of Metabolic Type Knee Osteoarthritis. Biomedicines 2021, 9, 1019. [Google Scholar] [CrossRef]
- Dumond, H.; Presle, N.; Terlain, B.; Mainard, D.; Loeuille, D.; Netter, P.; Pottie, P. Evidence for a Key Role of Leptin in Osteoarthritis. Arthritis Rheum. 2003, 48, 3118–3129. [Google Scholar] [CrossRef] [PubMed]
- Martel-Pelletier, J.; Raynauld, J.P.; Dorais, M.; Abram, F.; Pelletier, J.P. The levels of the adipokines adipsin and leptin are associated with knee osteoarthritis progression as assessed by MRI and incidence of total knee replacement in symptomatic osteoarthritis patients: A post hoc analysis. Rheumatology 2016, 55, 680–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stannus, O.P.; Cao, Y.; Antony, B.; Blizzard, L.; Cicuttini, F.; Jones, G.; Ding, C. Cross-sectional and longitudinal associations between circulating leptin and knee cartilage thickness in older adults. Ann. Rheum. Dis. 2015, 74, 82–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yusuf, E.; Ioan-Facsinay, A.; Bijsterbosch, J.; Klein-Wieringa, I.; Kwekkeboom, J.; Slagboom, P.E.; Huizinga, T.W.J.; Kloppenburg, M. Association between leptin, adiponectin and resistin and long-term progression of hand osteoarthritis. Ann. Rheum. Dis. 2011, 70, 1282–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massengale, M.; Reichmann, W.M.; Losina, E.; Solomon, D.H.; Katz, J.N. The relationship between hand osteoarthritis and serum leptin concentration in participants of the Third National Health and Nutrition Examination Survey. Arthritis Res. Ther. 2012, 14, R132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morales Abaunza, R.A.; Rojas, Á.P.; Rojas, C.; Motta, O.; Atuesta, J.; Alzate, J.P.; Rondón Herrera, F. Levels of serum leptin in patients with primary hand osteoarthritis. Rev. Colomb. Reumatol. 2020, 27, 20–25. [Google Scholar] [CrossRef]
- Xiong, H.; Li, W.; Ke, J.; Fang, W.; Li, B.; Wei, L. Leptin Levels in the Synovial Fluid of Patients With Temporomandibular Disorders. J. Oral Maxillofac. Surg. 2019, 77, 493–498. [Google Scholar] [CrossRef]
- Kroon, F.P.B.; Veenbrink, A.I.; de Mutsert, R.; Visser, A.W.; van Dijk, K.W.; le Cessie, S.; Rosendaal, F.R.; Kloppenburg, M. The role of leptin and adiponectin as mediators in the relationship between adiposity and hand and knee osteoarthritis. Osteoarthr. Cartil. 2019, 27, 1761–1767. [Google Scholar] [CrossRef]
- Cordero-Barreal, A.; González-Rodríguez, M.; Ruiz-Fernández, C.; Eldjoudi, D.A.; Abdelhafez, Y.R.F.; Lago, F.; Conde, J.; Gómez, R.; González-Gay, M.A.; Mobasheri, A.; et al. An update on the role of leptin in the immuno-metabolism of cartilage. Int. J. Mol. Sci. 2021, 22, 2411. [Google Scholar] [CrossRef]
- Hui, W.; Litherland, G.J.; Elias, M.S.; Kitson, G.I.; Cawston, T.E.; Rowan, A.D.; Young, D.A. Leptin produced by joint white adipose tissue induces cartilage degradation via upregulation and activation of matrix metalloproteinases. Ann. Rheum. Dis. 2012, 71, 455–462. [Google Scholar] [CrossRef]
- Koskinen, A.; Vuolteenaho, K.; Nieminen, R.; Moilanen, T.; Moilanen, E. Leptin enhances MMP-1, MMP-3 and MMP-13 production in human osteoarthritic cartilage and correlates with MMP-1 and MMP-3 in synovial fluid from oa patients. Clin. Exp. Rheumatol. 2011, 29, 57–64. [Google Scholar] [PubMed]
- Vuolteenaho, K.; Moilanen, E.; Koskinen, A.; Kukkonen, M.; Nieminen, R.; Pivrinta, U.; Moilanen, T. Leptin enhances synthesis of proinflammatory mediators in human osteoarthritic cartilage-Mediator role of NO in leptin-induced PGE 2, IL-6, and IL-8 Production. Mediat. Inflamm. 2009, 2009, 345838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otero, M.; Gomez Reino, J.J.; Gualillo, O. Synergistic induction of nitric oxide synthase type II: In vitro effect of leptin and interferon-γ in human chondrocytes and ATDC5 chondrogenic cells. Arthritis Rheum. 2003, 48, 404–409. [Google Scholar] [CrossRef] [PubMed]
- Jayakumar, T.; Bhavan, P.S.; Sheu, J.R. Molecular targets of natural products for chondroprotection in destructive joint diseases. Int. J. Mol. Sci. 2020, 21, 4931. [Google Scholar] [CrossRef]
- Otero, M.; Lago, R.; Lago, F.; Reino, J.J.G.; Gualillo, O. Signalling pathway involved in nitric oxide synthase type II activation in chondrocytes: Synergistic effect of leptin with interleukin-1. Arthritis Res. Ther. 2005, 7, 581–591. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Huang, P.; Li, G.; Zhendong, L.; Hu, G.; Xu, Q. Activation of the leptin pathway by high expression of the long form of the leptin receptor (Ob-Rb) accelerates chondrocyte senescence in osteoarthritis. Bone Jt. Res. 2019, 8, 425–436. [Google Scholar] [CrossRef]
- Samuels, J.; Krasnokutsky, S.; Abramson, S.B. Osteoarthritis: A tale of three tissues. Bull. NYU Hosp. Jt. Dis. 2008, 66, 244–250. [Google Scholar]
- Yang, W.H.; Liu, S.C.; Tsai, C.H.; Fong, Y.C.; Wang, S.J.; Chang, Y.S.; Tang, C.H. Leptin Induces IL-6 Expression through OBRl Receptor Signaling Pathway in Human Synovial Fibroblasts. PLoS ONE 2013, 8, 2–11. [Google Scholar] [CrossRef] [Green Version]
- Tong, K.M.; Shieh, D.C.; Chen, C.P.; Tzeng, C.Y.; Wang, S.P.; Huang, K.C.; Chiu, Y.C.; Fong, Y.C.; Tang, C.H. Leptin induces IL-8 expression via leptin receptor, IRS-1, PI3K, Akt cascade and promotion of NF-κB/p300 binding in human synovial fibroblasts. Cell. Signal. 2008, 20, 1478–1488. [Google Scholar] [CrossRef]
- Pearson, M.J.; Herndler-Brandstetter, D.; Tariq, M.A.; Nicholson, T.A.; Philp, A.M.; Smith, H.L.; Davis, E.T.; Jones, S.W.; Lord, J.M. IL-6 secretion in osteoarthritis patients is mediated by chondrocyte-synovial fibroblast cross-talk and is enhanced by obesity. Sci. Rep. 2017, 7, 3451. [Google Scholar] [CrossRef] [Green Version]
- Chou, C.H.; Wu, C.C.; Song, I.W.; Chuang, H.P.; Lu, L.S.; Chang, J.H.; Kuo, S.Y.; Lee, C.H.; Wu, J.Y.; Chen, Y.T.; et al. Genome-wide expression profiles of subchondral bone in osteoarthritis. Arthritis Res. Ther. 2013, 15, R190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mutabaruka, M.S.; Aoulad Aissa, M.; Delalandre, A.; Lavigne, M.; Lajeunesse, D. Local leptin production in osteoarthritis subchondral osteoblasts may be responsible for their abnormal phenotypic expression. Arthritis Res. Ther. 2010, 12, R20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Presle, N.; Pottie, P.; Dumond, H.; Guillaume, C.; Lapicque, F.; Pallu, S.; Mainard, D.; Netter, P.; Terlain, B. Differential distribution of adipokines between serum and synovial fluid in patients with osteoarthritis. Contribution of joint tissues to their articular production. Osteoarthr. Cartil. 2006, 14, 690–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conde, J.; Scotece, M.; López, V.; Abella, V.; Hermida, M.; Pino, J.; Lago, F.; Gómez-Reino, J.J.; Gualillo, O. Differential expression of adipokines in infrapatellar fat pad (IPFP) and synovium of osteoarthritis patients and healthy individuals. Ann. Rheum. Dis. 2014, 73, 631–633. [Google Scholar] [CrossRef]
- Liu, B.; Gao, Y.H.; Dong, N.; Zhao, C.W.; Huang, Y.F.; Liu, J.G.; Qi, X. Differential expression of adipokines in the synovium and infrapatellar fat pad of osteoarthritis patients with and without metabolic syndrome. Connect. Tissue Res. 2019, 60, 611–618. [Google Scholar] [CrossRef]
- Gross, J.-B.; Guillaume, C.; Gegout-Pottie, P.; Reboul, P.; Jouzeau, J.-Y.; Mainard, D.; Presle, N. The infrapatellar fat pad induces inflammatory and degradative effects in articular cells but not through leptin or adiponectin. Clin. Exp. Rheumatol. 2016, 35, 53–60. [Google Scholar]
- Raud, B.; Gay, C.; Guiguet-Auclair, C.; Bonnin, A.; Gerbaud, L.; Pereira, B.; Duclos, M.; Boirie, Y.; Coudeyre, E. Level of obesity is directly associated with the clinical and functional consequences of knee osteoarthritis. Sci. Rep. 2020, 10, 3601. [Google Scholar] [CrossRef]
- Griffin, T.M.; Huebner, J.L.; Kraus, V.B.; Guilak, F. Extreme obesity due to impaired leptin signaling in mice does not cause knee osteoarthritis. Arthritis Rheum. 2009, 60, 2935–2944. [Google Scholar] [CrossRef] [Green Version]
- Jiang, M.; He, J.; Sun, Y.; Dong, X.; Yao, J.; Gu, H.; Liu, L. Leptin Induced TLR4 Expression via the JAK2-STAT3 Pathway in Obesity-Related Osteoarthritis. Oxid. Med. Cell. Longev. 2021, 2021, 1–16. [Google Scholar] [CrossRef]
- Chung, I.M.; Ketharnathan, S.; Thiruvengadam, M.; Rajakumar, G. Rheumatoid arthritis: The stride from research to clinical practice. Int. J. Mol. Sci. 2016, 17, 900. [Google Scholar] [CrossRef]
- Alam, J.; Jantan, I.; Bukhari, S.N.A. Rheumatoid arthritis: Recent advances on its etiology, role of cytokines and pharmacotherapy. Biomed. Pharmacother. 2017, 92, 615–633. [Google Scholar] [CrossRef] [PubMed]
- Choy, E. Understanding the dynamics: Pathways involved in the pathogenesis of rheumatoid arthritis. Rheumatology 2012, 51, 3–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McInnes, I.B.; Schett, G. The Pathogenesis of Rheumatoid Arthritis. N. Engl. J. Med. 2011, 365, 2205–2219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bokarewa, M.; Bokarew, D.; Hultgren, O.; Tarkowski, A. Leptin consumption in the inflamed joints of patients with rheumatoid arthritis. Ann. Rheum. Dis. 2003, 62, 952–956. [Google Scholar] [CrossRef] [Green Version]
- Seven, A.; Güzel, S.; Aslan, M.; Hamuryudan, V. Serum and synovial fluid leptin levels and markers of inflammation in rheumatoid arthritis. Rheumatol. Int. 2009, 29, 743–747. [Google Scholar] [CrossRef]
- Lee, S.W.; Park, M.C.; Park, Y.B.; Lee, S.K. Measurement of the serum leptin level could assist disease activity monitoring in rheumatoid arthritis. Rheumatol. Int. 2007, 27, 537–540. [Google Scholar] [CrossRef]
- Abdalla, M.; Effat, D.; Sheta, M.; Hamed, W.E. Serum Leptin levels in Rheumatoid arthritis and relationship with disease activity. Egypt. Rheumatol. 2014, 36, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Hizmetli, S.; Kisa, M.; Gokalp, N.; Bakici, M.Z. Are plasma and synovial fluid leptin levels correlated with disease activity in rheumatoid arthritis? Rheumatol. Int. 2007, 27, 335–338. [Google Scholar] [CrossRef]
- Oner, S.Y.; Volkan, O.; Oner, C.; Mengi, A.; Direskeneli, H.; Tasan, D.A. Serum leptin levels do not correlate with disease activity in rheumatoid arthritis. Acta Reumatol. Port. 2015, 2015, 50–54. [Google Scholar]
- Popa, C.; Netea, M.G.; Radstake, T.R.D.S.; Van Riel, P.L.; Barrera, P.; Van Der Meer, J.W.M. Markers of inflammation are negatively correlated with serum leptin in rheumatoid arthritis. Ann. Rheum. Dis. 2005, 64, 1195–1198. [Google Scholar] [CrossRef]
- Anders, H.J.; Rihl, M.; Heufelder, A.; Loch, O.; Schattenkirchner, M. Leptin serum levels are not correlated with disease activity in patients with rheumatoid arthritis. Metabolism 1999, 48, 745–748. [Google Scholar] [CrossRef]
- Olama, S.M.; Senna, M.K.; Elarman, M. Synovial/Serum leptin ratio in rheumatoid arthritis: The association with activity and erosion. Rheumatol. Int. 2012, 32, 683–690. [Google Scholar] [CrossRef] [PubMed]
- Young, H.R.; Solus, J.; Sokka, T.; Oeser, A.; Chung, C.P.; Gebretsadik, T.; Shintani, A.; Pincus, T.; Stein, C.M. Adipocytokines are associated with radiographic joint damage in rheumatoid arthritis. Arthritis Rheum. 2009, 60, 1906–1914. [Google Scholar] [CrossRef] [Green Version]
- Juárez-rojop, I.E.; Antonio, J.; Batún-garrido, D.J.; Salas-maga, M.; Hernández-nú, E.; Olán, F. Relationship between leptin concentrations and disease activity in patients with rheumatoid arthritis. Med. Clínica 2018, 150, 341–344. [Google Scholar]
- Taylan, A.; Akinci, B.; Toprak, B.; Birlik, M.; Arslan, F.D.; Ekerbicer, H.; Gundogdu, B.; Colak, A.; Engin, B. Association of Leptin Levels and Disease Activity in Patients with Early Rheumatoid Arthritis. Arch. Med. Res. 2021, 52, 544–553. [Google Scholar] [CrossRef]
- Wisłowska, M.; Rok, M.; Jaszczyk, B.; Stȩpień, K.; Cicha, M. Serum leptin in rheumatoid arthritis. Rheumatol. Int. 2007, 27, 947–954. [Google Scholar] [CrossRef]
- Targońska-Stȩpniak, B.; Majdan, M.; Dryglewska, M. Leptin serum levels in rheumatoid arthritis patients: Relation to disease duration and activity. Rheumatol. Int. 2008, 28, 585–591. [Google Scholar] [CrossRef]
- Tian, G.; Liang, J.N.; Wang, Z.Y.; Zhou, D. Emerging role of leptin in rheumatoid arthritis. Clin. Exp. Immunol. 2014, 177, 557–570. [Google Scholar] [CrossRef]
- Petra, C.V.; Vonica, C.L.; Rahaian, R.; Berceanu, I.; Vesa, S.C.; Zdrenghea, M.; Rednic, S. Circulating leptin and resistin levels in a Romanian rheumatoid arthritis population. Rom. J. Rheumatol. 2020, 29, 79–83. [Google Scholar] [CrossRef]
- Otero, M.; Logo, R.; Gomez, R.; Logo, F.; Dieguez, C.; Gómez-Reino, J.J.; Gualillo, O. Changes in plasma levels of fat-derived hormones adiponectin, leptin, resistin and visfatin in patients with rheumatoid arthritis. Ann. Rheum. Dis. 2006, 65, 1198–1201. [Google Scholar] [CrossRef] [Green Version]
- Allam, A.; Radwan, A. The relationship of serum leptin levels with disease activity in Egyptian patients with rheumatoid arthritis. Egypt. Rheumatol. 2012, 34, 185–190. [Google Scholar] [CrossRef] [Green Version]
- Toussirot, É.; Grandclément, É.; Gaugler, B.; Michel, F.; Wendling, D.; Saas, P.; Dumoulin, G. Serum adipokines and adipose tissue distribution in rheumatoid arthritis and ankylosing spondylitis. A comparative study. Front. Immunol. 2013, 4, 453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toussirot, É.; Michel, F.; Binda, D.; Dumoulin, G. The role of leptin in the pathophysiology of rheumatoid arthritis. Life Sci. 2015, 140, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Busso, N.; So, A.; Chobaz-Péclat, V.; Morard, C.; Martinez-Soria, E.; Talabot-Ayer, D.; Gabay, C. Leptin Signaling Deficiency Impairs Humoral and Cellular Immune Responses and Attenuates Experimental Arthritis. J. Immunol. 2002, 168, 875–882. [Google Scholar] [CrossRef] [PubMed]
- Bernotiene, E.; Palmer, G.; Talabot-ayer, D.; Szalay-quinodoz, I.; Aubert, M.L.; Gabay, C. Delayed resolution of acute inflammation during zymosan-induced arthritis in Research article Delayed resolution of acute inflammation during zymosan-induced arthritis in leptin-deficient mice. Arthritis Res. Ther. 2004, 5, 93. [Google Scholar] [CrossRef]
- Otvos, L.; Shao, W.H.; Vanniasinghe, A.S.; Amon, M.A.; Holub, M.C.; Kovalszky, I.; Wade, J.D.; Doll, M.; Cohen, P.L.; Manolios, N.; et al. Toward understanding the role of leptin and leptin receptor antagonism in preclinical models of rheumatoid arthritis. Peptides 2011, 32, 1567–1574. [Google Scholar] [CrossRef]
- Sugioka, Y.; Tada, M.; Okano, T.; Nakamura, H.; Koike, T. Acquired leptin resistance by high-fat feeding reduces inflammation from collagen antibody-induced arthritis in mice. Clin. Exp. Rheumatol. 2012, 30, 707–713. [Google Scholar]
- Deng, J.; Liu, Y.; Yang, M.; Wang, S.; Zhang, M.; Wang, X.; Ko, K.H.; Hua, Z.; Sun, L.; Cao, X.; et al. Leptin exacerbates collagen-induced arthritis via enhancement of Th17 cell response. Arthritis Rheum. 2012, 64, 3564–3573. [Google Scholar] [CrossRef]
- Hultgren, O.H.; Tarkowski, A. Leptin in septic arthritis: Decreased levels during infection and amelioration of disease activity upon its adminstration. Arthritis Res. 2001, 3, 389–394. [Google Scholar] [CrossRef]
- Bartok, B.; Firestein, G.S. Fibroblast-like synoviocytes: Key effector cells in rheumatoid arthritis. Immunol. Rev. 2010, 233, 233–255. [Google Scholar] [CrossRef]
- Sun, X.; Wei, J.; Tang, Y.; Wang, B.; Zhang, Y.; Shi, L.; Guo, J.; Hu, F.; Li, X. Leptin-induced migration and angiogenesis in rheumatoid arthritis is mediated by reactive oxygen species. FEBS Open Bio 2017, 7, 1899–1908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holloway, W.R.; Collier, F.M.L.; Aitken, C.J.; Myers, D.E.; Hodge, J.M.; Malakellis, M.; Gough, T.J.; Collier, G.R.; Nicholson, G.C. Leptin inhibits osteoclast generation. J. Bone Miner. Res. 2002, 17, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Otero, M.; Lago, R.; Gómez, R.; Lago, F.; Gomez-Reino, J.J.; Gualillo, O. Phosphatidylinositol 3-kinase, MEK-1 and p38 mediate leptin/interferon-gamma synergistic NOS type II induction in chondrocytes. Life Sci. 2007, 81, 1452–1460. [Google Scholar] [CrossRef] [PubMed]
- Conde, J.; Scotece, M.; López, V.; Gómez, R.; Lago, F.; Pino, J.; Gómez-Reino, J.J.; Gualillo, O. Adiponectin and Leptin Induce VCAM-1 Expression in Human and Murine Chondrocytes. PLoS ONE 2012, 7, e52533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraser, D.A.; Thoen, J.; Reseland, J.E.; Førre, Ø.; Kjeldsen-Kragh, J. Decreased CD4+ lymphocyte activation and increased interleukin-4 production in peripheral blood of rheumatoid arthritis patients after acute starvation. Clin. Rheumatol. 1999, 18, 394–401. [Google Scholar] [CrossRef]
- Lord, G.M.; Matarese, G.; Howard, J.K.; Baker, R.J.; Bloom, S.R.; Lechler, R.I. Leptin modulates the T-cell immune response and reverses starvation- induced immunosuppression. Nature 1998, 394, 897–901. [Google Scholar] [CrossRef]
- Yang, P.; Qian, F.Y.; Zhang, M.F.; Xu, A.L.; Wang, X.; Jiang, B.P.; Zhou, L.L. Th17 cell pathogenicity and plasticity in rheumatoid arthritis. J. Leukoc. Biol. 2019, 106, 1233–1240. [Google Scholar] [CrossRef]
- Avdeeva, A.; Rubtsov, Y.; Dyikanov, D.; Popkova, T.; Nasonov, E. Regulatory T cells in patients with early untreated rheumatoid arthritis: Phenotypic changes in the course of methotrexate treatment. Biochimie 2020, 174, 9–17. [Google Scholar] [CrossRef]
- De Rosa, V.; Procaccini, C.; Calì, G.; Pirozzi, G.; Fontana, S.; Zappacosta, S.; La Cava, A.; Matarese, G. A Key Role of Leptin in the Control of Regulatory T Cell Proliferation. Immunity 2007, 26, 241–255. [Google Scholar] [CrossRef] [Green Version]
- Glyn-Jones, S.; Palmer, A.J.R.; Agricola, R.; Price, A.J.; Vincent, T.L.; Weinans, H.; Carr, A.J. Osteoarthritis. Lancet 2015, 386, 376–387. [Google Scholar] [CrossRef]
- Zhang, Y.; Vasheghani, F.; Li, Y.H.; Blati, M.; Simeone, K.; Fahmi, H.; Lussier, B.; Roughley, P.; Lagares, D.; Pelletier, J.P.; et al. Cartilage-specific deletion of mTOR upregulates autophagy and protects mice from osteoarthritis. Ann. Rheum. Dis. 2015, 74, 1432–1440. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.; Zhang, J.; Yang, H.; Sun, Y. The role of leptin in osteoarthritis. Medicine 2018, 97, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Peelman, F.; Van Beneden, K.; Zabeau, L.; Iserentant, H.; Ulrichts, P.; Defeau, D.; Verhee, A.; Catteeuw, D.; Elewaut, D.; Tavernier, J. Mapping of the leptin binding sites and design of a leptin antagonist. J. Biol. Chem. 2004, 279, 41038–41046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farooqi, I.S.; Jebb, S.A.; Langmack, G.; Lawrence, E.; Cheetham, C.H.; Prentice, A.M.; Hughes, I.A.; McCamish, M.A.; O’Rahilly, S. Effects of recombinant leptin therapy in a child with congenital leptin deficiency. Bone 1999, 341, 879–884. [Google Scholar] [CrossRef]
- Paz-Filho, G.; Mastronardi, C.A.; Licinio, J. Leptin treatment: Facts and expectations. Metabolism. 2015, 64, 146–156. [Google Scholar] [CrossRef]
- Koskinen-Kolasa, A.; Vuolteenaho, K.; Korhonen, R.; Moilanen, T.; Moilanen, E. Catabolic and proinflammatory effects of leptin in chondrocytes are regulated by suppressor of cytokine signaling-3. Arthritis Res. Ther. 2016, 18, 215. [Google Scholar] [CrossRef] [Green Version]
- Feng, G.S. Shp2 as a therapeutic target for leptin resistance and obesity. Expert Opin. Ther. Targets 2006, 10, 135–142. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ait Eldjoudi, D.; Cordero Barreal, A.; Gonzalez-Rodríguez, M.; Ruiz-Fernández, C.; Farrag, Y.; Farrag, M.; Lago, F.; Capuozzo, M.; Gonzalez-Gay, M.A.; Mera Varela, A.; et al. Leptin in Osteoarthritis and Rheumatoid Arthritis: Player or Bystander? Int. J. Mol. Sci. 2022, 23, 2859. https://doi.org/10.3390/ijms23052859
Ait Eldjoudi D, Cordero Barreal A, Gonzalez-Rodríguez M, Ruiz-Fernández C, Farrag Y, Farrag M, Lago F, Capuozzo M, Gonzalez-Gay MA, Mera Varela A, et al. Leptin in Osteoarthritis and Rheumatoid Arthritis: Player or Bystander? International Journal of Molecular Sciences. 2022; 23(5):2859. https://doi.org/10.3390/ijms23052859
Chicago/Turabian StyleAit Eldjoudi, Djedjiga, Alfonso Cordero Barreal, María Gonzalez-Rodríguez, Clara Ruiz-Fernández, Yousof Farrag, Mariam Farrag, Francisca Lago, Maurizio Capuozzo, Miguel Angel Gonzalez-Gay, Antonio Mera Varela, and et al. 2022. "Leptin in Osteoarthritis and Rheumatoid Arthritis: Player or Bystander?" International Journal of Molecular Sciences 23, no. 5: 2859. https://doi.org/10.3390/ijms23052859