
COVID-19: Immunohistochemical Analysis of TGF-β Signaling Pathways in Pulmonary Fibrosis

 , , , , , ,
, , , , , ,  and
and
Abstract
:1. Introduction
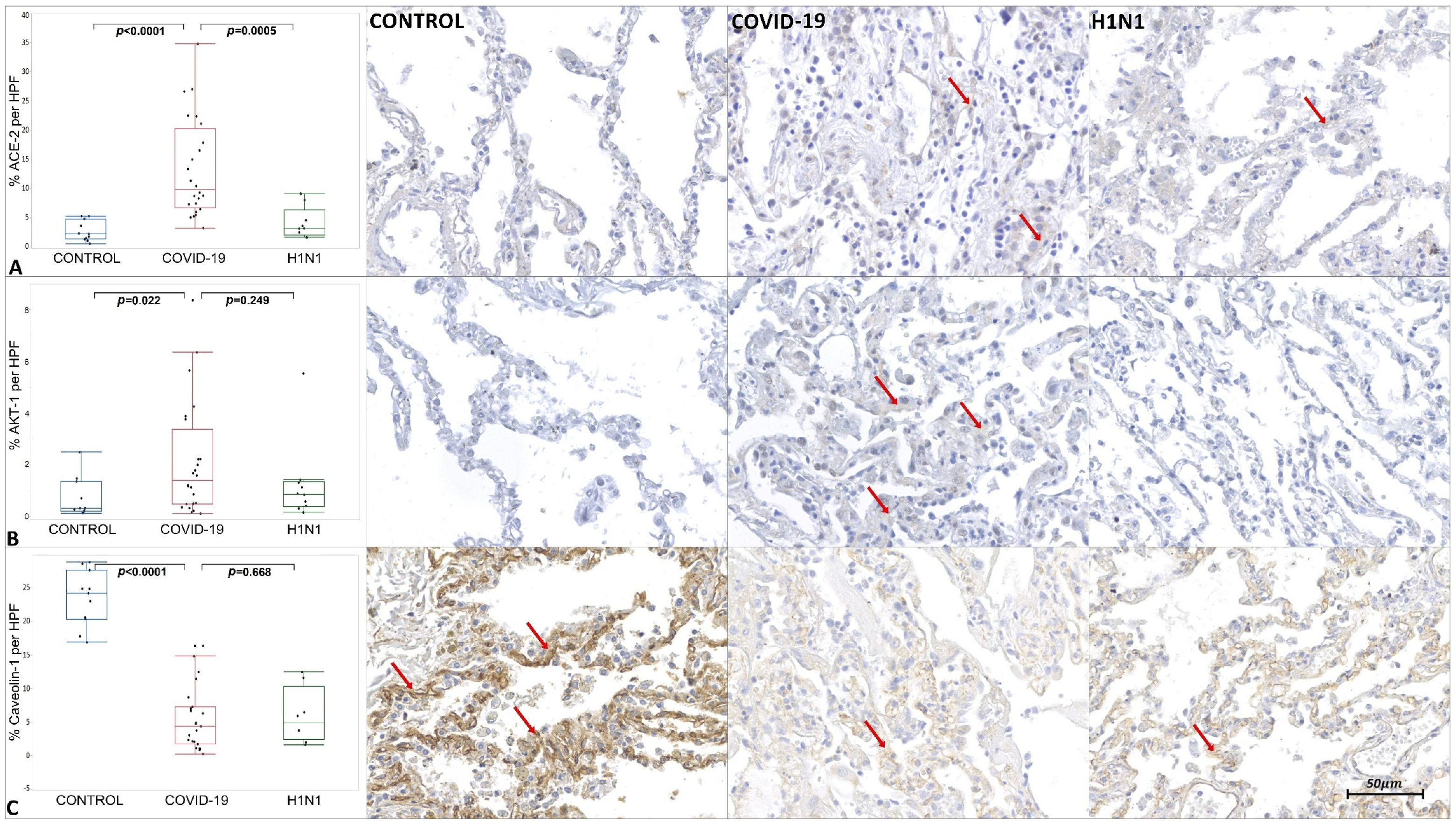
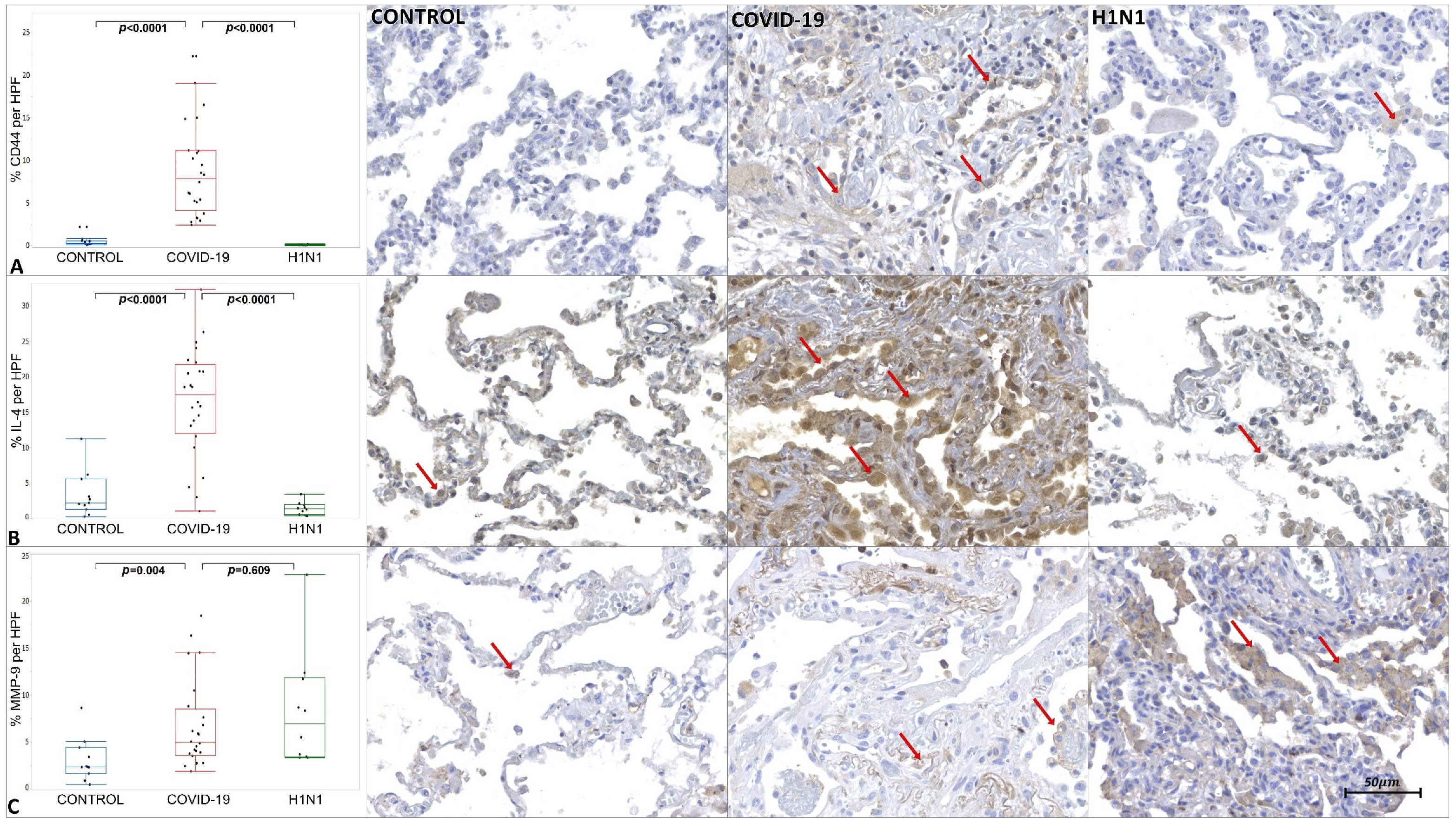
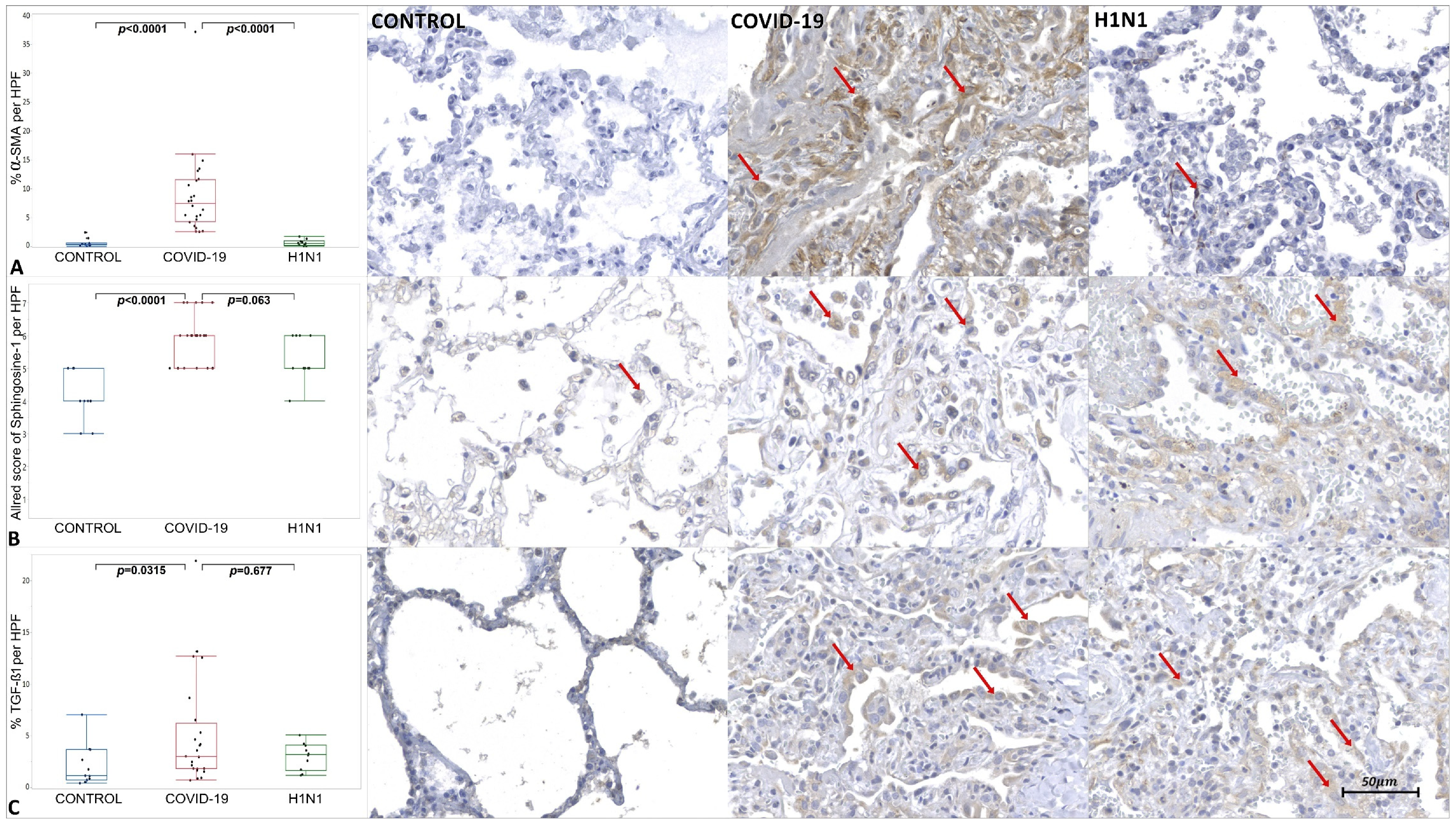
2. Results
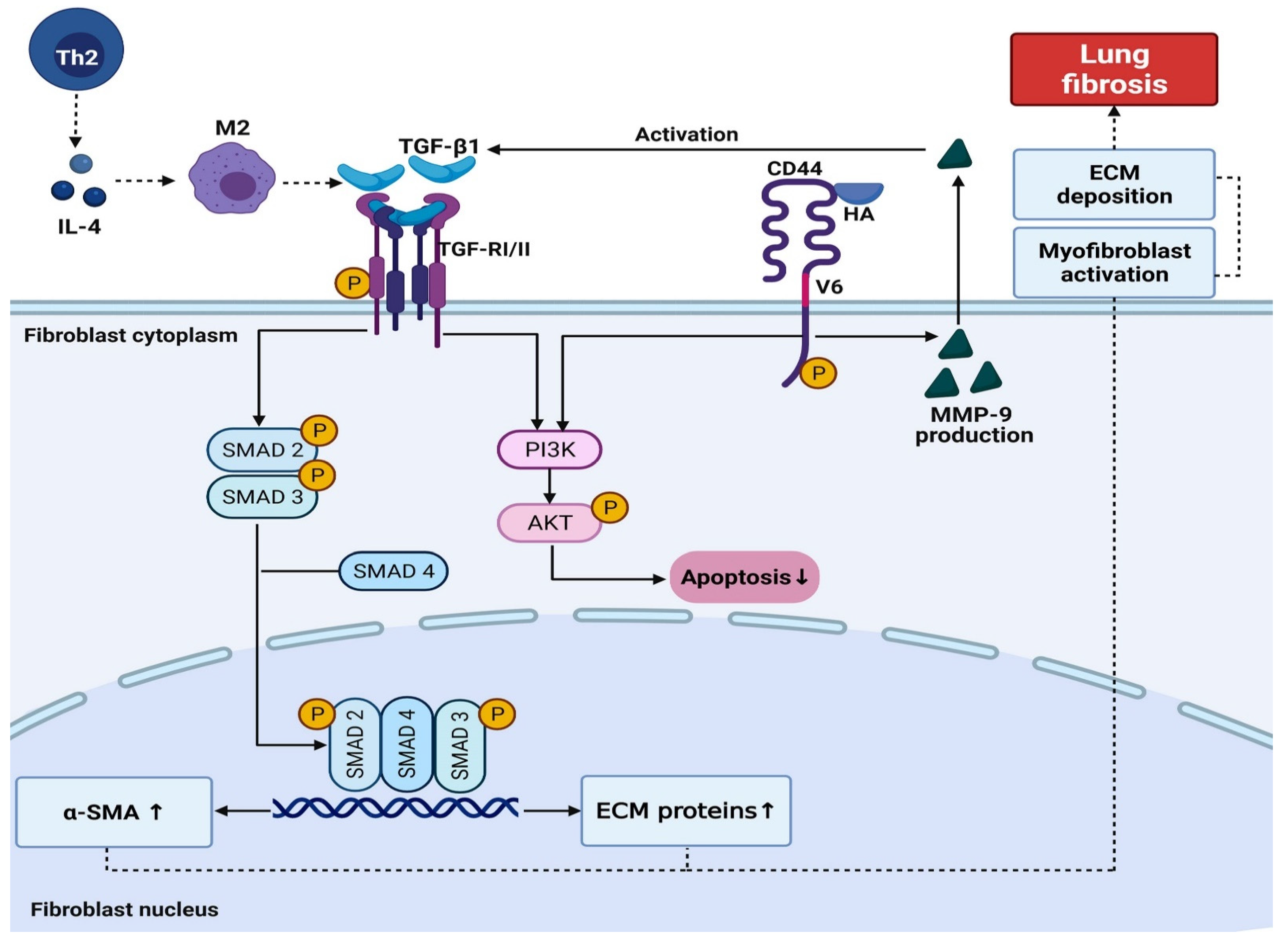
3. Discussion
3.1. Histopathological Findings and Collagens Analysis
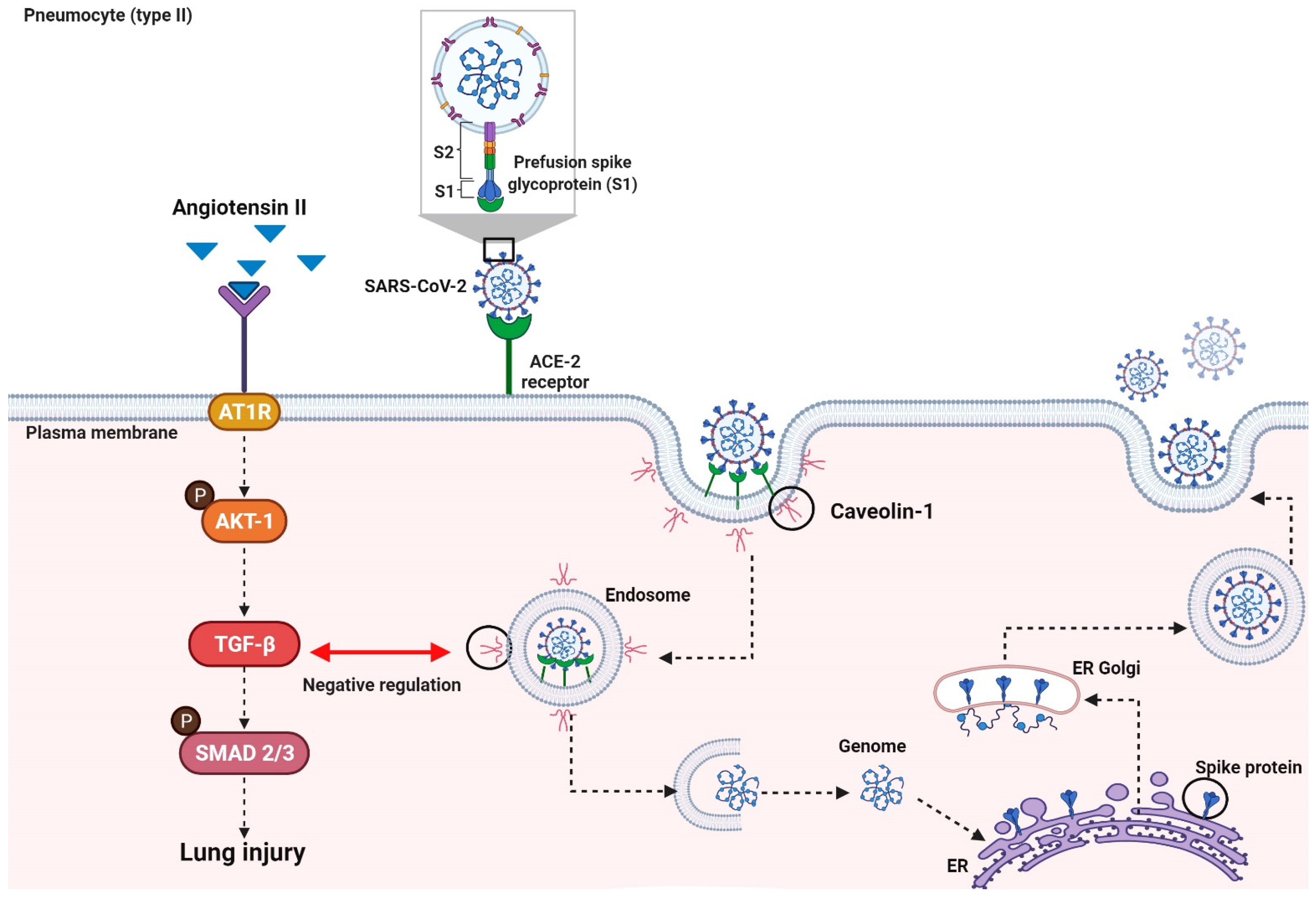
3.2. Virally Mediated Fibrogenic Pathways
3.3. TGF-β Signaling in Pulmonary Fibrosis
3.4. Corticosteroid and Pulmonary Fibrosis
3.5. Limitation of the Study
4. Materials and Methods
4.1. Ethical Approval
4.2. Samples
4.3. Histological and Morphometric Analysis
4.4. Immunohistochemical Analysis
4.5. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Petrilli, C.M.; Jones, S.A.; Yang, J.; Rajagopalan, H.; O’Donnell, L.; Chernyak, Y.; Tobin, K.A.; Cerfolio, R.J.; Francois, F.; Horwitz, L.I. Factors associated with hospital admission and critical illness among 5279 people with coronavirus disease 2019 in New York City: Prospective cohort study. BMJ 2020, 369, m1966. [Google Scholar] [CrossRef] [PubMed]
- De Paula, C.B.V.; De Azevedo, M.L.V.; Nagashima, S.; Martins, A.P.C.; Malaquias, M.A.S.; Miggiolaro, A.F.R.D.S.; Júnior, J.D.S.M.; Avelino, G.; Carmo, L.A.P.D.; Carstens, L.B.; et al. IL-4/IL-13 remodeling pathway of COVID-19 lung injury. Sci. Rep. 2020, 10, 4–11. [Google Scholar] [CrossRef]
- Lin, L.; Luo, S.; Qin, R.; Yang, M.; Wang, X.; Yang, Q.; Zhang, Y.; Wang, Q.; Zhu, R.; Fan, H.; et al. Long-term infection of SARS-CoV-2 changed the body’s immune status. Clin. Immunol. 2020, 218, 108524. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Sun, L.X.; Feng, R.E. Comparison of clinical and pathological features between severe acute respiratory syndrome and coronavirus disease 2019. Zhonghua Jie He He Hu Xi Za Zhi Zhonghua Jiehe He Huxi Zazhi Chin. J. Tuberc. Respir. Dis. 2020, 43, 496–502. [Google Scholar] [CrossRef]
- Ye, Q.; Wang, B.; Mao, J. The pathogenesis and treatment of the ‘Cytokine Storm’ in COVID-19’. J. Infect. 2020, 80, 607–613. [Google Scholar] [CrossRef]
- Bottasso, O.; Delpino, M.V.; Quarleri, J. SARS-CoV-2 Pathogenesis: Imbalance in the Renin-Angiotensin System Favors Lung Fibrosis. Front. Cell. Infect. Microbiol. 2020, 1, 340. [Google Scholar] [CrossRef]
- Abbul, K.A.; Andrew, H.; Lichtman, S.P. Imunologia Celular e Molecular, 8th ed.; Elsevier: Rio de Janeiro, Brazil, 2012; ISBN 9788535281644. [Google Scholar]
- Huang, X.; Xiu, H.; Zhang, S.; Zhang, G. The Role of Macrophages in the Pathogenesis of ALI/ARDS. Mediat. Inflamm. 2018, 2018, 1264913. [Google Scholar] [CrossRef]
- Jia, H. Pulmonary Angiotensin-Converting Enzyme 2 (ACE2) and Inflammatory Lung Disease. Shock 2016, 46, 239–248. [Google Scholar] [CrossRef]
- Tammi, M.I.; Day, A.J.; Turley, E.A. Hyaluronan and Homeostasis: A Balancing Act. J. Biol. Chem. 2002, 277, 4581–4584. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Dong, L.; Chang, P. CD44v6 engages in colorectal cancer progression. Cell Death Dis. 2019, 10, 30. [Google Scholar] [CrossRef]
- Pardo, A.; Cabrera, S.; Maldonado, M.; Selman, M. Role of matrix metalloproteinases in the pathogenesis of idiopathic pulmonary fibrosis. Respir. Res. 2016, 17, 23. [Google Scholar] [CrossRef] [Green Version]
- Yu, Q.; Stamenkovic, I. Cell Surface-Localized Matrix Metalloproteinase-9 Proteolytically Activates TGF-and Promotes Tumor Invasion and Angiogenesis. Genes Dev. 2000, 14, 163–176. [Google Scholar] [CrossRef]
- Rothan, H.A.; Byrareddy, S.N. The epidemeology and pathogensis of coronavirus (COVID-19) outbreak. J. Autoimmun. 2020, 109, 1–4. [Google Scholar] [CrossRef]
- Tang, T.; Bidon, M.; Jaimes, J.A.; Whittaker, G.R.; Daniel, S. Coronavirus membrane fusion mechanism offers a potential target for antiviral development. Antivir. Res. 2020, 178, 104792. [Google Scholar] [CrossRef]
- Li, X.; Zhu, W.; Fan, M.; Zhang, J.; Peng, Y.; Huang, F.; Wang, N.; He, L.; Zhang, L.; Holmdahl, R.; et al. Dependence of SARS-CoV-2 infection on cholesterol-rich lipid raft and endosomal acidification. Comput. Struct. Biotechnol. J. 2021, 19, 1933–1943. [Google Scholar] [CrossRef] [PubMed]
- Kruglikov, I.L.; Scherer, P.E. Caveolin-1 as a target in prevention and treatment of hypertrophic scarring. NPJ Regen. Med. 2019, 4, 9. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Akhter, H.; Jiang, C.; Macewen, M.; Ding, Q.; Antony, V.; John, V.; Liu, R. Plasminogen activator inhibitor 1, fibroblast apoptosis resistence, and aging-related susceptibility to lung fibrosis. Exp. Gerontol. 2015, 61, 62–75. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.; Wang, Y.; Zhu, T.; Xia, L. CT Features of Coronavirus Disease 2019 (COVID-19) Pneumonia in 62 Patients in Wuhan, China. Am. J. Roentgenol. 2020, 214, 1287–1294. [Google Scholar] [CrossRef]
- Azkur, A.K.; Akdis, M.; Azkur, D.; Sokolowska, M.; van de Veen, W.; Brüggen, M.C.; O’Mahony, L.; Gao, Y.; Nadeau, K.; Akdis, C.A. Immune response to SARS-CoV-2 and mechanisms of immunopathological changes in COVID-19. Allergy Eur. J. Allergy Clin. Immunol. 2020, 75, 1564–1581. [Google Scholar] [CrossRef]
- Murthy, K.; Sivashanmugam, K.; Kandasamy, M.; Subbiah, R.; Ravikumar, V. Repurposing of histone deacetylase inhibitors: A promising strategy to combat pulmonary fibrosis promoted by TGF-β signalling in COVID-19 survivors. Life Sci. 2021, 266, 118883. [Google Scholar] [CrossRef]
- Mehta, P.; McAuley, D.F.; Brown, M.; Sanchez, E.; Tattersall, R.S.; Manson, J.J.; HLH Across Speciality Collaboration UK. COVID-19: Consider cytokine storm syndromes and immunosuppression. Lancet 2020, 395, 1033–1034. [Google Scholar] [CrossRef]
- Bai, L.; Li, A.; Gong, C.; Ning, X.; Wang, Z. Protective effect of rutin against bleomycin induced lung fibrosis: Involvement of TGF-β1/α-SMA/Col I and III pathway. BioFactors 2020, 46, 637–644. [Google Scholar] [CrossRef] [PubMed]
- Snijder, J.; Peraza, J.; Padilla, M.; Capaccione, K.; Salvatore, M.M. Pulmonary fibrosis: A disease of alveolar collapse and collagen deposition. Expert Rev. Respir. Med. 2019, 13, 615–619. [Google Scholar] [CrossRef] [PubMed]
- Chesnutt, A.N.; Matthay, M.A.; Tibayan, F.A.; Clark, J.G. Early detection of type III procollagen peptide in acute lung injury. Pathogenetic and prognostic significance. Am. J. Respir. Crit. Care Med. 1997, 156, 840–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldstein, R.H. Control of type I collagen formation in the lung. Am. J. Physiol. 1991, 261. [Google Scholar] [CrossRef] [PubMed]
- Giménez, A.; Duch, P.; Puig, M.; Gabasa, M.; Xaubet, A.; Alcaraz, J. Dysregulated Collagen Homeostasis by Matrix Stiffening and TGF-β1 in Fibroblasts from Idiopathic Pulmonary Fibrosis Patients: Role of FAK/Akt. Int. J. Mol. Sci. 2017, 18, 2431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karsdal, M.A.; Nielsen, S.H.; Leeming, D.J.; Langholm, L.L.; Nielsen, M.J.; Manon-Jensen, T.; Siebuhr, A.; Gudmann, N.S.; Rønnow, S.; Sand, J.M.; et al. The good and the bad collagens of fibrosis–Their role in signaling and organ function. Adv. Drug Deliv. Rev. 2017, 121, 43–56. [Google Scholar] [CrossRef]
- McDonald, L.T. Healing after COVID-19: Are survivors at risk for pulmonary fibrosis? Am. J. Physiol. Lung Cell. Mol. Physiol. 2021, 320, L257–L265. [Google Scholar] [CrossRef] [PubMed]
- Antoniazzi, P.; Pereira Júnior, G.A.; Marson, F.; Abeid, M.; Baldisserotto, S.; Basile-Filho, A. Síndrome Da Angústia Respiratória Aguda (Sara). Medicina 1998, 31, 493. [Google Scholar] [CrossRef]
- Mueller, A.L.; McNamara, M.S.; Sinclair, D.A. Why does COVID-19 disproportionately affect older people? Aging 2020, 12, 9959–9981. [Google Scholar] [CrossRef] [PubMed]
- Uhal, B.D.; Dang, M.; Dang, V.; Llatos, R.; Cano, E.; Abdul-Hafez, A.; Markey, J.; Piasecki, C.C.; Molina-Molina, M. Cell cycle dependence of ACE-2 explains downregulation in idiopathic pulmonary fibrosis. Eur. Respir. J. 2013, 42, 198–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuki, K.; Fujiogi, M.; Koutsogiannaki, S. COVID-19 pathophysiology: A review. Clin. Immunol. 2020, 215, 108427. [Google Scholar] [CrossRef] [PubMed]
- Baker, S.A.; Kwok, S.; Berry, G.J.; Montine, T.J. Angiotensin-converting enzyme 2 (ACE2) expression increases with age in patients requiring mechanical ventilation. PLoS ONE 2021, 16, e0247060. [Google Scholar] [CrossRef]
- Wang, H.; Yang, P.; Liu, K.; Guo, F.; Zhang, Y.; Zhang, G.; Jiang, C. SARS coronavirus entry into host cells through a novel clathrin- and caveolae-independent endocytic pathway. Cell Res. 2008, 18, 290–301. [Google Scholar] [CrossRef] [Green Version]
- Kagami, S.; Border, W.A.; Miller, D.E.; Noble, N.A. Angiotensin II stimulates extracellular matrix protein synthesis through induction of transforming growth factor-beta expression in rat glomerular mesangial cells. J. Clin. Investig. 1994, 93, 2431–2437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, R.; Zhang, J.; Luo, D.; Yu, Y.; Chen, T.; Yang, Y.; Yu, F.; Li, M. Upregulation of Transient Receptor Potential Canonical Type 3 Channel via AT1R/TGF- β 1/Smad2/3 Induces Atrial Fibrosis in Aging and Spontaneously Hypertensive Rats. Oxid. Med. Cell. Longev. 2019, 2019, 4025496. [Google Scholar] [CrossRef]
- Glende, J.; Schwegmann-Wessels, C.; Al-Falah, M.; Pfefferle, S.; Qu, X.; Deng, H.; Drosten, C.; Naim, H.Y.; Herrler, G. Importance of cholesterol-rich membrane microdomains in the interaction of the S protein of SARS-coronavirus with the cellular receptor angiotensin-converting enzyme 2. Virology 2008, 381, 215–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walton, K.L.; Johnson, K.E.; Harrison, C.A. Targeting TGF-β Mediated SMAD Signaling for the Prevention of Fibrosis. Front. Pharmacol. 2017, 8, 461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, M.W.; Zhang, Y.; Hong, P.K.; Zhou, Z.; Feghali-Bostwick, C.A.; Liu, F.; Ifedigbo, E.; Xu, X.; Oury, T.D.; Kaminski, N.; et al. Caveolin-1: A critical regulator of lung fibrosis in idiopathic pulmonary fibrosis. J. Exp. Med. 2006, 203, 2895–2906. [Google Scholar] [CrossRef]
- Razani, B.; Zhang, X.L.; Bitzer, M.; Von Gersdorff, G.; Böttinger, E.P.; Lisanti, M.P. Caveolin-1 regulates transforming growth factor (TGF)-beta/SMAD signaling through an interaction with the TGF-beta type I receptor. J. Biol. Chem. 2001, 276, 6727–6738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ojo, A.S.; Balogun, S.A.; Williams, O.T.; Ojo, O.S. Predictive Factors and Risk Reduction Strategies. Pulm. Med. 2020, 2020, 1–10. [Google Scholar] [CrossRef]
- Roberts, A.B. Is Smad3 a major player in signal transduction pathways leading to fibrogenesis? Chest 2001, 120, 43S–47S. [Google Scholar] [CrossRef]
- Ragab, D.; Salah Eldin, H.; Taeimah, M.; Khattab, R.; Salem, R. The COVID-19 Cytokine Storm; What We Know So Far. Front. Immunol. 2020, 11, 1446. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Zhang, X.R.; Ju, Z.Y.; He, W.F. [Advances in the research of mechanism and related immunotherapy on the cytokine storm induced by coronavirus disease 2019]. Zhonghua Shao Shang Za Zhi 2020, 36, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Costela-Ruiz, V.J.; Illescas-Montes, R.; Puerta-Puerta, J.M.; Ruiz, C.; Melguizo-Rodríguez, L. SARS-CoV-2 infection: The role of cytokines in COVID-19 disease. Cytokine Growth Factor Rev. 2020, 54, 62. [Google Scholar] [CrossRef]
- Sgalla, G.; Iovene, B.; Calvello, M.; Ori, M.; Varone, F.; Richeldi, L. Idiopathic pulmonary fibrosis: Pathogenesis and management. Respir. Res. 2018, 19, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Hinz, B.; Gabbiani, G. Mechanisms of force generation and transmission by myofibroblasts. Curr. Opin. Biotechnol. 2003, 14, 538–546. [Google Scholar] [CrossRef] [PubMed]
- Paula, A.; Masseno, B.; Porto, C.D. Myofibroblasts: A review. Vet. E Zootec. 2010, 17, 177–190. [Google Scholar]
- Atkinson, J.J.; Senior, R.M. Matrix metalloproteinase-9 in lung remodeling. Am. J. Respir. Cell Mol. Biol. 2003, 28, 12–24. [Google Scholar] [CrossRef] [PubMed]
- Bazdyrev, E.; Rusina, P.; Panova, M.; Novikov, F.; Grishagin, I.; Nebolsin, V. Lung Fibrosis after COVID-19: Treatment Prospects. Pharmaceuticals 2021, 14, 807. [Google Scholar] [CrossRef] [PubMed]
- Alexaki, V.I.; Henneicke, H. The Role of Glucocorticoids in the Management of COVID-19. Horm. Metab. Res. 2021, 53, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Guo, F.; Song, X. Effects and mechanisms of pirfenidone, prednisone and acetylcysteine on pulmonary fibrosis in rat idiopathic pulmonary fibrosis models. Pharm. Biol. 2017, 55, 450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roach, K.M.; Sutcliffe, A.; Matthews, L.; Elliott, G.; Newby, C.; Amrani, Y.; Bradding, P. A model of human lung fibrogenesis for the assessment of anti-fibrotic strategies in idiopathic pulmonary fibrosis. Sci. Rep. 2018, 8, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuwano, K.; Hagimoto, N.; Nakanishi, Y. The role of apoptosis in pulmonary fibrosis. Histol. Histopathol. 2004, 19, 138–144. [Google Scholar] [CrossRef]
- Brasileiro, G.F. Bogliolo Patologia Geral, 9th ed.; Editora Guanabara Koogan S/A: Rio de Janeiro, Brazil, 2016. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data | CONTROL (n = 11) | COVID-19 (n = 24) | H1N1 (n = 10) |
|---|---|---|---|
| Gender a | Male (8) 72.7% | Male (15) 62.5% | Male (8) 80.0% |
| Female (3) 27.3% | Female (9) 37.5% | Female (2) 20.0% | |
| 0.709 * | 0.437 ** | ||
| Age (years) 1,b | 42.31 ± 4.4 | 71.96 ± 12.5 | 43.5 ± 14.0 |
| <0.001 * | <0.001 ** | ||
| Time from hospitalization to death (days) 1,b,c | 7.6/13.1 | 15.87 ± 10.2 | 4.70 ± 6.13 |
| 0.003 * | 0.003 ** | ||
| Mechanical ventilation 1,b | ----- | 12.0 ± 9.2 | 4.7 ± 6.13 |
| ----- | 0.028 ** | ||
| Previous pulmonary diseases | ----- | Bronchial Asthma (4/24) Interstitial Pulmonary Fibrosis (1/24) | ----- |
| Histological pattern of DAD | Normal septum | Interstitial pneumonitis with scarce septal neutrophils, hyaline membrane, type II pneumocyte hyperplasia, fibrosis, and micro thrombosis | Interstitial pneumonitis with high septal neutrophils infiltration and no micro thrombosis |
| Computed tomography chest at admission | ----- | “Opacities with ground-glass attenuation,” suggestive of viral pulmonary infection (24/24); Interstitial Pulmonary Fibrosis (1/24); Bronchial thickening (3/24); Bilateral pleural thickening (2/24); Pleural effusion (4/24); Parasseptal emphysema (2/24); Pulmonary consolidation (1/24); | ----- |
| Anti-inflammatory drugs | ----- | Dexamethasone 6 mg/day (12/24); Hydrocortisone 100 mg/day (1/24); Hydrocortisone 200 mg/day (3/24); Methylprednisolone 125 mg/day (2/24); Prednisone 60 mg/day (1/24); Prednisone 10 mg/day (1/24); | ----- |
| Data | Category | CONTROL (n = 11) | COVID-19 (n = 24) | H1N1 (n = 10) |
|---|---|---|---|---|
| DAD a | Absent | 11 (100.0%) | 0 (0%) | 0 (0%) |
| Initial | 0 (0%) | 10 (41.7%) | 5 (50.0%) | |
| Established | 0 (0%) | 14 (58.3%) | 5 (50.0%) | |
| <0.001 * | 0.718 ** | |||
| Hyaline membrane a | Absent | 11 (100%) | 3 (12.5%) | 6 (60.0%) |
| Present | 0 (0%) | 21 (87.5%) | 4 (40.0%) | |
| <0.001 * | 0.009 ** | |||
| Type II pneumocyte hyperplasia a | Absent | 6 (54.5%) | 1 (4.2%) | 0 (0%) |
| Present | 5 (45.5%) | 23 (95.8%) | 10 (100.0%) | |
| 0.0002 * | 1.000 ** | |||
| Fibrosis | Absent | 11 (100.0%) | 2 (8.3%) | 4 (40.0%) |
| Present | 0 (0%) | 22 (91.7%) | 6 (60.0%) | |
| <0.001 * | 0.048 ** | |||
| Collagen I (mature) b | - | 95.47 (60.32–99.51) 1 | 68.53 (6.93–99.38) 1 | 97.21 (70.88–99.51) 1 |
| 0.0015 * | 0.0006 ** | |||
| Collagen III (immature) b | - | 4.52 (0.49–39.68) 1 | 31.47 (0.62–93.06) 1 | 18.10 (8.95–44.35) 1 |
| 0.0015 * | 0.2863 ** | |||
| Fibrosis | Corticosteroid Use | No Corticosteroid Use |
|---|---|---|
| Absent | 0 (0%) | 2 (28.6%) |
| Present | 17 (100.0%) | 5 (50.0%) |
| 0.718 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vaz de Paula, C.B.; Nagashima, S.; Liberalesso, V.; Collete, M.; da Silva, F.P.G.; Oricil, A.G.G.; Barbosa, G.S.; da Silva, G.V.C.; Wiedmer, D.B.; da Silva Dezidério, F.; et al. COVID-19: Immunohistochemical Analysis of TGF-β Signaling Pathways in Pulmonary Fibrosis. Int. J. Mol. Sci. 2022, 23, 168. https://doi.org/10.3390/ijms23010168
Vaz de Paula CB, Nagashima S, Liberalesso V, Collete M, da Silva FPG, Oricil AGG, Barbosa GS, da Silva GVC, Wiedmer DB, da Silva Dezidério F, et al. COVID-19: Immunohistochemical Analysis of TGF-β Signaling Pathways in Pulmonary Fibrosis. International Journal of Molecular Sciences. 2022; 23(1):168. https://doi.org/10.3390/ijms23010168
Chicago/Turabian StyleVaz de Paula, Caroline Busatta, Seigo Nagashima, Vanessa Liberalesso, Mariana Collete, Felipe Paes Gomes da Silva, Alessandro Gonçalves Gomes Oricil, Giovanna Silva Barbosa, Guilherme Vieira Cavalcante da Silva, David Batista Wiedmer, Felipe da Silva Dezidério, and et al. 2022. "COVID-19: Immunohistochemical Analysis of TGF-β Signaling Pathways in Pulmonary Fibrosis" International Journal of Molecular Sciences 23, no. 1: 168. https://doi.org/10.3390/ijms23010168