
New Synthetic Lethality Re-Sensitizing Platinum-Refractory Cancer Cells to Cisplatin In Vitro: The Rationale to Co-Use PARP and ATM Inhibitors


Abstract
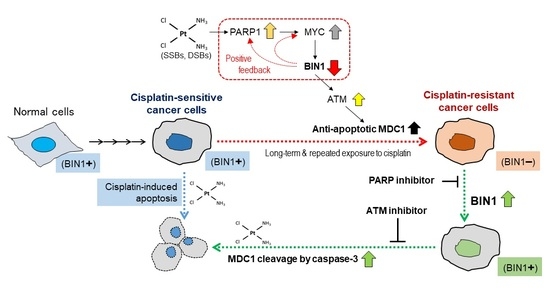
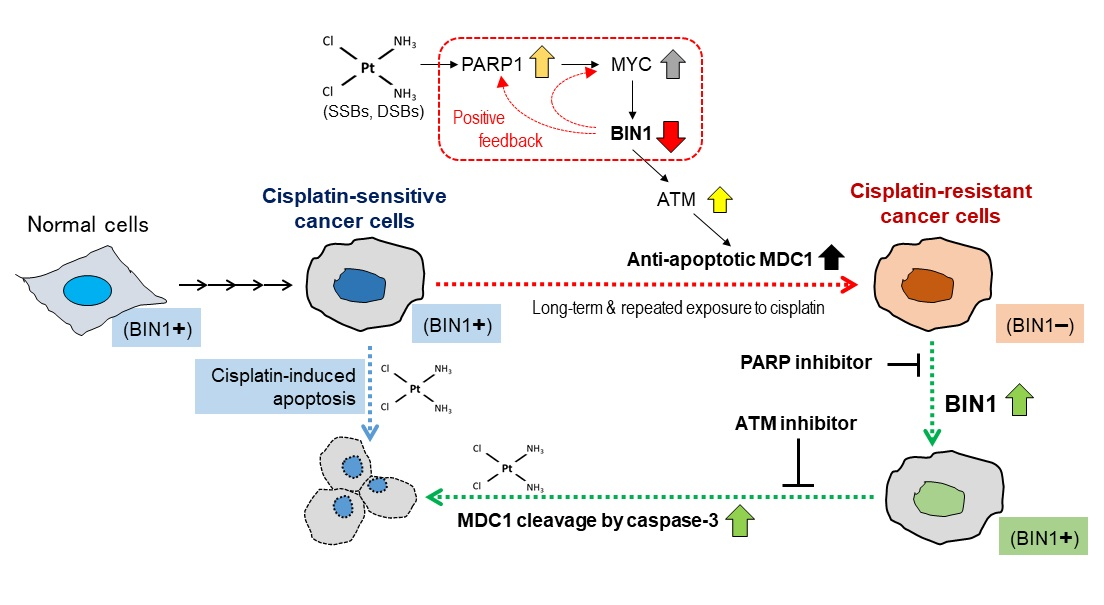
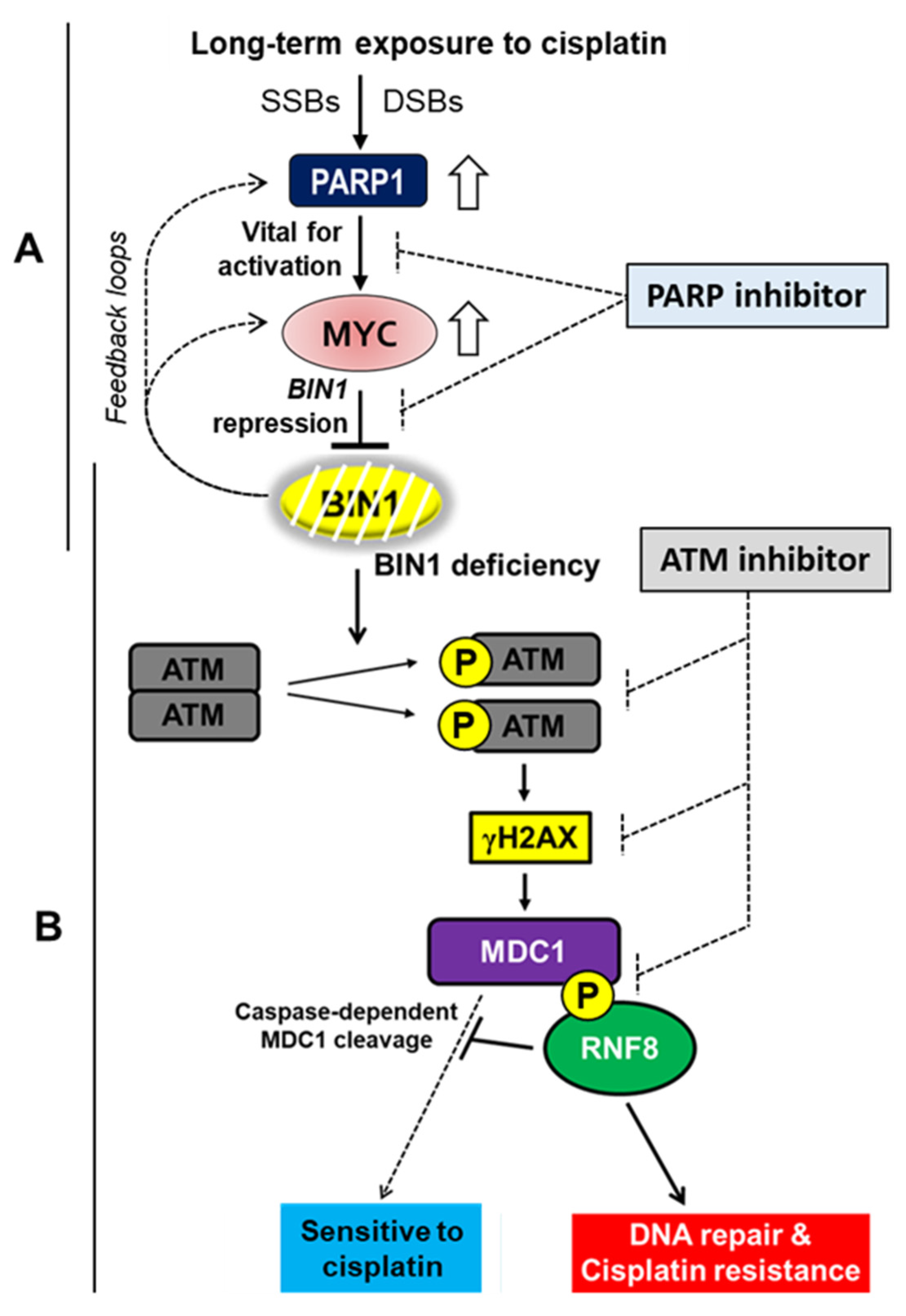
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
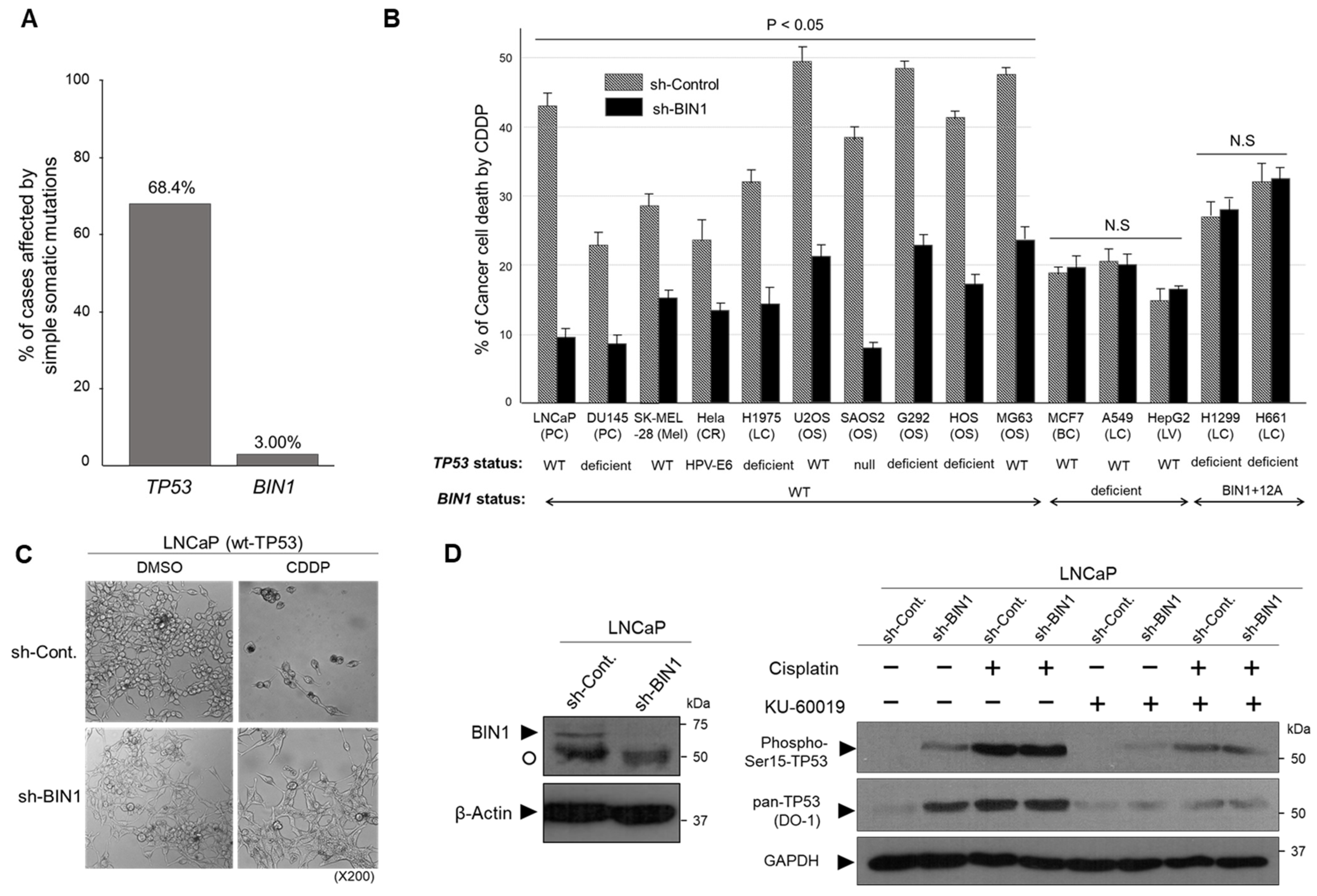
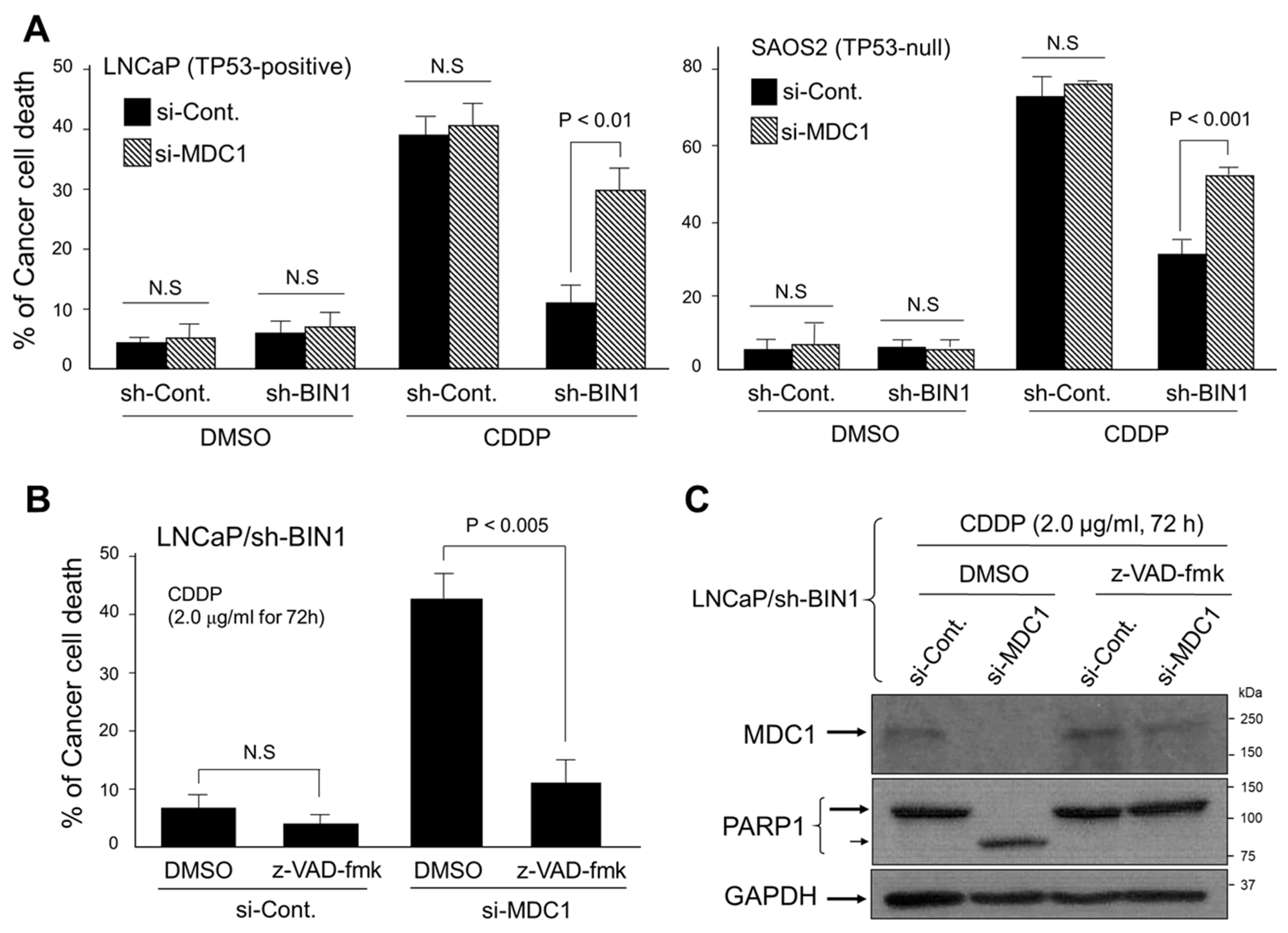
2.1. BIN1 Broadly Retains Cancer-Cell Susceptibility to Cisplatin Regardless of TP53 Statuses
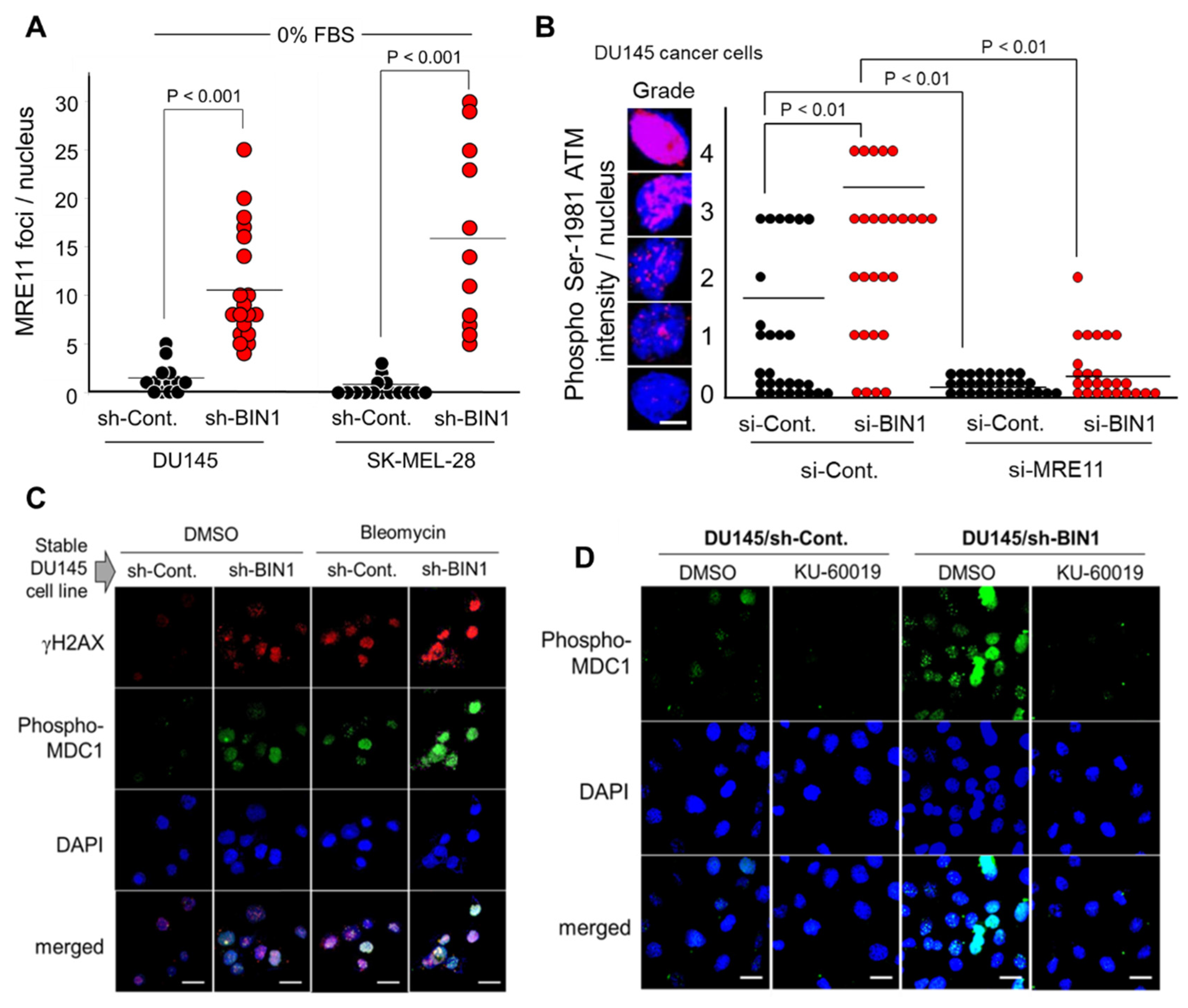
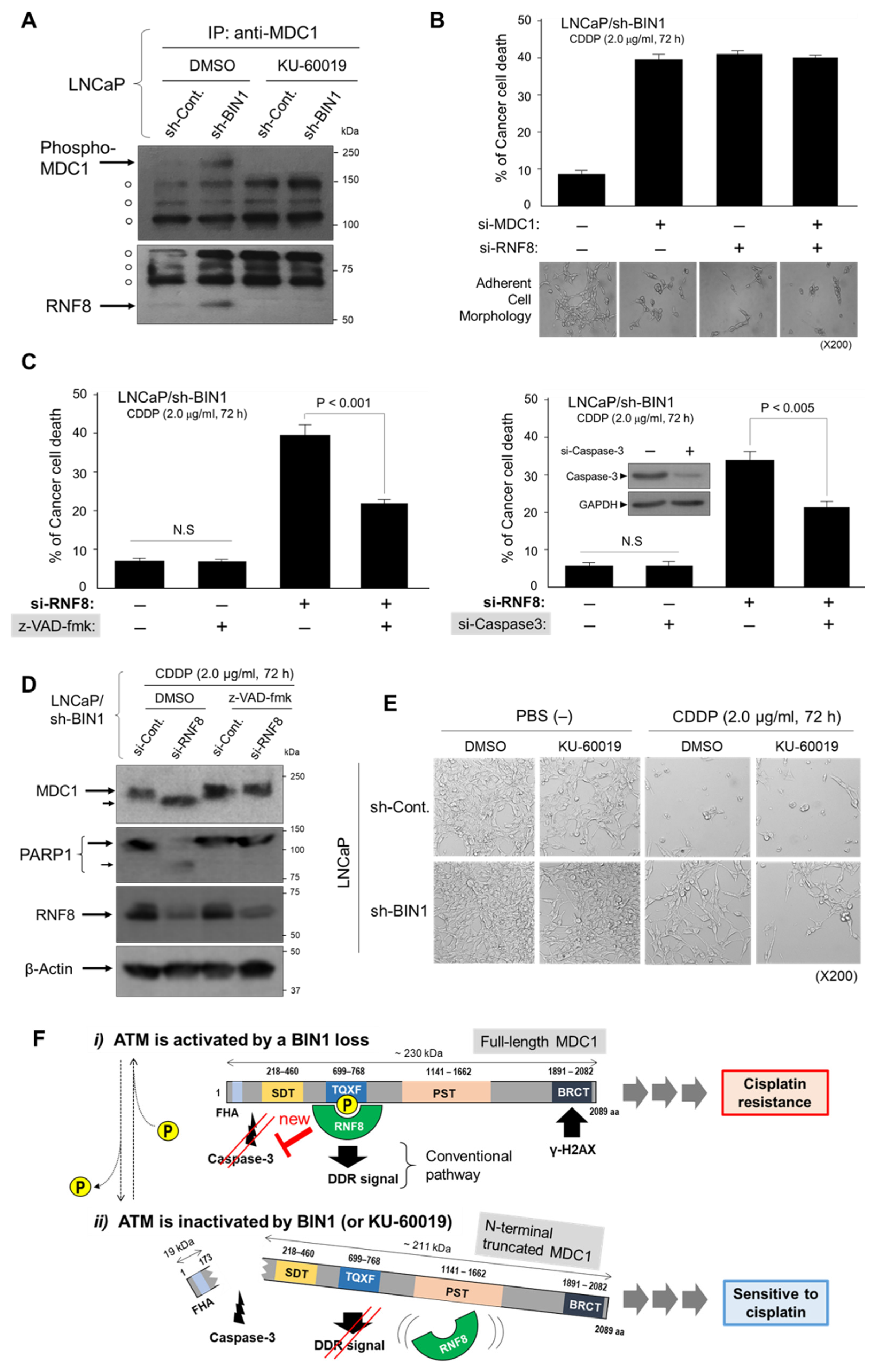
2.2. BIN1 Loss Is Adequate to Initiate and Promote ATM-Dependent DNA Damage Response (DDR)-Like Signals Even in the Absence of DSBs
2.3. MDC1 Sustains Cisplatin Resistance in BIN1-Deficient Cancer Cells Regardless of TP53
2.4. RNF8 Protects ATM-Phosphorylated MDC1 from Caspase-3 and Sustains Cisplatin Resistance in BIN1-Deficient Cancer Cells
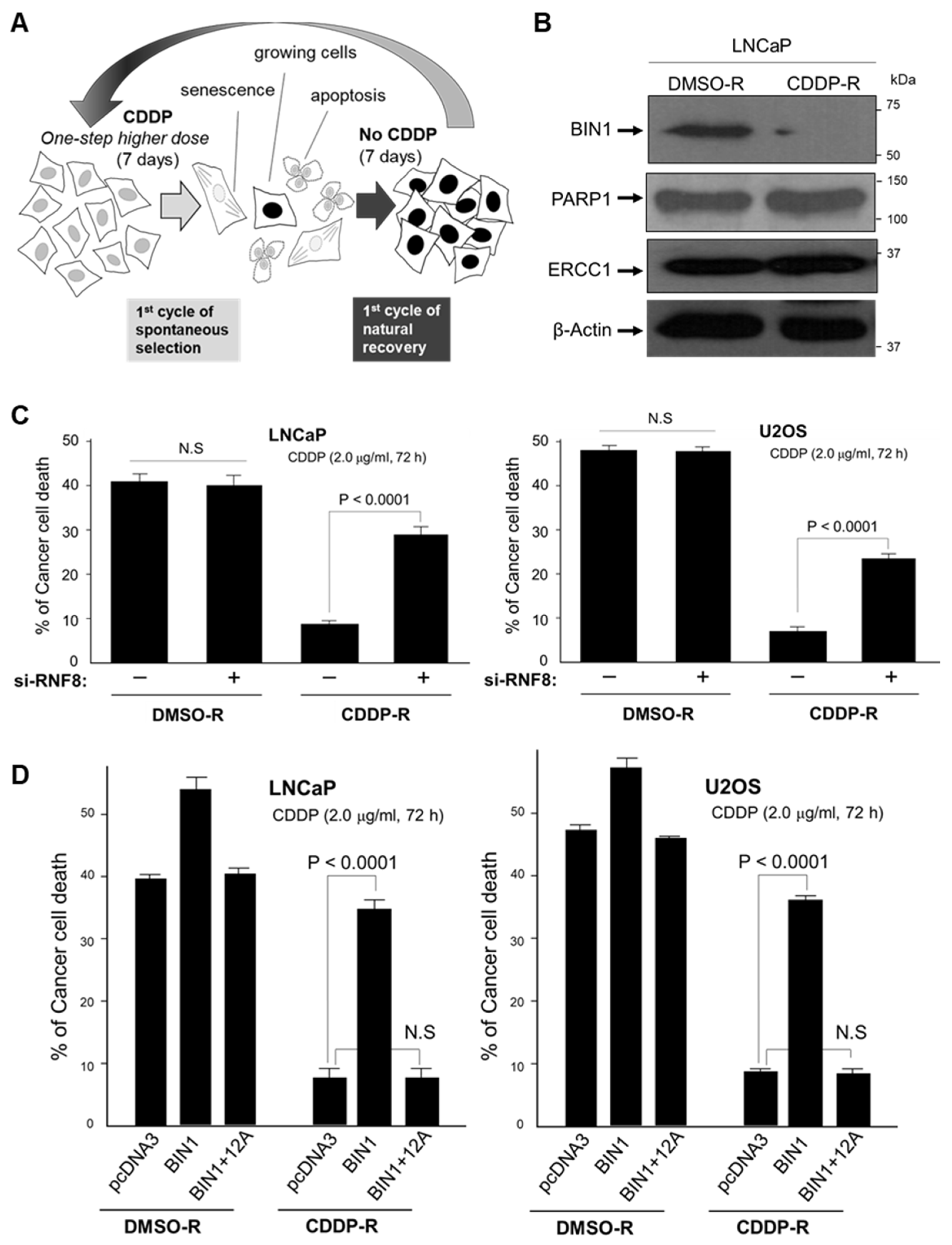
2.5. Long-Term and Repeated Exposure to Cisplatin Spontaneously Recapitulates a BIN1 Loss and Consequent RNF8-Dependent Cisplatin Resistance
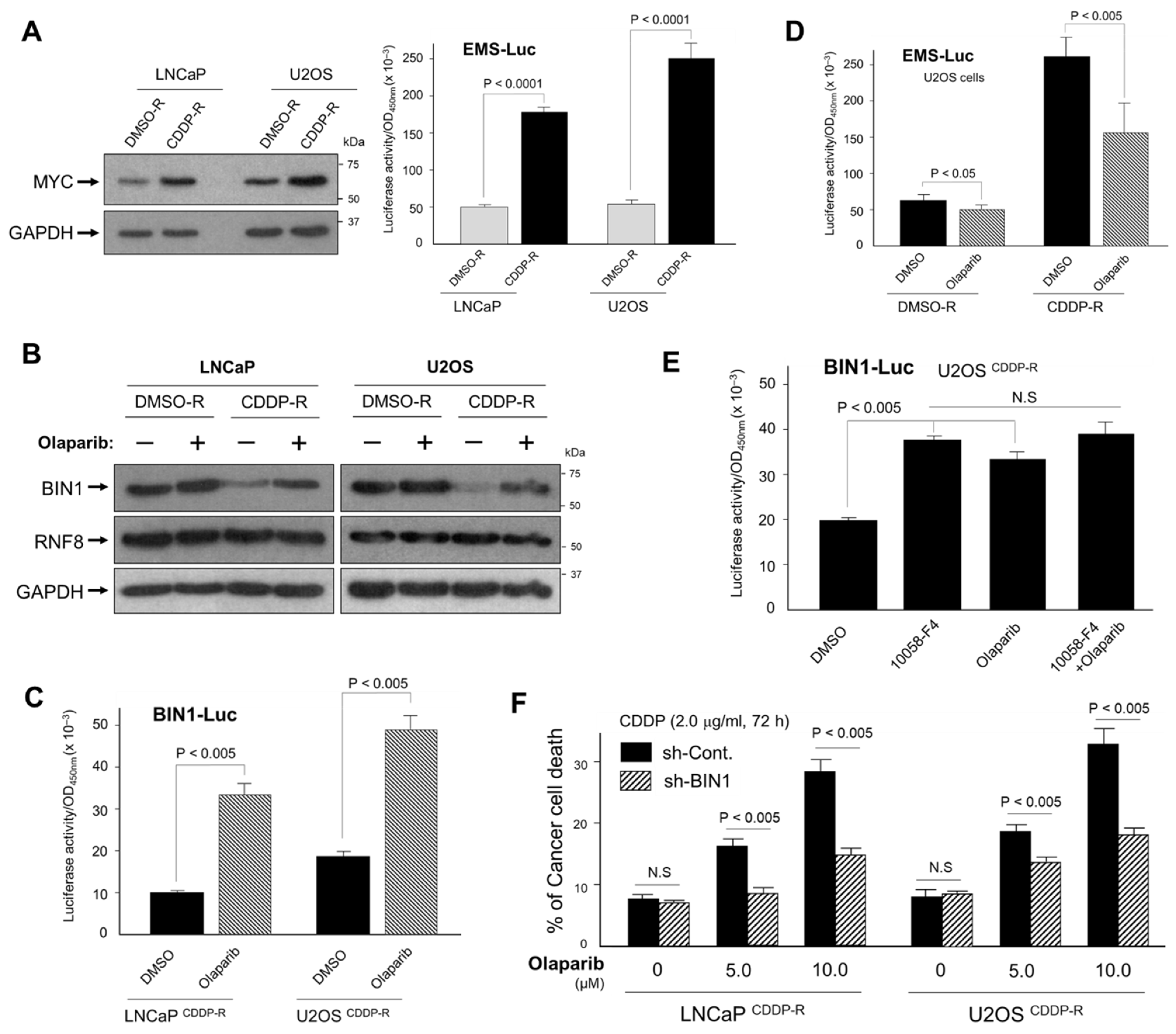
2.6. Inhibition of PARP Activity Reverses Hyperactivated MYC-Dependent BIN1 Gene Repression, Thereby Restoring Cisplatin Sensitivity in the CDDP-R Cell Lines
3. Discussion
4. Materials and Methods
4.1. Mammalian Cell Lines
4.2. Chemicals, Antibodies, and sh-RNAs/si-RNAs
4.3. Plasmid DNAs
4.4. DNA and siRNA Transfection
4.5. Recombinant Lentivirus Infection
4.6. Immunoprecipitation/Western Blot Analysis
4.7. Luciferase Reporter Assay
4.8. Trypan Blue Exclusion Assay
4.9. In Situ Immunofluorescence Microscopy
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Rosenberg, B.; Van Camp, L.; Krigas, T. Inhibition of cell division in Escherichia Coli by electrophoresis products from a platinum electrode. Nature 1965, 205, 698–699. [Google Scholar] [CrossRef]
- Rosenberg, B.; Van Camp, L.; Trosko, J.E.; Mansour, V.H. Platinum compounds: A new class of potent antitumour agents. Nature 1965, 222, 385–386. [Google Scholar] [CrossRef] [PubMed]
- Einhorn, L.H.; Donohue, J. Cis-diamminedichloroplatinum, vinblastine, and bleomycin combination chemotherapy in disseminated testicular cancer. Ann. Intern. Med. 1977, 87, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Einhorn, L.H.; Williams, S.D. The role of cis-platinum in solid-tumor therapy. N. Engl. J. Med. 1979, 300, 289–291. [Google Scholar] [CrossRef]
- Rottenberg, S.; Disler, C.; Perego, P. The rediscovery of platinum-based cancer therapy. Nat. Rev. Cancer 2021, 21, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S. Cisplatin: The first metal based anti-cancer drug. Bioorg. Chem. 2019, 88, 102925. [Google Scholar] [CrossRef]
- Duan, M.; Ulibarri, J.; Liu, K.J.; Mao, P. Role of nucleotide excision repair in cisplatin resistance. Int. J. Mol. Sci. 2021, 21, 9248. [Google Scholar] [CrossRef]
- Cleaver, J.E. Cancer in xeroderma pigmentosum and related disorders of DNA repair. Nat. Rev. Cancer 2005, 5, 564–573. [Google Scholar] [CrossRef]
- Sakamuro, D.; Elliott, K.J.; Wechsler-Reya, R.; Prendergast, G.C. BIN1 is a novel MYC-interacting protein with features of a tumour suppressor. Nat. Genet. 1996, 14, 69–77. [Google Scholar] [CrossRef]
- Elliott, K.; Sakamuro, D.; Basu, A.; Du, W.; Wunner, W.; Staller, P.; Gaubatz, S.; Zhang, H.; Prochownik, E.; Eilers, M.; et al. Bin1 functionally interacts with Myc and inhibits cell proliferation via multiple mechanisms. Oncogene 1999, 18, 3564–3573. [Google Scholar] [CrossRef] [Green Version]
- DuHadaway, J.B.; Sakamuro, D.; Ewert, D.L.; Prendergast, G.C. Bin1 mediates apoptosis by c-Myc in transformed primary cells. Cancer Res. 2001, 61, 3151–3156. [Google Scholar]
- Pineda-Lucena, A.; Ho, C.S.; Mao, D.Y.; Sheng, Y.; Laister, R.C.; Muhandiram, R.; Lu, Y.; Seet, B.T.; Katz, S.; Szyperski, T.; et al. A structure-based model of the c-Myc/Bin1 protein interaction shows alternative splicing of Bin1 and c-Myc phosphorylation are key binding determinants. J. Mol. Biol. 2005, 351, 182–194. [Google Scholar] [CrossRef]
- Kinney, E.L.; Tanida, S.; Rodrigue, A.A.; Johnson, J.K.; Tompkins, V.S.; Sakamuro, D. Adenovirus E1A oncoprotein liberates c-Myc activity to promote cell proliferation through abating Bin1 expression via an Rb/E2F1-dependent mechanism. J. Cell. Physiol. 2008, 216, 621–631. [Google Scholar] [CrossRef]
- Wechsler-Reya, R.; Sakamuro, D.; Zhang, J.; Duhadaway, J.; Prendergast, G.C. Structural analysis of the human BIN1 gene. Evidence for tissue-specific transcriptional regulation and alternate RNA splicing. J. Biol. Chem. 1997, 272, 31453–31458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lundgaard, G.L.; Daniels, N.E.; Pyndiah, S.; Cassimere, E.K.; Ahmed, K.M.; Rodrigue, A.; Kihara, D.; Post, C.B.; Sakamuro, D. Identification of a novel effector domain of BIN1 for cancer suppression. J. Cell. Biochem. 2011, 112, 2992–3001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, K.; Duhadaway, J.; Sakamuro, D.; Wechsler-Reya, R.; Reynolds, C.; Prendergast, G.C. Losses of the tumor suppressor BIN1 in breast carcinoma are frequent and reflect deficits in programmed cell death capacity. Int. J. Cancer 2000, 85, 376–383. [Google Scholar] [CrossRef] [PubMed]
- Ge, K.; Minhas, F.; Duhadaway, J.; Mao, N.C.; Wilson, D.; Buccafusca, R.; Sakamuro, D.; Nelson, P.; Malkowicz, S.B.; Tomaszewski, J.; et al. Loss of heterozygosity and tumor suppressor activity of Bin1 in prostate carcinoma. Int. J. Cancer 2000, 86, 155–161. [Google Scholar] [CrossRef]
- Cassimere, E.K.; Pyndiah, S.; Sakamuro, D. The c-MYC-interacting pro-apoptotic tumor suppressor BIN1 is a transcriptional target for E2F1 in response to DNA damage. Cell. Death Differ. 2009, 16, 1641–1653. [Google Scholar] [CrossRef] [Green Version]
- Pyndiah, S.; Tanida, S.; Ahmed, K.M.; Cassimere, E.K.; Choe, C.; Sakamuro, D. c-MYC suppresses BIN1 to release poly (ADP-ribose) polymerase 1: A mechanism by which cancer cells acquire cisplatin resistance. Sci. Signal 2011, 4, ra19. [Google Scholar] [CrossRef] [PubMed]
- Kumari, A.; Iwasaki, T.; Pyndiah, S.; Cassimere, E.K.; Palani, C.D.; Sakamuro, D. Regulation of E2F1-induced apoptosis by poly (ADP-ribosyl) ation. Cell. Death Differ. 2015, 22, 311–322. [Google Scholar] [CrossRef]
- Folk, W.P.; Kumari, A.; Iwasaki, T.; Pyndiah, S.; Johnson, J.C.; Cassimere, E.K.; Abdulovic-Cui, A.L.; Sakamuro, D. Loss of the tumor suppressor BIN1 enables ATM Ser/Thr kinase activation by the nuclear protein E2F1 and renders cancer cells resistant to cisplatin. J. Biol. Chem. 2019, 294, 5700–5719. [Google Scholar] [CrossRef]
- Vogelstein, B.; Kinzler, K.W. Cancer genes and the pathways they control. Nat. Med 2004, 10, 789–799. [Google Scholar] [CrossRef]
- Pfister, N.T.; Prives, C. Transcriptional regulation by wild-type and cancer-related mutant forms of p53. Cold Spring Harb. Perspect. Med. 2017, 7, a026054. [Google Scholar] [CrossRef] [Green Version]
- Ge, K.; DuHadaway, J.; Du, W.; Herlyn, M.; Rodeck, U.; Prendergast, G.C. Mechanism for elimination of a tumor suppressor: Aberrant splicing of a brain-specific exon causes loss of function of Bin1 in melanoma. Proc. Natl. Acad. Sci. USA 1999, 96, 9689–9694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golding, S.E.; Rosenberg, E.; Valerie, N.; Hussaini, I.; Frigerio, M.; Cockcroft, X.F.; Chong, W.Y.; Hummersone, M.; Rigoreau, L.; Menear, K.A.; et al. Improved ATM kinase inhibitor KU-60019 radiosensitizes glioma cells, compromises insulin, AKT and ERK prosurvival signaling, and inhibits migration and invasion. Mol. Cancer Ther. 2009, 8, 2894–2902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakanishi, M.; Ozaki, T.; Yamamoto, H.; Hanamoto, T.; Kikuchi, H.; Furuya, K.; Asaka, M.; Delia, D.; Nakagawara, A. NFBD1/MDC1 associates with p53 and regulates its function at the crossroad between cell survival and death in response to DNA damage. J. Biol. Chem. 2007, 282, 22993–23004. [Google Scholar] [CrossRef] [Green Version]
- Soldani, C.; Scovassi, A.I. Poly (ADP-ribose) polymerase-1 cleavage during apoptosis: An update. Apoptosis 2002, 7, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Jungmichel, S.; Stucki, M. MDC1: The art of keeping things in focus. Chromosoma 2010, 119, 337–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solier, S.; Pommier, Y. MDC1 cleavage by caspase-3: A novel mechanism for inactivating the DNA damage response during apoptosis. Cancer Res. 2011, 71, 906–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pines, A.; Vrouwe, M.G.; Marteijn, J.A.; Typas, D.; Luijsterburg, M.S.; Cansoy, M.; Hensbergen, P.; Deelder, A.; de Groot, A.; Matsumoto, S.; et al. PARP1 promotes nucleotide excision repair through DDB2 stabilization and recruitment of ALC1. J. Cell. Biol. 2012, 199, 235–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reed, E. Platinum-DNA adduct, nucleotide excision repair and platinum based anti-cancer chemotherapy. Cancer Treat. Rev. 1998, 24, 331–344. [Google Scholar] [CrossRef]
- Van Waardenburg, R.C.; Meijer, C.; Burger, H.; Nooter, K.; De Vries, E.G.; Mulder, N.H.; De Jong, S. Effects of an inducible anti-sense c-myc gene transfer in a drug-resistant human small-cell-lung-carcinoma cell line. Int. J. Cancer 1997, 73, 544–550. [Google Scholar] [CrossRef]
- Leonetti, C.; Biroccio, A.; Candiloro, A.; Citro, G.; Fornari, C.; Mottolese, M.; Del Bufalo, D.; Zupi, G. Increase of cisplatin sensitivity by c-myc anti-sense oligodeoxynucleotides in a human metastatic melanoma inherently resistant to cisplatin. Clin. Cancer Res. 1999, 5, 2588–2595. [Google Scholar] [PubMed]
- Biroccio, A.; Benassi, B.; Amodei, S.; Gabellini, C.; Del Bufalo, D.; Zupi, G. c-Myc down-regulation increases susceptibility to cisplatin through reactive oxygen species-mediated apoptosis in M14 human melanoma cells. Mol. Pharmacol. 2001, 60, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Reyes-González, J.M.; Armaiz-Peña, G.N.; Mangala, L.S.; Valiyeva, F.; Ivan, C.; Pradeep, S.; Echevarría-Vargas, I.M.; Rivera-Reyes, A.; Sood, A.K.; Vivas-Mejía, P.E. Targeting c-MYC in platinum-resistant ovarian cancer. Mol. Cancer Ther. 2015, 14, 2260–2269. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Cai, H.; Liang, Y.; Chen, L.; Wang, X.; Si, R.; Qu, K.; Jiang, Z.; Ma, B.; Miao, C.; et al. Inhibition of c-Myc by let-7b mimic reverses multidrug resistance in gastric cancer cells. Oncol. Rep. 2015, 33, 1723–1730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumari, A.; Folk, W.P.; Sakamuro, D. The dual roles of MYC in genomic instability and cancer chemoresistance. Genes 2017, 8, 158. [Google Scholar] [CrossRef] [Green Version]
- Zanellato, I.; Colangelo, D.; Osella, D. JQ1, a BET inhibitor, synergizes with cisplatin and induces apoptosis in highly chemoresistant malignant pleural mesothelioma cells. Curr. Cancer Drug Targets 2018, 18, 816–828. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.J.; Cheng, Y.C.; Liu, C.R.; Lin, S.; Liu, H.E. A small-molecule c-Myc inhibitor, 10058-F4, induces cell-cycle arrest, apoptosis, and myeloid differentiation of human acute myeloid leukemia. Exp. Hematol. 2006, 34, 1480–1489. [Google Scholar] [CrossRef]
- Wang, H.; Hammoudeh, D.I.; Follis, A.V.; Reese, B.E.; Lazo, J.S.; Metallo, S.J.; Prochownik, E.V. Improved low molecular weight Myc-Max inhibitors. Mol. Cancer Ther. 2007, 6, 2399–2408. [Google Scholar] [CrossRef] [Green Version]
- Michels, J.; Vitale, I.; Galluzzi, L.; Adam, J.; Olaussen, K.A.; Kepp, O.; Senovilla, L.; Talhaoui, I.; Guegan, J.; Enot, D.P.; et al. Cisplatin resistance associated with PARP hyperactivation. Cancer Res. 2013, 73, 2271–2280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michels, J.; Vitale, I.; Senovilla, L.; Enot, D.P.; Garcia, P.; Lissa, D.; Olaussen, K.A.; Brenner, C.; Soria, J.C.; Castedo, M.; et al. Synergistic interaction between cisplatin and PARP inhibitors in non-small cell lung cancer. Cell Cycle 2013, 12, 877–883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalkat, M.; De Melo, J.; Hickman, K.A.; Lourenco, C.; Redel, C.; Resetca, D.; Tamachi, A.; Tu, W.B.; Penn, L.Z. MYC deregulation in primary human cancers. Genes 2017, 8, 151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mota, J.M.; Barnett, E.; Nauseef, J.T.; Nguyen, B.; Stopsack, K.H.; Wibmer, A.; Flynn, J.R.; Heller, G.; Danila, D.C.; Rathkopf, D.; et al. Platinum-based chemotherapy in metastatic prostate cancer with DNA repair gene alterations. JCO Precis. Oncol. 2020, 4, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.; Tutt, A.; Ashworth, A. Hallmarks of ‘BRCAness’ in sporadic cancers. Nat. Rev. Cancer 2004, 4, 814–819. [Google Scholar] [CrossRef]
- Helleday, T. The underlying mechanism for the PARP and BRCA synthetic lethality: Clearing up the misunderstandings. Mol. Oncol. 2011, 5, 387–393. [Google Scholar] [CrossRef] [Green Version]
- Lord, C.J.; Tutt, A.N.; Ashworth, A. Synthetic lethality and cancer therapy: Lessons learned from the development of PARP inhibitors. Annu. Rev. Med. 2015, 66, 455–470. [Google Scholar] [CrossRef]
- Larsen, M.J.; Thomassen, M.; Gerdes, A.M.; Kruse, T.A. Hereditary breast cancer: Clinical, pathological and molecular characteristics. Breast Cancer 2014, 8, 145–155. [Google Scholar] [CrossRef] [Green Version]
- Ning, J.F.; Stanciu, M.; Humphrey, M.R.; Gorham, J.; Wakimoto, H.; Nishihara, R.; Lees, J.; Zou, L.; Martuza, R.L.; Wakimoto, H.; et al. Myc targeted CDK18 promotes ATR and homologous recombination to mediate PARP inhibitor resistance in glioblastoma. Nat. Commun. 2019, 10, 2910. [Google Scholar] [CrossRef]
- Wang, J.; Ding, Q.; Fujimori, H.; Motegi, A.; Miki, Y.; Masutani, M. Loss of CtIP disturbs homologous recombination repair and sensitizes breast cancer cells to PARP inhibitors. Oncotarget 2016, 7, 7701–7714. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Folk, W.P.; Kumari, A.; Iwasaki, T.; Cassimere, E.K.; Pyndiah, S.; Martin, E.; Homlar, K.; Sakamuro, D. New Synthetic Lethality Re-Sensitizing Platinum-Refractory Cancer Cells to Cisplatin In Vitro: The Rationale to Co-Use PARP and ATM Inhibitors. Int. J. Mol. Sci. 2021, 22, 13324. https://doi.org/10.3390/ijms222413324
Folk WP, Kumari A, Iwasaki T, Cassimere EK, Pyndiah S, Martin E, Homlar K, Sakamuro D. New Synthetic Lethality Re-Sensitizing Platinum-Refractory Cancer Cells to Cisplatin In Vitro: The Rationale to Co-Use PARP and ATM Inhibitors. International Journal of Molecular Sciences. 2021; 22(24):13324. https://doi.org/10.3390/ijms222413324
Chicago/Turabian StyleFolk, Watson P., Alpana Kumari, Tetsushi Iwasaki, Erica K. Cassimere, Slovénie Pyndiah, Elizabeth Martin, Kelly Homlar, and Daitoku Sakamuro. 2021. "New Synthetic Lethality Re-Sensitizing Platinum-Refractory Cancer Cells to Cisplatin In Vitro: The Rationale to Co-Use PARP and ATM Inhibitors" International Journal of Molecular Sciences 22, no. 24: 13324. https://doi.org/10.3390/ijms222413324