
Risk Factors for Retinal Ganglion Cell Distress in Glaucoma and Neuroprotective Potential Intervention


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
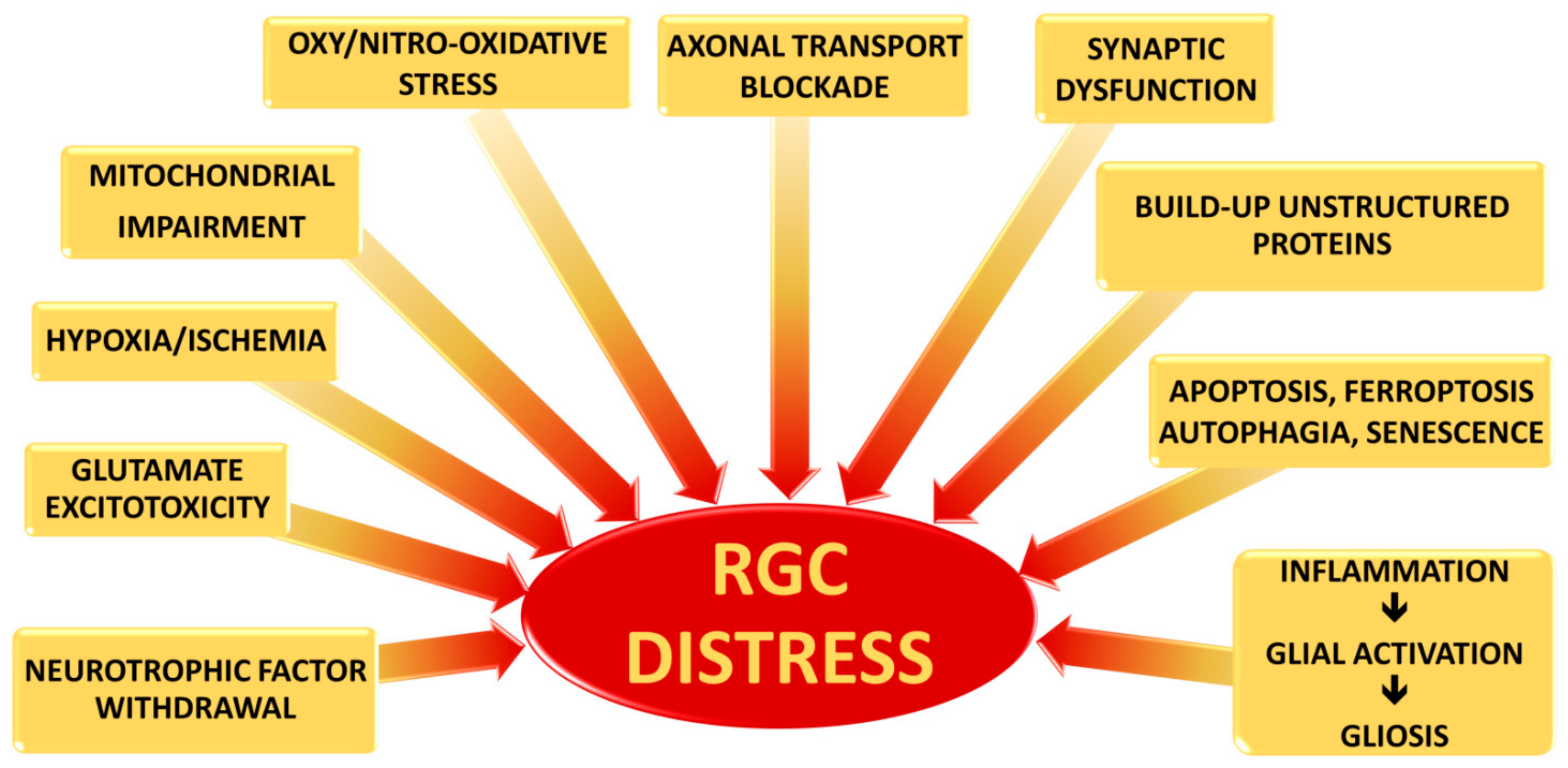
2. Retinal Ganglion Cells
2.1. Axonal Transport Blockade
2.2. Glutamate Excitotoxicity
2.3. Changes in Pro-Inflammatory Cytokine along the RGC Projection
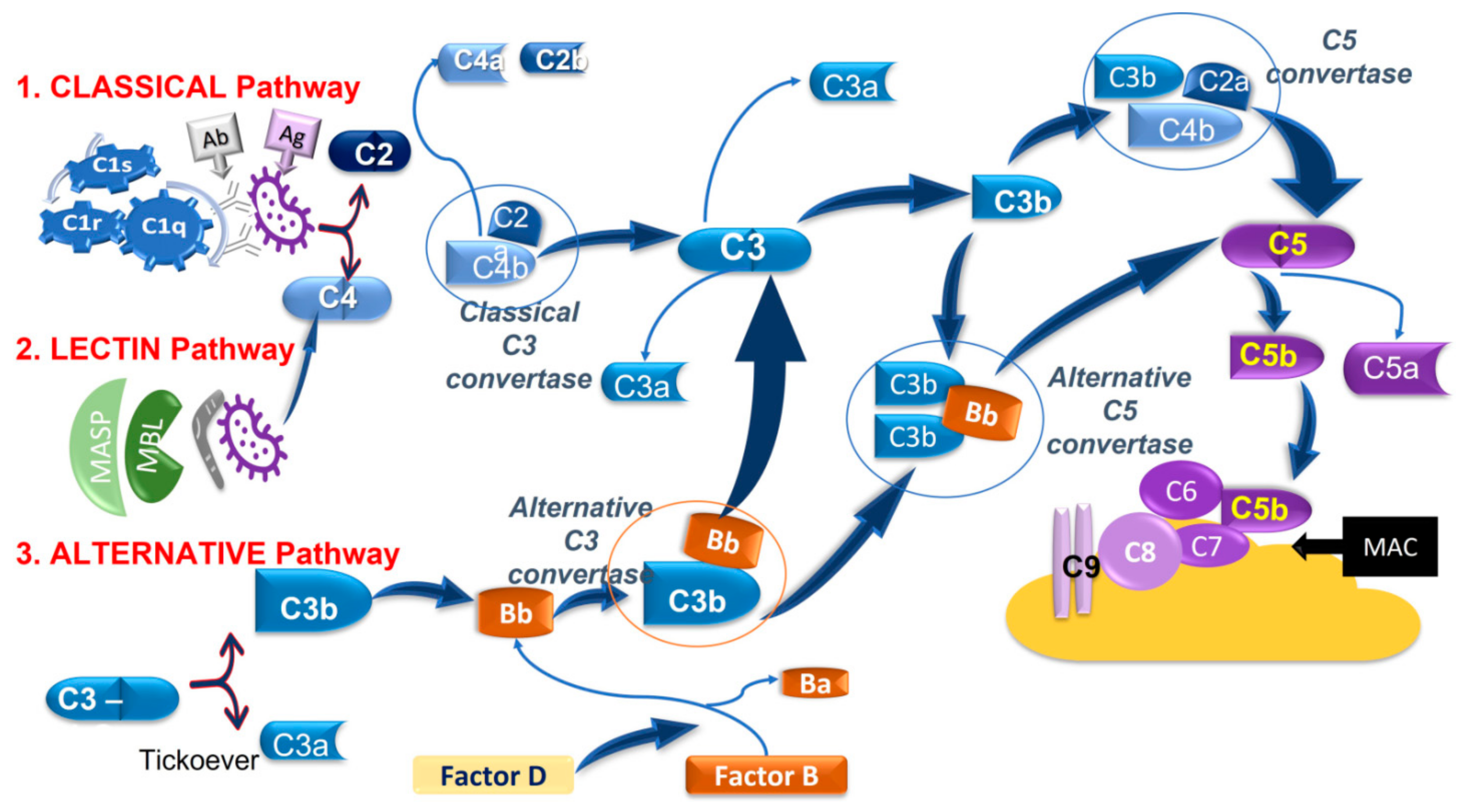
- (1)
- The classical pathwayis activated by the interaction between the antibody-antigen (Ab-Ag, respectively) and C1-complex, which consists of C1q, C1r, and C1s. This interaction leads to the cleavage of C4 and C2 and the complex C4bC2a to form the C3 convertase which cleaves C3 into C3b and C3a. Then C3b associates with C4bC2a to give rise to the C5 convertase which, in turn, cleaves C5 in C5a and C5b.C3a and C5a are anaphylatoxins, acting as vasoactive and chemotactic factors, while C3b is an opsonin inducing phagocytosis. C5b interacts and activates other complement components, namely C6, C7, C8, and C9 to form the membrane-attack-complex (MAC), which lyses targeted surfaces.
- (2)
- The lectinpathwayis activated by the mannose-binding lectin (MBL) recognition of pathogenic carbohydrate motifs. The complex MBL-associated serine protease (MASP) cleaves C2 and C4 and generates the C3 convertase which then merge at the subsequent step of the classical pathway.
- (3)
- The alternativepathwaystarts from the spontaneous hydrolysis of C3 to the C3b analog, C3(H2O), which, in the presence of Factors B and D, forms an alternative C3 convertase, namely C3(H2O)Bb, which converts C3 into C3b and C3a, acting as the C3 convertase of the classical and lectin pathways. The alternative pathway can also contribute to forming a C5 convertase (C3bBbC3b) merging at C5 of the classical and lectin pathways [129,130].
3. Neuroprotection
3.1. Müller Glia and Neuroprotection
3.2. Neuroprotective Agents in Glaucoma Management
3.2.1. Antioxidants
3.2.2. Neurotrophic Factors (NTFs)
3.2.3. Novel Neuroprotective Agents
4. Conclusions
Funding
Conflicts of Interest
References
- You, M.; Rong, R.; Zeng, Z.; Xia, X.; Ji, D. Transneuronal Degeneration in the Brain During Glaucoma. Front. Aging Neurosci. 2021, 13, 643685. [Google Scholar] [CrossRef]
- Quigley, H.A.; Broman, A.T. The Number of People with Glaucoma Worldwide in 2010 and 2020. Br. J. Ophthalmol. 2006, 90, 262–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kingman, S. Glaucoma Is Second Leading Cause of Blindness Globally. Bull. World Health Organ. 2004, 82, 887–888. [Google Scholar]
- Calkins, D.J. Adaptive Responses to Neurodegenerative Stress in Glaucoma. Prog. Retin. Eye Res. 2021, 100953. [Google Scholar] [CrossRef]
- Almasieh, M.; Wilson, A.M.; Morquette, B.; Cueva Vargas, J.L.; Di Polo, A. The Molecular Basis of Retinal Ganglion Cell Death in Glaucoma. Prog. Retin. Eye Res. 2012, 31, 152–181. [Google Scholar] [CrossRef] [PubMed]
- King, A.; Azuara-Blanco, A.; Tuulonen, A. Glaucoma. BMJ 2013, 346, f3518. [Google Scholar] [CrossRef]
- Quaranta, L.; Bruttini, C.; Micheletti, E.; Konstas, A.G.P.; Michelessi, M.; Oddone, F.; Katsanos, A.; Sbardella, D.; De Angelis, G.; Riva, I. Glaucoma and Neuroinflammation: An Overview. Surv. Ophthalmol. 2021, 66, 693–713. [Google Scholar] [CrossRef] [PubMed]
- Gordon, M.O. The Ocular Hypertension Treatment Study: Baseline Factors That Predict the Onset of Primary Open-Angle Glaucoma. Arch. Ophthalmol. 2002, 120, 714. [Google Scholar] [CrossRef] [PubMed]
- Friedman, D.S.; Wilson, M.R.; Liebmann, J.M.; Fechtner, R.D.; Weinreb, R.N. An Evidence-Based Assessment of Risk Factors for the Progression of Ocular Hypertension and Glaucoma. Am. J. Ophthalmol. 2004, 138, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Fahmy, I.A.; Amer, A.K.; EL-Ghaffar, N.A. The Role of T-Cell Subsets and Natural Killer Lymphocytes in the Pathogenesis of Primary Open Angle Glaucoma. Maced. J. Med. Sci. 2010, 3, 307–313. [Google Scholar] [CrossRef]
- Susanna, R.; Moraes, C.G.D.; Cioffi, G.A.; Ritch, R. Why Do People (Still) Go Blind from Glaucoma? Transl. Vis. Sci. Technol. 2015, 4. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y. Retinal Ganglion Cell-Derived Sonic Hedgehog Locally Controls Proliferation and the Timing of RGC Development in the Embryonic Mouse Retina. Development 2005, 132, 5103–5113. [Google Scholar] [CrossRef] [Green Version]
- Dakubo, G.D. Retinal Ganglion Cell-Derived Sonic Hedgehog Signaling Is Required for Optic Disc and Stalk Neuroepithelial Cell Development. Development 2003, 130, 2967–2980. [Google Scholar] [CrossRef] [Green Version]
- Miesfeld, J.B.; Ghiasvand, N.M.; Marsh-Armstrong, B.; Marsh-Armstrong, N.; Miller, E.B.; Zhang, P.; Manna, S.K.; Zawadzki, R.J.; Brown, N.L.; Glaser, T. The Atoh7 Remote Enhancer Provides Transcriptional Robustness during Retinal Ganglion Cell Development. Proc. Natl. Acad. Sci. USA 2020, 117, 21690–21700. [Google Scholar] [CrossRef]
- Wang, Y.P.; Dakubo, G.; Howley, P.; Campsall, K.D.; Mazarolle, C.J.; Shiga, S.A.; Lewis, P.M.; McMahon, A.P.; Wallace, V.A. Development of Normal Retinal Organization Depends on Sonic Hedgehog Signaling from Ganglion Cells. Nat. Neurosci. 2002, 5, 831–832. [Google Scholar] [CrossRef] [PubMed]
- Bisiak, F.; McCarthy, A.A. Structure and Function of Roundabout Receptors. In Macromolecular Protein Complexes II: Structure and Function; Harris, J.R., Marles-Wright, J., Eds.; Subcellular Biochemistry; Springer International Publishing: Cham, Switzerland, 2019; Volume 93, pp. 291–319. ISBN 978-3-030-28150-2. [Google Scholar]
- O’Sullivan, M.L.; Puñal, V.M.; Kerstein, P.C.; Brzezinski, J.A.; Glaser, T.; Wright, K.M.; Kay, J.N. Astrocytes Follow Ganglion Cell Axons to Establish an Angiogenic Template during Retinal Development. Glia 2017, 65, 1697–1716. [Google Scholar] [CrossRef]
- Mead, B.; Tomarev, S. Evaluating Retinal Ganglion Cell Loss and Dysfunction. Exp. Eye Res. 2016, 151, 96–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duarte, J.N. Neuroinflammatory Mechanisms of Mitochondrial Dysfunction and Neurodegeneration in Glaucoma. J. Ophthalmol. 2021, 2021, 4581909. [Google Scholar] [CrossRef]
- Ito, Y.A.; Di Polo, A. Mitochondrial Dynamics, Transport, and Quality Control: A Bottleneck for Retinal Ganglion Cell Viability in Optic Neuropathies. Mitochondrion 2017, 36, 186–192. [Google Scholar] [CrossRef]
- Saccà, S.C.; Paluan, F.; Gandolfi, S.; Manni, G.; Cutolo, C.A.; Izzotti, A. Common Aspects between Glaucoma and Brain Neurodegeneration. Mutat. Res. Mutat. Res. 2020, 786, 108323. [Google Scholar] [CrossRef]
- You, Y.; Gupta, V.K.; Li, J.C.; Klistorner, A.; Graham, S.L. Optic Neuropathies: Characteristic Features and Mechanisms of Retinal Ganglion Cell Loss. Rev. Neurosci. 2013, 24. [Google Scholar] [CrossRef]
- Sharif, N. Glaucomatous Optic Neuropathy Treatment Options: The Promise of Novel Therapeutics, Techniques and Tools to Help Preserve Vision. Neural Regen. Res. 2018, 13, 1145. [Google Scholar] [CrossRef]
- Millecamps, S.; Julien, J.-P. Axonal Transport Deficits and Neurodegenerative Diseases. Nat. Rev. Neurosci. 2013, 14, 161–176. [Google Scholar] [CrossRef]
- Perlson, E.; Maday, S.; Fu, M.; Moughamian, A.J.; Holzbaur, E.L.F. Retrograde Axonal Transport: Pathways to Cell Death? Trends Neurosci. 2010, 33, 335–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lasek, R.J.; Garner, J.A.; Brady, S.T. Axonal Transport of the Cytoplasmic Matrix. J. Cell Biol. 1984, 99, 212s–221s. [Google Scholar] [CrossRef] [PubMed]
- Morgan, J.E. Circulation and Axonal Transport in the Optic Nerve. Eye 2004, 18, 1089–1095. [Google Scholar] [CrossRef]
- De Vos, K.J.; Grierson, A.J.; Ackerley, S.; Miller, C.C.J. Role of Axonal Transport in Neurodegenerative Diseases. Annu. Rev. Neurosci. 2008, 31, 151–173. [Google Scholar] [CrossRef]
- Fahy, E.T.; Chrysostomou, V.; Crowston, J.G. Impaired Axonal Transport and Glaucoma. Curr. Eye Res. 2015, 1–11. [Google Scholar] [CrossRef]
- Hirokawa, N.; Noda, Y.; Tanaka, Y.; Niwa, S. Kinesin Superfamily Motor Proteins and Intracellular Transport. Nat. Rev. Mol. Cell Biol. 2009, 10, 682–696. [Google Scholar] [CrossRef]
- LaMonte, B.H.; Wallace, K.E.; Holloway, B.A.; Shelly, S.S.; Ascaño, J.; Tokito, M.; Van Winkle, T.; Howland, D.S.; Holzbaur, E.L.F. Disruption of Dynein/Dynactin Inhibits Axonal Transport in Motor Neurons Causing Late-Onset Progressive Degeneration. Neuron 2002, 34, 715–727. [Google Scholar] [CrossRef] [Green Version]
- Hafezparast, M. Mutations in Dynein Link Motor Neuron Degeneration to Defects in Retrograde Transport. Science 2003, 300, 808–812. [Google Scholar] [CrossRef]
- Saha, A.R. Parkinson’s Disease -Synuclein Mutations Exhibit Defective Axonal Transport in Cultured Neurons. J. Cell Sci. 2004, 117, 1017–1024. [Google Scholar] [CrossRef] [Green Version]
- Trushina, E.; Dyer, R.B.; Badger, J.D.; Ure, D.; Eide, L.; Tran, D.D.; Vrieze, B.T.; Legendre-Guillemin, V.; McPherson, P.S.; Mandavilli, B.S.; et al. Mutant Huntingtin Impairs Axonal Trafficking in Mammalian Neurons In Vivo and In Vitro. Mol. Cell. Biol. 2004, 24, 8195–8209. [Google Scholar] [CrossRef] [Green Version]
- Dengler-Crish, C.M.; Smith, M.A.; Inman, D.M.; Wilson, G.N.; Young, J.W.; Crish, S.D. Anterograde Transport Blockade Precedes Deficits in Retrograde Transport in the Visual Projection of the DBA/2J Mouse Model of Glaucoma. Front. Neurosci. 2014, 8. [Google Scholar] [CrossRef] [Green Version]
- Hirooka, K.; Yamamoto, T.; Kiuchi, Y. Dysfunction of Axonal Transport in Normal-Tension Glaucoma: A Biomarker of Disease Progression and a Potential Therapeutic Target. Neural Regen. Res. 2021, 16, 506. [Google Scholar] [CrossRef]
- Ip, N.Y.; Yancopoulos, G.D. The Neurotrophins and CNTF: Two Families of Collaborative Neurotrophic Factors. Annu. Rev. Neurosci. 1996, 19, 491–515. [Google Scholar] [CrossRef]
- Kimura, A.; Namekata, K.; Guo, X.; Harada, C.; Harada, T. Neuroprotection, Growth Factors and BDNF-TrkB Signalling in Retinal Degeneration. Int. J. Mol. Sci. 2016, 17, 1584. [Google Scholar] [CrossRef] [Green Version]
- Bibel, M. Neurotrophins: Key Regulators of Cell Fate and Cell Shape in the Vertebrate Nervous System. Genes Dev. 2000, 14, 2919–2937. [Google Scholar] [CrossRef] [Green Version]
- Platholi, J.; Lee, F.S. Neurotrophic Factors. In Handbook of Developmental Neurotoxicology; Elsevier: Amsterdam, The Netherlands, 2018; pp. 55–64. ISBN 978-0-12-809405-1. [Google Scholar]
- Levi-Montalcini, R. The Nerve Growth Factor: Its Mode of Action on Sensory and Sympathetic Nerve Cells. Harvey Lect. 1966, 60, 217–259. [Google Scholar] [PubMed]
- Levi-Montalcini, R.; Hamburger, V. A Diffusible Agent of Mouse Sarcoma, Producing Hyperplasia of Sympathetic Ganglia and Hyperneurotization of Viscera in the Chick Embryo. J. Exp. Zool. 1953, 123, 233–287. [Google Scholar] [CrossRef]
- Barde, Y.A.; Edgar, D.; Thoenen, H. Purification of a New Neurotrophic Factor from Mammalian Brain. EMBO J. 1982, 1, 549–553. [Google Scholar] [CrossRef]
- Maisonpierre, P.C.; Belluscio, L.; Friedman, B.; Alderson, R.F.; Wiegand, S.J.; Furth, M.E.; Lindsay, R.M.; Yancopoulos, G.D. NT-3, BDNF, and NGF in the Developing Rat Nervous System: Parallel as Well as Reciprocal Patterns of Expression. Neuron 1990, 5, 501–509. [Google Scholar] [CrossRef]
- Ip, N.Y.; Ibanez, C.F.; Nye, S.H.; McClain, J.; Jones, P.F.; Gies, D.R.; Belluscio, L.; Le Beau, M.M.; Espinosa, R.; Squinto, S.P. Mammalian Neurotrophin-4: Structure, Chromosomal Localization, Tissue Distribution, and Receptor Specificity. Proc. Natl. Acad. Sci. USA 1992, 89, 3060–3064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lessmann, V.; Gottmann, K.; Malcangio, M. Neurotrophin Secretion: Current Facts and Future Prospects. Prog. Neurobiol. 2003, 69, 341–374. [Google Scholar] [CrossRef]
- Chao, M.V. Neurotrophins and Their Receptors: A Convergence Point for Many Signalling Pathways. Nat. Rev. Neurosci. 2003, 4, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Hempstead, B. The Many Faces of P75NTR. Curr. Opin. Neurobiol. 2002, 12, 260–267. [Google Scholar] [CrossRef]
- Ibáñez, C.F.; Simi, A. P75 Neurotrophin Receptor Signaling in Nervous System Injury and Degeneration: Paradox and Opportunity. Trends Neurosci. 2012, 35, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Reichardt, L.F. Neurotrophin-Regulated Signalling Pathways. Philos. Trans. R. Soc. B Biol. Sci. 2006, 361, 1545–1564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fathy, M.; Darweesh, A.M.; Sharaf, S.; El-Hanafi, H.M.; Ghaleb, F.M.; Fahmy, I.A.; Hussein, S.M. Brain-Derived Neurotrophic Factor (BDNF) Gene Polymorphism in a Cohort of Egyptian Primary Open-Angle Glaucoma (POAG) Patients. Bull. Natl. Res. Cent. 2020, 44, 45. [Google Scholar] [CrossRef]
- Johnson, T.V.; Bull, N.D.; Martin, K.R. Neurotrophic Factor Delivery as a Protective Treatment for Glaucoma. Exp. Eye Res. 2011, 93, 196–203. [Google Scholar] [CrossRef]
- Martin, K.R.G.; Quigley, H.A.; Zack, D.J.; Levkovitch-Verbin, H.; Kielczewski, J.; Valenta, D.; Baumrind, L.; Pease, M.E.; Klein, R.L.; Hauswirth, W.W. Gene Therapy with Brain-Derived Neurotrophic Factor As a Protection: Retinal Ganglion Cells in a Rat Glaucoma Model. Investig. Opthalmol. Vis. Sci. 2003, 44, 4357. [Google Scholar] [CrossRef] [PubMed]
- Pease, M.E.; McKinnon, S.J.; Quigley, H.A.; Kerrigan-Baumrind, L.A.; Zack, D.J. Obstructed Axonal Transport of BDNF and Its Receptor TrkB in Experimental Glaucoma. Investig. Ophthalmol. Vis. Sci. 2000, 41, 764–774. [Google Scholar]
- Iwabe, S.; Moreno-Mendoza, N.A.; Trigo-Tavera, F.; Crowder, C.; García-Sánchez, G.A. Retrograde Axonal Transport Obstruction of Brain-Derived Neurotrophic Factor (BDNF) and Its TrkB Receptor in the Retina and Optic Nerve of American Cocker Spaniel Dogs with Spontaneous Glaucoma. Vet. Ophthalmol. 2007, 10, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Ko, M.-L.; Hu, D.-N.; Ritch, R.; Sharma, S.C.; Chen, C.-F. Patterns of Retinal Ganglion Cell Survival after Brain-Derived Neurotrophic Factor Administration in Hypertensive Eyes of Rats. Neurosci. Lett. 2001, 305, 139–142. [Google Scholar] [CrossRef]
- Di Polo, A.; Aigner, L.J.; Dunn, R.J.; Bray, G.M.; Aguayo, A.J. Prolonged Delivery of Brain-Derived Neurotrophic Factor by Adenovirus-Infected Muller Cells Temporarily Rescues Injured Retinal Ganglion Cells. Proc. Natl. Acad. Sci. USA 1998, 95, 3978–3983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coassin, M.; Lambiase, A.; Sposato, V.; Micera, A.; Bonini, S.; Aloe, L. Retinal P75 and Bax Overexpression Is Associated with Retinal Ganglion Cells Apoptosis in a Rat Model of Glaucoma. Graefes Arch. Clin. Exp. Ophthalmol. 2008, 246, 1743–1749. [Google Scholar] [CrossRef]
- Sposato, V.; Bucci, M.G.; Coassin, M.; Russo, M.A.; Lambiase, A.; Aloe, L. Reduced NGF Level and TrkA Protein and TrkA Gene Expression in the Optic Nerve of Rats with Experimentally Induced Glaucoma. Neurosci. Lett. 2008, 446, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Johnson, E.; Cepurna, W.; Jia, L.; Dyck, J.; Morrison, J.C. Does Elevated Intraocular Pressure Reduce Retinal TRKB-Mediated Survival Signaling in Experimental Glaucoma? Exp. Eye Res. 2009, 89, 921–933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walton, K.M. GDNF: A Novel Factor with Therapeutic Potential for Neurodegenerative Disorders. Mol. Neurobiol. 1999, 19, 43–59. [Google Scholar] [CrossRef]
- Kramer, E.R.; Liss, B. GDNF-Ret Signaling in Midbrain Dopaminergic Neurons and Its Implication for Parkinson Disease. FEBS Lett. 2015, 589, 3760–3772. [Google Scholar] [CrossRef] [PubMed]
- Budni, J.; Bellettini-Santos, T.; Mina, F.; Lima Garcez, M.; Ioppi Zugno, A. The Involvement of BDNF, NGF and GDNF in Aging and Alzheimer’s Disease. Aging Dis. 2015, 6, 331. [Google Scholar] [CrossRef] [Green Version]
- Frasson, M.; Picaud, S.; Léveillard, T.; Simonutti, M.; Mohand–Said, S.; Dreyfus, H.; Hicks, D.; Sahel, J. Glial Cell Line–Derived Neurotrophic Factor Induces Histologic and Functional Protection of Rod Photoreceptors in the Rd/Rd Mouse. Investig. Ophthalmol. Vis. Sci. 1999, 40, 2724–2734. [Google Scholar]
- Harada, C.; Harada, T.; Quah, H.-M.A.; Maekawa, F.; Yoshida, K.; Ohno, S.; Wada, K.; Parada, L.F.; Tanaka, K. Potential Role of Glial Cell Line-Derived Neurotrophic Factor Receptors in Müller Glial Cells during Light-Induced Retinal Degeneration. Neuroscience 2003, 122, 229–235. [Google Scholar] [CrossRef]
- Ward, M.S.; Khoobehi, A.; Lavik, E.B.; Langer, R.; Young, M.J. Neuroprotection of Retinal Ganglion Cells in DBA/2J Mice With GDNF-Loaded Biodegradable Microspheres. J. Pharm. Sci. 2007, 96, 558–568. [Google Scholar] [CrossRef]
- Checa-Casalengua, P.; Jiang, C.; Bravo-Osuna, I.; Tucker, B.A.; Molina-Martínez, I.T.; Young, M.J.; Herrero-Vanrell, R. Retinal Ganglion Cells Survival in a Glaucoma Model by GDNF/Vit E PLGA Microspheres Prepared According to a Novel Microencapsulation Procedure. J. Control. Release 2011, 156, 92–100. [Google Scholar] [CrossRef]
- Koeberle, P.D.; Bähr, M. The Upregulation of GLAST-1 Is an Indirect Antiapoptotic Mechanism of GDNF and Neurturin in the Adult CNS. Cell Death Differ. 2008, 15, 471–483. [Google Scholar] [CrossRef] [PubMed]
- Fischer, D.; Leibinger, M. Promoting Optic Nerve Regeneration. Prog. Retin. Eye Res. 2012, 31, 688–701. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Rhee, K.-D.; Pellegrini, M.; Yang, X.-J. Impacts of Ciliary Neurotrophic Factor on the Retinal Transcriptome in a Mouse Model of Photoreceptor Degeneration. Sci. Rep. 2020, 10, 6593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ernst, M. Acquiring Signalling Specificity from the Cytokine Receptor Gp130. Trends Genet. 2004, 20, 23–32. [Google Scholar] [CrossRef]
- Shpak, A.A.; Guekht, A.B.; Druzhkova, T.A.; Kozlova, K.I.; Gulyaeva, N.V. Ciliary Neurotrophic Factor in Patients with Primary Open-Angle Glaucoma and Age-Related Cataract. Mol. Vis. 2017, 23, 799–809. [Google Scholar]
- Valter, K.; Bisti, S.; Gargini, C.; Di Loreto, S.; Maccarone, R.; Cervetto, L.; Stone, J. Time Course of Neurotrophic Factor Upregulation and Retinal Protection against Light-Induced Damage after Optic Nerve Section. Investig. Opthalmol. Vis. Sci. 2005, 46, 1748. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Cheng, J.; Huang, X.; Li, Y.; Ma, X.; Li, Y.; Wei, R. Pathologic Changes in Chronic Intraorbital Optic Nerve Damage in Rabbits. Brain Res. 2009, 1267, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Bouvier, M.; Szatkowski, M.; Amato, A.; Attwell, D. The Glial Cell Glutamate Uptake Carrier Countertransports PH-Changing Anions. Nature 1992, 360, 471–474. [Google Scholar] [CrossRef] [PubMed]
- Clements, J.; Lester, R.; Tong, G.; Jahr, C.; Westbrook, G. The Time Course of Glutamate in the Synaptic Cleft. Science 1992, 258, 1498–1501. [Google Scholar] [CrossRef]
- Atoji, Y.; Sarkar, S. Localization of AMPA, Kainate, and NMDA Receptor MRNAs in the Pigeon Cerebellum. J. Chem. Neuroanat. 2019, 98, 71–79. [Google Scholar] [CrossRef]
- Girling, K.D.; Demers, M.-J.; Laine, J.; Zhang, S.; Wang, Y.T.; Graham, R.K. Activation of Caspase-6 and Cleavage of Caspase-6 Substrates Is an Early Event in NMDA Receptor-Mediated Excitotoxicity. J. Neurosci. Res. 2018, 96, 391–406. [Google Scholar] [CrossRef] [PubMed]
- Riedel, G. Glutamate Receptor Function in Learning and Memory. Behav. Brain Res. 2003, 140, 1–47. [Google Scholar] [CrossRef]
- Hardingham, G.E. Pro-Survival Signalling from the NMDA Receptor. Biochem. Soc. Trans. 2006, 34, 936–938. [Google Scholar] [CrossRef]
- Hetman, M.; Kharebava, G. Survival Signaling Pathways Activated by NMDA Receptors. Curr. Top. Med. Chem. 2006, 6, 787–799. [Google Scholar] [CrossRef]
- Casson, R.J. Possible Role of Excitotoxicity in the Pathogenesis of Glaucoma. Clin. Experiment. Ophthalmol. 2006, 34, 54–63. [Google Scholar] [CrossRef]
- Lalo, U. NMDA Receptors Mediate Neuron-to-Glia Signaling in Mouse Cortical Astrocytes. J. Neurosci. 2006, 26, 2673–2683. [Google Scholar] [CrossRef] [Green Version]
- Salter, M.G.; Fern, R. NMDA Receptors Are Expressed in Developing Oligodendrocyte Processes and Mediate Injury. Nature 2005, 438, 1167–1171. [Google Scholar] [CrossRef]
- Bylicky, M.A.; Mueller, G.P.; Day, R.M. Mechanisms of Endogenous Neuroprotective Effects of Astrocytes in Brain Injury. Oxid. Med. Cell. Longev. 2018, 2018, 6501031. [Google Scholar] [CrossRef]
- Volterra, A.; Trotti, D.; Racagni, G. Glutamate Uptake Is Inhibited by Arachidonic Acid and Oxygen Radicals via Two Distinct and Additive Mechanisms. Mol. Pharmacol. 1994, 46, 986–992. [Google Scholar]
- Harvey, B.K.; Airavaara, M.; Hinzman, J.; Wires, E.M.; Chiocco, M.J.; Howard, D.B.; Shen, H.; Gerhardt, G.; Hoffer, B.J.; Wang, Y. Targeted Over-Expression of Glutamate Transporter 1 (GLT-1) Reduces Ischemic Brain Injury in a Rat Model of Stroke. PLoS ONE 2011, 6, e22135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.-N.; Hao, L.; Guo, Y.-S.; Wang, H.-Y.; Li, L.; Liu, L.-Z.; Li, W.-B. Are Glutamate Transporters Neuroprotective or Neurodegenerative during Cerebral Ischemia? J. Mol. Med. 2019, 97, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Milewski, K.; Bogacińska-Karaś, M.; Hilgier, W.; Albrecht, J.; Zielińska, M. TNFα Increases STAT3-Mediated Expression of Glutaminase Isoform KGA in Cultured Rat Astrocytes. Cytokine 2019, 123, 154774. [Google Scholar] [CrossRef] [PubMed]
- Kemp, J.A.; McKernan, R.M. NMDA Receptor Pathways as Drug Targets. Nat. Neurosci. 2002, 5, 1039–1042. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Reddy, P.H. Role of Glutamate and NMDA Receptors in Alzheimer’s Disease. J. Alzheimers Dis. 2017, 57, 1041–1048. [Google Scholar] [CrossRef] [Green Version]
- Kumagai, A.; Sasaki, T.; Matsuoka, K.; Abe, M.; Tabata, T.; Itoh, Y.; Fuchino, H.; Wugangerile, S.; Suga, M.; Yamaguchi, T.; et al. Monitoring of Glutamate-Induced Excitotoxicity by Mitochondrial Oxygen Consumption: XXXX. Synapse 2019, 73, e22067. [Google Scholar] [CrossRef] [Green Version]
- Vernazza, S.; Tirendi, S.; Bassi, A.M.; Traverso, C.E.; Saccà, S.C. Neuroinflammation in Primary Open-Angle Glaucoma. J. Clin. Med. 2020, 9, 3172. [Google Scholar] [CrossRef]
- Dreyer, E.B. Elevated Glutamate Levels in the Vitreous Body of Humans and Monkeys with Glaucoma. Arch. Ophthalmol. 1996, 114, 299. [Google Scholar] [CrossRef] [PubMed]
- Dreyer, E.B. A Proposed Role for Excitotoxicity in Glaucoma. J. Glaucoma 1998, 7, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Honkanen, R.A. Vitreous Amino Acid Concentrations in Patients with Glaucoma Undergoing Vitrectomy. Arch. Ophthalmol. 2003, 121, 183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wamsley, S. Vitreous Glutamate Concentration and Axon Loss in Monkeys with Experimental Glaucoma. Arch. Ophthalmol. 2005, 123, 64. [Google Scholar] [CrossRef] [Green Version]
- Belov Kirdajova, D.; Kriska, J.; Tureckova, J.; Anderova, M. Ischemia-Triggered Glutamate Excitotoxicity from the Perspective of Glial Cells. Front. Cell. Neurosci. 2020, 14, 51. [Google Scholar] [CrossRef] [Green Version]
- Dallas, M.; Boycott, H.E.; Atkinson, L.; Miller, A.; Boyle, J.P.; Pearson, H.A.; Peers, C. Hypoxia Suppresses Glutamate Transport in Astrocytes. J. Neurosci. 2007, 27, 3946–3955. [Google Scholar] [CrossRef]
- Kaur, C. Hypoxia-Ischemia and Retinal Ganglion Cell Damage. Clin. Ophthalmol. 2008, 879. [Google Scholar] [CrossRef] [Green Version]
- Medzhitov, R. Origin and Physiological Roles of Inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef]
- Kimura, A.; Namekata, K.; Guo, X.; Noro, T.; Harada, C.; Harada, T. Targeting Oxidative Stress for Treatment of Glaucoma and Optic Neuritis. Oxid. Med. Cell. Longev. 2017, 2017, 2817252. [Google Scholar] [CrossRef]
- Qi, Y.; Zhao, M.; Bai, Y.; Huang, L.; Yu, W.; Bian, Z.; Zhao, M.; Li, X. Retinal Ischemia/Reperfusion Injury Is Mediated by Toll-like Receptor 4 Activation of NLRP3 Inflammasomes. Investig. Opthalmol. Vis. Sci. 2014, 55, 5466. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, S.; Tanihara, H.; Kido, N.; Honda, Y.; Goto, W.; Hara, H.; Miyawaki, N. Interleukin-1β Mediates Ischemic Injury in the Rat Retina. Exp. Eye Res. 2001, 73, 661–667. [Google Scholar] [CrossRef]
- Cohen, L. Relationships between Visual Function and Metabolism. In Biochemistry of the Retina; Graymore, C.N., Ed.; Academic Press: New York, NY, USA, 1965; pp. 36–50. [Google Scholar]
- Tielsch, J.M.; Katz, J.; Sommer, A.; Quigley, H.A.; Javitt, J.C. Hypertension, Perfusion Pressure, and Primary Open-Angle Glaucoma: A Population-Based Assessment. Arch. Ophthalmol. 1995, 113, 216–221. [Google Scholar] [CrossRef]
- Flammer, J. The Vascular Concept of Glaucoma. Surv. Ophthalmol. 1994, 38, S3–S6. [Google Scholar] [CrossRef]
- Chung, H.S.; Harris, A.; Evans, D.W.; Kagemann, L.; Garzozi, H.J.; Martin, B. Vascular Aspects in the Pathophysiology of Glaucomatous Optic Neuropathy. Surv. Ophthalmol. 1999, 43, S43–S50. [Google Scholar] [CrossRef]
- Osborne, N.N.; Casson, R.J.; Wood, J.P.; Chidlow, G.; Graham, M.; Melena, J. Retinal Ischemia: Mechanisms of Damage and Potential Therapeutic Strategies. Prog. Retin. Eye Res. 2004, 23, 91–147. [Google Scholar] [CrossRef] [PubMed]
- Abramov, A.Y.; Scorziello, A.; Duchen, M.R. Three Distinct Mechanisms Generate Oxygen Free Radicals in Neurons and Contribute to Cell Death during Anoxia and Reoxygenation. J. Neurosci. 2007, 27, 1129–1138. [Google Scholar] [CrossRef] [PubMed]
- Vohra, R.; Tsai, J.C.; Kolko, M. The Role of Inflammation in the Pathogenesis of Glaucoma. Surv. Ophthalmol. 2013, 58, 311–320. [Google Scholar] [CrossRef]
- Casson, R.J.; Chidlow, G.; Crowston, J.G.; Williams, P.A.; Wood, J.P.M. Retinal Energy Metabolism in Health and Glaucoma. Prog. Retin. Eye Res. 2020, 100881. [Google Scholar] [CrossRef]
- Yang, X.; Yu, X.-W.; Zhang, D.-D.; Fan, Z.-G. Blood-Retinal Barrier as a Converging Pivot in Understanding the Initiation and Development of Retinal Diseases. Chin. Med. J. 2020, 133, 2586–2594. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Kametani, M.; Chen, D.F. Adaptive Immunity: New Aspects of Pathogenesis Underlying Neurodegeneration in Glaucoma and Optic Neuropathy. Front. Immunol. 2020, 11, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, X. Matrix Metalloproteinases and Tumor Necrosis Factor α in Glaucomatous Optic Nerve Head. Arch. Ophthalmol. 2000, 118, 666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tezel, G.; Li, L.Y.; Patil, R.V.; Wax, M.B. TNF-α and TNF-α Receptor-1 in the Retina of Normal and Glaucomatous Eyes. Investig. Ophthalmol. Vis. Sci. 2001, 42, 1787–1794. [Google Scholar]
- Tezel, G. TNF-α signaling in glaucomatous neurodegeneration. In Progress in Brain Research; Elsevier: Amsterdam, The Netherlands, 2008; Volume 173, pp. 409–421. ISBN 978-0-444-53256-5. [Google Scholar]
- Yang, X.; Luo, C.; Cai, J.; Powell, D.W.; Yu, D.; Kuehn, M.H.; Tezel, G. Neurodegenerative and Inflammatory Pathway Components Linked to TNF-α/TNFR1 Signaling in the Glaucomatous Human Retina. Investig. Opthalmol. Vis. Sci. 2011, 52, 8442. [Google Scholar] [CrossRef]
- Luna, J.D.; Chan, C.-C.; Derevjanik, N.L.; Mahlow, J.; Chiu, C.; Peng, B.; Tobe, T.; Campochiaro, P.A.; Vinores, S.A. Blood-Retinal Barrier (BRB) Breakdown in Experimental Autoimmune Uveoretinitis: Comparison with Vascular Endothelial Growth Factor, Tumor Necrosis Factor α, and Interleukin-1β-Mediated Breakdown. J. Neurosci. Res. 1997, 49, 268–280. [Google Scholar] [CrossRef]
- Rolle, T.; Ponzetto, A.; Malinverni, L. The Role of Neuroinflammation in Glaucoma: An Update on Molecular Mechanisms and New Therapeutic Options. Front. Neurol. 2021, 11, 612422. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Cho, K.-S.; Vu, T.H.K.; Shen, C.-H.; Kaur, M.; Chen, G.; Mathew, R.; McHam, M.L.; Fazelat, A.; Lashkari, K.; et al. Commensal Microflora-Induced T Cell Responses Mediate Progressive Neurodegeneration in Glaucoma. Nat. Commun. 2018, 9, 3209. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zeng, Q.; Göktas, E.; Gopal, K.; Al-Aswad, L.; Blumberg, D.M.; Cioffi, G.A.; Liebmann, J.M.; Tezel, G. T-Lymphocyte Subset Distribution and Activity in Patients with Glaucoma. Investig. Opthalmol. Vis. Sci. 2019, 60, 877. [Google Scholar] [CrossRef] [Green Version]
- Schlereth, S.L.; Kremers, S.; Schrödl, F.; Cursiefen, C.; Heindl, L.M. Characterization of Antigen-Presenting Macrophages and Dendritic Cells in the Healthy Human Sclera. Investig. Ophthalmol. Vis. Sci. 2016, 57, 4878–4885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howell, G.R.; Soto, I.; Zhu, X.; Ryan, M.; Macalinao, D.G.; Sousa, G.L.; Caddle, L.B.; MacNicoll, K.H.; Barbay, J.M.; Porciatti, V. Radiation Treatment Inhibits Monocyte Entry into the Optic Nerve Head and Prevents Neuronal Damage in a Mouse Model of Glaucoma. J. Clin. Investig. 2012, 122, 1246–1261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, P.A.; Marsh-Armstrong, N.; Howell, G.R.; Bosco, A.; Danias, J.; Simon, J.; Di Polo, A.; Kuehn, M.H.; Przedborski, S.; Raff, M.; et al. Neuroinflammation in Glaucoma: A New Opportunity. Exp. Eye Res. 2017, 157, 20–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubens, W.H.G.; Beckers, H.J.M.; Gorgels, T.G.M.F.; Webers, C.A.B. Increased Ratios of Complement Factors C3a to C3 in Aqueous Humor and Serum Mark Glaucoma Progression. Exp. Eye Res. 2021, 204, 108460. [Google Scholar] [CrossRef]
- Samelska, K.; Zaleska-Żmijewska, A.; Bałan, B.; Grąbczewski, A.; Szaflik, J.; Kubiak, A.; Skopiński, P. Immunological and Molecular Basics of the Primary Open Angle Glaucoma Pathomechanism. Cent. Eur. J. Immunol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Kuehn, M.H.; Kim, C.Y.; Ostojic, J.; Bellin, M.; Alward, W.L.M.; Stone, E.M.; Sakaguchi, D.S.; Grozdanic, S.D.; Kwon, Y.H. Retinal Synthesis and Deposition of Complement Components Induced by Ocular Hypertension. Exp. Eye Res. 2006, 83, 620–628. [Google Scholar] [CrossRef] [PubMed]
- Noris, M.; Remuzzi, G. Overview of Complement Activation and Regulation. Semin. Nephrol. 2013, 33, 479–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling, M.; Murali, M. Analysis of the Complement System in the Clinical Immunology Laboratory. Clin. Lab. Med. 2019, 39, 579–590. [Google Scholar] [CrossRef] [PubMed]
- Chang, E.E.; Goldberg, J.L. Glaucoma 2.0: Neuroprotection, Neuroregeneration, Neuroenhancement. Ophthalmology 2012, 119, 979–986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.W.; Wang, N.; Cai, S.; Fang, Z.; Yu, M.; Wu, Q.; Tang, L.; Guo, B.; Feng, Y.; Jonas, J.B.; et al. Structural Brain Abnormalities in Patients with Primary Open-Angle Glaucoma: A Study with 3T MR Imaging. Investig. Ophthalmol. Vis. Sci. 2013, 54, 545–554. [Google Scholar] [CrossRef]
- Shen, J.; Wang, Y.; Yao, K. Protection of Retinal Ganglion Cells in Glaucoma: Current Status and Future. Exp. Eye Res. 2021, 205, 108506. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Wang, L.; Zhang, X. Neuroprotection in Glaucoma: Present and Future. Chin. Med. J. 2013, 126, 1567–1577. [Google Scholar] [CrossRef]
- Naik, S.; Pandey, A.; Lewis, S.A.; Rao, B.S.S.; Mutalik, S. Neuroprotection: A Versatile Approach to Combat Glaucoma. Eur. J. Pharmacol. 2020, 881, 173208. [Google Scholar] [CrossRef] [PubMed]
- Pardue, M.T.; Allen, R.S. Neuroprotective Strategies for Retinal Disease. Prog. Retin. Eye Res. 2018, 65, 50–76. [Google Scholar] [CrossRef] [PubMed]
- Osborne, N.N.; Núñez-Álvarez, C.; Joglar, B.; Del Olmo-Aguado, S. Glaucoma: Focus on Mitochondria in Relation to Pathogenesis and Neuroprotection. Eur. J. Pharmacol. 2016, 787, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Ju, W.-K.; Kim, K.-Y.; Lindsey, J.D.; Angert, M.; Patel, A.; Scott, R.T.; Liu, Q.; Crowston, J.G.; Ellisman, M.H.; Perkins, G.A.; et al. Elevated Hydrostatic Pressure Triggers Release of OPA1 and Cytochrome C, and Induces Apoptotic Cell Death in Differentiated RGC-5 Cells. Mol. Vis. 2009, 15, 120–134. [Google Scholar] [PubMed]
- Wang, Y.X.; Jonas, J.B.; Wang, N.; You, Q.S.; Yang, D.; Xie, X.B.; Xu, L. Intraocular Pressure and Estimated Cerebrospinal Fluid Pressure. The Beijing Eye Study 2011. PLoS ONE 2014, 9, e104267. [Google Scholar] [CrossRef] [Green Version]
- Jonas, J.B.; Yang, D.; Wang, N. Einfluss des Liquordrucks auf die glaukomatöse Schädigung des Nervus opticus. Ophthalmol. 2014, 111, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Izzotti, A.; Bagnis, A.; Saccà, S.C. The Role of Oxidative Stress in Glaucoma. Mutat. Res. Mutat. Res. 2006, 612, 105–114. [Google Scholar] [CrossRef]
- Costa, V.P.; Harris, A.; Stefánsson, E.; Flammer, J.; Krieglstein, G.K.; Orzalesi, N.; Heijl, A.; Renard, J.-P.; Serra, L.M. The Effects of Antiglaucoma and Systemic Medications on Ocular Blood Flow. Prog. Retin. Eye Res. 2003, 22, 769–805. [Google Scholar] [CrossRef]
- García-Bermúdez, M.Y.; Freude, K.K.; Mouhammad, Z.A.; van Wijngaarden, P.; Martin, K.K.; Kolko, M. Glial Cells in Glaucoma: Friends, Foes, and Potential Therapeutic Targets. Front. Neurol. 2021, 12, 624983. [Google Scholar] [CrossRef] [PubMed]
- Chao, T.I.; Grosche, J.; Friedrich, K.J.; Biedermann, B.; Francke, M.; Pannicke, T.; Reichelt, W.; Wulst, M.; Mühle, C.; Pritz-Hohmeier, S.; et al. Comparative Studies on Mammalian Müller (Retinal Glial) Cells. J. Neurocytol. 1997, 26, 439–454. [Google Scholar] [CrossRef] [PubMed]
- Eastlake, K.; Luis, J.; Limb, G.A. Potential of Müller Glia for Retina Neuroprotection. Curr. Eye Res. 2020, 45, 339–348. [Google Scholar] [CrossRef] [Green Version]
- Taylor, S.; Srinivasan, B.; Wordinger, R.J.; Roque, R.S. Glutamate Stimulates Neurotrophin Expression in Cultured Müller Cells. Mol. Brain Res. 2003, 111, 189–197. [Google Scholar] [CrossRef]
- Wahlin, K.J.; Campochiaro, P.A.; Zack, D.J.; Adler, R. Neurotrophic Factors Cause Activation of Intracellular Signaling Pathways in Muller Cells and Other Cells of the Inner Retina, but Not Photoreceptors. Investig. Ophthalmol. Vis. Sci. 2000, 41, 927–936. [Google Scholar]
- Boss, J.D.; Singh, P.K.; Pandya, H.K.; Tosi, J.; Kim, C.; Tewari, A.; Juzych, M.S.; Abrams, G.W.; Kumar, A. Assessment of Neurotrophins and Inflammatory Mediators in Vitreous of Patients with Diabetic Retinopathy. Investig. Opthalmol. Vis. Sci. 2017, 58, 5594. [Google Scholar] [CrossRef]
- Huster, D.; Reichenbach, A.; Reichelt, W. The Glutathione Content of Retinal Müller (Glial) Cells: Effect of Pathological Conditions. Neurochem. Int. 2000, 36, 461–469. [Google Scholar] [CrossRef]
- Pow, D.V.; Crook, D.K. Immunocytochemical Evidence for the Presence of High Levels of Reduced Glutathione in Radial Glial Cells and Horizontal Cells in the Rabbit Retina. Neurosci. Lett. 1995, 193, 25–28. [Google Scholar] [CrossRef]
- Inoue, Y.; Shimazawa, M.; Noda, Y.; Nagano, R.; Otsuka, T.; Kuse, Y.; Nakano, Y.; Tsuruma, K.; Nakagami, Y.; Hara, H. RS9, a Novel Nrf2 Activator, Attenuates Light-Induced Death of Cells of Photoreceptor Cells and Müller Glia Cells. J. Neurochem. 2017, 141, 750–765. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Wei, Y.; Gong, J.; Cho, H.; Park, J.K.; Sung, E.-R.; Huang, H.; Wu, L.; Eberhart, C.; Handa, J.T.; et al. NRF2 Plays a Protective Role in Diabetic Retinopathy in Mice. Diabetologia 2014, 57, 204–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, S.; Ishita, S.; Sugawara, K.; Mawatari, K. Cystine/Glutamate Antiporter Expression in Retinal Mu\” Ller Glial Cells: Implications Fordl-Alpha-Aminoadipate Toxicity. Neuroscience 1993, 57, 473–482. [Google Scholar] [CrossRef]
- Tomi, M.; Funaki, T.; Abukawa, H.; Katayama, K.; Kondo, T.; Ohtsuki, S.; Ueda, M.; Obinata, M.; Terasaki, T.; Hosoya, K.-I. Expression and Regulation of L-Cystine Transporter, System Xc-, in the Newly Developed Rat Retinal Müller Cell Line (TR-MUL). Glia 2003, 43, 208–217. [Google Scholar] [CrossRef]
- Lewerenz, J.; Klein, M.; Methner, A. Cooperative Action of Glutamate Transporters and Cystine/Glutamate Antiporter System Xc–Protects from Oxidative Glutamate Toxicity. J. Neurochem. 2006, 98, 916–925. [Google Scholar] [CrossRef] [PubMed]
- Martis, R.M.; Knight, L.J.; Donaldson, P.J.; Lim, J.C. Identification, Expression, and Roles of the Cystine/Glutamate Antiporter in Ocular Tissues. Oxid. Med. Cell. Longev. 2020, 2020, 4594606. [Google Scholar] [CrossRef]
- Albrecht, P.; Lewerenz, J.; Dittmer, S.; Noack, R.; Maher, P.; Methner, A. Mechanisms of Oxidative Glutamate Toxicity: The Glutamate/Cystine Antiporter System Xc¯ as a Neuroprotective Drug Target. CNS Neurol. Disord.-Drug Targets 2010, 9, 373–382. [Google Scholar] [CrossRef]
- Derouiche, A.; Rauen, T. Coincidence of L-Glutamate/L-Aspartate Transporter (GLAST) and Glutamine Synthetase (GS) Immunoreactions in Retinal Glia: Evidence for Coupling of GLAST and GS in Transmitter Clearance. J. Neurosci. Res. 1995, 42, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Biedermann, B.; Bringmann, A.; Reichenbach, A. High-Affinity GABA Uptake in Retinal Glial (Müller) Cells of the Guinea Pig: Electrophysiological Characterization, Immunohistochemical Localization, and Modeling of Efficiency: Gaba Uptake in Müller Glial Cells. Glia 2002, 39, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Bringmann, A.; Grosche, A.; Pannicke, T.; Reichenbach, A. GABA and Glutamate Uptake and Metabolism in Retinal Glial (Müller) Cells. Front. Endocrinol. 2013, 4. [Google Scholar] [CrossRef] [Green Version]
- Uga, S.; Smelser, G.K. Comparative Study of the Fine Structure of Retinal Müller Cells in Various Vertebrates. Investig. Ophthalmol. 1973, 12, 434–448. [Google Scholar]
- Tsacopoulos, M.; Poitry-Yamate, C.L.; MacLeish, P.R.; Poitry, S. Trafficking of Molecules and Metabolic Signals in the Retina. Prog. Retin. Eye Res. 1998, 17, 429–442. [Google Scholar] [CrossRef]
- Poitry-Yamate, C.L.; Poitry, S.; Tsacopoulos, M. Lactate Released by Müller Glial Cells Is Metabolized by Photoreceptors from Mammalian Retina. J. Neurosci. Off. J. Soc. Neurosci. 1995, 15, 5179–5191. [Google Scholar] [CrossRef]
- Poitry-Yamate, C.L.; Tsacopoulos, M. Glucose Metabolism in Freshly Isolated Müller Glial Cells from a Mammalian Retina. J. Comp. Neurol. 1992, 320, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Reichenbach, A.; Bringmann, A. New Functions of Müller Cells. Glia 2013, 61, 651–678. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, C.; Su, G. Cellular Signaling in Müller Glia: Progenitor Cells for Regenerative and Neuroprotective Responses in Pharmacological Models of Retinal Degeneration. J. Ophthalmol. 2019, 2019, 5743109. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zheng, Y.; Wang, C.; Liu, Y. Glutathione Depletion Induces Ferroptosis, Autophagy, and Premature Cell Senescence in Retinal Pigment Epithelial Cells. Cell Death Dis. 2018, 9, 753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paasche, G.; Huster, D.; Reichenbach, A. The Glutathione Content of Retinal Müller (Glial) Cells: The Effects of Aging and of Application of Free-Radical Scavengers. Ophthalmic Res. 1998, 30, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Aydemir, O.; Celebi, S.; Yilmaz, T.; Yekeler, H.; Kükner, A.S. Protective Effects of Vitamin E Forms (Alpha-Tocopherol, Gamma-Tocopherol and d-Alpha-Tocopherol Polyethylene Glycol 1000 Succinate) on Retinal Edema during Ischemia-Reperfusion Injury in the Guinea Pig Retina. Int. Ophthalmol. 2004, 25, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Nucci, C.; Martucci, A.; Giannini, C.; Morrone, L.A.; Bagetta, G.; Mancino, R. Neuroprotective Agents in the Management of Glaucoma. Eye 2018, 32, 938–945. [Google Scholar] [CrossRef] [PubMed]
- Russo, R.; Cavaliere, F.; Rombolà, L.; Gliozzi, M.; Cerulli, A.; Nucci, C.; Fazzi, E.; Bagetta, G.; Corasaniti, M.T.; Morrone, L.A. Rational Basis for the Development of Coenzyme Q10 as a Neurotherapeutic Agent for Retinal Protection. Prog. Brain Res. 2008, 173, 575–582. [Google Scholar]
- Nebbioso, M.; Scarsella, G.; Tafani, M.; Pescosolido, N. Mechanisms of Ocular Neuroprotection by Antioxidant Molecules in Animal Models. J. Biol. Regul. Homeost. Agents 2013, 27, 197–209. [Google Scholar] [PubMed]
- Jiang, W.; Tang, L.; Zeng, J.; Chen, B. Adeno-Associated Virus Mediated SOD Gene Therapy Protects the Retinal Ganglion Cells from Chronic Intraocular Pressure Elevation Induced Injury via Attenuating Oxidative Stress and Improving Mitochondrial Dysfunction in a Rat Model. Am. J. Transl. Res. 2016, 8, 799–810. [Google Scholar] [PubMed]
- Perez, C.I.; Singh, K.; Lin, S. Relationship of Lifestyle, Exercise, and Nutrition with Glaucoma. Curr. Opin. Ophthalmol. 2019, 30, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Ramdas, W.D. The Relation between Dietary Intake and Glaucoma: A Systematic Review. Acta Ophthalmol. 2018, 96, 550–556. [Google Scholar] [CrossRef] [PubMed]
- Braakhuis, A.; Raman, R.; Vaghefi, E. The Association between Dietary Intake of Antioxidants and Ocular Disease. Diseases 2017, 5, 3. [Google Scholar] [CrossRef] [Green Version]
- Ramdas, W.D.; Schouten, J.S.A.G.; Webers, C.A.B. The Effect of Vitamins on Glaucoma: A Systematic Review and Meta-Analysis. Nutrients 2018, 10, 359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, F.-J.; Zhang, S.-H.; Xu, P.; Yang, B.-Q.; Zhang, R.; Cheng, Y.; Zhou, X.-J.; Huang, W.-J.; Wang, M.; Chen, J.-Y.; et al. Quercetin Declines Apoptosis, Ameliorates Mitochondrial Function and Improves Retinal Ganglion Cell Survival and Function in In Vivo Model of Glaucoma in Rat and Retinal Ganglion Cell Culture In Vitro. Front. Mol. Neurosci. 2017, 10, 285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shim, S.H.; Kim, J.M.; Choi, C.Y.; Kim, C.Y.; Park, K.H. Ginkgo Biloba Extract and Bilberry Anthocyanins Improve Visual Function in Patients with Normal Tension Glaucoma. J. Med. Food 2012, 15, 818–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, S.; Liu, S.; Yan, G. Lycium Barbarum Exerts Protection against Glaucoma-Like Injury Via Inhibition of MMP-9 Signaling In Vitro. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2019, 25, 9794–9800. [Google Scholar] [CrossRef]
- Liu, L.; Sha, X.-Y.; Wu, Y.-N.; Chen, M.-T.; Zhong, J.-X. Lycium Barbarum Polysaccharides Protects Retinal Ganglion Cells against Oxidative Stress Injury. Neural Regen. Res. 2020, 15, 1526–1531. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, P.S.; Li, B.; Vachali, P.P.; Gorusupudi, A.; Shyam, R.; Henriksen, B.S.; Nolan, J.M. Lutein, Zeaxanthin, and Meso-Zeaxanthin: The Basic and Clinical Science Underlying Carotenoid-Based Nutritional Interventions against Ocular Disease. Prog. Retin. Eye Res. 2016, 50, 34–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammond, B.R.; Fletcher, L.M.; Roos, F.; Wittwer, J.; Schalch, W. A Double-Blind, Placebo-Controlled Study on the Effects of Lutein and Zeaxanthin on Photostress Recovery, Glare Disability, and Chromatic Contrast. Investig. Ophthalmol. Vis. Sci. 2014, 55, 8583–8589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Age-Related Eye Disease Study 2 Research Group Lutein + Zeaxanthin and Omega-3 Fatty Acids for Age-Related Macular Degeneration: The Age-Related Eye Disease Study 2 (AREDS2) Randomized Clinical Trial. JAMA 2013, 309, 2005–2015. [CrossRef]
- Davey, P.G.; Henderson, T.; Lem, D.W.; Weis, R.; Amonoo-Monney, S.; Evans, D.W. Visual Function and Macular Carotenoid Changes in Eyes with Retinal Drusen-An Open Label Randomized Controlled Trial to Compare a Micronized Lipid-Based Carotenoid Liquid Supplementation and AREDS-2 Formula. Nutrients 2020, 12, 3271. [Google Scholar] [CrossRef] [PubMed]
- Lem, D.W.; Gierhart, D.L.; Davey, P.G. Management of Diabetic Eye Disease Using Carotenoids and Nutrients; IntechOpen: London, UK, 2021; ISBN 978-1-83968-865-2. [Google Scholar]
- Choi, J.-S.; Kim, D.; Hong, Y.-M.; Mizuno, S.; Joo, C.-K. Inhibition of NNOS and COX-2 Expression by Lutein in Acute Retinal Ischemia. Nutr. Burbank Los Angel. Cty. Calif 2006, 22, 668–671. [Google Scholar] [CrossRef] [PubMed]
- Fung, F.K.C.; Law, B.Y.K.; Lo, A.C.Y. Lutein Attenuates Both Apoptosis and Autophagy upon Cobalt (II) Chloride-Induced Hypoxia in Rat Műller Cells. PLoS ONE 2016, 11, e0167828. [Google Scholar] [CrossRef] [PubMed]
- Li, S.-Y.; Fung, F.K.C.; Fu, Z.J.; Wong, D.; Chan, H.H.L.; Lo, A.C.Y. Anti-Inflammatory Effects of Lutein in Retinal Ischemic/Hypoxic Injury: In Vivo and in Vitro Studies. Investig. Ophthalmol. Vis. Sci. 2012, 53, 5976–5984. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Wang, Z.; Zhao, J.; Li, Q.; Huang, C.; Zhu, L.; Lu, D. Neuroprotective Effect of Lutein on NMDA-Induced Retinal Ganglion Cell Injury in Rat Retina. Cell. Mol. Neurobiol. 2016, 36, 531–540. [Google Scholar] [CrossRef]
- Szeto, H.H. Cell-Permeable, Mitochondrial-Targeted, Peptide Antioxidants. AAPS J. 2006, 8, E277–E283. [Google Scholar] [CrossRef] [PubMed]
- Rocha, M.; Hernandez-Mijares, A.; Garcia-Malpartida, K.; Bañuls, C.; Bellod, L.; Victor, V.M. Mitochondria-Targeted Antioxidant Peptides. Curr. Pharm. Des. 2010, 16, 3124–3131. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K.; Zhao, G.-M.; Wu, D.; Soong, Y.; Birk, A.V.; Schiller, P.W.; Szeto, H.H. Cell-Permeable Peptide Antioxidants Targeted to Inner Mitochondrial Membrane Inhibit Mitochondrial Swelling, Oxidative Cell Death, and Reperfusion Injury. J. Biol. Chem. 2004, 279, 34682–34690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Pang, Y.; Zhang, Z.; Li, X.; Wang, C.; Lei, Y.; Li, A.; Yu, L.; Ye, J. Mitochondria-Targeted Antioxidant Peptide SS-31 Mediates Neuroprotection in a Rat Experimental Glaucoma Model. Acta Biochim. Biophys. Sin. 2019, 51, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Vasudevan, S.K.; Gupta, V.; Crowston, J.G. Neuroprotection in Glaucoma. Indian J. Ophthalmol. 2011, 59, S102–S113. [Google Scholar] [CrossRef] [PubMed]
- Sposato, V.; Parisi, V.; Manni, L.; Antonucci, M.T.; Fausto, V.D.; Sornelli, F.; Aloe, L. Glaucoma Alters the Expression of NGF and NGF Receptors in Visual Cortex and Geniculate Nucleus of Rats: Effect of Eye NGF Application. Vis. Res. 2009, 49, 54–63. [Google Scholar] [CrossRef] [Green Version]
- Lambiase, A.; Aloe, L.; Centofanti, M.; Parisi, V.; Mantelli, F.; Colafrancesco, V.; Manni, G.L.; Bucci, M.G.; Bonini, S.; Levi-Montalcini, R. Experimental and Clinical Evidence of Neuroprotection by Nerve Growth Factor Eye Drops: Implications for Glaucoma. Proc. Natl. Acad. Sci. USA 2009, 106, 13469–13474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Wang, R.; Thrimawithana, T.; Little, P.J.; Xu, J.; Feng, Z.-P.; Zheng, W. The Nerve Growth Factor Signaling and Its Potential as Therapeutic Target for Glaucoma. BioMed Res. Int. 2014, 2014, 759473. [Google Scholar] [CrossRef] [Green Version]
- Capsoni, S.; Tiveron, C.; Vignone, D.; Amato, G.; Cattaneo, A. Dissecting the Involvement of Tropomyosin-Related Kinase A and P75 Neurotrophin Receptor Signaling in NGF Deficit-Induced Neurodegeneration. Proc. Natl. Acad. Sci. USA 2010, 107, 12299–12304. [Google Scholar] [CrossRef] [Green Version]
- Ko, M.-L.; Hu, D.-N.; Ritch, R.; Sharma, S.C. The Combined Effect of Brain-Derived Neurotrophic Factor and a Free Radical Scavenger in Experimental Glaucoma. Investig. Ophthalmol. Vis. Sci. 2000, 41, 2967–2971. [Google Scholar]
- Ji, J.-Z.; Elyaman, W.; Yip, H.K.; Lee, V.W.; Yick, L.-W.; Hugon, J.; So, K.-F. CNTF Promotes Survival of Retinal Ganglion Cells after Induction of Ocular Hypertension in Rats: The Possible Involvement of STAT3 Pathway. Eur. J. Neurosci. 2004, 19, 265–272. [Google Scholar] [CrossRef]
- Jiang, C.; Moore, M.J.; Zhang, X.; Klassen, H.; Langer, R.; Young, M. Intravitreal Injections of GDNF-Loaded Biodegradable Microspheres Are Neuroprotective in a Rat Model of Glaucoma. Mol. Vis. 2007, 13, 1783–1792. [Google Scholar] [PubMed]
- Schmeer, C.; Straten, G.; Kügler, S.; Gravel, C.; Bähr, M.; Isenmann, S. Dose-Dependent Rescue of Axotomized Rat Retinal Ganglion Cells by Adenovirus-Mediated Expression of Glial Cell-Line Derived Neurotrophic Factorin Vivo. Eur. J. Neurosci. 2002, 15, 637–643. [Google Scholar] [CrossRef]
- Bhattacharya, A. Lipid Metabolism in Plants Under High Temperature. In Effect of High Temperature on Crop Productivity and Metabolism of Macro Molecules; Elsevier: Amsterdam, The Netherlands, 2019; pp. 311–389. ISBN 978-0-12-817562-0. [Google Scholar]
- Oddone, F.; Rossetti, L.; Parravano, M.; Sbardella, D.; Coletta, M.; Ziccardi, L.; Roberti, G.; Carnevale, C.; Romano, D.; Manni, G.; et al. Citicoline in Ophthalmological Neurodegenerative Disease: A Comprehensive Review. Pharmaceuticals 2021, 14, 281. [Google Scholar] [CrossRef]
- Matteucci, A.; Varano, M.; Gaddini, L.; Mallozzi, C.; Villa, M.; Pricci, F.; Malchiodi-Albedi, F. Neuroprotective Effects of Citicoline in in Vitro Models of Retinal Neurodegeneration. Int. J. Mol. Sci. 2014, 15, 6286–6297. [Google Scholar] [CrossRef]
- Skopiński, P.; Magdalena Radomska-Leśniewska, D.; Izdebska, J.; Kamińska, A.; Kupis, M.; Kubiak, A.; Samelska, K. New Perspectives of Immunomodulation and Neuroprotection in Glaucoma. Cent. Eur. J. Immunol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Lazaridis, K.N.; Gores, G.J.; Lindor, K.D. Ursodeoxycholic Acid “Mechanisms of Action and Clinical Use in Hepatobiliary Disorders”. J. Hepatol. 2001, 35, 134–146. [Google Scholar] [CrossRef]
- Duan, W.-M.; Rodrigues, C.M.P.; Zhao, L.-R.; Steer, C.J.; Low, W.C.; Rodrigures, C.M.P. Tauroursodeoxycholic Acid Improves the Survival and Function of Nigral Transplants in a Rat Model of Parkinson’s Disease. Cell Transplant. 2002, 11, 195–205. [Google Scholar] [CrossRef] [Green Version]
- Keene, C.D.; Rodrigues, C.M.P.; Eich, T.; Chhabra, M.S.; Steer, C.J.; Low, W.C. Tauroursodeoxycholic Acid, a Bile Acid, Is Neuroprotective in a Transgenic Animal Model of Huntington’s Disease. Proc. Natl. Acad. Sci. USA 2002, 99, 10671–10676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Win, A.; Delgado, A.; Jadeja, R.N.; Martin, P.M.; Bartoli, M.; Thounaojam, M.C. Pharmacological and Metabolic Significance of Bile Acids in Retinal Diseases. Biomolecules 2021, 11, 292. [Google Scholar] [CrossRef] [PubMed]
- Yoon, Y.M.; Lee, J.H.; Yun, S.P.; Han, Y.-S.; Yun, C.W.; Lee, H.J.; Noh, H.; Lee, S.-J.; Han, H.J.; Lee, S.H. Tauroursodeoxycholic Acid Reduces ER Stress by Regulating of Akt-Dependent Cellular Prion Protein. Sci. Rep. 2016, 6, 39838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soares, R.; Ribeiro, F.F.; Xapelli, S.; Genebra, T.; Ribeiro, M.F.; Sebastião, A.M.; Rodrigues, C.M.P.; Solá, S. Tauroursodeoxycholic Acid Enhances Mitochondrial Biogenesis, Neural Stem Cell Pool, and Early Neurogenesis in Adult Rats. Mol. Neurobiol. 2018, 55, 3725–3738. [Google Scholar] [CrossRef]
- Vang, S.; Longley, K.; Steer, C.J.; Low, W.C. The Unexpected Uses of Urso- and Tauroursodeoxycholic Acid in the Treatment of Non-Liver Diseases. Glob. Adv. Health Med. 2014, 3, 58–69. [Google Scholar] [CrossRef] [Green Version]
- Omura, T.; Asari, M.; Yamamoto, J.; Oka, K.; Hoshina, C.; Maseda, C.; Awaya, T.; Tasaki, Y.; Shiono, H.; Yonezawa, A.; et al. Sodium Tauroursodeoxycholate Prevents Paraquat-Induced Cell Death by Suppressing Endoplasmic Reticulum Stress Responses in Human Lung Epithelial A549 Cells. Biochem. Biophys. Res. Commun. 2013, 432, 689–694. [Google Scholar] [CrossRef]
- Gaspar, J.M.; Martins, A.; Cruz, R.; Rodrigues, C.M.P.; Ambrósio, A.F.; Santiago, A.R. Tauroursodeoxycholic Acid Protects Retinal Neural Cells from Cell Death Induced by Prolonged Exposure to Elevated Glucose. Neuroscience 2013, 253, 380–388. [Google Scholar] [CrossRef]
- Oveson, B.C.; Iwase, T.; Hackett, S.F.; Lee, S.Y.; Usui, S.; Sedlak, T.W.; Snyder, S.H.; Campochiaro, P.A.; Sung, J.U. Constituents of Bile, Bilirubin and TUDCA, Protect against Oxidative Stress-Induced Retinal Degeneration. J. Neurochem. 2011, 116, 144–153. [Google Scholar] [CrossRef] [Green Version]
- Noailles, A.; Fernández-Sánchez, L.; Lax, P.; Cuenca, N. Microglia Activation in a Model of Retinal Degeneration and TUDCA Neuroprotective Effects. J. Neuroinflam. 2014, 11. [Google Scholar] [CrossRef] [Green Version]
- Romero-Ramírez, L.; Nieto-Sampedro, M.; Barreda-Manso, M.A. Integrated Stress Response as a Therapeutic Target for CNS Injuries. BioMed Res. Int. 2017, 2017, 6953156. [Google Scholar] [CrossRef] [PubMed]
- Howell, G.R.; MacNicoll, K.H.; Braine, C.E.; Soto, I.; Macalinao, D.G.; Sousa, G.L.; John, S.W.M. Combinatorial Targeting of Early Pathways Profoundly Inhibits Neurodegeneration in a Mouse Model of Glaucoma. Neurobiol. Dis. 2014, 71, 44–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, S.; Stankowska, D.L.; Ellis, D.Z.; Krishnamoorthy, R.R.; Yorio, T. Targets of Neuroprotection in Glaucoma. J. Ocul. Pharmacol. Ther. Off. J. Assoc. Ocul. Pharmacol. Ther. 2018, 34, 85–106. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez Deniselle, M.C.; Garay, L.; Gonzalez, S.; Guennoun, R.; Schumacher, M.; De Nicola, A.F. Progesterone Restores Retrograde Labeling of Cervical Motoneurons in Wobbler Mouse Motoneuron Disease. Exp. Neurol. 2005, 195, 518–523. [Google Scholar] [CrossRef] [PubMed]
- González, S.L.; Coronel, M.F.; Raggio, M.C.; Labombarda, F. Progesterone Receptor-Mediated Actions and the Treatment of Central Nervous System Disorders: An up-Date of the Known and the Challenge of the Unknown. Steroids 2020, 153, 108525. [Google Scholar] [CrossRef]
- Drew, P.D.; Chavis, J.A. Female Sex Steroids: Effects upon Microglial Cell Activation. J. Neuroimmunol. 2000, 111, 77–85. [Google Scholar] [CrossRef]
- Allen, R.S.; Sayeed, I.; Oumarbaeva, Y.; Morrison, K.C.; Choi, P.H.; Pardue, M.T.; Stein, D.G. Progesterone Treatment Shows Greater Protection in Brain vs. Retina in a Rat Model of Middle Cerebral Artery Occlusion: Progesterone Receptor Levels May Play an Important Role. Restor. Neurol. Neurosci. 2016, 34, 947–963. [Google Scholar] [CrossRef]
- Ishrat, T.; Sayeed, I.; Atif, F.; Hua, F.; Stein, D.G. Progesterone and Allopregnanolone Attenuate Blood-Brain Barrier Dysfunction Following Permanent Focal Ischemia by Regulating the Expression of Matrix Metalloproteinases. Exp. Neurol. 2010, 226, 183–190. [Google Scholar] [CrossRef] [Green Version]
- Guo, L.; Salt, T.E.; Maass, A.; Luong, V.; Moss, S.E.; Fitzke, F.W.; Cordeiro, M.F. Assessment of Neuroprotective Effects of Glutamate Modulation on Glaucoma-Related Retinal Ganglion Cell Apoptosis in vivo. Investig. Ophthalmol. Vis. Sci. 2006, 47, 626–633. [Google Scholar] [CrossRef]
- González, S.L.; Labombarda, F.; González Deniselle, M.C.; Guennoun, R.; Schumacher, M.; De Nicola, A.F. Progesterone Up-Regulates Neuronal Brain-Derived Neurotrophic Factor Expression in the Injured Spinal Cord. Neuroscience 2004, 125, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Kokona, D.; Georgiou, P.-C.; Kounenidakis, M.; Kiagiadaki, F.; Thermos, K. Endogenous and Synthetic Cannabinoids as Therapeutics in Retinal Disease. Neural Plast. 2016, 2016, 8373020. [Google Scholar] [CrossRef] [Green Version]
- Rapino, C.; Tortolani, D.; Scipioni, L.; Maccarrone, M. Neuroprotection by (Endo)Cannabinoids in Glaucoma and Retinal Neurodegenerative Diseases. Curr. Neuropharmacol. 2018, 16, 959–970. [Google Scholar] [CrossRef] [PubMed]
- Nucci, C.; Tartaglione, R.; Cerulli, A.; Mancino, R.; Spanò, A.; Cavaliere, F.; Rombolà, L.; Bagetta, G.; Corasaniti, M.T.; Morrone, L.A. Retinal Damage Caused by High Intraocular Pressure–Induced Transient Ischemia is Prevented by Coenzyme Q10 in Rat. In International Review of Neurobiology; Neuroinflammation in Neuronal Death and Repair; Academic Press: Cambridge, MA, USA, 2007; Volume 82, pp. 397–406. [Google Scholar]
- Drew, P.D.; Xu, J.; Storer, P.D.; Chavis, J.A.; Racke, M.K. Peroxisome Proliferator-Activated Receptor Agonist Regulation of Glial Activation: Relevance to CNS Inflammatory Disorders. Neurochem. Int. 2006, 49, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Araújo, D.S.M.; Miya-Coreixas, V.S.; Pandolfo, P.; Calaza, K.C. Cannabinoid Receptors and TRPA1 on Neuroprotection in a Model of Retinal Ischemia. Exp. Eye Res. 2017, 154, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Hohmann, U.; Pelzer, M.; Kleine, J.; Hohmann, T.; Ghadban, C.; Dehghani, F. Opposite Effects of Neuroprotective Cannabinoids, Palmitoylethanolamide, and 2-Arachidonoylglycerol on Function and Morphology of Microglia. Front. Neurosci. 2019, 13, 1180. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Choi, J.Y.; Seo, J.; Choi, I.S. Neuroprotective Effect of Cannabidiol Against Hydrogen Peroxide in Hippocampal Neuron Culture. Cannabis Cannabinoid Res. 2020, 6, 40–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharif, N.A. IDrugs and IDevices Discovery Research: Preclinical Assays, Techniques, and Animal Model Studies for Ocular Hypotensives and Neuroprotectants. J. Ocul. Pharmacol. Ther. 2018, 34, 7–39. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vernazza, S.; Oddone, F.; Tirendi, S.; Bassi, A.M. Risk Factors for Retinal Ganglion Cell Distress in Glaucoma and Neuroprotective Potential Intervention. Int. J. Mol. Sci. 2021, 22, 7994. https://doi.org/10.3390/ijms22157994
Vernazza S, Oddone F, Tirendi S, Bassi AM. Risk Factors for Retinal Ganglion Cell Distress in Glaucoma and Neuroprotective Potential Intervention. International Journal of Molecular Sciences. 2021; 22(15):7994. https://doi.org/10.3390/ijms22157994
Chicago/Turabian StyleVernazza, Stefania, Francesco Oddone, Sara Tirendi, and Anna Maria Bassi. 2021. "Risk Factors for Retinal Ganglion Cell Distress in Glaucoma and Neuroprotective Potential Intervention" International Journal of Molecular Sciences 22, no. 15: 7994. https://doi.org/10.3390/ijms22157994