
In Vitro and In Vivo Pharmaco-Toxicological Characterization of 1-Cyclohexyl-x-methoxybenzene Derivatives in Mice: Comparison with Tramadol and PCP


 , , , , ,
, , , , , 
Abstract
:1. Introduction
2. Results
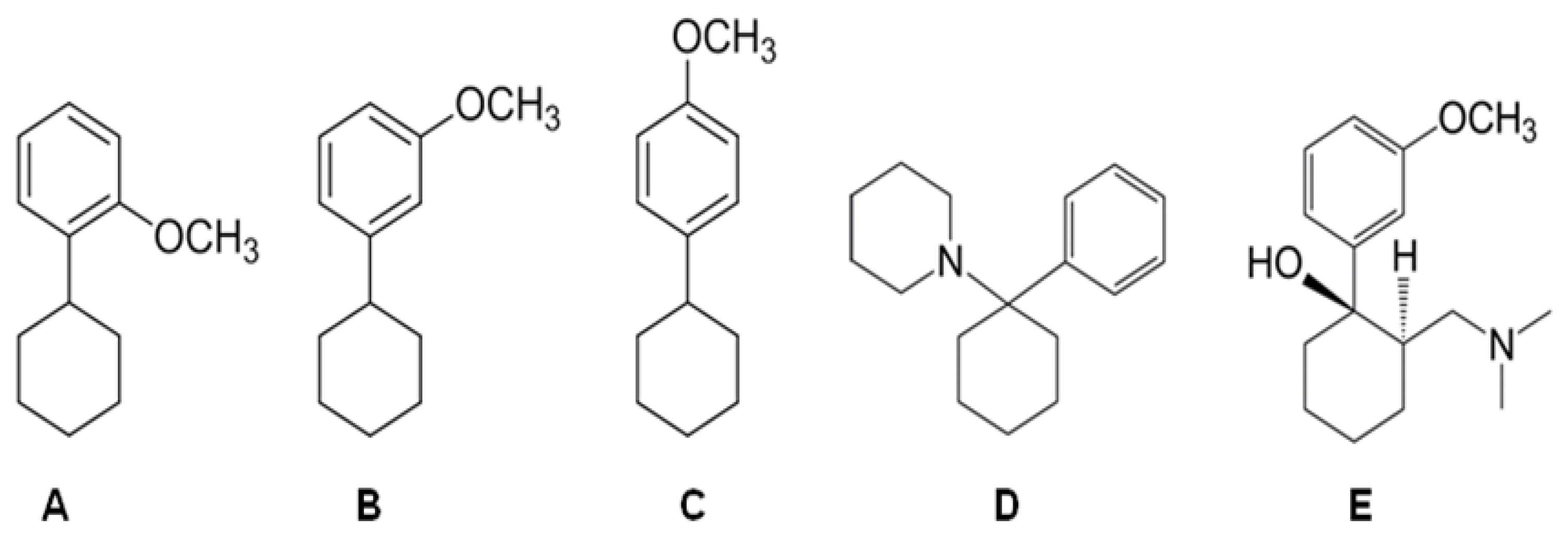
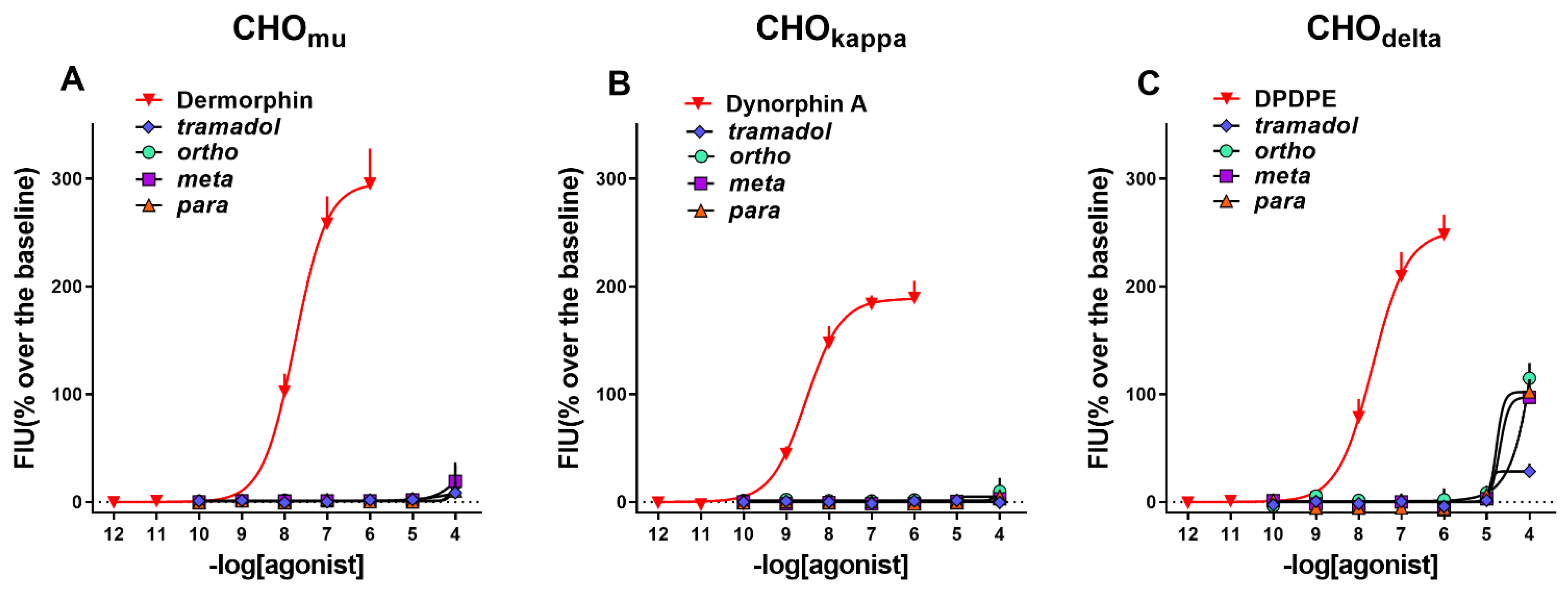
2.1. In Vitro
2.2. In Vivo
2.2.1. Major Neurological Changes
2.2.2. Sensorimotor Studies
Evaluation of the Visual Object Response
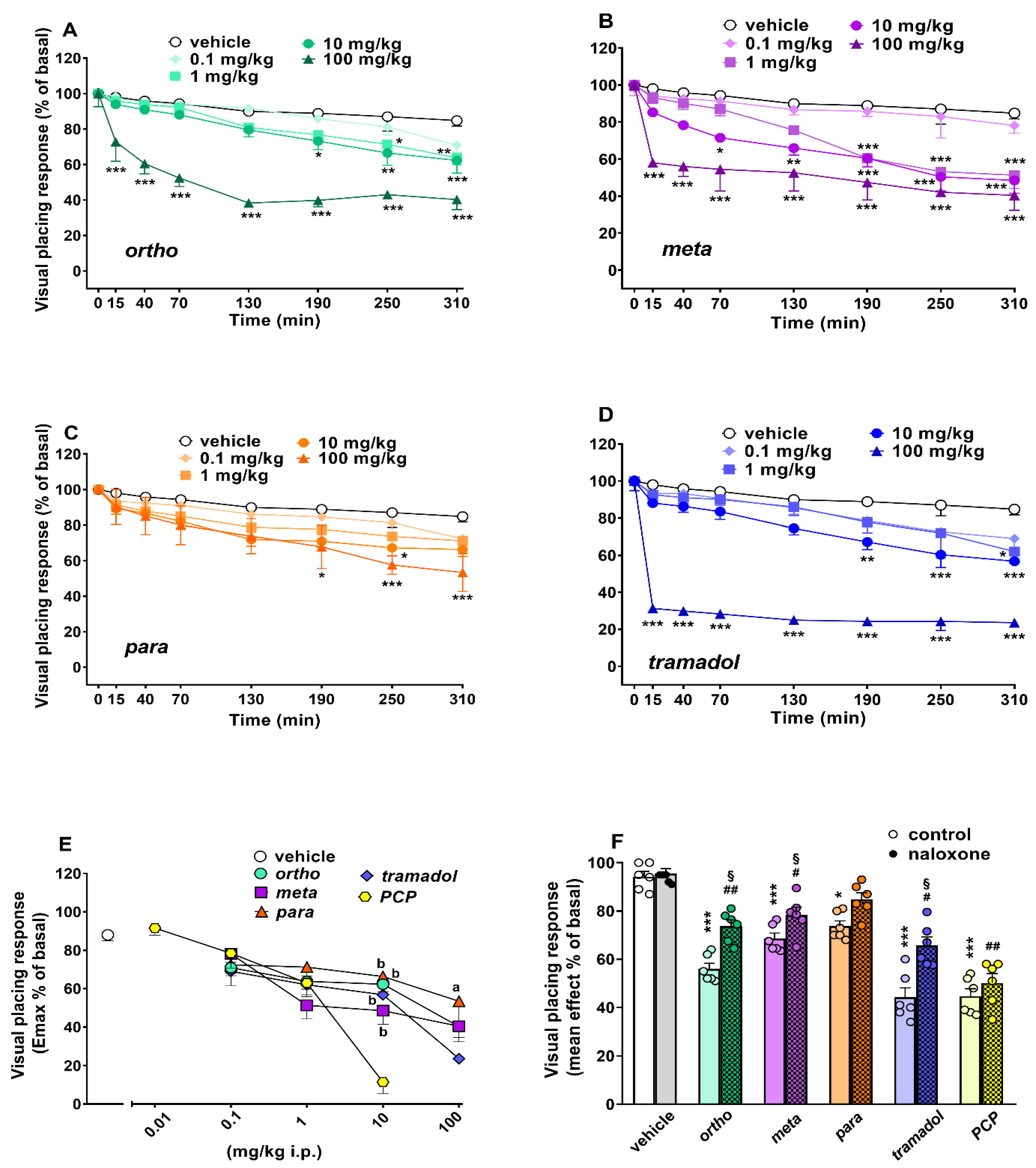
Evaluation of the Visual Placing Response
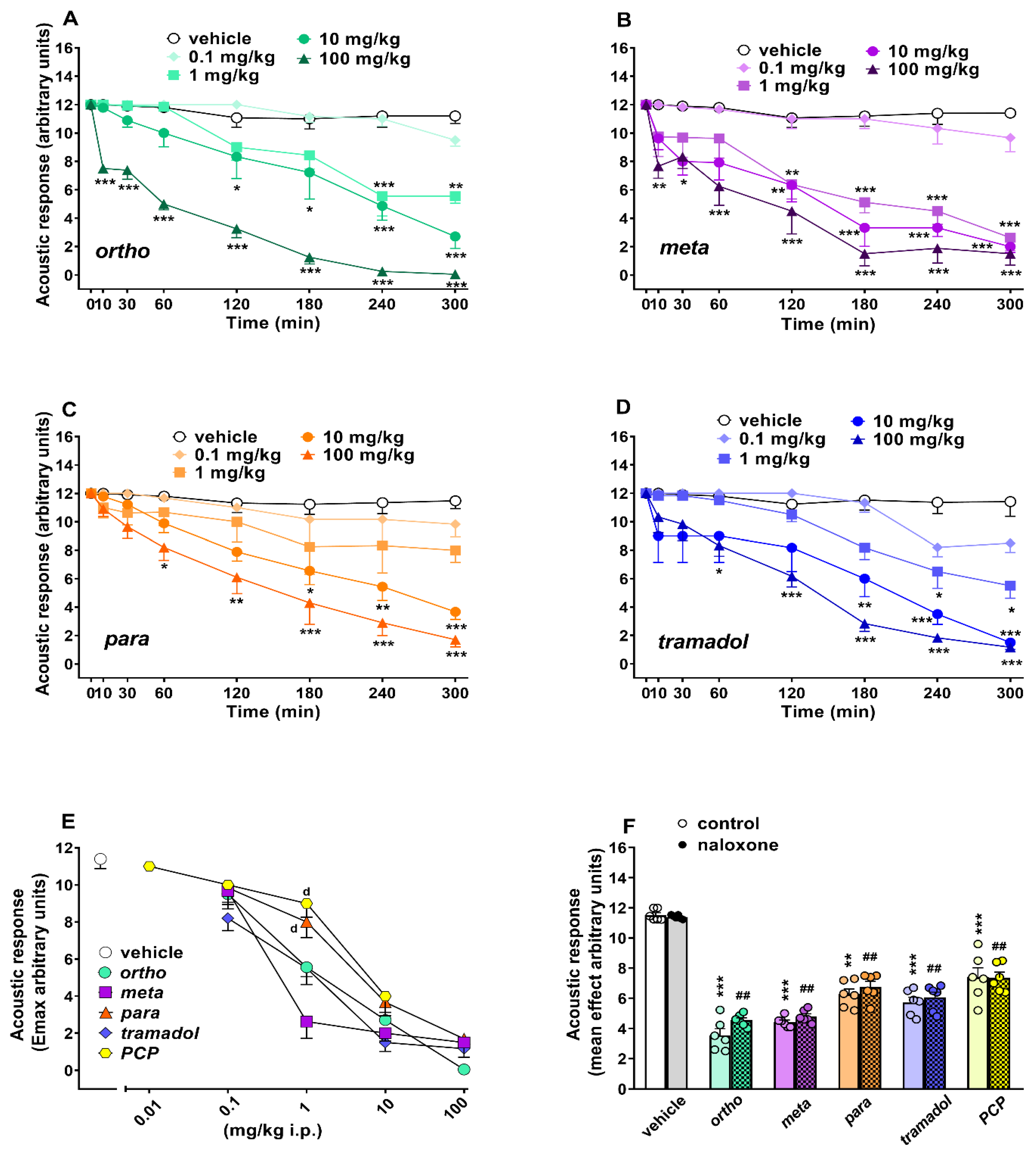
Evaluation of the Acoustic Response
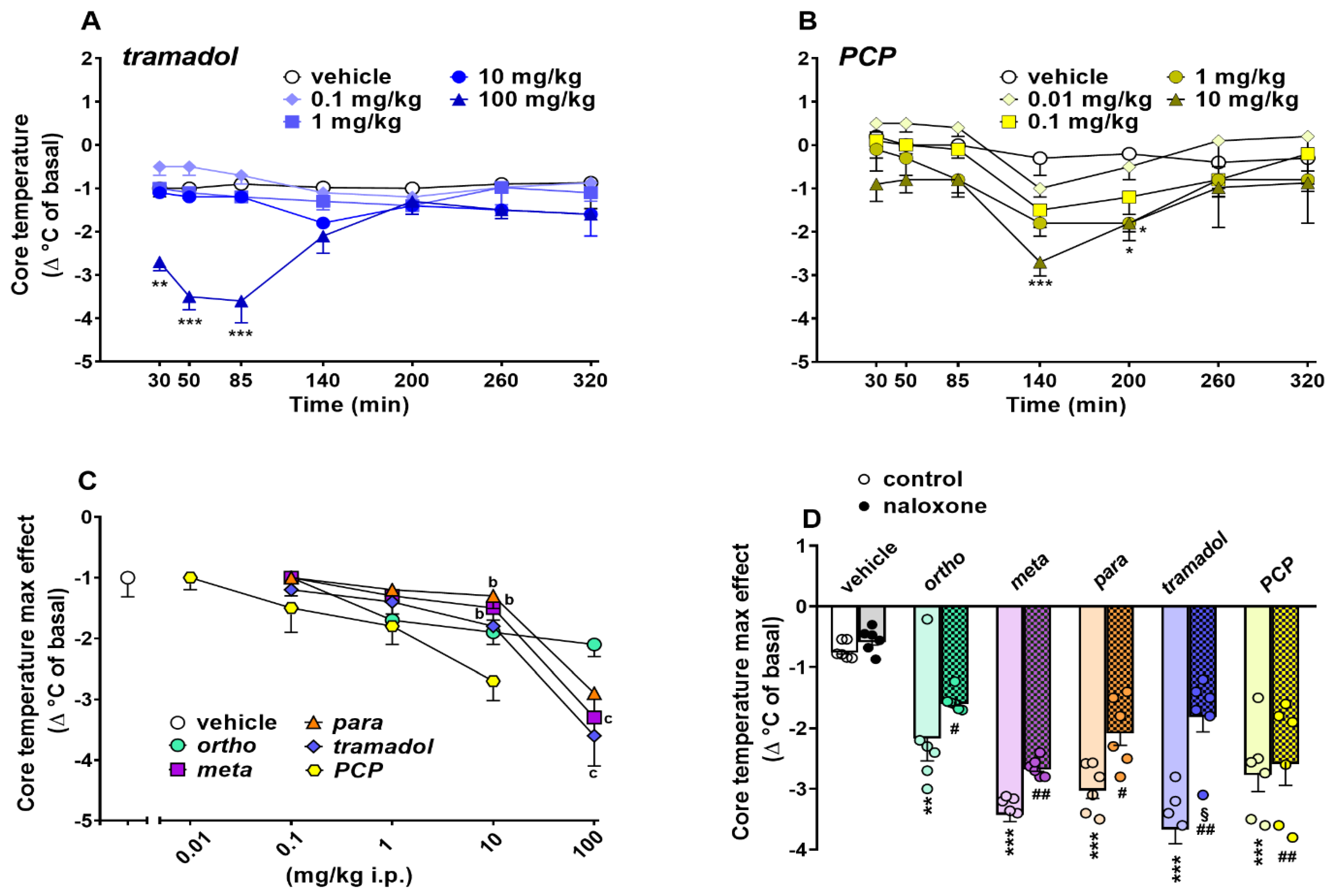
Evaluation of the Core Body Temperature
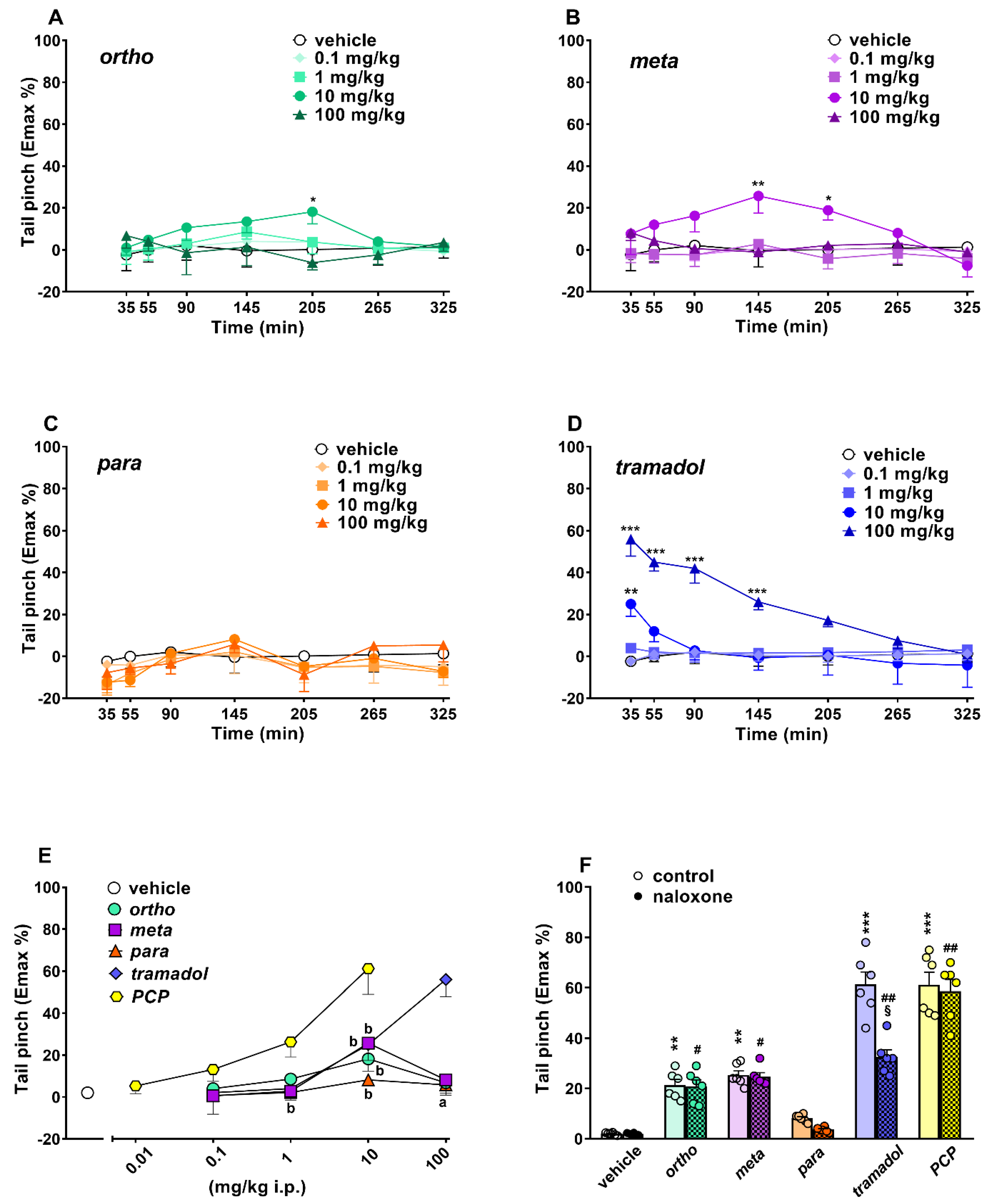
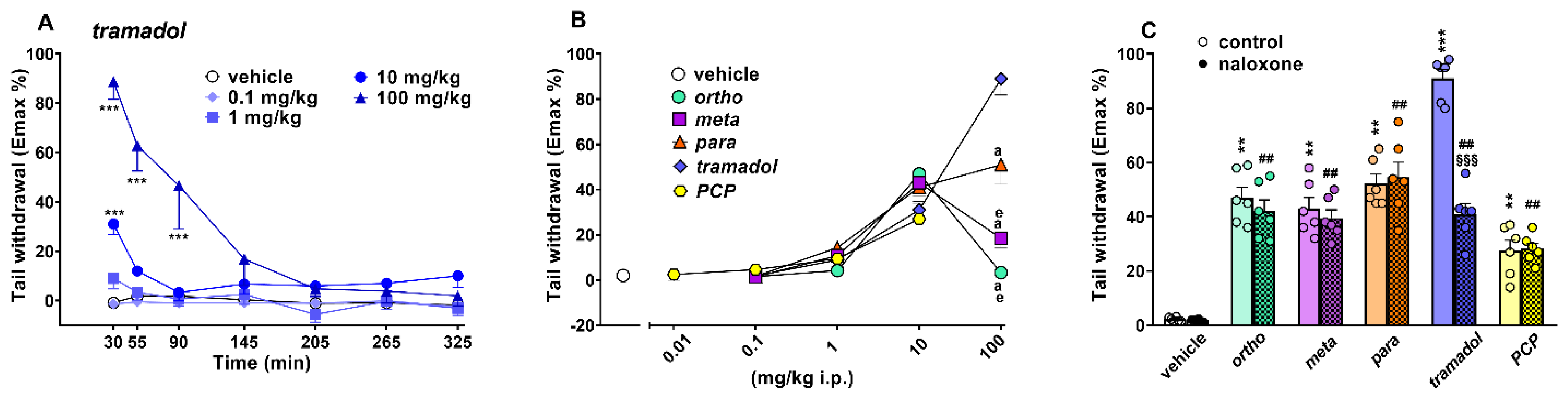
Evaluation of Pain Induced by Mechanical and Thermal Stimuli
Bar Test
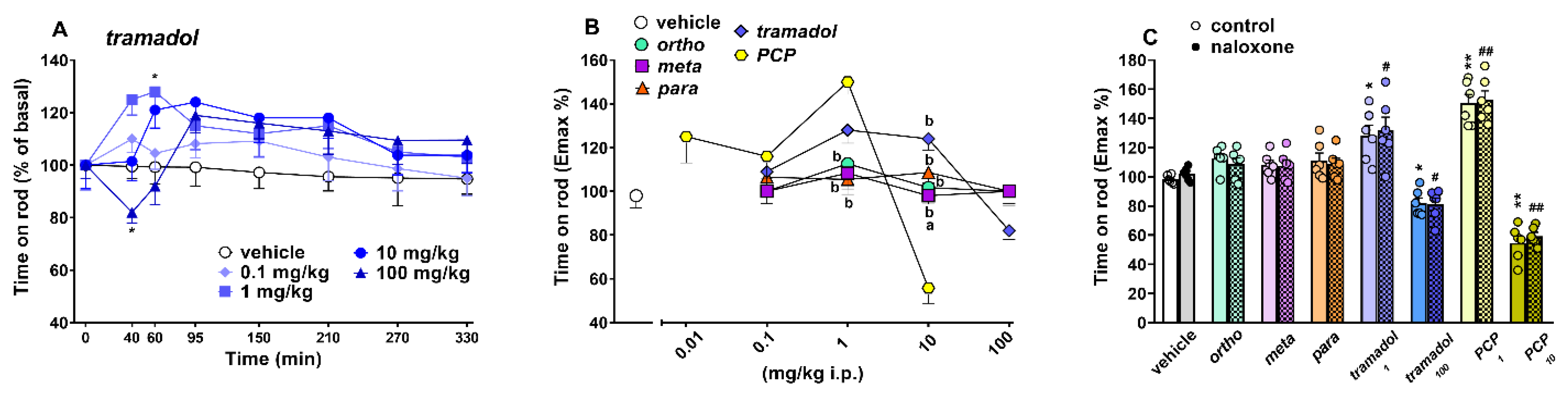
Accelerod Test
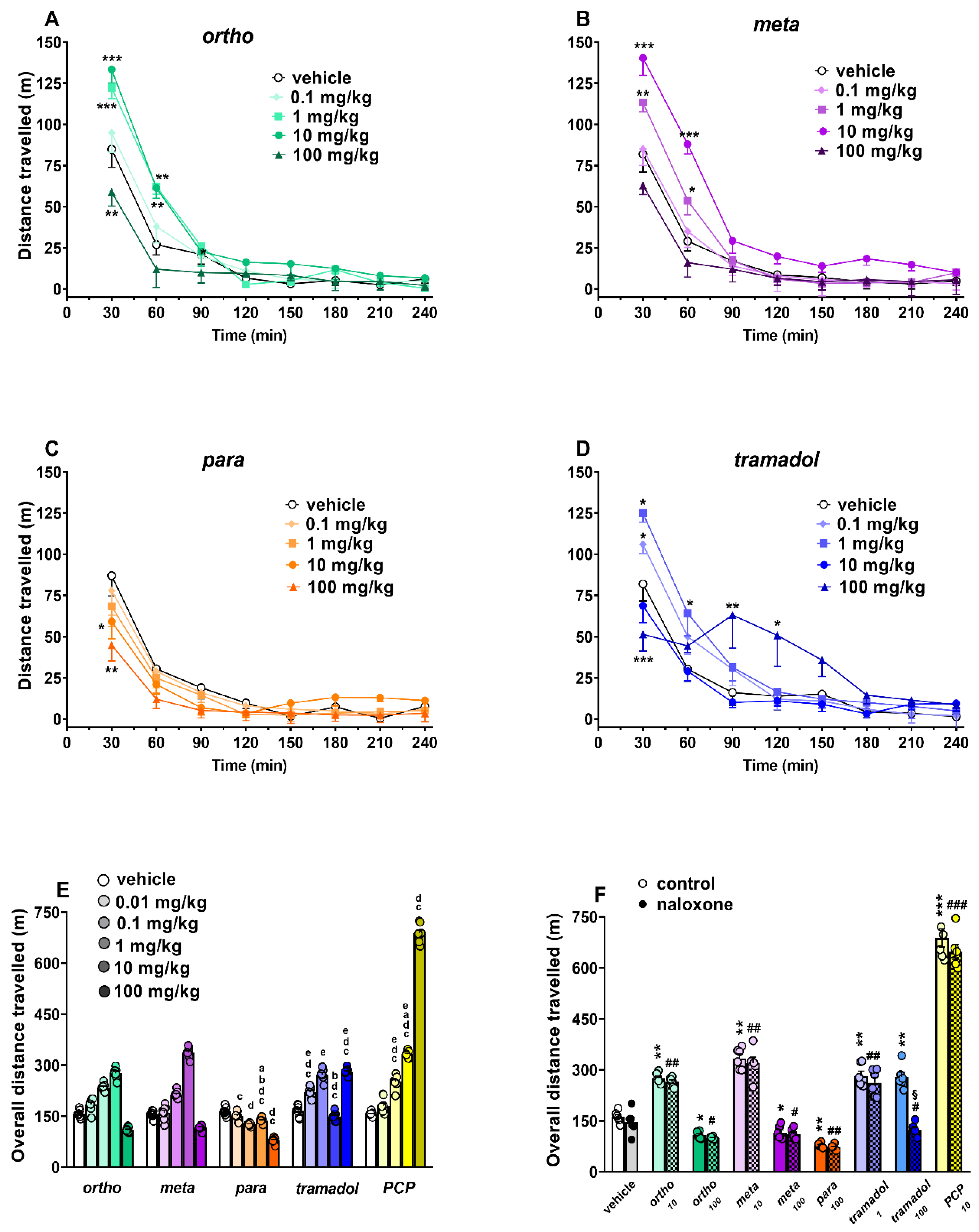
Spontaneous Locomotion Test
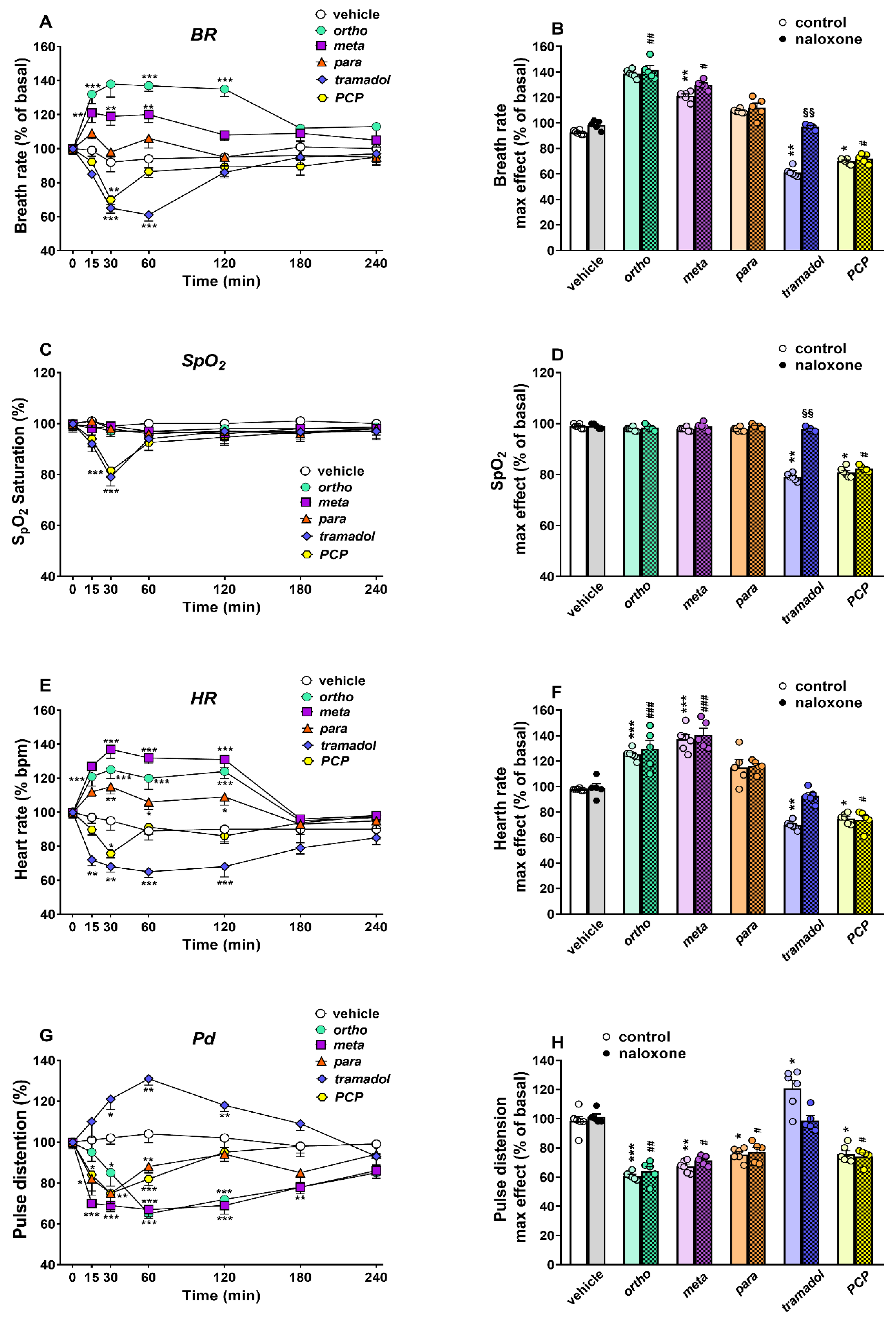
Cardiorespiratory Analysis
3. Discussion
3.1. Major Neurological Changes
3.2. Effect on Sensorimotor Responses
3.3. Effect on Spontaneous and Stimulated Motor Activity
3.4. Effect on Core Body Thermoregulation
3.5. Effect on Acute Mechanical and Thermal Analgesia
3.6. Cardiorespiratory Effects
4. Materials and Methods
4.1. In Vitro Studies
4.1.1. Drugs and Reagents
4.1.2. Cells
4.1.3. Data Analysis and Terminology
4.2. In Vivo Studies
4.2.1. Animals
4.2.2. Drug Preparation and Dose Selection
4.3. Behavioral Studies
4.3.1. Major Neurological Changes
4.3.2. Sensorimotor Studies
Evaluation of the Visual Response
Evaluation of Acoustic Response
Evaluation of Core Body Temperature
Evaluation of Pain Induced by a Mechanical and a Thermal Stimulus
Motor Activity Assessment
4.4. Cardiorespiratory Analysis
4.5. Data and Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- European Monitoring Centre for Drugs and Drug Addiction (EMCDDA). EU Drug Markets Report: In-Depth Analysis. 2016. Available online: https://www.emcdda.europa.eu/publications/joint-publications/eu-drug-markets-2016-in-depth-analysis_en (accessed on 31 March 2021).
- Baumeister, D.; Tojo, L.M.; Tracy, D.K. Legal highs: Staying on top of the flood of novel psychoactive substances. Ther. Adv. Psychopharmacol. 2015, 5, 97–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- European Monitoring Centre for Drugs and Drug Addiction (EMCDDA). Annual Report on the State of the Drugs Problem in Europe. 2012. Available online: https://www.emcdda.europa.eu/publications/annual-report/2012_en (accessed on 31 March 2021).
- Serpelloni, G.; Macchia, T.; Locatelli, C.; Rimondo, C. Seri Nuove Sostanze Psicoattive (NSP): Schede Tecniche Relative alle Molecole Registrate dal Sistema Nazionale di Allerta Precoce. 2013. Available online: http://www.en.npsalert.it/modules/pubbdetails/690/Nuove+Sostanze+Psicoattive+(NSP):+schede+tec.html (accessed on 16 July 2021).
- Fantinati, A.; Ossato, A.; Bianco, S.; Canazza, I.; de Giorgio, F.; Trapella, C.; Marti, M. 1-cyclohexyl-x-methoxybenzene derivatives, novel psychoactive substances seized on the internet market. Synthesis and in vivo pharmacological studies in mice. Hum. Psychopharmacol. 2017, 32, e2560. [Google Scholar] [CrossRef]
- Lanier, R.K.; Lofwall, M.R.; Mintzer, M.Z.; Bigelow, G.E.; Strain, E.C. Physical dependence potential of daily tramadol dosing in humans. Psychopharmacology 2010, 211, 457–466. [Google Scholar] [CrossRef] [Green Version]
- United Nations Office on Drugs and Crime (UNDOC). Annual Report: Covering Activities during 2017. 2017. Available online: https://www.unodc.org/documents/AnnualReport/Annual-Report_2017.pdf (accessed on 31 March 2021).
- European Monitoring Centre for Drugs and Drug Addiction (EMCDDA). European Drug Report. 2019. Available online: https://www.emcdda.europa.eu/edr2019_en (accessed on 31 March 2021).
- Finnegan, K.T.; Kanner, M.I.; Meltzer, H.Y. Phencyclidine-induced rotational behavior in rats with nigrostriatal lesions and its modulation by dopaminergic and cholinergic agents. Pharmacol. Biochem. Behav. 1976, 5, 651–660. [Google Scholar] [CrossRef]
- Simonsen, K.W.; Christoffersen, D.J.; Banner, J.; Linnet, K.; Andersen, L.V. Fatal poisoning among patients with drug addiction. Dan. Med. J. 2015, 62, A5147. [Google Scholar]
- Shadnia, S.; Soltaninejad, K.; Heydari, K.; Sasanian, G.; Abdollahi, M. Tramadol intoxication: A review of 114 cases. Hum. Exp. Toxicol. 2008, 27, 201–205. [Google Scholar] [CrossRef]
- Ryan, N.M.; Isbister, G.K. Tramadol overdose causes seizures and respiratory depression but serotonin toxicity appears unlikely. Clin. Toxicol. 2015, 53, 545–550. [Google Scholar] [CrossRef] [PubMed]
- Brents, L.K.; Gallus-Zawada, A.; Radominska-Pandya, A.; Vasiljevik, T.; Prisinzano, T.E.; Fantegrossi, W.E.; Moran, J.H.; Prather, P.L. Monohydroxylated metabolites of the K2 synthetic cannabinoid JWH-073 retain intermediate to high cannabinoid 1 receptor (CB1R) affinity and exhibit neutral antagonist to partial agonist activity. Biochem. Pharm. 2012, 83, 952–961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiley, J.L.; Marusich, J.A.; Huffman, J.W. Moving around the molecule: Relationship between chemical structure and in vivo activity of synthetic cannabinoids. Life Sci. 2014, 97, 55–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camarda, V.; Fischetti, C.; Anzellotti, N.; Molinari, P.; Ambrosio, C.; Kostenis, E.; Regoli, D.; Trapella, C.; Guerrini, R.; Severo, S.; et al. Pharmacological profile of NOP receptors coupled with calcium signaling via the chimeric protein G alpha qi5. Naunyn-Schmiedeberg’s Arch. Pharm. 2009, 379, 599–607. [Google Scholar] [CrossRef]
- Camarda, V.; Calo, G. Chimeric G proteins in fluorimetric calcium assays: Experience with opioid receptors. Methods Mol. Biol. 2013, 937, 293–306. [Google Scholar] [CrossRef] [PubMed]
- Ossato, A.; Bilel, S.; Gregori, A.; Talarico, A.; Trapella, C.; Gaudio, R.M.; De-Giorgio, F.; Tagliaro, F.; Neri, M.; Fattore, L.; et al. Neurological, sensorimotor and cardiorespiratory alterations induced by methoxetamine, ketamine and phencyclidine in mice. Neuropharmacology 2018, 141, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Spiller, H.A.; Gorman, S.E.; Villalobos, D.; Benson, B.E.; Ruskosky, D.R.; Stancavage, M.M.; Anderson, D.L. Prospective multicenter evaluation of tramadol exposure. J. Toxicol. Clin. Toxicol. 1997, 35, 361–364. [Google Scholar] [CrossRef]
- Tobias, J.D. Seizure after overdose of tramadol. South. Med. J. 1997, 90, 826–827. [Google Scholar] [CrossRef] [PubMed]
- Matthiesen, T.; Wöhrmann, T.; Coogan, T.P.; Uragg, H. The experimental toxicology of tramadol: An overview. Toxicol. Lett. 1998, 16, 63–71. [Google Scholar] [CrossRef]
- Marquardt, K.A.; Alsop, J.A.; Albertson, T.E. Tramadol exposures reported to statewide poison control system. Ann. Pharm. 2005, 39, 1039–1044. [Google Scholar] [CrossRef]
- Raffa, R.B.; Stone, D.J., Jr. Unexceptional seizure potential of tramadol or its enantiomers or metabolites in mice. J. Pharmacol. Exp. 2008, 325, 500–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujimoto, Y.; Funao, T.; Suehiro, K.; Takahashi, R.; Mori, T.; Nishikawa, K. Brain serotonin content regulates the manifestation of tramadol-induced seizures in rats: Disparity between tramadol-induced seizure and serotonin syndrome. Anesthesiology 2015, 122, 178–189. [Google Scholar] [CrossRef]
- Raffa, R.B.; Friderichs, E.; Reimann, W.; Shank, R.P.; Codd, E.E.; Vaught, J.L. Opioid and nonopioid components independently contribute to the mechanism of action of tramadol, an ‘atypical’ opioid analgesic. J. Pharmacol. Exp. Ther. 1992, 260, 275–285. [Google Scholar] [PubMed]
- Gillen, C.; Haurand, M.; Kobelt, D.J.; Wnendt, S. Affinity, potency and efficacy of tramadol and its metabolites at the cloned human mu-opioid receptor. Naunyn-Schmiedeberg’s Arch. Pharm. 2000, 362, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Hara, K.; Minami, K.; Sata, T. The effects of tramadol and its metabolite on glycine, gamma-aminobutyric acidA, and N-methyl-D-aspartate receptors expressed in Xenopus oocytes. Anesth. Analg. 2005, 100, 1400–1405. [Google Scholar] [CrossRef]
- Tao, Q.; Stone, D.J.; Borenstein, M.R.; Codd, E.E.; Coogan, T.P.; Desai-Krieger, D.; Liao, S.; Raffa, R.B. Differential tramadol and O-desmethyl metabolite levels in brain vs. plasma of mice and rats administered tramadol hydrochloride orally. J. Clin. Pharm. Ther. 2002, 27, 99–106. [Google Scholar] [CrossRef]
- Bilel, S.; Azevedo, N.J.; Arfè, R.; Tirri, M.; Gregori, A.; Serpelloni, G.; De-Giorgio, F.; Frisoni, P.; Neri, M.; Calò, G.; et al. In vitro and in vivo pharmacological characterization of the synthetic opioid MT-45. Neuropharmacology 2020, 171, 108110. [Google Scholar] [CrossRef]
- Bilel, S.; Arfè, R.; Tirri, M.; Trapella, C.; Frisoni, P.; Neri, M.; Marti, M. The novel fentanyl-analog “Acrylofentanyl” impairs motor, sensorimotor and cardiovascular functions in mice. Pharmadvances 2020, 2. [Google Scholar] [CrossRef]
- Canal, C.E.; Morgan, D. Head-twitch response in rodents induced by the hallucinogen 2,5-dimethoxy-4-iodoamphetamine: A comprehensive history, a re-evaluation of mechanisms, and its utility as a model. Drug Test. Anal. 2012, 4, 556–576. [Google Scholar] [CrossRef] [PubMed]
- Halberstadt, A.L.; Geyer, M.A. Characterization of the head-twitch response induced by hallucinogens in mice: Detection of the behavior based on the dynamics of head movement. Psychopharmacology 2013, 227, 727–739. [Google Scholar] [CrossRef] [PubMed]
- Bilel, S.; Tirri, M.; Arfè, R.; Stopponi, S.; Soverchia, L.; Ciccocioppo, R.; Frisoni, P.; Strano-Rossi, S.; Miliano, C.; De-Giorgio, F.; et al. Pharmacological and behavioral effects of the synthetic cannabinoid AKB48 in rats. Front. Neurosci. 2019, 13, 1163. [Google Scholar] [CrossRef]
- Tirri, M.; Ponzoni, L.; Bilel, S.; Arfè, R.; Braida, D.; Sala, M.; Marti, M. Acute DOB and PMA administration impairs motor and sensorimotor responses in mice and causes hallucinogenic effects in adult zebrafish. Brain Sci. 2020, 10, 586. [Google Scholar] [CrossRef]
- Miliano, C.; Marti, M.; Pintori, N.; Castelli, M.P.; Tirri, M.; Arfè, R.; de Luca, M.A. Neurochemical and behavioral profiling in male and female rats of the psychedelic agent 25I-NBOMe. Front. Pharm. 2019, 12, 1406. [Google Scholar] [CrossRef] [Green Version]
- Morbiato, E.; Bilel, S.; Tirri, M.; Arfè, R.; Fantinati, A.; Savchuk, S.; Appolonova, S.; Frisoni, P.; Tagliaro, F.; Neri, M.; et al. Potential of the zebrafish model for the forensic toxicology screening of NPS: A comparative study of the effects of APINAC and methiopropamine on the behavior of zebrafish larvae and mice. Neurotoxicology 2020, 78, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Wakita, R.; Tanabe, S.; Tabei, K.; Funaki, A.; Inoshita, T.; Hirano, T. Differential regulations of vestibulo-ocular reflex and optokinetic response by β- and α2-adrenergic receptors in the cerebellar flocculus. Sci. Rep. 2017, 7, 3944. [Google Scholar] [CrossRef] [Green Version]
- Papesh, M.A.; Hurley, L.M. Modulation of auditory brainstem responses by serotonin and specific serotonin receptors. Hear. Res. 2016, 332, 121–136. [Google Scholar] [CrossRef] [PubMed]
- Felix, R.A., 2nd; Elde, C.J.; Nevue, A.A.; Portfors, C.V. Serotonin modulates response properties of neurons in the dorsal cochlear nucleus of the mouse. Hear. Res. 2017, 344, 13–23. [Google Scholar] [CrossRef] [Green Version]
- Tang, Z.Q.; Trussell, L.O. Serotonergic modulation of sensory representation in a central multisensory circuit is pathway specific. Cell Rep. 2017, 20, 1844–1854. [Google Scholar] [CrossRef] [Green Version]
- Baumann, M.H.; Ayestas, M.A., Jr.; Partilla, J.S.; Sink, J.R.; Shulgin, A.T.; Daley, P.F.; Brandt, S.D.; Rothman, R.B.; Ruoho, A.E.; Cozzi, N.V. The designer methcathinone analogs, mephedrone and methylone, are substrates for monoamine transporters in brain tissue. Neuropsychopharmacology 2012, 37, 1192–1203. [Google Scholar] [CrossRef]
- Giannotti, G.; Canazza, I.; Caffino, L.; Bilel, S.; Ossato, A.; Fumagalli, F.; Marti, M. The cathinones MDPV and α-PVP elicit different behavioral and molecular effects following acute exposure. Neurotox. Res. 2017, 32, 594–602. [Google Scholar] [CrossRef]
- De-Giorgio, F.; Bilel, S.; Tirri, M.; Arfè, R.; Trapella, C.; Camuto, C.; Foti, F.; Frisoni, P.; Neri, M.; Botrè, F.; et al. Methiopropamine and its acute behavioral effects in mice: Is there a gray zone in new psychoactive substances users? Int. J. Leg. Med. 2020, 134, 1695–1711. [Google Scholar] [CrossRef] [PubMed]
- Luethi, D.; Liechti, M.E. Designer drugs: Mechanism of action and adverse effects. Arch. Toxicol. 2020, 94, 1085–1133. [Google Scholar] [CrossRef] [Green Version]
- Castañé, A.; Santana, N.; Artigas, F. PCP-based mice models of schizophrenia: Differential behavioral, neurochemical and cellular effects of acute and subchronic treatments. Psychopharmacology 2015, 232, 4085–4097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giannini, A.J.; Nageotte, C.; Loiselle, R.H.; Malone, D.A.; Price, W.A. Comparison of chlorpromazine, haloperidol and pimozide in the treatment of phencyclidine psychosis: DA-2 receptor specificity. J. Toxicol. Clin. Toxicol. 1984, 22, 573–579. [Google Scholar] [CrossRef] [PubMed]
- Ogren, S.O.; Goldstein, M. Phencyclidine- and dizocilpine-induced hyperlocomotion are differentially mediated. Neuropsychopharmacology 1994, 11, 167–177. [Google Scholar] [CrossRef] [Green Version]
- Seeman, P.; Lasaga, M. Dopamine agonist action of phencyclidine. Synapse 2005, 58, 275–277. [Google Scholar] [CrossRef] [PubMed]
- Seeman, P.; Guan, H.C. Phencyclidine and glutamate agonist LY379268 stimulate dopamine D2High receptors: D2 basis for schizophrenia. Synapse 2008, 62, 819–828. [Google Scholar] [CrossRef] [PubMed]
- Jodo, E. The role of the hippocampo-prefrontal cortex system in phencyclidine-induced psychosis: A model for schizophrenia. J. Physiol. 2013, 107, 434–440. [Google Scholar] [CrossRef] [PubMed]
- Hiramatsu, M.; Nabeshima, T.; Kameyama, T. Involvement of opioid receptors in hypo- and hyperthermic effects induced by phencyclidine in mice. J. Pharm. 1986, 9, 466–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, A.K.; Meert, T.F. Functional effects of systemically administered agonists and antagonists of mu, delta, and kappa opioid receptor subtypes on body temperature in mice. J. Pharmacol. Exp. 2002, 302, 1253–1264. [Google Scholar] [CrossRef]
- Wolfe, A.M.; Kennedy, L.H.; Na, J.J.; Nemzek-Hamlin, J.A. Efficacy of tramadol as a sole analgesic for postoperative pain in male and female mice. J. Am. Assoc. Lab. Anim. Sci. 2015, 54, 411–419. [Google Scholar]
- Kiyatkin, E.A. Brain temperature and its role in physiology and pathophysiology: Lessons from 20 years of thermorecording. Temperature 2019, 6, 271–333. [Google Scholar] [CrossRef] [Green Version]
- Itoh, Y.; Oishi, R.; Nishibori, M.; Saeki, K. Comparison of effects of phencyclidine and methamphetamine on body temperature in mice: A possible role for histamine neurons in thermoregulation. Naunyn-Schmiedeberg’s Arch. Pharm. 1986, 332, 293–296. [Google Scholar] [CrossRef]
- Evangelista Vaz, R.; Draganov, D.I.; Rapp, C.; Avenel, F.; Steiner, G.; Arras, M.; Bergadano, A. Preliminary pharmacokinetics of tramadol hydrochloride after administration via different routes in male and female B6 mice. Vet. Anaesth. Analg. 2018, 45, 111–122. [Google Scholar] [CrossRef]
- Sevcik, J.; Nieber, K.; Driessen, B.; Illes, P. Effects of the central analgesic tramadol and its main metabolite, O-desmethyltramadol, on rat locus coeruleus neurones. Br. J. Pharmacol. 1993, 110, 169–176. [Google Scholar] [CrossRef] [Green Version]
- Shen, Q.; Qian, Y.Y.; Xu, X.J.; Li, W.; Liu, J.G.; Fu, W. Design, synthesis and biological evaluation of N-phenylalkyl-substituted tramadol derivatives as novel μ opioid receptor ligands. Acta Pharmacol. Sin. 2015, 36, 887–894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ide, S.; Minami, M.; Ishihara, K.; Uhl, G.R.; Sora, I.; Ikeda, K. Mu opioid receptor-dependent and independent components in effects of tramadol. Neuropharmacology 2006, 51, 651–658. [Google Scholar] [CrossRef]
- Aydin, O.N.; Ek, R.O.; Temoçin, S.; Uğur, B.; Alaçam, B.; Şen, S. The antinociceptive effects of systemic administration of tramadol, gabapentin and their combination on mice model of acute pain. Agri 2012, 24, 49–55. [Google Scholar] [CrossRef]
- Yanarates, O.; Dogrul, A.; Yildirim, V.; Sahin, A.; Sizlan, A.; Seyrek, M.; Akgül, O.; Kozak, O.; Kurt, E.; Aypar, U. Spinal 5-HT7 receptors play an important role in the antinociceptive and antihyperalgesic effects of tramadol and its metabolite, O-Desmethyltramadol, via activation of descending serotonergic pathways. Anesthesiology 2010, 112, 696–710. [Google Scholar] [CrossRef] [Green Version]
- Müller, B.; Wilsmann, K. Cardiac and hemodynamic effects of the centrally acting analgesics tramadol and pentazocine in anaesthetized rabbits and isolated guinea-pig atria and papillary muscles. Arzneimittel-forschung 1984, 34, 430–433. [Google Scholar]
- Itami, T.; Tamaru, N.; Kawase, K.; Ishizuka, T.; Tamura, J.; Miyoshi, K.; Umar, M.A.; Inoue, H.; Yamashita, K. Cardiovascular effects of tramadol in dogs anesthetized with sevoflurane. J. Vet. Med. Sci. 2011, 73, 1603–1609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagaoka, E.; Minami, K.; Shiga, Y.; Uezono, Y.; Shiraishi, M.; Aoyama, K.; Shigematsu, A. Tramadol has no effect on cortical renal blood flow—despite increased serum catecholamine levels—in anesthetized rats: Implications for analgesia in renal insufficiency. Anesth. Analg. 2002, 94, 619–625. [Google Scholar] [CrossRef]
- Egger, C.M.; Souza, M.J.; Greenacre, C.B.; Cox, S.K.; Rohrbach, B.W. Effect of intravenous administration of tramadol hydrochloride on the minimum alveolar concentration of isoflurane in rabbits. Am. J. Vet. Res. 2009, 70, 945–949. [Google Scholar] [CrossRef] [PubMed]
- Nishioka, K. The effect of non-narcotic analgesic, tramadol, on cardiac contractility in dog. Tohoku J. Exp. Med. 1979, 128, 401–402. [Google Scholar] [PubMed]
- Raimundo, J.M.; Sudo, R.T.; Pontes, L.B.; Antunes, F.; Trachez, M.M.; Zapata-Sudo, G. In vitro and in vivo vasodilator activity of racemic tramadol and its enantiomers in Wistar rats. Eur. J. Pharm. 2006, 530, 117–123. [Google Scholar] [CrossRef]
- Kaya, T.; Gursoy, S.; Karadas, B.; Sarac, B.; Fafali, H.; Soydan, A.S. High-concentration tramadol-induced vasodilation in rabbit aorta is mediated by both endothelium-dependent and -independent mechanisms. Acta Pharmacol. Sin. 2003, 24, 385–389. [Google Scholar] [PubMed]
- Close, B.R. Tramadol: Does it have a role in emergency medicine? Emerg. Med. Australas. 2005, 17, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Müller, H.; Stoyanov, M.; Brähler, A.; Hempelmann, G. Hämodynamische und respiratorische effekte von tramadol bei lachgas-sauerstoff-beatmung und in der postoperativen phase [Hemodynamic and respiratory effects of tramadol during nitrous oxide-oxygen-artificial respiration and in the postoperative period]. Anaesthesist 1982, 31, 604–610. [Google Scholar]
- Elkalioubie, A.; Allorge, D.; Robriquet, L.; Wiart, J.F.; Garat, A.; Broly, F.; Fourrier, F. Near-fatal tramadol cardiotoxicity in a CYP2D6 ultrarapid metabolizer. Eur. J. Clin. Pharm. 2011, 67, 855–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Decker, K.; Cordonnier, J.; Jacobs, W.; Coucke, V.; Schepens, P.; Jorens, P.G. Fatal intoxication due to tramadol alone: Case report and review of the literature. Forensic Sci. Int. 2008, 175, 79–82. [Google Scholar] [CrossRef]
- Daubin, C.; Quentin, C.; Goullé, J.P.; Guillotin, D.; Lehoux, P.; Lepage, O.; Charbonneau, P. Refractory shock and asystole related to tramadol overdose. Clin. Toxicol. 2007, 45, 961–964. [Google Scholar] [CrossRef]
- Lagard, C.; Malissin, I.; Indja, W.; Risède, P.; Chevillard, L.; Mégarbane, B. Is naloxone the best antidote to reverse tramadol-induced neuro-respiratory toxicity in overdose? An experimental investigation in the rat. Clin. Toxicol. 2018, 56, 737–743. [Google Scholar] [CrossRef]
- Hassanian-Moghaddam, H.; Farajidana, H.; Sarjami, S.; Owliaey, H. Tramadol-induced apnea. Am. J. Emerg. Med. 2013, 31, 26–31. [Google Scholar] [CrossRef]
- Hondebrink, L.; Zwartsen, A.; Westerink, R.H.S. Effect fingerprinting of new psychoactive substances (NPS): What can we learn from in vitro data? Pharmacology 2018, 182, 193–224. [Google Scholar] [CrossRef]
- Zhang, H.; Cuevas, J. Sigma receptor activation blocks potassium channels and depresses neuroexcitability in rat intracardiac neurons. J. Pharmacol. Exp. 2005, 313, 1387–1396. [Google Scholar] [CrossRef] [Green Version]
- Polakowski, J.S.; Segreti, J.A.; Cox, B.F.; Hsieh, G.C.; Kolasa, T.; Moreland, R.B.; Brioni, J.D. Effects of selective dopamine receptor subtype agonists on cardiac contractility and regional haemodynamics in rats. Clin. Exp. Pharmacol. Physiol. 2004, 31, 837–841. [Google Scholar] [CrossRef]
- D’Amico, G.A.; Kline, R.P.; Maayani, S.; Weinstein, H.; Kupersmith, J. Effects of phencyclidine on cardiac action potential: pH dependence and structure-activity relationships. Eur. J. Pharm. 1983, 88, 283–290. [Google Scholar] [CrossRef]
- Vigolo, A.; Ossato, A.; Trapella, C.; Vincenzi, F.; Rimondo, C.; Seri, C.; Varani, K.; Serpelloni, G.; Marti, M. Novel halogenated derivates of JWH-018: Behavioral and binding studies in mice. Neuropharmacology 2015, 95, 68–82. [Google Scholar] [CrossRef] [PubMed]
- Ossato, A.; Vigolo, A.; Trapella, C.; Seri, C.; Rimondo, C.; Serpelloni, G.; Marti, M. JWH-018 impairs sensorimotor functions in mice. Neuroscience 2015, 300, 174–188. [Google Scholar] [CrossRef] [PubMed]
- Canazza, I.; Ossato, A.; Trapella, C.; Fantinati, A.; De Luca, M.A.; Margiani, G.; Vincenzi, F.; Rimondo, C.; di Rosa, F.; Gregori, A.; et al. Effect of the novel synthetic cannabinoids AKB48 and 5F-AKB48 on “tetrad”, sensorimotor, neurological and neurochemical responses in mice. In vitro and in vivo pharmacological studies. Psychopharmacology 2016, 233, 3685–3709. [Google Scholar] [CrossRef] [PubMed]
- Ossato, A.; Canazza, I.; Trapella, C.; Vincenzi, F.; de Luca, M.A.; Rimondo, C.; Varani, K.; Borea, P.A.; Serpelloni, G.; Marti, M. Effect of JWH-250, JWH-073 and their interaction on “tetrad”, sensorimotor, neurological and neurochemical responses in mice. Prog. Neuropsychopharmacol. Biol. Psychiatry 2016, 67, 31–50. [Google Scholar] [CrossRef]
- Foti, F.; Marti, M.; Ossato, A.; Bilel, S.; Sangiorgi, E.; Botrè, F.; Cerbelli, B.; Baldi, A.; De-Giorgio, F. Phenotypic effects of chronic and acute use of methiopropamine in a mouse model. Int. J. Leg. Med. 2019, 133, 811–820. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mu | Kappa | Delta | ||||
|---|---|---|---|---|---|---|
| Compounds | pEC50 (CL95%) | Emax ± sem % | pEC50 (CL95%) | Emax ± sem % | pEC50 (CL95%) | Emax ± sem % |
| Standard agonists | 7.76 (7.53–7.99) | 295 ± 33% | 8.50 (8.32–8.68) | 189 ± 16% | 7.65 (7.36–7.94) | 248 ± 19% |
| Tramadol | Inactive | Inactive | Crc incomplete, at 100 mM 102 ± 9% | |||
| Ortho | Inactive | Inactive | Crc incomplete, at 100 mM 115 ± 7% | |||
| Meta | Inactive | Inactive | Crc incomplete, at 100 mM 115 ± 7% | |||
| Para | Inactive | Inactive | Crc incomplete, at 100 mM 97 ± 8% | |||
| 1-Cyclohexyl-x-Methoxybenzene (0.1–100 mg/kg i.p) | Tramadol (0.1–100 mg/kg i.p) | PCP (0.01–10 mg/kg i.p) | |||
|---|---|---|---|---|---|
| ortho | meta | para | |||
| Neurological changes | no effect | tail elevation and convulsion NLX insensitive | no effect | ||
| Visual object response | dose-dependent inhibition NLX partially sensitive | dose-dependent inhibition NLX partially sensitive | dose-dependent inhibition NLX insensitive | ||
| Visual placing response | dose-dependent inhibition NLX partially sensitive | dose-dependent inhibition NLX partially sensitive | dose-dependent inhibition NLX sensitive | dose-dependent inhibition NLX sensitive | dose-dependant inhibition NLX insensitive |
| Acoustic response | dose-dependent inhibition NLX insensitive | dose-dependent inhibition NLX insensitive | dose-dependent inhibition NLX insensitive | ||
| Core body temperature | dose-dependent inhibition NLX insensitive | dose-dependent inhibition NLX partially sensitive | dose-dependent inhibition NLX insensitive | ||
| Mechanical analgesia | mild analgesia NLX insensitive | mild analgesia NLX insensitive | no effect | dose-dependent analgesia NLX partially sensitive | dose-dependent analgesia NLX insensitive |
| Thermal analgesia | dose-dependent inhibition NLX insensitive | dose-dependent analgesia NLX partially sensitive | dose-dependent analgesia NLX insensitive | ||
| Bar test | no effect | no effect | no effect | ||
| Stimulated locomotion | no effect | biphasic effect NLX insensitive | biphasic effect NLX insensitive | ||
| Sponotaneous locomotion | biphasic effect NLX insensitive | biphasic effect NLX insensitive | inhibitory effect NLX insensitive | biphasic effect NLX partially sensitive with low dosage | biphasic effect NLX insensitive |
| Breath rate | increased NLX insensitive | increased NLX insensitive | no effect | decreased NLX sensitive | decreased NLX insensitive |
| SpO2 saturation | no effect | no effect | no effect | decreased NLX sensitive | decreased NLX insensitive |
| Heart rate | increased NLX insensitive | increased NLX insensitive | increased NLX insensitive | decreased NLX sensitive | decreased NLX insensitive |
| Pulse distention | decreased NLX sensitive | decreased NLX sensitive | decreased NLX sensitive | increased NLX sensitive | decreased NLX insensitive |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bilel, S.; Tirri, M.; Arfè, R.; Sturaro, C.; Fantinati, A.; Cristofori, V.; Bernardi, T.; Boccuto, F.; Cavallo, M.; Cavalli, A.; et al. In Vitro and In Vivo Pharmaco-Toxicological Characterization of 1-Cyclohexyl-x-methoxybenzene Derivatives in Mice: Comparison with Tramadol and PCP. Int. J. Mol. Sci. 2021, 22, 7659. https://doi.org/10.3390/ijms22147659
Bilel S, Tirri M, Arfè R, Sturaro C, Fantinati A, Cristofori V, Bernardi T, Boccuto F, Cavallo M, Cavalli A, et al. In Vitro and In Vivo Pharmaco-Toxicological Characterization of 1-Cyclohexyl-x-methoxybenzene Derivatives in Mice: Comparison with Tramadol and PCP. International Journal of Molecular Sciences. 2021; 22(14):7659. https://doi.org/10.3390/ijms22147659
Chicago/Turabian StyleBilel, Sabrine, Micaela Tirri, Raffaella Arfè, Chiara Sturaro, Anna Fantinati, Virginia Cristofori, Tatiana Bernardi, Federica Boccuto, Marco Cavallo, Alessandro Cavalli, and et al. 2021. "In Vitro and In Vivo Pharmaco-Toxicological Characterization of 1-Cyclohexyl-x-methoxybenzene Derivatives in Mice: Comparison with Tramadol and PCP" International Journal of Molecular Sciences 22, no. 14: 7659. https://doi.org/10.3390/ijms22147659