
Cellular Senescence and Inflammaging in the Skin Microenvironment


1
Department of Dermatology, Severance Hospital, Cutaneous Biology Research Institute, Yonsei University College of Medicine, Seoul 107-11, Korea
2
Scar Laser and Plastic Surgery Center, Yonsei Cancer Hospital, Yonsei University College of Medicine, Seoul 107-11, Korea
*
Authors to whom correspondence should be addressed.
Int. J. Mol. Sci. 2021, 22(8), 3849; https://doi.org/10.3390/ijms22083849
Submission received: 19 March 2021
/
Revised: 5 April 2021
/
Accepted: 6 April 2021
/
Published: 8 April 2021
(This article belongs to the Special Issue Skin Inflammation Aging and Diseases)
{kind=link}
Abstract
:Cellular senescence and aging result in a reduced ability to manage persistent types of inflammation. Thus, the chronic low-level inflammation associated with aging phenotype is called “inflammaging”. Inflammaging is not only related with age-associated chronic systemic diseases such as cardiovascular disease and diabetes, but also skin aging. As the largest organ of the body, skin is continuously exposed to external stressors such as UV radiation, air particulate matter, and human microbiome. In this review article, we present mechanisms for accumulation of senescence cells in different compartments of the skin based on cell types, and their association with skin resident immune cells to describe changes in cutaneous immunity during the aging process.
1. Introduction
As the aging process occurs, the ability of the human body to resolve inflammation becomes significantly reduced, resulting in an imbalance between proinflammation and anti-inflammation. This results in a chronic low-grade pro-inflammatory state known as “inflammaging”, which accelerates age-related diseases, such as diabetes, heart diseases, and even certain types of cancer [1]. It is driven by a systemic increase in multiple pro-inflammatory cytokines and is significantly influenced by several extrinsic factors, including UV radiation (UVR), air particulate matter (PM), and the microbiome [1]. Notably, cellular senescence arises from intrinsic (proliferative exhaustion and telomere shortening) and extrinsic stresses and by the activation of oncogenes [2]. Senescent cells exhibit an altered secretome referred to as a senescence-associated secretory phenotype (SASP), which secretes pro-inflammatory cytokines that considerably alter the skin microenvironment [3]. Cellular senescence can transiently occur in several physiological conditions during embryogenesis and wound healing; by contrast, senescence due to the process of skin aging is mainly a persisting permanent state [4]. In this review, we discussed the recent updates on cellular senescence and inflammaging during the aging process of the skin. The emphasis of this review lies on the comprehensive understanding of the cellular cross-talks among the components of skin microenvironment with resident immune cells, in association with external environmental stressors that cause skin aging (Figure 1).
2. Fibroblast Senescence
An emerging hypothesis postulates that fibroblast senescence is the main driver of the skin aging process as the release of SASPs increase and the proliferation of cells is arrested irreversibly [4]. Fibroblasts are the major type of cells that constitute the dermis layer of the skin. They produce integral components of the extracellular matrix (ECM), such as collagen [5]. During the skin aging process, the ECM undergoes dramatic structural alterations and degradation, resulting in aging phenotypes of dermal thinning and loss of elasticity that eventually cause wrinkle formation [6]. Permanent senescence or skin aging can be induced in nonreplicating (senescent) fibroblasts by both intrinsic and extrinsic stressors that lead to the shortening of telomeres, mitochondrial dysfunction, and subsequent activation of the DNA damage response signaling pathway, leading to cell cycle arrest [7,8,9,10,11]. The subsequent increase in the number of senescent fibroblasts results in the production of SASPs rich in pro-inflammatory cytokines, including interleukin (IL)-1, IL-6, IL-8, IL-18, matrix metalloproteinases (MMPs), and a variety of other inflammatory chemokines [12]. These cytokines can induce c-Jun N-terminal kinase (JNK) and the transcription factor activator protein (AP-1), which subsequently activate MMPs, resulting in the breakdown of collagen and loss of elasticity, and consequently, wrinkle formation [4,13,14,15].
The acquired senescence-like phenotype in fibroblasts is indicated by the elevated levels of the cyclin-dependent kinases, p16INK4a and p21, and the tumor protein p53, and the expression of senescence-associated β-galactosidase (SA-β-gal), which are critically influenced by UVR [2]. In vitro, ultraviolet B light (UVB)-exposed skin cell types, including fibroblasts and keratinocytes, exhibit DNA damage and cell cycle arrest evidenced by expression of senescence markers, such as increased SASPs, and decreased Lamin B1 [16,17,18]. On the contrary, in vivo experiments showed that chronic low-dose exposure to UVB resulted in the accumulation of DNA damage and reduced Lamin B1 expression in senescent cells within the mouse epidermis, explicitly in the basal and suprabasal layers, but not the dermis [19].
UVR also downregulates tumor growth factor (TGF)-β signaling pathway by activating AP-1 and p53 [20]. TGF-β is a cytokine that promotes ECM formation via the Smad pathway, leading to the downregulation of MMPs and enhancement of ECM gene expression. TGF-β signaling is one of the major pathways that induce type I procollagen synthesis and secretion through connective tissue growth factor (CTGF). The impaired TGF-β/SMAD3 signaling results in the reduction of CTGF-dependent type I collagen synthesis while increasing MMP1-induced collagen degradation, leading to dermal thinning. Thus, decreased TGF-β signaling by UVR critically contributes to wrinkle formation [21].
Moreover, TGF-β regulates the activation/induction of autophagy by increasing the mRNA expression of Beclin1, autophagy-related gene (ATG)5, and ATG7 in a Smad- and JNK-dependent manner [22]. Autophagy in dermal fibroblasts plays a crucial role in the aging process and skin homeostasis under damaged conditions induced by external stimuli, such as UVR and PM, by repairing cellular machineries [23]. A lack of autophagy results in a hyper-inflammatory skin reaction caused by inflammasome activation, leading to increased aging phenotypes. Interestingly, the autophagy system and the circadian clock counteract tissue degeneration and support longevity in many organisms [24]. Hence, the aged dermal fibroblasts exhibit impaired autophagic responses because of the reduced expression of the period circadian regulator 2 and mini-chromosome maintenance 7 helicase, a transcriptional repressor component of the circadian oscillator [23,24,25].
Dermal fibroblasts also release insulin-like growth factor (IGF)-1, which is essential for the balanced regulation of epidermal cell proliferation and differentiation [4]. IGF-1 signaling in senescent fibroblasts is significantly decreased due to the enhanced production of superoxide anions in the dysfunctional mitochondria of aging dermal fibroblasts [26]. The inhibition of the IGF-1 pathway not only suppresses collagen synthesis in the dermis, but also leads to epidermal atrophy due to the increased accumulation of the DNA damage-induced phosphorylated histone protein, γH2AX, and p16INK4a-positive epidermal cells [4].
In addition, mammalian target of rapamycin (mTOR) signaling in dermal fibroblasts has been implicated in senescence through the regulation of SASPs. The inhibition of mTOR suppresses SASPs by downregulating mitogen-activated protein kinase (MAPK)-activated protein kinase 2 translation through the binding protein 4EBP1 [27]. Rapamycin, an mTOR inhibitor, suppresses translation of the membrane-bound cytokine IL-1a, resulting in the downregulation of IL-1a-induced secretion of inflammatory SASPs [28,29]. Moreover, mTOR signaling pathway regulates inflammaging by activating the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), which in turn increases SASPs [1]. Reduced IL-1a by rapamycin diminishes NF-κB transcriptional activity, resulting in decreased SASPs [29].
Mitochondrial dysfunction is another major driver in both intrinsic and extrinsic skin aging [30]. In aging fibroblasts increasing concentrations of reactive oxygen species (ROS) either drive DNA damage in the skin or enhance AP-1 and NF-κB-dependent signaling pathways, leading to inflammaging [31]. A previous study in a superoxide dismutase-deficient mouse model indicated that mitochondrial superoxide anions could not be detoxified, hence they accumulated in the mitochondria of the fibroblasts resulting in accelerated skin aging phenotypes [32]. These data showed that mitochondrial dysfunction and enhanced superoxide anion concentrations lead to fibroblast senescence, thus, accelerating aging in connective tissue-rich organs, such as skin [4].
Persistent ROS-induced DNA damage drives cellular senescence and can be identified by markers for DNA double-strand breaks, such as γH2AX, that are co-localized with DNA damage checkpoint factors including p53-binding protein 1 (53BP1), mediator of DNA damage checkpoint protein 1, and Nijmegen breakage syndrome 1 [33]. In addition, the occurrence of telomere-associated DNA damage foci (TAF) has been used to detect senescent cells and quantify tissue aging in situ [34,35]. Replicative senescent baboon skin fibroblasts and skin tissues from aged baboons revealed increased TAF as indicated by co-localization of 53BP1 and γH2AX on telomeric DNA [16,36]. Among the extrinsic factors that cause ROS-induced oxidative DNA damage, tobacco extract triggered inflammaging on skin fibroblasts in vitro, resulting in premature cell cycle arrest, secretion of cytokines and MMPs, and downregulation of cell junction proteins [16]. The tobacco extract-exposed fibroblasts also caused keratinocytes to lose their expression of E-cadherin, tight junction proteins, ZO-1, and involucrin [37].
3. Keratinocyte Senescence
Due to the higher turnover rate of skin keratinocytes compared to fibroblasts, it has been postulated that the impact of a senescent phenotype is limited in the epidermis. In senescent keratinocytes, ECM production and cell adhesions are significantly downregulated [38]. Due to the chronic low-level inflammation in cellular aging, IL-1α secretion is increased in keratinocytes from aged skin [39]. A previous study on age-dependent variation in cytokines, chemokines, and biological analytes rinsed from the surface of healthy human skin showed significantly decreased levels of epidermal growth factor, fibroblast growth factor 2, IFN-α2, IL-1 receptor antagonist, HSA, keratin-6, and involucrin in aged skin, whereas cortisol levels were increased [40]. Moreover, several novel stratum corneum proteins related to chronic inflammation were upregulated in aged skin, including ORM1, a plasma-binding protein involved in the transport of inflammatory mediators [41].
Several studies reported the expression of senescence markers (p16INK4a, p21, and p53), SA-β-gal activity, and the loss of lamin B1 expression in human keratinocytes after UVB exposure [28]. Nonetheless, skin tissues from the sun-protected areas of young and aged donors showed that p16INK4a-positive cells were mainly melanocytes and not keratinocytes in the epidermis [42]. Lamin B1, an intermediate filament protein expressed in all somatic cells, is downregulated in UV-induced senescent cells in vitro [19,43,44]. Wang et al. described that the co-staining of lamin B1 with keratinocyte differentiation markers revealed the accumulation and clearance of senescent cells in epidermal compartments after the UV exposure [19].
The generation of ROS by UVR can lead to the phosphorylation and subsequent activation of MAPKs; extracellular signal-regulated kinase (ERK), p38, and JNK [28]. These molecules subsequently activate the c-Jun and c-fos components of AP-1. AP-1 increases the expression of MMPs, which are highly expressed in UV-induced senescent keratinocytes in culture and in the epidermis of irradiated human skin tissues [45]. Other SASPs elevated in UV-induced senescent keratinocytes in culture include proinflammatory cytokines such as TNF-α, IL-1α, IL-1β, and IL-6 [28]. SASPs from keratinocytes are not only under canonical transcriptional control of transcription factors including NF-κB, but also under translational control of the RNA binding protein YBX1, which is significantly decreased in senescent cells. This, in turn, promotes translation of SASP cytokines [46].
Air PM induces senescence in human skin cells, especially keratinocytes, via production of ROS. PM is a mixture of particles suspended in the air, and a recent study on PM showed that it could penetrate into the barrier disrupted skin, producing increased IL-8 and MMP-1 from PM-treated keratinocytes [47]. Especially, PM2.5 with the aerodynamic diameter less than 2.5μm, has been studied as the predictive indicator of cellular stress [48]. PM2.5-induced senescence involves aryl hydrocarbon receptor (AhR)-induced ROS production. Ryu et al. showed that PM2.5-AhR-ROS pathway results in keratinocyte senescence by the epigenetic regulation of the expression of the senescence-associated p16INK4a gene [49]. PM2.5-induced ROS results in a decrease in DNA methyltransferase expression and an increase in DNA demethylase expression, leading to hypomethylation of the p16INK4A promotor region, thus accelerating skin senescence [49].
Changes in the skin microbiome depend on environmental challenges, including cumulative UV exposure, smoking, and pollution, which result in subsequent immunosenescence. These factors can influence skin physiology, including sebum production, immune homeostasis, and alterations in pH and lipid composition [50,51]. Shibagaki et al. established that diversification and alterations in the skin microbiome of older and younger women in a healthy Japanese population have a significant effect on the aging process [52]. Wu et al. also confirmed that age-related alterations in skin microbiomes are body site-dependent [53]. These authors found an age-associated decrease in Propionibacterium abundance in the forehead, cheek, and forearm skin, which is probably related to the decrease in sebaceous gland activity with aging [51,52]. Kim et al. studied age-related changes in the skin microbiota of Korean women on the forehead and hands, and it was revealed that the overall microbial distribution varied on the forehead, but was similar on the hands across the age groups [54]. Their results showed that Firmicutes was more abundant in the younger age group, while Bacteroidetes and Proteobacteria increased linearly with aging. Notably, the alpha diversity indices increased significantly with age on the forehead skin, indicating that the older people may be more susceptible to pathogenic invasions due to the altered diversity of skin microbiota [54]. Thus, the imbalance of commensal microbiome and increased abundance of pathogenic bacteria such as Proteobacteria might lead to the aggravation of age-related skin disorders.
Additionally, the regulatory role of autophagy in the aging of fibroblasts and keratinocytes has been thoroughly studied. A recent study suggested that the modulation of autophage flux could regulate the skin pigmentation via melanosome degradation in keratinocytes and melanocytes [54]. Melatonin, which plays a central role in cell apoptosis and survival, induces autophagy, and protects keratinocytes from oxidative stress-induced cell damage via the Sirtuin 1 pathway, whereas an autophagy inhibitor or small interfering RNA-mediated ATG5 knockdown inhibits melatonin-mediated cell survival [55]. Caffeine, for example, is known to protect keratinocytes from stress-triggered inflammaging by activation of autophagy regulated by the adenosine A2a receptor, Sirtuin 3, and adenosine monophosphate-activated protein kinase [56]. Hence, aging is closely associated with a decline in autophagic capacity, which results in intracellular protein aggregation and accumulation of dysfunctional mitochondria and ROS production [57]. This increase in ROS production triggers intracellular danger-sensing multiprotein platforms called inflammasome, which regulates the secretion of pro-inflammatory IL-1β and IL-18 via activation of caspase-1 [58].
4. Aging-Associated Pigmentation and Melanocyte Senescence
Keratinocytes are the main type of cells that signal the need for melanogenesis [28]. Upon UVR, keratinocytes secrete factors such as α-melanocyte stimulating hormone (α-MSH), endothelin (ET)-1, and stem cell factor (SCF) in a p53-proopiomelanocortin-dependent manner, which bind melanocortin 1, ETB, and c-kit receptors, respectively [28,59,60]. Microphthalmia-associated transcription factor (MITF) plays a critical role in the cascade process for the induction of melanogenesis. UVR-induced DNA damage in keratinocytes activates p53 which initiates the transcription of proopiomelanocortin, which is subsequently cleaved into peptides including α-MSH. Its binding to MC1R on melanocytes induces MITF which activates melanogenesis [61]. ET-1 and SCF binding activates ERK/MAPK signaling, thus stimulating melanogenesis [59,62]. While UVR strongly stimulates the production of ET-1 and SCF in the epidermis, the fibroblasts isolated from photoaged skin produce a greater amount of pro-melanogenic growth factors, including hepatocyte growth factor and SCF [63]. Aging-associated pigmentation has also been reported to be driven by fibroblast senescence through the repression of stromal cell-derived factor-1 expression [64]. A recent study showed that the growth differentiation factor 15 increased in UVA-induced senescent fibroblasts, and it increased melanocyte pigmentation through β-catenin [65].
In melanocytes, the α-MSH-induced protein kinase A/cyclic adenosine monophosphate response element-binding protein pathway and UVB-triggered mTOR signaling (which subsequently inhibits autophagy) participate in upregulation of microphthalmia-associated transcription factor activity, resulting in melanin production [23]. Interestingly, autophagy is able to induce as well as to inhibit melanogenesis. The knockdown studies of cellular autophagy proteins, such as microtubule-associated protein 1A/1B-light chain 3 (LC3), Beclin-1, and ATG5, in melanoma reversibly showed that the inhibition of LC3 can reduce melanin and tyrosinase activity via decreased ERK activity [66]. Melanogenesis is a tightly regulated process involving many intracellular molecules, including p53, which reduces the level of tyrosinase when inhibited [28]. UVB irradiation of melanocytes elevates the levels of p53, p21, and c-Fos and inhibits retinoblastoma phosphorylation. Thus, repeated exposure of human melanocytes to UVB leads to increased pigmentation through prolonged p53 expression [67].
A recent study in melanocyte senescence reported that melanocytes are the only epidermal cells that truly express p16INK4A during aging on sun-protected skin [42]. Senescent melanocytes also express markers of inflammaging, such as reduced high-mobility group box 1 and dysfunctional telomeres [68]. Additionally, senescent melanocyte SASPs induce telomere dysfunction in a paracrine manner and limit the proliferation of the surrounding cells via activation of CXC chemokine receptor 3-dependent mitochondrial ROS [68]. Hence, senescent melanocytes affect and impair basal keratinocyte proliferation and contribute to epidermal atrophy [68].
Several dermatological diseases are associated with the accumulation of senescent cells in the skin. Melasma, also known as melanotic hypermelanosis, is a chronic relapsing hyperpigmentary disorder with a symmetric distribution primarily localized on sun-exposed sites, generally the face, therefore presenting large dark patches [69]. Notably, the demonstration of this chronic, acquired pigmentary disorder shows central molecular traits, including evidence of oxidative stress, subclinical inflammation, and the presence of numerous premature senescence markers in the skin [63,70,71]. The modification of dermal senescent fibroblasts on melasma through their autocrine and paracrine activity has been investigated. The damaged senescent p38/MAPK-positive cells in the upper dermis of lesional hyperpigmented skin mediates stress-responses, showing its role in inflammaging in melasma [63,72]. Moreover, the expression of MMP2 and MMP9 is significantly increased when exposed to chronic UVR; this is possibly responsible for the disruption of the basement membrane in melasma [73]. Interestingly, the rise in MMP2 and a reduction in collagen IV correlates with the increase in melanocytes that protrude into the dermis [74,75]. These changes may enhance the transfer and accumulation of dermis-derived factors into the epidermis, leading to excessive activation of dermal–epidermal interaction resulting in melanogenesis [63].
The pathophysiology of hair graying is also been related to inflammaging of the hair follicle, causing a combination of reduced ability to handle oxidative stress as well as the loss of follicular melanocyte activity [76]. The impact from ROS and environmental stressors is important in the aging process of melanocytes, and which may cause a direct melanocyte apoptosis [77]. The generation of melanin is itself an oxidative reaction; hence, the accumulation of ROS and depletion of antioxidative enzymes, such as tyrosinase-related protein-2 and catalase, have been reported as the contributing factors for hair graying [78].
5. Association between Skin Resident Immune Cells and Inflammaging
The skin is the outermost barrier organ which provides the first physical and immunological defense for our body. As an active immuno-protective organ, the skin contains several types of immune cells including mononuclear phagocytes (MNPs) such as Langerhans cells (LCs), dendritic cells, macrophages, monocytes, and T cells [79]. Functional cutaneous immune surveillance is gradually decreased as people age, which leads to an increased susceptibility to skin infections and cancers [50]. Although the precise association between the senescence of skin-resident immune cells and inflammaging has not been properly understood thus far, recent growing evidence suggests that cellular crosstalk between senescent stromal cells and immune cells results in the senescent phenotype of the skin.
Among MNPs, LCs are the sole epidermal dendritic cells with self-renewal properties [80]. LCs are reduced in number and show a decreased cellular migration toward the regional lymph nodes due to the lower expression level of IL-1β in aged skin [81,82]. The decreased migration of LCs potentially results in both suboptimal antigen-specific T cell priming against pathogenic antigens and activation of regulatory T cells, which contribute to the reduced immune surveillance and tolerance in older individuals [83]. In addition, LCs in aged skin express a low amount of human beta-defensin-3, which is an important antimicrobial peptide produced in response to a microbial challenge or skin dysbiosis [82]. Our group has demonstrated that the homeostatic cellular network of LCs is crucial for maintaining epidermal barrier integrity and function, which provides a possible mechanism for the decreased skin barrier function in the elderly skin [84]. Consequently, a reduced skin barrier function further promotes the chronic low-level inflammation in response to environmental pollutants and triggers [47]. Macrophages and monocytes are another major subset of MNPs in the skin which are recruited to the photoaged skin induced by repeated exposure to UV [85]. Senescent fibroblasts produce several SASPs including C-C motif chemokine ligand 2 (CCL2) which subsequently recruits prostaglandin E2-producing monocytes and suppresses T cell immune responses [86]. Under the inflamed conditions, skin-infiltrating monocytes are guided to differentiate into macrophages by cytokine milieu containing a monocyte colony-stimulating factor [87], and these macrophages release high levels of MMPs and ROS to degrade the dermal ECM and cause chronic inflammation [88]. These results strongly suggest that MNPs of the skin actively contribute to inflammaging and promote the senescent phenotypes of the skin.
Quiescent skin harbors twice as many T cells as T cells in the circulation [89]. Those skin-resident cells express memory and skin-resident phenotypes, so-called skin-resident memory T cells [90]. It has been demonstrated that the aged skin has an increased CD4+ to CD8+ T cell ratio and regulatory T cell frequencies and decreased proliferation by prostaglandin E2-producing monocytes [86,91,92]. In addition, repeated antigen stimulation throughout aging leads to exhaustion of T cells, which are characterized by higher expression of programmed cell death protein 1 (PD-1) and limits the effector function of T cells in elderly [93]. It has been also proposed that senescent T cells produce a higher level of Th2 and Th17 cytokines which might contribute to the higher incidence of bullous pemphigoid in elderly [94,95,96]. However, one elegant study has shown no decline in T cell density and the number of CD49A+CD8+ resident memory T cells in the aged skin [97]. Furthermore, antigen-specific proliferation to the microbial antigens and cytokine production of IFN-γ and IL-17A was comparable between T cells derived from young and old skins. Thus, further studies are necessary to conclude the cellular nature and functional characteristics of senescent T cells of the skin to understand their role in inflammaging.
6. Concluding Remark
Throughout a lifetime, skin is consistently exposed to inflammatory changes as a result of external and internal stressors, such as UVR, air PM, and microorganisms. As local inflammation and secretion of SASPs from skin microenvironment (consisted of keratinocytes, melanocytes, and fibroblasts) increase, a shift toward cellular senescence trigger inflammaging and the subsequent expressions of clinical skin aging phenotypes, such as wrinkles. In this review, we summarized the most recent updates on the pathophysiology of inflammaging of the skin, emphasizing on the cellular cross-talks among keratinocytes, melanocytes, and fibroblasts, as well as their association with external environmental stressors. We further discussed the association of these cells with skin resident immunes cells, and showed the impact of inflammaging on impairment of adaptive immunity and breakdown of skin ECM compartments. The development of novel strategies for blocking the cross-talk between skin senescent cells and the immune cells will bring clinical benefits of delaying inflammaging and promoting skin health and appearance.
Author Contributions
Conceptualization, Y.I.L. and T.G.K.; methodology, T.-G.K.; investigation, J.H.L.; resources, W.S.R. and S.C.; writing—original draft preparation, Y.I.L. and T.-G.K.; writing—review and editing, Y.I.L. and T.-G.K.; visualization, Y.I.L.; supervision, T.-G.K.; project administration, J.H.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by a Basic Science Research Program through the National Research Foundation of Republic of Korea funded by the Ministry of Education (2019R1A6A1A03032869) and Ministry of Science and Information and Communications Technology (2018R1A5A2025079, 2019M3A9E8022135 and 2020R1C1C1014513), by a Korea Health Technology R&D Project through the Korea Health Industry Development Institute funded by the Ministry of Health and Welfare (HP20C0019 and HP20C0171) and Korea Centers for Disease Control and Prevention (2020-ER6714-00), and by the Yonsei University Faculty Research Grant (6-2020-0081).
Acknowledgments
The authors thank Medical Illustration & Design, part of the Medical Research Support Services of Yonsei University College of Medicine, for the artistic support related to this work.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| 4EBP | 4E-binding protein 1 |
| 53BP1 | p53-binding protein 1 |
| α-MSH | α-melanocyte stimulating hormone |
| AhR | Aryl hydrocarbon receptor |
| AP-1 | Activator protein 1 |
| ATG | Autophagy related |
| CCL2 | C-C motif chemokine ligand 2 |
| CREB | Cyclic AMP-responsive element-binding protein |
| CTGF | Connective tissue growth factor |
| DC | Dendritic cell |
| DNMT | DNA methyltransferase |
| ECM | Extracellular matrix |
| ERK | Extracellular signal-regulated kinase |
| ET | Endothelin |
| GF | Growth factor |
| γH2AX | Gamma H2A histone family member X |
| IGF | Insulin-like growth factor |
| IL | Interleukin |
| JNK | c-Jun N-terminal kinase |
| LC3 | microtubule-associated protein 1A/1B-light chain 3 |
| LCs | Langerhans cells |
| MAPKAPK2 | MAP kinase Activated Protein Kinase 2 |
| MAPKs | Mitogen-activated protein kinases |
| MDC1 | Mediator of DNA checkpoint protein 1 |
| MITF | Microphthalmia-associated transcription factor |
| MMPs | Matrix metalloproteinases |
| MNPs | Mononuclear phagocytes |
| mTOR | Mammalian target of rapamycin |
| NBS1 | Nijmegen breakage syndrome 1 protein |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| ORM1 | Orosomucoid 1 protein |
| PD-1 | Programmed cell death protein 1 |
| PKA | Protein kinase A |
| PM | Particulate matter |
| POMC | Pro-opiomelanocortin |
| ROS | Reactive oxygen species |
| SA-β-gal | Senescence-associated β-galactosidase |
| SASP | Senescence-associate secretory phenotype |
| SCF | Stem cell factor |
| SDF-1 | Stromal cell-derived factor-1 |
| TAF | Telomere-associated DNA damage foci |
| TGF | Tumor growth factor |
| Th cell | T helper cell |
| TNF-α | Tumor necrosis factor- α |
| TRM | Tissue-resident memory T cell |
| UVB | Ultraviolet B light |
| UVR | UV radiation |
| YBX-1 | Y-Box Binding Protein 1 |
| ZO-1 | Zonula occludens-1 |
References
- Xia, S.; Zhang, X.; Zheng, S.; Khanabdali, R.; Kalionis, B.; Wu, J.; Wan, W.; Tai, X. An update on inflamm-aging: Mechanisms, prevention, and treatment. J. Immunol. Res. 2016, 2016, 8426874. [Google Scholar] [CrossRef] [PubMed]
- Coppe, J.P.; Patil, C.K.; Rodier, F.; Sun, Y.; Munoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.Y.; Campisi, J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic ras and the p53 tumor suppressor. PLoS Biol. 2008, 6, e301. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J. Aging, cellular senescence, and cancer. Annu. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wlaschek, M.; Maity, P.; Makrantonaki, E.; Scharffetter-Kochanek, K. Connective tissue and fibroblast senescence in skin aging. J. Investig. Dermatol. 2021, 141, 985–992. [Google Scholar] [CrossRef]
- Debacq-Chainiaux, F.; Leduc, C.; Verbeke, A.; Toussaint, O. Uv, stress and aging. Dermato-Endocrinol. 2012, 4, 236–240. [Google Scholar] [CrossRef] [Green Version]
- Shuster, S.; Black, M.M.; McVitie, E. The influence of age and sex on skin thickness, skin collagen and density. Br. J. Dermatol. 1975, 93, 639–643. [Google Scholar] [CrossRef]
- Harley, C.B.; Futcher, A.B.; Greider, C.W. Telomeres shorten during ageing of human fibroblasts. Nature 1990, 345, 458–460. [Google Scholar] [CrossRef] [PubMed]
- Birch-Machin, M.A.; Tindall, M.; Turner, R.; Haldane, F.; Rees, J.L. Mitochondrial DNA deletions in human skin reflect photo- rather than chronologic aging. J. Investig. Dermatol. 1998, 110, 149–152. [Google Scholar] [CrossRef] [Green Version]
- Passos, J.F.; Saretzki, G.; von Zglinicki, T. DNA damage in telomeres and mitochondria during cellular senescence: Is there a connection? Nucleic Acids Res. 2007, 35, 7505–7513. [Google Scholar] [CrossRef] [PubMed]
- Hoeijmakers, J.H. DNA damage, aging, and cancer. N. Engl. J. Med. 2009, 361, 1475–1485. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, P.F.L.; Ogrodnik, M.; Kucheryavenko, O.; Glibert, J.; Miwa, S.; Cameron, K.; Ishaq, A.; Saretzki, G.; Nagaraja-Grellscheid, S.; Nelson, G.; et al. The bystander effect contributes to the accumulation of senescent cells in vivo. Aging Cell 2019, 18, e12848. [Google Scholar] [CrossRef] [PubMed]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O.; et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef] [Green Version]
- Schneider, L.A.; Raizner, K.; Wlaschek, M.; Brenneisen, P.; Gethoffer, K.; Scharffetter-Kochanek, K. Uva-1 exposure in vivo leads to an il-6 surge within the skin. Exp. Dermatol. 2017, 26, 830–832. [Google Scholar] [CrossRef] [Green Version]
- Fisher, G.J.; Quan, T.; Purohit, T.; Shao, Y.; Cho, M.K.; He, T.; Varani, J.; Kang, S.; Voorhees, J.J. Collagen fragmentation promotes oxidative stress and elevates matrix metalloproteinase-1 in fibroblasts in aged human skin. Am. J. Pathol. 2009, 174, 101–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wlaschek, M.; Heinen, G.; Poswig, A.; Schwarz, A.; Krieg, T.; Scharffetter-Kochanek, K. Uva-induced autocrine stimulation of fibroblast-derived collagenase/mmp-1 by interrelated loops of interleukin-1 and interleukin-6. Photochem. Photobiol. 1994, 59, 550–556. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.S.; Dreesen, O. Biomarkers of cellular senescence and skin aging. Front. Genet. 2018, 9, 247. [Google Scholar] [CrossRef]
- Debacq-Chainiaux, F.; Erusalimsky, J.D.; Campisi, J.; Toussaint, O. Protocols to detect senescence-associated beta-galactosidase (sa-betagal) activity, a biomarker of senescent cells in culture and in vivo. Nat. Protoc. 2009, 4, 1798–1806. [Google Scholar] [CrossRef]
- McCart, E.A.; Thangapazham, R.L.; Lombardini, E.D.; Mog, S.R.; Panganiban, R.A.M.; Dickson, K.M.; Mansur, R.A.; Nagy, V.; Kim, S.Y.; Selwyn, R.; et al. Accelerated senescence in skin in a murine model of radiation-induced multi-organ injury. J. Radiat. Res. 2017, 58, 636–646. [Google Scholar] [CrossRef] [Green Version]
- Wang, A.S.; Ong, P.F.; Chojnowski, A.; Clavel, C.; Dreesen, O. Loss of lamin b1 is a biomarker to quantify cellular senescence in photoaged skin. Sci. Rep. 2017, 7, 15678. [Google Scholar] [CrossRef] [Green Version]
- Ke, Y.; Wang, X.J. Tgfbeta signaling in photoaging and uv-induced skin cancer. J. Investig. Dermatol. 2020, 141, 1104–1110. [Google Scholar] [CrossRef]
- Quan, T.; Shao, Y.; He, T.; Voorhees, J.J.; Fisher, G.J. Reduced expression of connective tissue growth factor (ctgf/ccn2) mediates collagen loss in chronologically aged human skin. J. Investig. Dermatol. 2010, 130, 415–424. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, H.I.; Kiyono, K.; Miyazono, K. Regulation of autophagy by transforming growth factor-beta (tgf-beta) signaling. Autophagy 2010, 6, 645–647. [Google Scholar] [CrossRef] [Green Version]
- Jeong, D.; Qomaladewi, N.P.; Lee, J.; Park, S.H.; Cho, J.Y. The role of autophagy in skin fibroblasts, keratinocytes, melanocytes, and epidermal stem cells. J. Investig. Dermatol. 2020, 140, 1691–1697. [Google Scholar] [CrossRef] [PubMed]
- Kalfalah, F.; Janke, L.; Schiavi, A.; Tigges, J.; Ix, A.; Ventura, N.; Boege, F.; Reinke, H. Crosstalk of clock gene expression and autophagy in aging. Aging (Albany NY) 2016, 8, 1876–1895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.S.; Park, S.Y.; Moon, S.H.; Lee, J.D.; Kim, S. Autophagy in human skin fibroblasts: Impact of age. Int. J. Mol. Sci. 2018, 19, 2254. [Google Scholar] [CrossRef] [Green Version]
- Singh, K.; Maity, P.; Krug, L.; Meyer, P.; Treiber, N.; Lucas, T.; Basu, A.; Kochanek, S.; Wlaschek, M.; Geiger, H.; et al. Superoxide anion radicals induce igf-1 resistance through concomitant activation of ptp1b and pten. EMBO Mol. Med. 2015, 7, 59–77. [Google Scholar] [CrossRef] [PubMed]
- Herranz, N.; Gallage, S.; Mellone, M.; Wuestefeld, T.; Klotz, S.; Hanley, C.J.; Raguz, S.; Acosta, J.C.; Innes, A.J.; Banito, A.; et al. Mtor regulates mapkapk2 translation to control the senescence-associated secretory phenotype. Nat. Cell Biol. 2015, 17, 1205–1217. [Google Scholar] [CrossRef] [Green Version]
- Fitsiou, E.; Pulido, T.; Campisi, J.; Alimirah, F.; Demaria, M. Cellular senescence and the senescence-associated secretory phenotype as drivers of skin photoaging. J. Investig. Dermatol. 2020, 141, 1119–1126. [Google Scholar] [CrossRef]
- Laberge, R.M.; Sun, Y.; Orjalo, A.V.; Patil, C.K.; Freund, A.; Zhou, L.; Curran, S.C.; Davalos, A.R.; Wilson-Edell, K.A.; Liu, S.; et al. Mtor regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting il1a translation. Nat. Cell Biol. 2015, 17, 1049–1061. [Google Scholar] [CrossRef]
- Krutmann, J.; Schroeder, P. Role of mitochondria in photoaging of human skin: The defective powerhouse model. J. Investig. Dermatol. Symp. Proc. 2009, 14, 44–49. [Google Scholar] [CrossRef] [Green Version]
- Meyer, P.; Maity, P.; Burkovski, A.; Schwab, J.; Mussel, C.; Singh, K.; Ferreira, F.F.; Krug, L.; Maier, H.J.; Wlaschek, M.; et al. A model of the onset of the senescence associated secretory phenotype after DNA damage induced senescence. PLoS Comput. Biol. 2017, 13, e1005741. [Google Scholar] [CrossRef] [Green Version]
- Treiber, N.; Maity, P.; Singh, K.; Kohn, M.; Keist, A.F.; Ferchiu, F.; Sante, L.; Frese, S.; Bloch, W.; Kreppel, F.; et al. Accelerated aging phenotype in mice with conditional deficiency for mitochondrial superoxide dismutase in the connective tissue. Aging Cell 2011, 10, 239–254. [Google Scholar] [CrossRef] [PubMed]
- D’Adda di Fagagna, F.; Reaper, P.M.; Clay-Farrace, L.; Fiegler, H.; Carr, P.; Von Zglinicki, T.; Saretzki, G.; Carter, N.P.; Jackson, S.P. A DNA damage checkpoint response in telomere-initiated senescence. Nature 2003, 426, 194–198. [Google Scholar] [CrossRef] [PubMed]
- Waaijer, M.E.C.; Gunn, D.A.; van Heemst, D.; Slagboom, P.E.; Sedivy, J.M.; Dirks, R.W.; Tanke, H.J.; Westendorp, R.G.J.; Maier, A.B. Do senescence markers correlate in vitro and in situ within individual human donors? Aging (Albany NY) 2018, 10, 278–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hewitt, G.; Jurk, D.; Marques, F.D.; Correia-Melo, C.; Hardy, T.; Gackowska, A.; Anderson, R.; Taschuk, M.; Mann, J.; Passos, J.F. Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nat. Commun. 2012, 3, 708. [Google Scholar] [CrossRef] [PubMed]
- Herbig, U.; Ferreira, M.; Condel, L.; Carey, D.; Sedivy, J.M. Cellular senescence in aging primates. Science 2006, 311, 1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coppe, J.P.; Boysen, M.; Sun, C.H.; Wong, B.J.; Kang, M.K.; Park, N.H.; Desprez, P.Y.; Campisi, J.; Krtolica, A. A role for fibroblasts in mediating the effects of tobacco-induced epithelial cell growth and invasion. Mol. Cancer Res. 2008, 6, 1085–1098. [Google Scholar] [CrossRef] [Green Version]
- Sprenger, A.; Weber, S.; Zarai, M.; Engelke, R.; Nascimento, J.M.; Gretzmeier, C.; Hilpert, M.; Boerries, M.; Has, C.; Busch, H.; et al. Consistency of the proteome in primary human keratinocytes with respect to gender, age, and skin localization. Mol. Cell Proteom. 2013, 12, 2509–2521. [Google Scholar] [CrossRef] [Green Version]
- Okazaki, M.; Yoshimura, K.; Uchida, G.; Harii, K. Correlation between age and the secretions of melanocyte-stimulating cytokines in cultured keratinocytes and fibroblasts. Br. J. Dermatol. 2005, 153 (Suppl. S2), 23–29. [Google Scholar] [CrossRef]
- Kinn, P.M.; Holdren, G.O.; Westermeyer, B.A.; Abuissa, M.; Fischer, C.L.; Fairley, J.A.; Brogden, K.A.; Brogden, N.K. Age-dependent variation in cytokines, chemokines, and biologic analytes rinsed from the surface of healthy human skin. Sci. Rep. 2015, 5, 10472. [Google Scholar] [CrossRef]
- Ma, J.; Liu, M.; Wang, Y.; Xin, C.; Zhang, H.; Chen, S.; Zheng, X.; Zhang, X.; Xiao, F.; Yang, S. Quantitative proteomics analysis of young and elderly skin with dia mass spectrometry reveals new skin aging-related proteins. Aging (Albany NY) 2020, 12, 13529–13554. [Google Scholar] [CrossRef] [PubMed]
- Waaijer, M.E.; Gunn, D.A.; Adams, P.D.; Pawlikowski, J.S.; Griffiths, C.E.; van Heemst, D.; Slagboom, P.E.; Westendorp, R.G.; Maier, A.B. P16ink4a positive cells in human skin are indicative of local elastic fiber morphology, facial wrinkling, and perceived age. J. Gerontol. A Biol. Sci. Med. Sci. 2016, 71, 1022–1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, P.P.; Donahue, G.; Otte, G.L.; Capell, B.C.; Nelson, D.M.; Cao, K.; Aggarwala, V.; Cruickshanks, H.A.; Rai, T.S.; McBryan, T.; et al. Lamin b1 depletion in senescent cells triggers large-scale changes in gene expression and the chromatin landscape. Genes Dev. 2013, 27, 1787–1799. [Google Scholar] [CrossRef] [Green Version]
- Sadaie, M.; Salama, R.; Carroll, T.; Tomimatsu, K.; Chandra, T.; Young, A.R.; Narita, M.; Perez-Mancera, P.A.; Bennett, D.C.; Chong, H.; et al. Redistribution of the lamin b1 genomic binding profile affects rearrangement of heterochromatic domains and sahf formation during senescence. Genes Dev. 2013, 27, 1800–1808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quan, T.; Little, E.; Quan, H.; Qin, Z.; Voorhees, J.J.; Fisher, G.J. Elevated matrix metalloproteinases and collagen fragmentation in photodamaged human skin: Impact of altered extracellular matrix microenvironment on dermal fibroblast function. J. Investig. Dermatol. 2013, 133, 1362–1366. [Google Scholar] [CrossRef] [Green Version]
- Kwon, E.; Todorova, K.; Wang, J.; Horos, R.; Lee, K.K.; Neel, V.A.; Negri, G.L.; Sorensen, P.H.; Lee, S.W.; Hentze, M.W.; et al. The rna-binding protein ybx1 regulates epidermal progenitors at a posttranscriptional level. Nat. Commun. 2018, 9, 1734. [Google Scholar] [CrossRef] [Green Version]
- Jin, S.P.; Li, Z.; Choi, E.K.; Lee, S.; Kim, Y.K.; Seo, E.Y.; Chung, J.H.; Cho, S. Urban particulate matter in air pollution penetrates into the barrier-disrupted skin and produces ros-dependent cutaneous inflammatory response in vivo. J. Dermatol. Sci. 2018, 91, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Crobeddu, B.; Aragao-Santiago, L.; Bui, L.C.; Boland, S.; Baeza Squiban, A. Oxidative potential of particulate matter 2.5 as predictive indicator of cellular stress. Environ. Pollut. 2017, 230, 125–133. [Google Scholar] [CrossRef]
- Ryu, Y.S.; Kang, K.A.; Piao, M.J.; Ahn, M.J.; Yi, J.M.; Bossis, G.; Hyun, Y.M.; Park, C.O.; Hyun, J.W. Particulate matter-induced senescence of skin keratinocytes involves oxidative stress-dependent epigenetic modifications. Exp. Mol. Med. 2019, 51, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chambers, E.S.; Vukmanovic-Stejic, M. Skin barrier immunity and ageing. Immunology 2020, 160, 116–125. [Google Scholar] [CrossRef] [Green Version]
- Santoro, A.; Zhao, J.; Wu, L.; Carru, C.; Biagi, E.; Franceschi, C. Microbiomes other than the gut: Inflammaging and age-related diseases. Semin Immunopathol. 2020, 42, 589–605. [Google Scholar] [CrossRef] [PubMed]
- Shibagaki, N.; Suda, W.; Clavaud, C.; Bastien, P.; Takayasu, L.; Iioka, E.; Kurokawa, R.; Yamashita, N.; Hattori, Y.; Shindo, C.; et al. Aging-related changes in the diversity of women’s skin microbiomes associated with oral bacteria. Sci. Rep. 2017, 7, 10567. [Google Scholar] [CrossRef]
- Wu, L.; Zeng, T.; Deligios, M.; Milanesi, L.; Langille, M.G.I.; Zinellu, A.; Rubino, S.; Carru, C.; Kelvin, D.J. Age-related variation of bacterial and fungal communities in different body habitats across the young, elderly, and centenarians in sardinia. mSphere 2020, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.; Park, T.; Yun, J.I.; Lim, H.W.; Han, N.R.; Lee, S.T. Investigation of age-related changes in the skin microbiota of korean women. Microorganisms 2020, 8, 1581. [Google Scholar] [CrossRef]
- Lee, J.H.; Moon, J.H.; Nazim, U.M.; Lee, Y.J.; Seol, J.W.; Eo, S.K.; Lee, J.H.; Park, S.Y. Melatonin protects skin keratinocyte from hydrogen peroxide-mediated cell death via the sirt1 pathway. Oncotarget 2016, 7, 12075–12088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.F.; Ouyang, S.H.; Tu, L.F.; Wang, X.; Yuan, W.L.; Wang, G.E.; Wu, Y.P.; Duan, W.J.; Yu, H.M.; Fang, Z.Z.; et al. Caffeine protects skin from oxidative stress-induced senescence through the activation of autophagy. Theranostics 2018, 8, 5713–5730. [Google Scholar] [CrossRef]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. Inflammaging: Disturbed interplay between autophagy and inflammasomes. Aging (Albany NY) 2012, 4, 166–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awad, F.; Assrawi, E.; Louvrier, C.; Jumeau, C.; Giurgea, I.; Amselem, S.; Karabina, S.A. Photoaging and skin cancer: Is the inflammasome the missing link? Mech. Ageing Dev. 2018, 172, 131–137. [Google Scholar] [CrossRef]
- Murase, D.; Hachiya, A.; Amano, Y.; Ohuchi, A.; Kitahara, T.; Takema, Y. The essential role of p53 in hyperpigmentation of the skin via regulation of paracrine melanogenic cytokine receptor signaling. J. Biol. Chem. 2009, 284, 4343–4353. [Google Scholar] [CrossRef] [Green Version]
- Niwano, T.; Terazawa, S.; Nakajima, H.; Wakabayashi, Y.; Imokawa, G. Astaxanthin and withaferin a block paracrine cytokine interactions between uvb-exposed human keratinocytes and human melanocytes via the attenuation of endothelin-1 secretion and its downstream intracellular signaling. Cytokine 2015, 73, 184–197. [Google Scholar] [CrossRef]
- Hida, T.; Kamiya, T.; Kawakami, A.; Ogino, J.; Sohma, H.; Uhara, H.; Jimbow, K. Elucidation of melanogenesis cascade for identifying pathophysiology and therapeutic approach of pigmentary disorders and melanoma. Int. J. Mol. Sci. 2020, 21, 6129. [Google Scholar] [CrossRef] [PubMed]
- Hyter, S.; Coleman, D.J.; Ganguli-Indra, G.; Merrill, G.F.; Ma, S.; Yanagisawa, M.; Indra, A.K. Endothelin-1 is a transcriptional target of p53 in epidermal keratinocytes and regulates ultraviolet-induced melanocyte homeostasis. Pigment Cell Melanoma Res. 2013, 26, 247–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellei, B.; Picardo, M. Premature cell senescence in human skin: Dual face in chronic acquired pigmentary disorders. Ageing Res. Rev. 2020, 57, 100981. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.E.; Kim, Y.; Kwon, S.; Kim, M.; Kim, Y.H.; Kim, J.H.; Park, T.J.; Kang, H.Y. Senescent fibroblasts drive ageing pigmentation: A potential therapeutic target for senile lentigo. Theranostics 2018, 8, 4620–4632. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Kang, B.; Kim, J.C.; Park, T.J.; Kang, H.Y. Senescent fibroblast-derived gdf15 induces skin pigmentation. J. Investig. Dermatol. 2020, 140, 2478–2486. [Google Scholar] [CrossRef] [PubMed]
- Yun, W.J.; Kim, E.Y.; Park, J.E.; Jo, S.Y.; Bang, S.H.; Chang, E.J.; Chang, S.E. Microtubule-associated protein light chain 3 is involved in melanogenesis via regulation of mitf expression in melanocytes. Sci. Rep. 2016, 6, 19914. [Google Scholar] [CrossRef]
- Choi, S.Y.; Bin, B.H.; Kim, W.; Lee, E.; Lee, T.R.; Cho, E.G. Exposure of human melanocytes to uvb twice and subsequent incubation leads to cellular senescence and senescence-associated pigmentation through the prolonged p53 expression. J. Dermatol. Sci. 2018, 90, 303–312. [Google Scholar] [CrossRef] [Green Version]
- Victorelli, S.; Lagnado, A.; Halim, J.; Moore, W.; Talbot, D.; Barrett, K.; Chapman, J.; Birch, J.; Ogrodnik, M.; Meves, A.; et al. Senescent human melanocytes drive skin ageing via paracrine telomere dysfunction. EMBO J. 2019, 38, e101982. [Google Scholar] [CrossRef]
- Kwon, S.H.; Hwang, Y.J.; Lee, S.K.; Park, K.C. Heterogeneous pathology of melasma and its clinical implications. Int. J. Mol. Sci 2016, 17, 824. [Google Scholar] [CrossRef]
- Seckin, H.Y.; Kalkan, G.; Bas, Y.; Akbas, A.; Onder, Y.; Ozyurt, H.; Sahin, M. Oxidative stress status in patients with melasma. Cutan. Ocul. Toxicol. 2014, 33, 212–217. [Google Scholar] [CrossRef]
- Rodriguez-Arambula, A.; Torres-Alvarez, B.; Cortes-Garcia, D.; Fuentes-Ahumada, C.; Castanedo-Cazares, J.P. Cd4, il-17, and cox-2 are associated with subclinical inflammation in malar melasma. Am. J. Dermatopathol. 2015, 37, 761–766. [Google Scholar] [CrossRef]
- Esposito, A.C.C.; Brianezi, G.; de Souza, N.P.; Miot, L.D.B.; Marques, M.E.A.; Miot, H.A. Exploring pathways for sustained melanogenesis in facial melasma: An immunofluorescence study. Int. J. Cosmet. Sci. 2018, 40, 420–424. [Google Scholar] [CrossRef]
- Inomata, S.; Matsunaga, Y.; Amano, S.; Takada, K.; Kobayashi, K.; Tsunenaga, M.; Nishiyama, T.; Kohno, Y.; Fukuda, M. Possible involvement of gelatinases in basement membrane damage and wrinkle formation in chronically ultraviolet b-exposed hairless mouse. J. Investig. Dermatol. 2003, 120, 128–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, D.J.; Park, K.C.; Ortonne, J.P.; Kang, H.Y. Pendulous melanocytes: A characteristic feature of melasma and how it may occur. Br. J. Dermatol. 2012, 166, 684–686. [Google Scholar] [CrossRef] [PubMed]
- Torres-Alvarez, B.; Mesa-Garza, I.G.; Castanedo-Cazares, J.P.; Fuentes-Ahumada, C.; Oros-Ovalle, C.; Navarrete-Solis, J.; Moncada, B. Histochemical and immunohistochemical study in melasma: Evidence of damage in the basal membrane. Am. J. Dermatopathol. 2011, 33, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Arck, P.C.; Overall, R.; Spatz, K.; Liezman, C.; Handjiski, B.; Klapp, B.F.; Birch-Machin, M.A.; Peters, E.M. Towards a “free radical theory of graying”: Melanocyte apoptosis in the aging human hair follicle is an indicator of oxidative stress induced tissue damage. FASEB J. 2006, 20, 1567–1569. [Google Scholar] [CrossRef]
- Ji, J.; Ho, B.S.; Qian, G.; Xie, X.M.; Bigliardi, P.L.; Bigliardi-Qi, M. Aging in hair follicle stem cells and niche microenvironment. J. Dermatol. 2017, 44, 1097–1104. [Google Scholar] [CrossRef]
- Denat, L.; Kadekaro, A.L.; Marrot, L.; Leachman, S.A.; Abdel-Malek, Z.A. Melanocytes as instigators and victims of oxidative stress. J. Investig. Dermatol. 2014, 134, 1512–1518. [Google Scholar] [CrossRef] [Green Version]
- Nestle, F.O.; Di Meglio, P.; Qin, J.Z.; Nickoloff, B.J. Skin immune sentinels in health and disease. Nat. Rev. Immunol. 2009, 9, 679–691. [Google Scholar] [CrossRef] [Green Version]
- Merad, M.; Ginhoux, F.; Collin, M. Origin, homeostasis and function of langerhans cells and other langerin-expressing dendritic cells. Nat. Rev. Immunol. 2008, 8, 935–947. [Google Scholar] [CrossRef]
- Cumberbatch, M.; Dearman, R.J.; Kimber, I. Influence of ageing on langerhans cell migration in mice: Identification of a putative deficiency of epidermal interleukin-1beta. Immunology 2002, 105, 466–477. [Google Scholar] [CrossRef]
- Pilkington, S.M.; Dearman, R.J.; Kimber, I.; Griffiths, C.E.M. Langerhans cells express human beta-defensin 3: Relevance for immunity during skin ageing. Br. J. Dermatol. 2018, 179, 1170–1171. [Google Scholar] [CrossRef] [Green Version]
- Seneschal, J.; Clark, R.A.; Gehad, A.; Baecher-Allan, C.M.; Kupper, T.S. Human epidermal langerhans cells maintain immune homeostasis in skin by activating skin resident regulatory t cells. Immunity 2012, 36, 873–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.J.; Kim, T.G.; Kim, S.H.; Park, J.Y.; Lee, M.; Lee, J.W.; Lee, S.H.; Lee, M.G. Epidermal barrier function is impaired in langerhans cell-depleted mice. J. Investig. Dermatol. 2019, 139, 1182–1185. [Google Scholar] [CrossRef]
- Meunier, L.; Bata-Csorgo, Z.; Cooper, K.D. In human dermis, ultraviolet radiation induces expansion of a cd36+ cd11b+ cd1- macrophage subset by infiltration and proliferation; cd1+ langerhans-like dendritic antigen-presenting cells are concomitantly depleted. J. Investig. Dermatol. 1995, 105, 782–788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chambers, E.S.; Akbar, A.N. Can blocking inflammation enhance immunity during aging? J. Allergy Clin. Immunol. 2020, 145, 1323–1331. [Google Scholar] [CrossRef]
- Ushach, I.; Zlotnik, A. Biological role of granulocyte macrophage colony-stimulating factor (gm-csf) and macrophage colony-stimulating factor (m-csf) on cells of the myeloid lineage. J. Leukoc. Biol. 2016, 100, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Takahara, M.; Kang, K.; Liu, L.; Yoshida, Y.; McCormick, T.S.; Cooper, K.D. Ic3b arrests monocytic cell differentiation into cd1c-expressing dendritic cell precursors: A mechanism for transiently decreased dendritic cells in vivo after human skin injury by ultraviolet b. J. Investig. Dermatol. 2003, 120, 802–809. [Google Scholar] [CrossRef] [Green Version]
- Clark, R.A.; Chong, B.; Mirchandani, N.; Brinster, N.K.; Yamanaka, K.; Dowgiert, R.K.; Kupper, T.S. The vast majority of cla+ t cells are resident in normal skin. J. Immunol. 2006, 176, 4431–4439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, R.A. Resident memory t cells in human health and disease. Sci. Transl. Med. 2015, 7, 269rv261. [Google Scholar] [CrossRef] [Green Version]
- Zuelgaray, E.; Boccara, D.; Ly Ka So, S.; Boismal, F.; Mimoun, M.; Bagot, M.; Bensussan, A.; Bouaziz, J.D.; Michel, L. Increased expression of pd1 and cd39 on cd3(+) cd4(+) skin t cells in the elderly. Exp. Dermatol. 2019, 28, 80–82. [Google Scholar] [CrossRef] [PubMed]
- Lages, C.S.; Suffia, I.; Velilla, P.A.; Huang, B.; Warshaw, G.; Hildeman, D.A.; Belkaid, Y.; Chougnet, C. Functional regulatory t cells accumulate in aged hosts and promote chronic infectious disease reactivation. J. Immunol. 2008, 181, 1835–1848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vukmanovic-Stejic, M.; Sandhu, D.; Seidel, J.A.; Patel, N.; Sobande, T.O.; Agius, E.; Jackson, S.E.; Fuentes-Duculan, J.; Suarez-Farinas, M.; Mabbott, N.A.; et al. The characterization of varicella zoster virus-specific t cells in skin and blood during aging. J. Investig. Dermatol. 2015, 135, 1752–1762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakievska, L.; Holtsche, M.M.; Kunstner, A.; Goletz, S.; Petersen, B.S.; Thaci, D.; Ibrahim, S.M.; Ludwig, R.J.; Franke, A.; Sadik, C.D.; et al. Il-17a is functionally relevant and a potential therapeutic target in bullous pemphigoid. J. Autoimmun. 2019, 96, 104–112. [Google Scholar] [CrossRef]
- Le Jan, S.; Plee, J.; Vallerand, D.; Dupont, A.; Delanez, E.; Durlach, A.; Jackson, P.L.; Edwin Blalock, J.; Bernard, P.; Antonicelli, F. Innate immune cell-produced il-17 sustains inflammation in bullous pemphigoid. J. Investig. Dermatol. 2014, 134, 2908–2917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gounni Abdelilah, S.; Wellemans, V.; Agouli, M.; Guenounou, M.; Hamid, Q.; Beck, L.A.; Lamkhioued, B. Increased expression of th2-associated chemokines in bullous pemphigoid disease. Role of eosinophils in the production and release of these chemokines. Clin. Immunol. 2006, 120, 220–231. [Google Scholar] [CrossRef]
- Koguchi-Yoshioka, H.; Hoffer, E.; Cheuk, S.; Matsumura, Y.; Vo, S.; Kjellman, P.; Grema, L.; Ishitsuka, Y.; Nakamura, Y.; Okiyama, N.; et al. Skin t cells maintain their diversity and functionality in the elderly. Commun. Biol. 2021, 4, 13. [Google Scholar] [CrossRef]
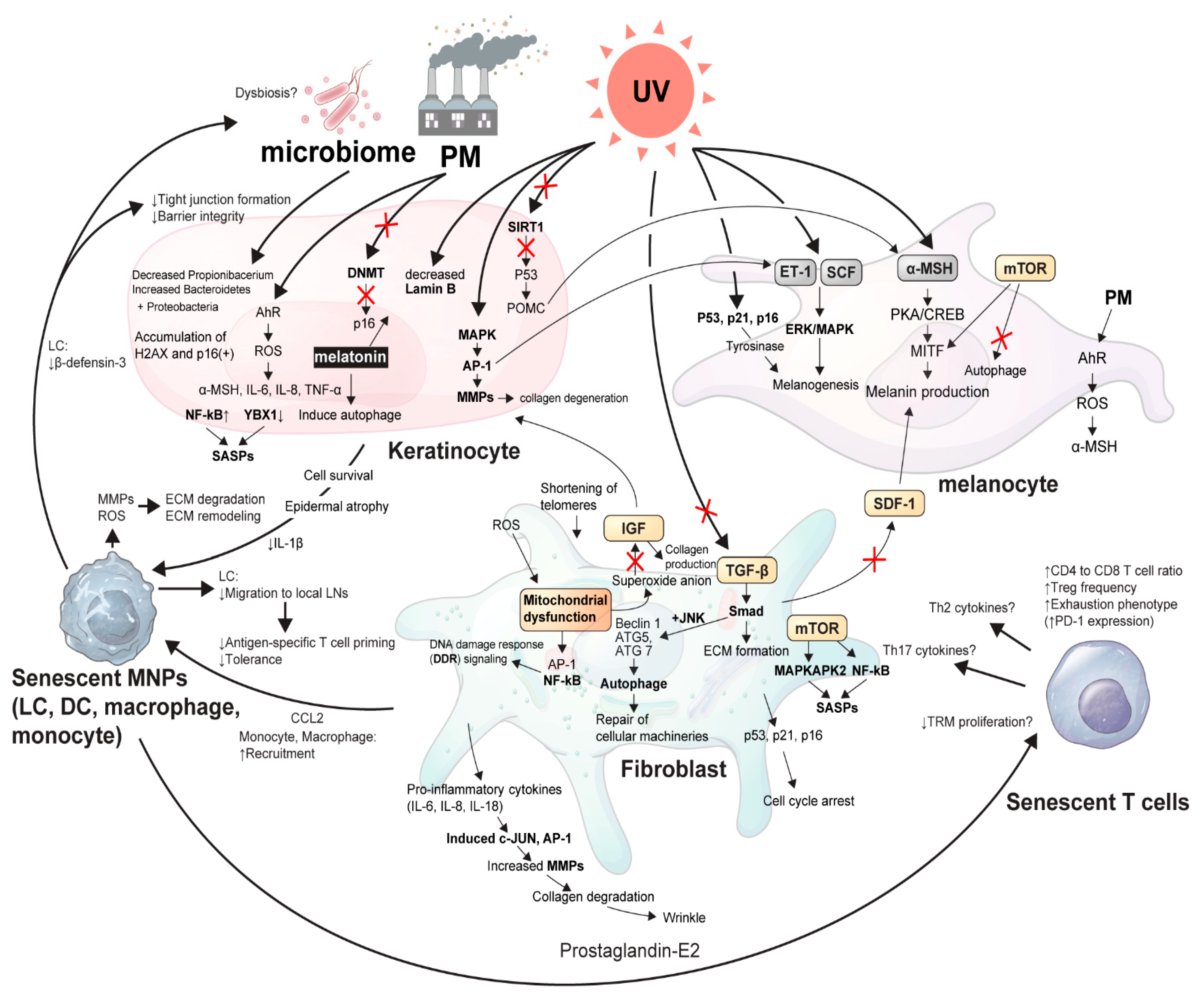
Figure 1.
The schematic view of cellular cross-talk between skin microenvironment and inflammaging. An illustrative overview of the article showing the delicate signaling pathways from senescent skin microenvironments in association with external stressors, such as UVR, microbiome, and PM. α-MSH, α-melanocyte stimulating hormone; AhR, Aryl hydrocarbon receptor; AP-1, Activator protein 1; ATG, Autophagy related; CCL2, C-C motif chemokine ligand 2; CREB, Cyclic AMP-responsive element-binding protein; DCs, Dendritic cells; DNMT, DNA methyltransferase; ECM, Extracellular matrix; ERK, Extracellular signal-regulated kinase; ET, Endothelin; H2AX, H2A histone family member X; IGF, Insulin-like growth factor; IL, Interleukin; JNK, c-Jun N-terminal kinase; LCs, Langerhans cells; LN, Lymph node; MAPKAPK2, MAP kinase Activated Protein Kinase 2; MAPKs, Mitogen-activated protein kinases; MITF, Microphthalmia-associated transcription factor; MMPs, Matrix metalloproteinases; MNPs, Mononuclear phagocytes; mTOR, Mammalian target of rapamycin; NF-κB, Nuclear factor kappa-light-chain-enhancer of activated B cells; PD-1, Programmed cell death protein 1; PKA, Protein kinase A; PM, Particulate matter; POMC, Pro-opiomelanocortin; ROS, Reactive oxygen species; SASP, Senescence-associate secretory phenotype; SCF, Stem cell factor; SDF-1, Stromal cell-derived factor-1; TGF, Tumor growth factor; Th cells, T helper cells; TNF-α, Tumor necrosis factor- α; TRM, Tissue-resident memory T cell; UV, Ultraviolet light; YBX-1, Y-Box Binding Protein 1.
Figure 1.
The schematic view of cellular cross-talk between skin microenvironment and inflammaging. An illustrative overview of the article showing the delicate signaling pathways from senescent skin microenvironments in association with external stressors, such as UVR, microbiome, and PM. α-MSH, α-melanocyte stimulating hormone; AhR, Aryl hydrocarbon receptor; AP-1, Activator protein 1; ATG, Autophagy related; CCL2, C-C motif chemokine ligand 2; CREB, Cyclic AMP-responsive element-binding protein; DCs, Dendritic cells; DNMT, DNA methyltransferase; ECM, Extracellular matrix; ERK, Extracellular signal-regulated kinase; ET, Endothelin; H2AX, H2A histone family member X; IGF, Insulin-like growth factor; IL, Interleukin; JNK, c-Jun N-terminal kinase; LCs, Langerhans cells; LN, Lymph node; MAPKAPK2, MAP kinase Activated Protein Kinase 2; MAPKs, Mitogen-activated protein kinases; MITF, Microphthalmia-associated transcription factor; MMPs, Matrix metalloproteinases; MNPs, Mononuclear phagocytes; mTOR, Mammalian target of rapamycin; NF-κB, Nuclear factor kappa-light-chain-enhancer of activated B cells; PD-1, Programmed cell death protein 1; PKA, Protein kinase A; PM, Particulate matter; POMC, Pro-opiomelanocortin; ROS, Reactive oxygen species; SASP, Senescence-associate secretory phenotype; SCF, Stem cell factor; SDF-1, Stromal cell-derived factor-1; TGF, Tumor growth factor; Th cells, T helper cells; TNF-α, Tumor necrosis factor- α; TRM, Tissue-resident memory T cell; UV, Ultraviolet light; YBX-1, Y-Box Binding Protein 1.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Lee, Y.I.; Choi, S.; Roh, W.S.; Lee, J.H.; Kim, T.-G. Cellular Senescence and Inflammaging in the Skin Microenvironment. Int. J. Mol. Sci. 2021, 22, 3849. https://doi.org/10.3390/ijms22083849
AMA Style
Lee YI, Choi S, Roh WS, Lee JH, Kim T-G. Cellular Senescence and Inflammaging in the Skin Microenvironment. International Journal of Molecular Sciences. 2021; 22(8):3849. https://doi.org/10.3390/ijms22083849
Chicago/Turabian StyleLee, Young In, Sooyeon Choi, Won Seok Roh, Ju Hee Lee, and Tae-Gyun Kim. 2021. "Cellular Senescence and Inflammaging in the Skin Microenvironment" International Journal of Molecular Sciences 22, no. 8: 3849. https://doi.org/10.3390/ijms22083849
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.