Overview of Epstein–Barr-Virus-Associated Gastric Cancer Correlated with Prognostic Classification and Development of Therapeutic Options

 , , , , , , , , and
, , , , , , , , and
Abstract
:
1. Introduction
2. Molecular Classification and Prognosis of EBVaGC Subtypes
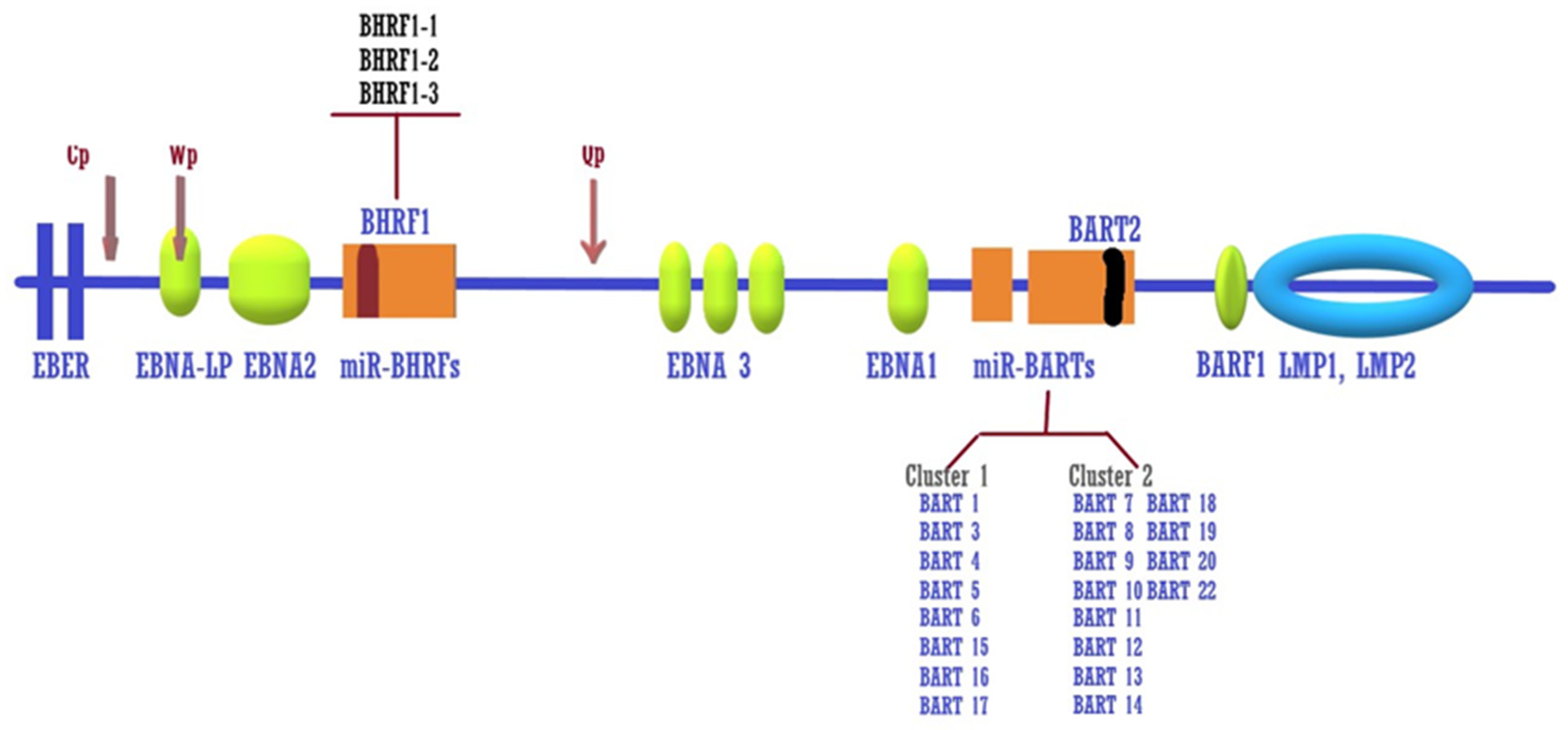
3. Variations in the EBV Genome Based on Geographical Differences
4. EBV Latency and Epigenetic Activity
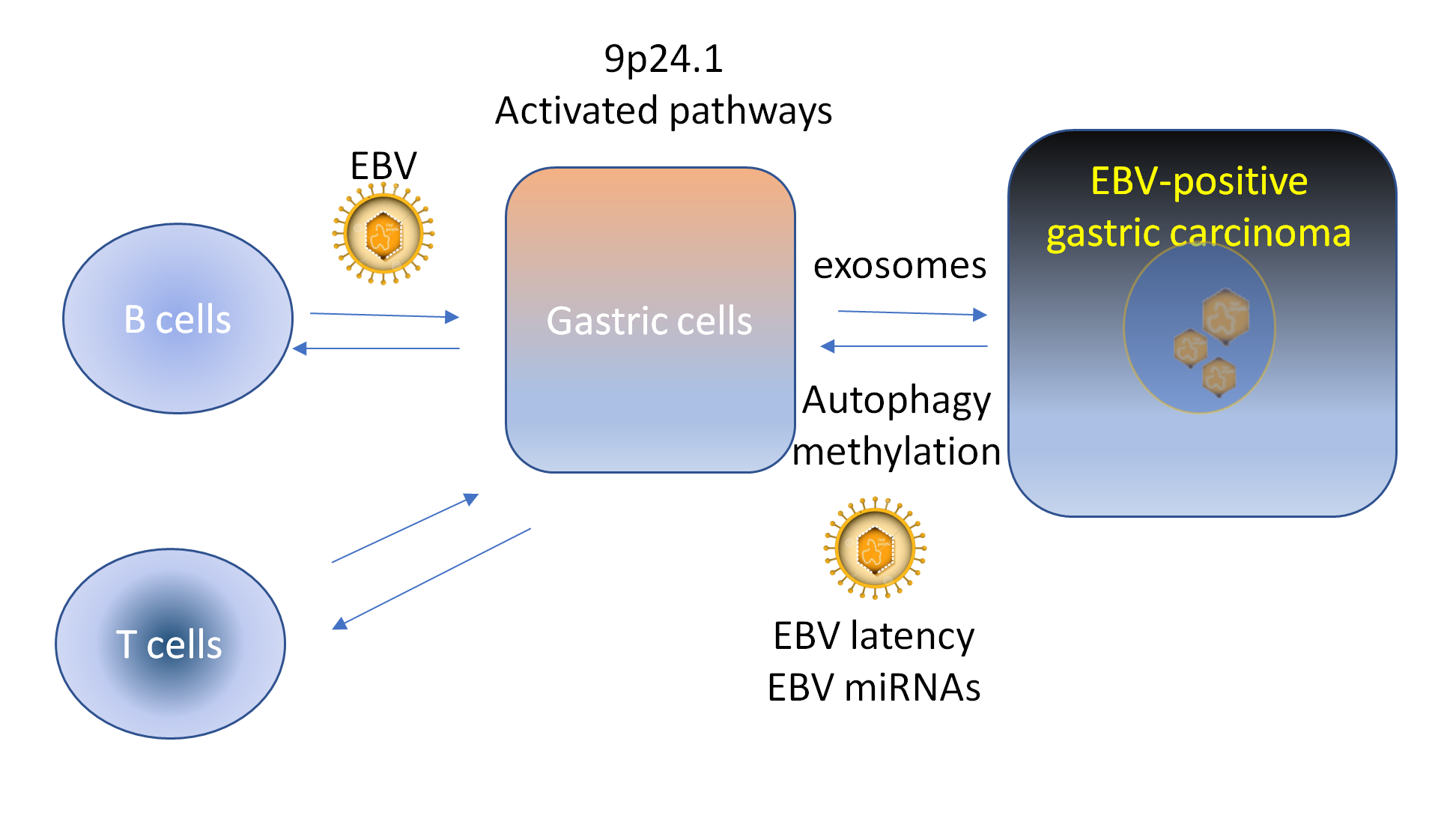
5. Exosomes and Autophagy in EBVaGC
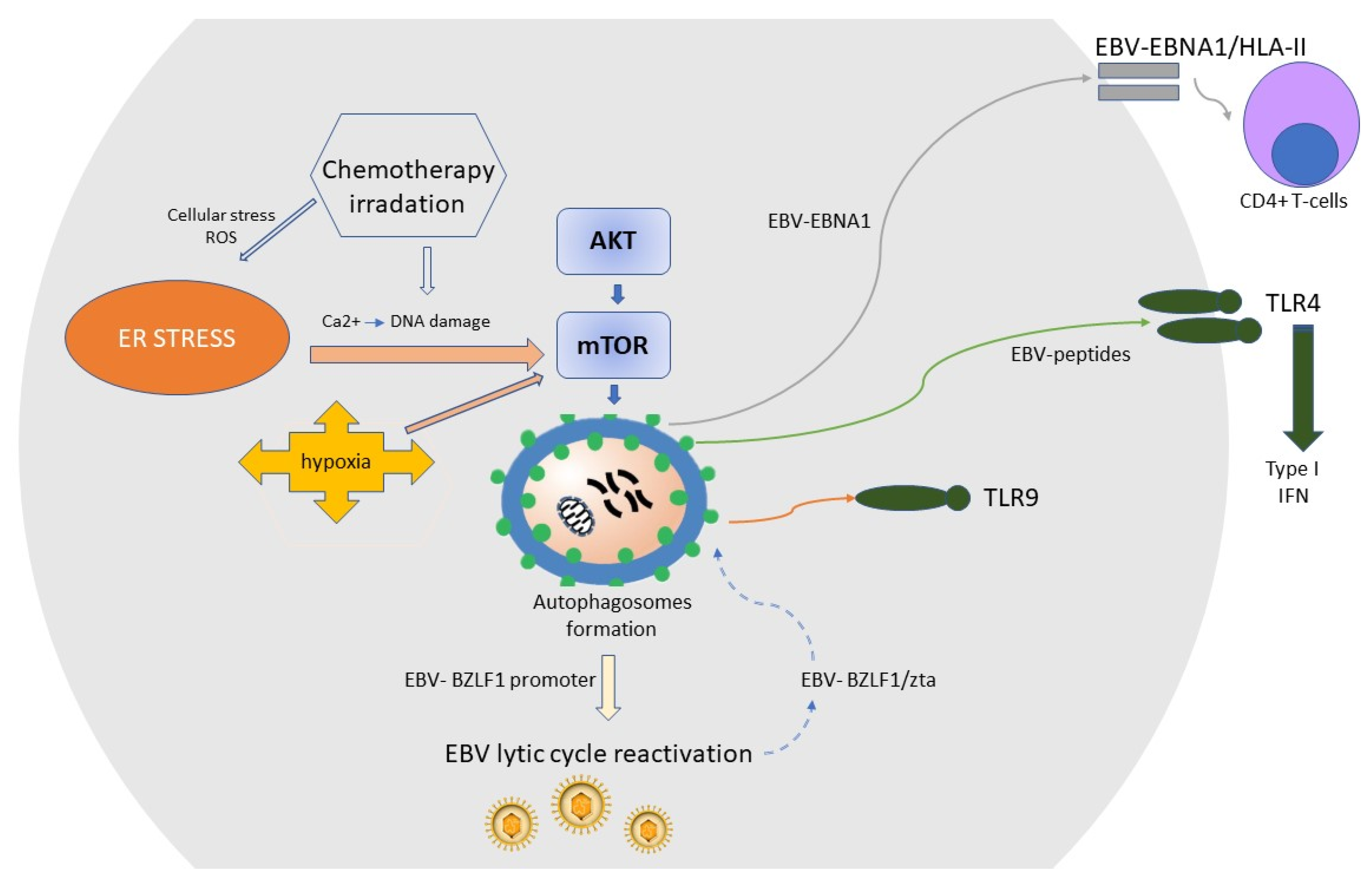
6. EBV Lytic Reactivation
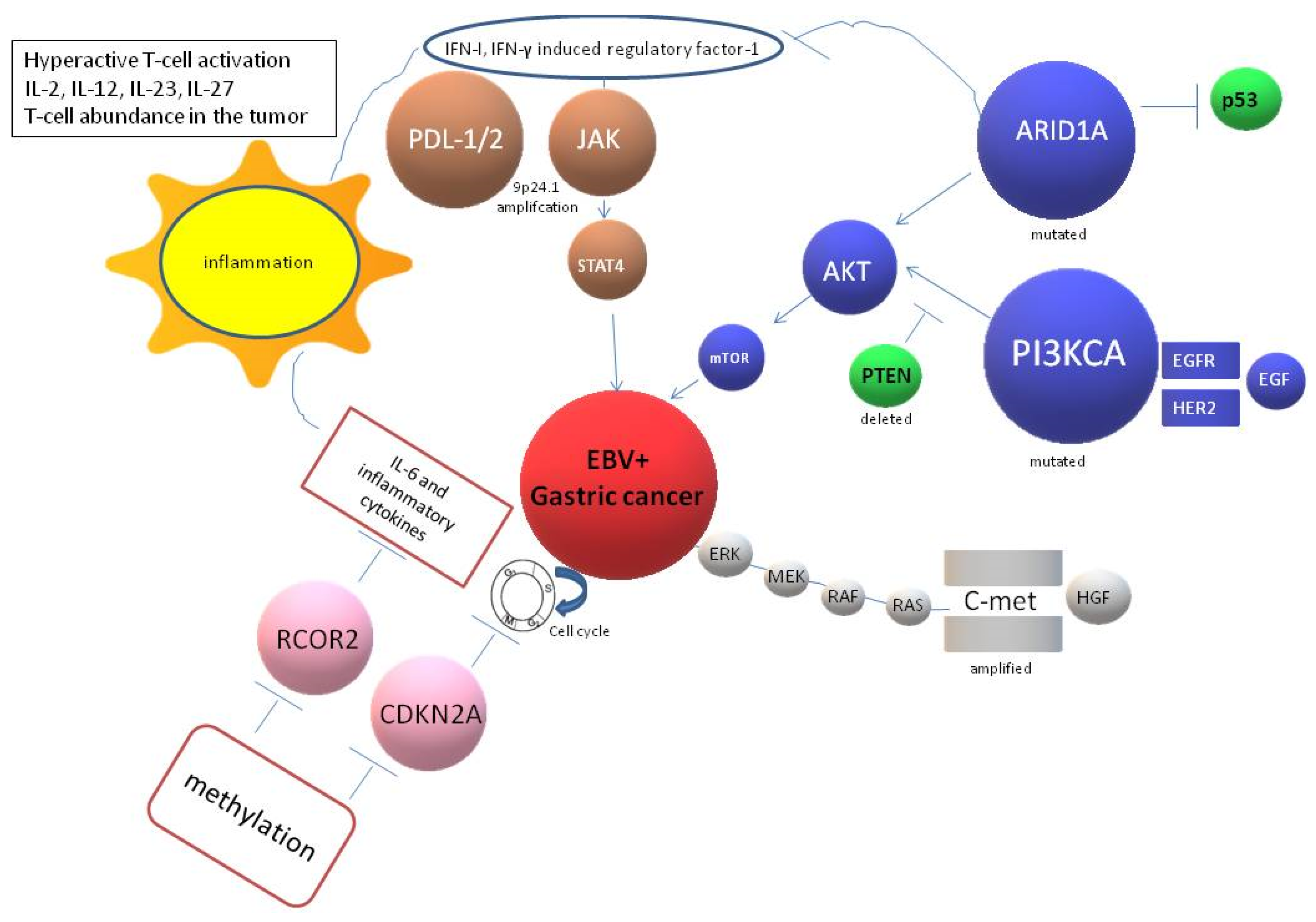
7. EBV Interference with Host Immune Responses
8. Targeted Therapies for EBV + GC
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| EBV | Epstein-Barr virus |
| GC | Gastric cancer |
| EBVaGC | Epstein-Barr-positive gastric cancer |
| H. pylori | Helicobacter pylori |
| EBNA | Nuclear antigen proteins |
| LMP | latent membrane proteins |
| BART | miR-BamH1 A rightwards transcripts |
| EBERs | Epstein–Barr-encoded RNAs |
| MHC | Major histocompatibility complex |
| NPC | nasopharyngeal cancer |
| IL | Interleukine |
| IFN | Interferon |
| TCGA | Cancer Genome Atlas |
| ACRG | Asian Cancer Research Group |
| MSI | Microsatellite unstable |
| MSS | Microsatellite stable |
| EMT | Epithelial-mesenchymal transition |
| PD-(L1) | Programmed Death-(Ligand 1) |
References
- Murphy, G.; Pfeiffer, R.; Camargo, M.C.; Rabkin, C.S. Meta-analysis shows that prevalence of Epstein–Barr virus-positive gastric cancer differs based on sex and anatomic location. Gastroenterology 2009, 137, 824–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, J.L.; Glaser, S.L. Epstein–Barr virus-associated malignancies: Epidemiologic patterns and etiologic implications. Crit. Rev. Oncol. Hematol. 2000, 34, 27–53. [Google Scholar] [CrossRef]
- Shannon-Lowe, C.; Adland, E.; Bell, A.I.; Delecluse, H.-J.; Rickinson, A.B.; Rowe, M. Features Distinguishing Epstein–Barr Virus Infections of Epithelial Cells and B Cells: Viral Genome Expression, Genome Maintenance, and Genome Amplification. J. Virol. 2009, 83, 7749–7760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshiyama, H.; Imai, S.; Shimizu, N.; Takada, K. Epstein–Barr virus infection of human gastric carcinoma cells: Implication of the existence of a new virus receptor different from CD21. J. Virol. 1997, 71, 5688–5691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Sathiyamoorthy, K.; Zhang, X.; Schaller, S.; Perez White, B.E.; Jardetzky, T.S.; Longnecker, R. Ephrin receptor A2 is a functional entry receptor for Epstein–Barr virus. Nat. Microbiol. 2018, 3, 172–180. [Google Scholar] [CrossRef]
- Xiao, J.; Palefsky, J.M.; Herrera, R.; Berline, J.; Tugizov, S.M. The Epstein–Barr Virus BMRF-2 Protein Facilitates Virus Attachment to Oral Epithelial Cells. Virology 2008, 370, 430–442. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Hutt-Fletcher, L.M. Epstein–Barr virus lacking glycoprotein gp42 can bind to B cells but is not able to infect. J. Virol. 1998, 72, 158–163. [Google Scholar] [CrossRef] [Green Version]
- Chesnokova, L.S.; Ahuja, M.K.; Hutt-Fletcher, L.M. Epstein–Barr virus glycoprotein gB and gHgL can mediate fusion and entry in trans, and heat can act as a partial surrogate for gHgL and trigger a conformational change in gB. J. Virol. 2014, 88, 12193–12201. [Google Scholar] [CrossRef] [Green Version]
- Miller, N.; Hutt-Fletcher, L.M. Epstein–Barr virus enters B cells and epithelial cells by different routes. J. Virol. 1992, 66, 3409–3414. [Google Scholar] [CrossRef] [Green Version]
- Ni, C.; Chen, Y.; Zeng, M.; Pei, R.; Du, Y.; Tang, L.; Wang, M.; Hu, Y.; Zhu, H.; He, M.; et al. In-cell infection: A novel pathway for Epstein–Barr virus infection mediated by cell-in-cell structures. Cell Res. 2015, 25, 785–800. [Google Scholar] [CrossRef] [Green Version]
- Shannon-Lowe, C.; Rowe, M. Epstein–Barr virus infection of polarized epithelial cells via the basolateral surface by memory B cell-mediated transfer infection. PLoS Pathog. 2011, 7, e1001338. [Google Scholar] [CrossRef] [Green Version]
- Yue, W.; Zhu, M.; Zuo, L.; Xin, S.; Zhang, J.; Liu, L.; Li, S.; Dang, W.; Zhang, S.; Xie, Y.; et al. Early Pattern of Epstein–Barr Virus Infection in Gastric Epithelial Cells by “Cell-in-cell”. Virol Sin. 2019, 34, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Ludigs, K.; Parfenov, V.; Du Pasquier, R.A.; Guarda, G. Type I IFN-mediated regulation of IL-1 production in inflammatory disorders. Cell. Mol. Life Sci. 2012, 69, 3395–3418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukayama, M.; Hayashi, Y.; Iwasaki, Y.; Chong, J.; Ooba, T.; Takizawa, T.; Koike, M.; Mizutani, S.; Miyaki, M.; Hirai, K. Epstein–Barr virus-associated gastric carcinoma and Epstein–Barr virus infection of the stomach. Lab. Investig. 1994, 71, 73–81. [Google Scholar] [PubMed]
- Ahmad, S.A.; Xia, B.T.; Bailey, C.E.; Abbott, D.E.; Helmink, B.A.; Daly, M.C.; Thota, R.; Schlegal, C.; Winer, L.K.; Ahmad, S.A.; et al. An update on gastric cancer. Curr. Probl. Surg. 2016, 53, 449–490. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Ye, Y.; Fu, M.; Zheng, B.; Qiu, Q.; Huang, Z. Implication of viral microRNAs in the genesis and diagnosis of Epstein–Barr virus-associated tumors. Oncol. Lett. 2019, 18, 3433–3442. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.-Y.; Liu, Y.Y.; Liu, K.-H.; Hsu, J.-T.; Chen, T.-C.; Chiu, C.-T.; Yeh, T.-S. Comprehensive profiling of virus microRNAs of Epstein–Barr virus-associated gastric carcinoma: Highlighting the interactions of ebv-Bart9 and host tumor cells. J. Gastroenterol. Hepatol. 2017, 32, 82–91. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Fachko, D.; Ivanov, N.S.; Skinner, C.M.; Skalsky, R.L. Epstein–Barr virus microRNAs regulate B cell receptor signal transduction and lytic reactivation. PLoS Pathog. 2019, 15, e1007535. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Yu, F.; Wu, W.; Wang, Y.; Ding, H.; Qian, L. Epstein–Barr virus-encoded microRNAs as regulators in host immune responses. Int. J. Biol. Sci. 2018, 14, 565–576. [Google Scholar] [CrossRef] [Green Version]
- Plummer, M.; Franceschi, S.; Vignat, J.; Forman, D.; de Martel, C. Global burden of gastric cancer attributable to Helicobacter pylori. Int. J. Cancer 2015, 136, 487–490. [Google Scholar] [CrossRef]
- Martín-de-Argila, C.; Boixeda, D.; Redondo, C.; Alvarez, I.; Gisbert, J.P.; García Plaza, A.; Cantón, R. Relation between histologic subtypes and location of gastric cancer and Helicobacter pylori. Scand. J. Gastroenterol. 1997, 32, 303–307. [Google Scholar] [CrossRef] [PubMed]
- Alessandrini, L.; Manchi, M.; De Re, V.; Dolcetti, R.; Canzonieri, V. Proposed Molecular and miRNA Classification of Gastric Cancer. Int. J. Mol. Sci. 2018, 19, 1683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farrell, P. Pathogenesis: Infections causing gastric cancer. Nat. Microbiol. 2016, 1, 16038. [Google Scholar] [CrossRef] [PubMed]
- Saju, P.; Murata-Kamiya, N.; Hayashi, T.; Senda, Y.; Nagase, L.; Noda, S.; Matsusaka, K.; Funata, S.; Kunita, A.; Urabe, M.; et al. Host SHP1 phosphatase antagonizes Helicobacter pylori CagA and can be downregulated by Epstein–Barr virus. Nat. Microbiol. 2016, 1, 16026. [Google Scholar] [CrossRef] [PubMed]
- Dursun, N.; Hacıhasanoğlu, E.; Okçu, O.; Paşaoğlu, E.; Leblebici, C. Epstein–Barr virus infection in patients with chronic gastritis without Helicobacter pylori infection. Turk. J. Gastroenterol. 2020, 31, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Zur Hausen, A.; van Rees, B.P.; van Beek, J.; Craanen, M.E.; Bloemena, E.; Offerhaus, G.J.A.; Meijer, C.J.L.M.; van den Brule, A.J.C. Epstein–Barr virus in gastric carcinomas and gastric stump carcinomas: A late event in gastric carcinogenesis. J. Clin. Pathol. 2004, 57, 487–491. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.-H.; Kim, S.-H.; Han, S.-H.; An, J.-S.; Lee, E.-S.; Kim, Y.-S. Clinicopathological and molecular characteristics of Epstein–Barr virus-associated gastric carcinoma: A meta-analysis. J. Gastroenterol. Hepatol. 2009, 24, 354–365. [Google Scholar] [CrossRef]
- Camargo, M.C.; Kim, K.-M.; Matsuo, K.; Torres, J.; Liao, L.M.; Morgan, D.R.; Michel, A.; Waterboer, T.; Zabaleta, J.; Dominguez, R.L.; et al. Anti-Helicobacter pylori Antibody Profiles in Epstein–Barr virus (EBV)-Positive and EBV-Negative Gastric Cancer. Helicobacter 2016, 21, 153–157. [Google Scholar] [CrossRef] [Green Version]
- Minoura-Etoh, J.; Gotoh, K.; Sato, R.; Ogata, M.; Kaku, N.; Fujioka, T.; Nishizono, A. Helicobacter pylori-associated oxidant monochloramine induces reactivation of Epstein–Barr virus (EBV) in gastric epithelial cells latently infected with EBV. J. Med. Microbiol. 2006, 55, 905–911. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.; Shun, C.; Wu, C.; Hsu, T.; Lin, M.; Chang, M.; Wang, H.; Lin, J. Epstein–Barr virus—Associated gastric carcinomas: Relation to H. pylori infection and genetic alterations. Gastroenterology 2000, 118, 1031–1038. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014, 513, 202–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genitsch, V.; Novotny, A.; Seiler, C.A.; Kroell, D.; Walch, A.; Langer, R. Epstein Barr virus in gastro-esophageal adenocarcinomas—Single center experiences in the context of current literature. Front. Oncol. 2015, 5, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.-C.; Ng, K.-F.; Chen, K.-H.; Hsu, J.-T.; Liu, K.-H.; Yeh, T.-S.; Chen, T.-C. Prognostic factors in Epstein–Barr virus-associated stage I-III gastric carcinoma: Implications for a unique type of carcinogenesis. Oncol. Rep. 2014, 32, 530–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.-C.; Ng, K.-F.; Yeh, T.-S.; Cheng, C.-T.; Lin, J.-S.; Liu, Y.-J.; Chuang, H.-C.; Chen, T.-C. Subtraction of Epstein–Barr virus and microsatellite instability genotypes from the Lauren histotypes: Combined molecular and histologic subtyping with clinicopathological and prognostic significance validated in a cohort of 1248 cases. Int. J. Cancer 2019, 145, 3218–3230. [Google Scholar] [CrossRef] [PubMed]
- Corallo, S.; Fucà, G.; Morano, F.; Salati, M.; Spallanzani, A.; Gloghini, A.; Volpi, C.C.; Trupia, D.V.; Lobefaro, R.; Guarini, V.; et al. Clinical Behavior and Treatment Response of Epstein–Barr Virus-Positive Metastatic Gastric Cancer: Implications for the Development of Future Trials. Oncologist 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohlruss, M.; Grosser, B.; Krenauer, M.; Slotta-Huspenina, J.; Jesinghaus, M.; Blank, S.; Novotny, A.; Reiche, M.; Schmidt, T.; Ismani, L.; et al. Prognostic implication of molecular subtypes and response to neoadjuvant chemotherapy in 760 gastric carcinomas: Role of Epstein–Barr virus infection and high- and low-microsatellite instability. J. Pathol. Clin. Res. 2019, 5, 227–239. [Google Scholar] [CrossRef] [Green Version]
- Ghasemi, F.; Gameiro, S.F.; Tessier, T.M.; Maciver, A.H.; Mymryk, J.S. High Levels of Class I Major Histocompatibility Complex mRNA Are Present in Epstein–Barr Virus-Associated Gastric Adenocarcinomas. Cells 2020, 9, 499. [Google Scholar] [CrossRef] [Green Version]
- Saiki, Y.; Ohtani, H.; Naito, Y.; Miyazawa, M.; Nagura, H. Immunophenotypic characterization of Epstein–Barr virus-associated gastric carcinoma: Massive infiltration by proliferating CD8+ T-lymphocytes. Lab. Investig. 1996, 75, 67–76. [Google Scholar]
- Cristescu, R.; Lee, J.; Nebozhyn, M.; Kim, K.-M.; Ting, J.C.; Wong, S.S.; Liu, J.; Yue, Y.G.; Wang, J.; Yu, K.; et al. Molecular analysis of gastric cancer identifies subtypes associated with distinct clinical outcomes. Nat. Med. 2015, 21, 449–456. [Google Scholar] [CrossRef]
- David, L.; Seruca, R.; Nesland, J.M.; Soares, P.; Sansonetty, F.; Holm, R.; Børresen, A.L.; Sobrinho-Simões, M. c-erbB-2 expression in primary gastric carcinomas and their metastases. Mod. Pathol. 1992, 5, 384–390. [Google Scholar] [CrossRef]
- Barros-Silva, J.D.; Leitão, D.; Afonso, L.; Vieira, J.; Dinis-Ribeiro, M.; Fragoso, M.; Bento, M.J.; Santos, L.; Ferreira, P.; Rêgo, S.; et al. Association of ERBB2 gene status with histopathological parameters and disease-specific survival in gastric carcinoma patients. Br. J. Cancer 2009, 100, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Kubota, Y.; Kawazoe, A.; Sasaki, A.; Mishima, S.; Sawada, K.; Nakamura, Y.; Kotani, D.; Kuboki, Y.; Taniguchi, H.; Kojima, T.; et al. The Impact of Molecular Subtype on Efficacy of Chemotherapy and Checkpoint Inhibition in Advanced Gastric Cancer. Clin. Cancer Res. 2020, 26, 3784–3790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shenoy, S. CDH1 (E-Cadherin) Mutation and Gastric Cancer: Genetics, Molecular Mechanisms and Guidelines for Management. Cancer Manag. Res. 2019, 11, 10477–10486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falchetti, M.; Saieva, C.; Lupi, R.; Masala, G.; Rizzolo, P.; Zanna, I.; Ceccarelli, K.; Sera, F.; Mariani-Costantini, R.; Nesi, G.; et al. Gastric cancer with high-level microsatellite instability: Target gene mutations, clinicopathologic features, and long-term survival. Hum. Pathol. 2008, 39, 925–932. [Google Scholar] [CrossRef]
- Pietrantonio, F.; Miceli, R.; Raimondi, A.; Kim, Y.W.; Kang, W.K.; Langley, R.E.; Choi, Y.Y.; Kim, K.-M.; Nankivell, M.G.; Morano, F.; et al. Individual patient data meta-analysis of the value of microsatellite instability as a biomarker in gastric cancer. J. Clin. Oncol. 2019, 37, 3392–3400. [Google Scholar] [CrossRef]
- Smyth, E.C.; Wotherspoon, A.; Peckitt, C.; Gonzalez, D.; Hulkki-Wilson, S.; Eltahir, Z.; Fassan, M.; Rugge, M.; Valeri, N.; Okines, A.; et al. Mismatch Repair Deficiency, Microsatellite Instability, and Survival An Exploratory Analysis of the Medical Research Council Adjuvant Gastric Infusional Chemotherapy (MAGIC) Trial. JAMA Oncol. 2017, 3, 1197–1203. [Google Scholar] [CrossRef] [Green Version]
- Meyers, M.; Wagner, M.W.; Mazurek, A.; Schmutte, C.; Fishel, R.; Boothman, D.A. DNA mismatch repair-dependent response to fluoropyrimidine-generated damage. J. Biol. Chem. 2005, 280, 5516–5526. [Google Scholar] [CrossRef] [Green Version]
- Sowitt, B.E.; Shukla, S.A.; Sholl, L.M.; Ritterhouse, L.L.; Watkins, J.C.; Rodig, S.; Stover, E.; Strickland, K.C.; D’Andrea, A.D.; Wu, C.J.; et al. Association of Polymerase e-Mutated and Microsatellite-Instable Endometrial Cancers with Neoantigen Load, Number of Tumor-Infiltrating Lymphocytes, and Expression of PD-1 and PD-L1. JAMA Oncol. 2015, 1, 1319–1323. [Google Scholar] [CrossRef]
- Camargo, M.C.; Kim, W.-H.; Chiaravalli, A.M.; Kim, K.-M.; Corvalan, A.H.; Matsuo, K.; Yu, J.; Sung, J.J.Y.; Herrera-Goepfert, R.; Meneses-Gonzalez, F.; et al. Improved survival of gastric cancer with tumour Epstein–Barr virus positivity: An international pooled analysis. Gut 2014, 63, 236–243. [Google Scholar] [CrossRef]
- van Beek, J.; zur Hausen, A.; Kranenbarg, E.K.; van de Velde, C.J.H.; Middeldorp, J.M.; van den Brule, A.J.C.; Meijer, C.; Bloemena, E. EBV-positive gastric adenocarcinomas: A distinct clinicopathologic entity with a low frequency of lymph node involvement. J. Clin. Oncol. 2004, 22, 664–670. [Google Scholar] [CrossRef]
- Nishikawa Nishikawa, J.; Iizasa, H.; Yoshiyama, H.; Shimokuri, K.; Kobayashi, Y.; Sasaki, S.; Nakamura, M.; Yanai, H.; Sakai, K.; Suehiro, Y.; et al. Clinical Importance of Epstein–Barr Virus-Associated Gastric Cancer. Cancers 2018, 10, 167. [Google Scholar] [CrossRef] [Green Version]
- Yanagi, A.; Nishikawa, J.; Shimokuri, K.; Shuto, T.; Takagi, T.; Takagi, F.; Kobayashi, Y.; Yamamoto, M.; Miura, O.; Yanai, H.; et al. Clinicopathologic Characteristics of Epstein–Barr Virus-Associated Gastric Cancer Over the Past Decade in Japan. Microorganisms 2019, 7, 305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, A.; Hikri, E.; Goshen-Lago, T.; Barkan, T.; Morgenstern, S.; Brook, E.; Maderer, A.; Roth, W.; Gordon, N.; Kashtan, H.; et al. Young-onset gastric cancer and Epstein-Barr Virus (EBV)—A major player in the pathogenesis? BMC Cancer 2020, 20, 34. [Google Scholar] [CrossRef] [PubMed]
- Lauren, P. The two histological main types of gastric carcinoma: Diffuse and so-called intestinal-type carcinoma. An attempt at a histo-clinical classification. Acta Pathol. Microbiol. Scand. 1965, 64, 31–49. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.; Lee, S.-J.; Kim, Y.; Kim, A.; Shin, N.; Choi, K.U.; Lee, C.-H.; Huh, G.Y.; Kim, K.-M.; Setia, N.; et al. High-throughput Protein and mRNA Expression-based Classification of Gastric Cancers Can Identify Clinically Distinct Subtypes, Concordant With Recent Molecular Classifications. Am. J. Surg. Pathol. 2017, 41, 106–115. [Google Scholar] [CrossRef]
- Abe, H.; Kaneda, A.; Fukayama, M. Epstein-Barr Virus-Associated Gastric Carcinoma: Use of Host Cell Machineries and Somatic Gene Mutations. Pathobiology 2015, 82, 212–223. [Google Scholar] [CrossRef]
- Barua, R.R.; Uozaki, H.; Chong, J.-M.; Ushiku, T.; Hino, R.; Chang, M.-S.; Nagai, H.; Fukayama, M. Phenotype analysis by MUC2, MUC5AC, MUC6, and CD10 expression in Epstein–Barr virus-associated gastric carcinoma. J. Gastroenterol. 2006, 41, 733–739. [Google Scholar] [CrossRef]
- Shinozaki, A.; Ushiku, T.; Morikawa, T.; Hino, R.; Sakatani, T.; Uozaki, H.; Fukayama, M. Epstein–Barr virus-associated gastric carcinoma: A distinct carcinoma of gastric phenotype by claudin expression profiling. J. Histochem. Cytochem. 2009, 57, 775–785. [Google Scholar] [CrossRef] [Green Version]
- Kawachi, H.; Takizawa, T.; Eishi, Y.; Shimizu, S.; Kumagai, J.; Funata, N.; Koike, M. Absence of either gastric or intestinal phenotype in microscopic differentiated gastric carcinomas. J. Pathol. 2003, 199, 436–446. [Google Scholar] [CrossRef]
- Tsukashita, S.; Kushima, R.; Bamba, M.; Sugihara, H.; Hattori, T. MUC gene expression and histogenesis of adenocarcinoma of the stomach. Int. J. Cancer 2001, 94, 166–170. [Google Scholar] [CrossRef] [Green Version]
- Dottermusch Dottermusch, M.; Krüger, S.; Behrens, H.-M.; Halske, C.; Röcken, C. Expression of the potential therapeutic target claudin-18.2 is frequently decreased in gastric cancer: Results from a large Caucasian cohort study. Virchows Arch. 2019, 475, 563–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coati, I.; Lotz, G.; Fanelli, G.N.; Brignola, S.; Lanza, C.; Cappellesso, R.; Pellino, A.; Pucciarelli, S.; Spolverato, G.; Guzzardo, V.; et al. Claudin-18 expression in oesophagogastric adenocarcinomas: A tissue microarray study of 523 molecularly profiled cases. Br. J. Cancer 2019, 121, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, J.; Malta, M.; Galaghar, A.; Afonso, L.P.; Libânio, D.; Medeiros, R.; Dinis-Ribeiro, M.; Pimentel-Nunes, P.; Sousa, H. Epstein–Barr virus is absent in gastric superficial neoplastic lesions. Virchows Arch. 2019, 475, 757–762. [Google Scholar] [CrossRef] [PubMed]
- De Re, V.; De Vita, S.; Dolcetti, R.; Boiocchi, M. Association between B-type Epstein–Barr virus and Hodgkin’s disease in immunocompromised patients. Blood 1993, 82, 328–330. [Google Scholar] [CrossRef] [Green Version]
- Zanella, L.; Riquelme, I.; Buchegger, K.; Abanto, M.; Ili, C.; Brebi, P. A reliable Epstein–Barr Virus classification based on phylogenomic and population analyses. Sci. Rep. 2019, 9, 9829. [Google Scholar] [CrossRef] [Green Version]
- Borozan, I.; Zapatka, M.; Frappier, L.; Ferretti, V. Analysis of Epstein–Barr Virus Genomes and Expression Profiles in Gastric Adenocarcinoma. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [Green Version]
- Corvalán, A.H.; Ruedlinger, J.; de Mayo, T.; Polakovicova, I.; Gonzalez-Hormazabal, P.; Aguayo, F. The Phylogeographic Diversity of EBV and Admixed Ancestry in the Americas−Another Model of Disrupted Human-Pathogen Co-Evolution. Cancers 2019, 11, 217. [Google Scholar] [CrossRef] [Green Version]
- Bonequi, P.; Meneses-González, F.; Correa, P.; Rabkin, C.S.; Camargo, M.C. Risk factors for gastric cancer in Latin-America: A meta-analysis. Cancer Causes Control 2013, 24, 217–231. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Xu, M.; Zhang, X.; Chu, F.; Zhou, T. MAPK/c-Jun signaling pathway contributes to the upregulation of the anti-apoptotic proteins Bcl-2 and Bcl-xL induced by Epstein–Barr virus-encoded BARF1 in gastric carcinoma cells. Oncol. Lett. 2018, 15, 7537–7544. [Google Scholar] [CrossRef] [Green Version]
- El-Sharkawy, A.; Al Zaidan, L.; Malki, A. Epstein–Barr Virus-Associated Malignancies: Roles of Viral Oncoproteins in Carcinogenesis. Front. Oncol. 2018, 8. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.H.; Chang, M.S.; Yoon, C.J.; Middeldorp, J.M.; Martinez, O.M.; Byeon, S.; Rha, S.Y.; Kim, S.H.; Kim, Y.S.; Woo, J.H. Epstein–Barr virus BARF1-induced NF kappa B/miR-146a/SMAD4 alterations in stomach cancer cells. Oncotarget 2016, 7, 82213–82227. [Google Scholar] [CrossRef] [PubMed]
- Hoebe, E.K.; Le Large, T.Y.S.; Greijer, A.E.; Middeldorp, J.M. BamHI-A rightward frame 1, an Epstein–Barr virus-encoded oncogene and immune modulator. Rev. Med. Virol. 2013, 23, 367–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinozaki-Ushiku, A.; Kunita, A.; Fukayama, M. Update on Epstein-Barr virus and gastric cancer (Review). Int. J. Oncol. 2015, 46, 1421–1434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasui, M.; Kunita, A.; Numakura, S.; Uozaki, H.; Ushiku, T.; Fukayama, M. Cancer stem cells in Epstein-Barr virus-associated gastric carcinoma. Cancer Sci. 2020, 111, 2598–2607. [Google Scholar] [CrossRef]
- Gong, L.; Chen, J.; Dong, M.; Xiao, Z.; Feng, Z.; Pan, Y.; Zhang, Y.; Du, Y.; Zhang, J.; Bi, Y.; et al. Epstein-Barr virus-derived circularRNA LMP2A induces stemness inEBV-associated gastric cancer. EMBO Rep. 2020, 21, e49689. [Google Scholar] [CrossRef] [PubMed]
- Luo, B.; Wang, Y.; Wang, X.-F.; Liang, H.; Yan, L.-P.; Huang, B.-H.; Zhao, P. Expression of Epstein-Barr virus genes in EBV-associated gastric carcinomas. World J. Gastroenterol. 2005, 11, 629–633. [Google Scholar] [CrossRef]
- Iwakiri, D. Multifunctional non-coding Epstein–Barr virus encoded RNAs (EBERs) contribute to viral pathogenesis. Virus Res. 2016, 212, 30–38. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhang, J.; Cheng, A.S.L.; Yu, J.; To, K.F.; Kang, W. Gastric cancer: Genome damaged by bugs. Oncogene 2020, 39, 3427–3442. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Burassakarn, A.; Kang, Y.; Iizasa, H.; Yoshiyama, H. A single nucleotide polymorphism in the BART promoter region of Epstein–Barr virus isolated from nasopharyngeal cancer cells. Biochem. Biophys. Res. Commun. 2019, 520, 373–378. [Google Scholar] [CrossRef]
- Kim, D.N.; Chae, H.-S.; Oh, S.T.; Kang, J.-H.; Park, C.H.; Park, W.S.; Takada, K.; Lee, J.M.; Lee, W.-K.; Lee, S.K. Expression of viral microRNAs in Epstein-Barr virus-associated gastric carcinoma. J. Virol. 2007, 81, 1033–1036. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, J.; Malta, M.; Galaghar, A.; Silva, F.; Afonso, L.P.; Medeiros, R.; Sousa, H. P53 deregulation in Epstein–Barr virus-associated gastric cancer. Cancer Lett. 2017, 404, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; He, C.; Wu, J.; Yang, D.; Yi, W. Epstein barr virus encodes miRNAs to assist host immune escape. J. Cancer 2020, 11, 2091–2100. [Google Scholar] [CrossRef]
- Choi, H.; Lee, H.; Kim, S.R.; Gho, Y.S.; Lee, S.K. Epstein–Barr virus-encoded microRNA BART15-3p promotes cell apoptosis partially by targeting BRUCE. J. Virol. 2013, 87, 8135–8144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, B.; Li, W.; Wu, Y.; Wei, F.; Gong, Z.; Bo, H.; Wang, Y.; Li, X.; Xiang, B.; Guo, C.; et al. Epstein–Barr virus-encoded miR-BART6-3p inhibits cancer cell metastasis and invasion by targeting long non-coding RNA LOC553103. Cell Death Dis. 2016, 7, e2353. [Google Scholar] [CrossRef] [PubMed]
- Edwards, R.H.; Dekroon, R.; Raab-Traub, N. Alterations in cellular expression in EBV infected epithelial cell lines and tumors. PLoS Pathog. 2019, 15, e1008071. [Google Scholar] [CrossRef] [PubMed]
- Ryan, J.L.; Jones, R.J.; Kenney, S.C.; Rivenbark, A.G.; Tang, W.; Knight, E.R.; Coleman, W.B.; Gulley, M.L. Epstein–Barr virus-specific methylation of human genes in gastric cancer cells. Infect. Agents Cancer 2010, 5, 27. [Google Scholar] [CrossRef] [Green Version]
- Vo, Q.N.; Geradts, J.; Gulley, M.L.; Boudreau, D.A.; Bravo, J.C.; Schneider, B.G. Epstein–Barr virus in gastric adenocarcinomas: Association with ethnicity and CDKN2A promoter methylation. J. Clin. Pathol. 2002, 55, 669–675. [Google Scholar] [CrossRef] [Green Version]
- Fang, W.-L.; Chen, M.-H.; Huang, K.-H.; Chang, S.-C.; Lin, C.-H.; Chao, Y.; Lo, S.-S.; Li, A.F.-Y.; Wu, C.-W.; Shyr, Y.-M. Analysis of the clinical significance of DNA methylation in gastric cancer based on a genome-wide high-resolution array. Clin. Epigenet. 2019, 11, 154. [Google Scholar] [CrossRef]
- Anderson, B.W.; Suh, Y.-S.; Choi, B.; Lee, H.-J.; Yab, T.C.; Taylor, W.R.; Dukek, B.A.; Berger, C.K.; Cao, X.; Foote, P.H.; et al. Detection of Gastric Cancer with Novel Methylated DNA Markers: Discovery, Tissue Validation, and Pilot Testing in Plasma. Clin. Cancer Res. 2018, 24, 5724–5734. [Google Scholar] [CrossRef] [Green Version]
- Ghadami, E.; Nikbakhsh, N.; Fattahi, S.; Kosari-Monfared, M.; Ranaee, M.; Taheri, H.; Amjadi-Moheb, F.; Godazandeh, G.; Shafaei, S.; Nosrati, A.; et al. Epigenetic alterations of CYLD promoter modulate its expression in gastric adenocarcinoma: A footprint of infections. J. Cell. Physiol. 2019, 234, 4115–4124. [Google Scholar] [CrossRef]
- Miao, J.; Liu, Y.; Zhao, G.; Liu, X.; Ma, Y.; Li, H.; Li, S.; Zhu, Y.; Xiong, S.; Zheng, M.; et al. Feasibility of Plasma-Methylated SFRP2 for Early Detection of Gastric Cancer. Cancer Control. 2020, 27, 107327482092255. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Lung, H.L.; Huang, T.; Lan, R.; Zha, S.; Chan, L.S.; Thor, W.; Tsoi, T.-H.; Chau, H.-F.; Boreström, C.; et al. Reactivation of Epstein–Barr virus by a dual-responsive fluorescent EBNA1-targeting agent with Zn2+-chelating function. Proc. Natl. Acad. Sci. USA 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.-M.; Hur, D.Y.; Hong, S.-W.; Kim, J.H. EBV-encoded EBNA1 regulates cell viability by modulating miR34a-NOX2-ROS signaling in gastric cancer cells. Biochem. Biophys. Res. Commun. 2017, 494, 550–555. [Google Scholar] [CrossRef] [PubMed]
- Incrocci, R.; McAloon, J.; Montesano, M.; Bardahl, J.; Vagvala, S.; Stone, A.; Swanson-Mungerson, M. Epstein-Barr virus LMP2A utilizes Syk and PI3K to activate NF-κB in B-cell lymphomas to increase MIP-1α production. J. Med. Virol. 2019, 91, 845–855. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.-H.; Lin, J.-Y.; Chou, Y.-C.; Chen, M.-R.; Yeh, T.-H.; Lin, C.-W.; Lin, S.-J.; Tsai, C.-H. Epstein-Barr virus LMP2A suppresses MHC class II expression by regulating the B-cell transcription factors E47 and PU.1. Blood 2015, 125, 2228–2238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Q.; Zhang, Y.; Liu, W.; Xiao, H.; Qi, Y.; Li, J.; Luo, B. Latent membrane protein 2A inhibits expression level of Smad2 through regulating miR-155-5p in EBV-associated gastric cancer cell lines. J. Med. Virol. 2020, 92, 96–106. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, Y.; Liu, W.; Zhang, Q.; Xiao, H.; Song, H.; Luo, B. MiR-BART1-5p targets core 2β-1,6-acetylglucosaminyltransferase GCNT3 to inhibit cell proliferation and migration in EBV-associated gastric cancer. Virology 2020, 541, 63–74. [Google Scholar] [CrossRef]
- Lei, T.; Yuen, K.-S.; Xu, R.; Tsao, S.W.; Chen, H.; Li, M.; Kok, K.-H.; Jin, D.-Y. Targeting of DICE1 tumor suppressor by Epstein-Barr virus-encoded miR-BART3* microRNA in nasopharyngeal carcinoma. Int. J. Cancer 2013, 133, 79–87. [Google Scholar] [CrossRef]
- Choy, E.Y.-W.; Siu, K.-L.; Kok, K.-H.; Lung, R.W.-M.; Tsang, C.M.; To, K.-F.; Kwong, D.L.-W.; Tsao, S.W.; Jin, D.-Y. An Epstein-Barr virus-encoded microRNA targets PUMA to promote host cell survival. J. Exp. Med. 2008, 205, 2551–2560. [Google Scholar] [CrossRef] [Green Version]
- Marquitz, A.R.; Mathur, A.; Nam, C.S.; Raab-Traub, N. The Epstein-Barr Virus BART microRNAs target the pro-apoptotic protein Bim. Virology 2011, 412, 392–400. [Google Scholar] [CrossRef]
- Shinozaki-Ushiku, A.; Kunita, A.; Isogai, M.; Hibiya, T.; Ushiku, T.; Takada, K.; Fukayama, M. Profiling of Virus-Encoded MicroRNAs in Epstein-Barr Virus-Associated Gastric Carcinoma and Their Roles in Gastric Carcinogenesis. J. Virol. 2015, 89, 5581–5591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.; Choi, H.; Lee, S.K. Epstein-Barr Virus MicroRNA miR-BART20-5p Suppresses Lytic Induction by Inhibiting BAD-Mediated caspase-3-Dependent Apoptosis. J. Virol. 2016, 90, 1359–1368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.; Qin, Z.; Wang, J.; Zheng, X.; Lu, J.; Zhang, X.; Wei, L.; Peng, Q.; Zheng, Y.; Ou, C.; et al. Epstein-Barr Virus miR-BART6-3p Inhibits the RIG-I Pathway. J. Innate Immun. 2017, 9, 574–586. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Huang, J.; Wu, F.Y.; Liao, G.; Hutt-Fletcher, L.; Hayward, S.D. Regulation of expression of the Epstein-Barr virus BamHI-A rightward transcripts. J. Virol. 2005, 79, 1724–1733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Re, V.; Caggiari, L.; De Zorzi, M.; Fanotto, V.; Miolo, G.; Puglisi, F.; Cannizzaro, R.; Canzonieri, V.; Steffan, A.; Farruggia, P.; et al. Epstein-Barr virus BART microRNAs in EBV- associated Hodgkin lymphoma and gastric cancer. Infect. Agent Cancer 2020, 15. [Google Scholar] [CrossRef]
- Vallhov, H.; Gutzeit, C.; Johansson, S.M.; Nagy, N.; Paul, M.; Li, Q.; Friend, S.; George, T.C.; Klein, E.; Scheynius, A.; et al. Exosomes containing glycoprotein 350 released by EBV-transformed B cells selectively target B cells through CD21 and block EBV infection in vitro. J. Immunol. 2011, 186, 73–82. [Google Scholar] [CrossRef] [Green Version]
- Paludan, C.; Schmid, D.; Landthaler, M.; Vockerodt, M.; Kube, D.; Tuschl, T.; Münz, C. Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science 2005, 307, 593–596. [Google Scholar] [CrossRef]
- Severa, M.; Giacomini, E.; Gafa, V.; Anastasiadou, E.; Rizzo, F.; Corazzari, M.; Romagnoli, A.; Trivedi, P.; Fimia, G.M.; Coccia, E.M. EBV stimulates TLR- and autophagy-dependent pathways and impairs maturation in plasmacytoid dendritic cells: Implications for viral immune escape. Eur. J. Immunol. 2013, 43, 147–158. [Google Scholar] [CrossRef]
- Fathallah, I.; Parroche, P.; Gruffat, H.; Zannetti, C.; Johansson, H.; Yue, J.; Manet, E.; Tommasino, M.; Sylla, B.S.; Hasan, U.A. EBV latent membrane protein 1 is a negative regulator of TLR9. J. Immunol. 2010, 185, 6439–6447. [Google Scholar] [CrossRef] [Green Version]
- Yiu, S.P.T.; Hui, K.F.; Münz, C.; Lo, K.-W.; Tsao, S.W.; Kao, R.Y.T.; Yang, D.; Chiang, A.K.S. Autophagy-Dependent Reactivation of Epstein-Barr Virus Lytic Cycle and Combinatorial Effects of Autophagy-Dependent and Independent Lytic Inducers in Nasopharyngeal Carcinoma. Cancers 2019, 11, 1871. [Google Scholar] [CrossRef] [Green Version]
- Yiu, S.P.T.; Hui, K.F.; Choi, C.K.; Kao, R.Y.T.; Ma, C.W.; Yang, D.; Chiang, A.K.S. Intracellular Iron Chelation by a Novel Compound, C7, Reactivates Epstein–Barr Virus (EBV) Lytic Cycle via the ERK-Autophagy Axis in EBV-Positive Epithelial Cancers. Cancers 2018, 10, 505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nandakumar, A.; Uwatoko, F.; Yamamoto, M.; Tomita, K.; Majima, H.J.; Akiba, S.; Koriyama, C. Radiation-induced Epstein-Barr virus reactivation in gastric cancer cells with latent EBV infection. Tumour Biol. 2017, 39, 1010428317717718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraus, R.J.; Yu, X.; Cordes, B.-L.A.; Sathiamoorthi, S.; Iempridee, T.; Nawandar, D.M.; Ma, S.; Romero-Masters, J.C.; McChesney, K.G.; Lin, Z.; et al. Hypoxia-inducible factor-1α plays roles in Epstein-Barr virus’s natural life cycle and tumorigenesis by inducing lytic infection through direct binding to the immediate-early BZLF1 gene promoter. PLoS Pathog. 2017, 13, e1006404. [Google Scholar] [CrossRef] [PubMed]
- Ghasemi, F.; Tessier, T.M.; Gameiro, S.F.; Maciver, A.H.; Cecchini, M.J.; Mymryk, J.S. High MHC-II expression in Epstein–Barr virus-associated gastric cancers suggests that tumor cells serve an important role in antigen presentation. Sci. Rep. 2020, 10. [Google Scholar] [CrossRef]
- Kim, S.Y.; Park, C.; Kim, H.-J.; Park, J.; Hwang, J.; Kim, J.-I.; Choi, M.G.; Kim, S.; Kim, K.-M.; Kang, M.-S. Deregulation of immune response genes in patients with Epstein-Barr virus-associated gastric cancer and outcomes. Gastroenterology 2015, 148, 137–147. [Google Scholar] [CrossRef]
- Ichimura, T.; Abe, H.; Morikawa, T.; Yamashita, H.; Ishikawa, S.; Ushiku, T.; Seto, Y.; Fukayama, M. Low density of CD204-positive M2-type tumor-associated macrophages in Epstein-Barr virus-associated gastric cancer: A clinicopathologic study with digital image analysis. Hum. Pathol. 2016, 56, 74–80. [Google Scholar] [CrossRef]
- Hooykaas, M.J.G.; van Gent, M.; Soppe, J.A.; Kruse, E.; Boer, I.G.J.; van Leenen, D.; Koerkamp, M.J.A.G.; Holstege, F.C.P.; Ressing, M.E.; Wiertz, E.J.H.J.; et al. EBV MicroRNA BART16 Suppresses Type I IFN Signaling. J. Immunol. 2017, 198, 4062–4073. [Google Scholar] [CrossRef] [Green Version]
- Nachmani, D.; Stern-Ginossar, N.; Sarid, R.; Mandelboim, O. Diverse herpesvirus microRNAs target the stress-induced immune ligand MICB to escape recognition by natural killer cells. Cell Host Microbe 2009, 5, 376–385. [Google Scholar] [CrossRef] [Green Version]
- Ross, N.; Gandhi, M.K.; Nourse, J.P. The Epstein-Barr virus microRNA BART11-5p targets the early B-cell transcription factor EBF1. Am. J. Blood Res. 2013, 3, 210–224. [Google Scholar]
- Haneklaus, M.; Gerlic, M.; Kurowska-Stolarska, M.; Rainey, A.-A.; Pich, D.; McInnes, I.B.; Hammerschmidt, W.; O’Neill, L.A.J.; Masters, S.L. Cutting edge: miR-223 and EBV miR-BART15 regulate the NLRP3 inflammasome and IL-1β production. J. Immunol. 2012, 189, 3795–3799. [Google Scholar] [CrossRef] [Green Version]
- Hooykaas, M.J.G.; Kruse, E.; Wiertz, E.J.H.J.; Lebbink, R.J. Comprehensive profiling of functional Epstein-Barr virus miRNA expression in human cell lines. BMC Genom. 2016, 17, 644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, W.; Morgan, D.R.; Meyers, M.O.; Dominguez, R.L.; Martinez, E.; Kakudo, K.; Kuan, P.F.; Banet, N.; Muallem, H.; Woodward, K.; et al. Epstein-barr virus infected gastric adenocarcinoma expresses latent and lytic viral transcripts and has a distinct human gene expression profile. Infect. Agent Cancer 2012, 7, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skinner, C.M.; Ivanov, N.S.; Barr, S.A.; Chen, Y.; Skalsky, R.L. An Epstein-Barr Virus MicroRNA Blocks Interleukin-1 (IL-1) Signaling by Targeting IL-1 Receptor 1. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Cao, W.; Zhu, Y. Immunoregulatory Functions of the IL-12 Family of Cytokines in Antiviral Systems. Viruses 2019, 11, 772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balan, N.; Osborn, K.; Sinclair, A.J. Repression of CIITA by the Epstein-Barr virus transcription factor Zta is independent of its dimerization and DNA binding. J. Gen. Virol. 2016, 97, 725–732. [Google Scholar] [CrossRef] [PubMed]
- Zuo, J.; Thomas, W.A.; Haigh, T.A.; Fitzsimmons, L.; Long, H.M.; Hislop, A.D.; Taylor, G.S.; Rowe, M. Epstein-Barr virus evades CD4+ T cell responses in lytic cycle through BZLF1-mediated downregulation of CD74 and the cooperation of vBcl-2. PLoS Pathog. 2011, 7, e1002455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magalhaes, I.; Yogev, O.; Mattsson, J.; Schurich, A. The Metabolic Profile of Tumor and Virally Infected Cells Shapes Their Microenvironment Counteracting T Cell Immunity. Front. Immunol. 2019, 10, 2309. [Google Scholar] [CrossRef]
- Shin, E.-C.; Park, S.-H.; Nascimbeni, M.; Major, M.; Caggiari, L.; de Re, V.; Feinstone, S.M.; Rice, C.M.; Rehermann, B. The frequency of CD127(+) hepatitis C virus (HCV)-specific T cells but not the expression of exhaustion markers predicts the outcome of acute HCV infection. J. Virol. 2013, 87, 4772–4777. [Google Scholar] [CrossRef] [Green Version]
- Yoon, S.J.; Kim, J.Y.; Long, N.P.; Min, J.E.; Kim, H.M.; Yoon, J.H.; Anh, N.H.; Park, M.C.; Kwon, S.W.; Lee, S.K. Comprehensive Multi-Omics Analysis Reveals Aberrant Metabolism of Epstein–Barr-Virus-Associated Gastric Carcinoma. Cells 2019, 8, 1220. [Google Scholar] [CrossRef] [Green Version]
- Ambinder, R.F.; Mann, R.B. Epstein-Barr-encoded RNA in situ hybridization: Diagnostic applications. Hum. Pathol. 1994, 25, 602–605. [Google Scholar] [CrossRef]
- Qiu, M.-Z.; He, C.-Y.; Lu, S.-X.; Guan, W.-L.; Wang, F.; Wang, X.-J.; Jin, Y.; Wang, F.-H.; Li, Y.-H.; Shao, J.-Y.; et al. Prospective observation: Clinical utility of plasma Epstein–Barr virus DNA load in EBV-associated gastric carcinoma patients. Int. J. Cancer 2020, 146, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Ponnappan, S.; Ponnappan, U. Aging and Immune Function: Molecular Mechanisms to Interventions. Antioxid Redox Signal. 2011, 14, 1551–1585. [Google Scholar] [CrossRef] [Green Version]
- Broomfield, S.; Currie, A.; van der Most, R.G.; Brown, M.; van Bruggen, I.; Robinson, B.W.S.; Lake, R.A. Partial, but not complete, tumor-debulking surgery promotes protective antitumor memory when combined with chemotherapy and adjuvant immunotherapy. Cancer Res. 2005, 65, 7580–7584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noguchi, A.; Kaneko, T.; Naitoh, K.; Saito, M.; Iwai, K.; Maekawa, R.; Kamigaki, T.; Goto, S. Impaired and imbalanced cellular immunological status assessed in advanced cancer patients and restoration of the T cell immune status by adoptive T-cell immunotherapy. Int. Immunopharmacol. 2014, 18, 90–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, R.; Abe, H.; Kunita, A.; Yamashita, H.; Seto, Y.; Fukayama, M. Overexpression and gene amplification of PD-L1 in cancer cells and PD-L1(+) immune cells in Epstein-Barr virus-associated gastric cancer: The prognostic implications. Mod. Pathol. 2017, 30, 427–439. [Google Scholar] [CrossRef]
- Thompson, E.D.; Zahurak, M.; Murphy, A.; Cornish, T.; Cuka, N.; Abdelfatah, E.; Yang, S.; Duncan, M.; Ahuja, N.; Taube, J.M.; et al. Patterns of PD-L1 expression and CD8 T cell infiltration in gastric adenocarcinomas and associated immune stroma. Gut 2017, 66, 794–801. [Google Scholar] [CrossRef] [PubMed]
- Cristino, A.S.; Nourse, J.; West, R.A.; Sabdia, M.B.; Law, S.C.; Gunawardana, J.; Vari, F.; Mujaj, S.; Thillaiyampalam, G.; Snell, C.; et al. EBV microRNA-BHRF1-2-5p targets the 3’UTR of immune checkpoint ligands PD-L1 and PD-L2. Blood 2019, 134, 2261–2270. [Google Scholar] [CrossRef] [PubMed]
- De Re, V.; Caggiari, L.; Repetto, O.; Mussolin, L.; Mascarin, M. Classical Hodgkin’s Lymphoma in the Era of Immune Checkpoint Inhibition. J. Clin. Med. 2019, 8, 1596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mimura, K.; Teh, J.L.; Okayama, H.; Shiraishi, K.; Kua, L.-F.; Koh, V.; Smoot, D.T.; Ashktorab, H.; Oike, T.; Suzuki, Y.; et al. PD-L1 expression is mainly regulated by interferon gamma associated with JAK-STAT pathway in gastric cancer. Cancer Sci. 2018, 109, 43–53. [Google Scholar] [CrossRef]
- Kim, S.T.; Cristescu, R.; Bass, A.J.; Kim, K.-M.; Odegaard, J.I.; Kim, K.; Liu, X.Q.; Sher, X.; Jung, H.; Lee, M.; et al. Comprehensive molecular characterization of clinical responses to PD-1 inhibition in metastatic gastric cancer. Nat. Med. 2018, 24, 1449–1458. [Google Scholar] [CrossRef]
- Kawazoe, A.; Fukuoka, S.; Nakamura, Y.; Kuboki, Y.; Wakabayashi, M.; Nomura, S.; Mikamoto, Y.; Shima, H.; Fujishiro, N.; Higuchi, T.; et al. Lenvatinib plus pembrolizumab in patients with advanced gastric cancer in the first-line or second-line setting (EPOC1706): An open-label, single-arm, phase 2 trial. Lancet Oncol. 2020, 21, 1057–1065. [Google Scholar] [CrossRef]
- Bang, Y.-J.; Van Cutsem, E.; Feyereislova, A.; Chung, H.C.; Shen, L.; Sawaki, A.; Lordick, F.; Ohtsu, A.; Omuro, Y.; Satoh, T.; et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): A phase 3, open-label, randomised controlled trial. Lancet 2010, 376, 687–697. [Google Scholar] [CrossRef]
- Shitara, K.; Bang, Y.-J.; Iwasa, S.; Sugimoto, N.; Ryu, M.-H.; Sakai, D.; Chung, H.-C.; Kawakami, H.; Yabusaki, H.; Lee, J.; et al. Trastuzumab Deruxtecan in Previously Treated HER2-Positive Gastric Cancer. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Wilke, H.; Muro, K.; Van Cutsem, E.; Oh, S.-C.; Bodoky, G.; Shimada, Y.; Hironaka, S.; Sugimoto, N.; Lipatov, O.; Kim, T.-Y.; et al. Ramucirumab plus paclitaxel versus placebo plus paclitaxel in patients with previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (RAINBOW): A double-blind, randomised phase 3 trial. Lancet Oncol. 2014, 15, 1224–1235. [Google Scholar] [CrossRef]
- Sunakawa, Y.; Lenz, H.-J. Molecular classification of gastric adenocarcinoma: Translating new insights from the cancer genome atlas research network. Curr. Treat. Options Oncol. 2015, 16, 17. [Google Scholar] [CrossRef]
- Fang, W.-L.; Huang, K.-H.; Lan, Y.-T.; Lin, C.-H.; Chang, S.-C.; Chen, M.-H.; Chao, Y.; Lin, W.-C.; Lo, S.-S.; Li, A.F.-Y.; et al. Mutations in PI3K/AKT pathway genes and amplifications of PIK3CA are associated with patterns of recurrence in gastric cancers. Oncotarget 2016, 7, 6201–6220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J. Roles of the PI3K/Akt pathway in Epstein-Barr virus-induced cancers and therapeutic implications. World J. Virol. 2012, 1, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Seo, A.N.; Kang, B.W.; Bae, H.I.; Kwon, O.K.; Park, K.B.; Lee, S.S.; Chung, H.Y.; Yu, W.; Jeon, S.W.; Kang, H.; et al. Exon 9 Mutation of PIK3CA Associated With Poor Survival in Patients With Epstein-Barr Virus-associated Gastric Cancer. Anticancer Res. 2019, 39, 2145–2154. [Google Scholar] [CrossRef]
- Ito, C.; Nishizuka, S.S.; Ishida, K.; Uesugi, N.; Sugai, T.; Tamura, G.; Koeda, K.; Sasaki, A. Analysis of PIK3CA mutations and PI3K pathway proteins in advanced gastric cancer. J. Surg. Res. 2017, 212, 195–204. [Google Scholar] [CrossRef]
- Cheng, C.; Qin, Y.; Zhi, Q.; Wang, J.; Qin, C. Knockdown of long non-coding RNA HOTAIR inhibits cisplatin resistance of gastric cancer cells through inhibiting the PI3K/Akt and Wnt/β-catenin signaling pathways by up-regulating miR-34a. Int. J. Biol. Macromol. 2018, 107, 2620–2629. [Google Scholar] [CrossRef]
- Du, D.-X.; Lian, D.-B.; Amin, B.-H.; Yan, W. Long non-coding RNA CRNDE is a novel tumor promoter by modulating PI3K/AKT signal pathways in human gastric cancer. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 5392–5398. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Dang, Y.; Liu, S.; Zhang, Y.; Zhang, G. LncRNA HOTAIR promotes cisplatin resistance in gastric cancer by targeting miR-126 to activate the PI3K/AKT/MRP1 genes. Tumour Biol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Janku, F.; Hong, D.S.; Fu, S.; Piha-Paul, S.A.; Naing, A.; Falchook, G.S.; Tsimberidou, A.M.; Stepanek, V.M.; Moulder, S.L.; Lee, J.J.; et al. Assessing PIK3CA and PTEN in early-phase trials with PI3K/AKT/mTOR inhibitors. Cell Rep. 2014, 6, 377–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inada, R.; Sekine, S.; Taniguchi, H.; Tsuda, H.; Katai, H.; Fujiwara, T.; Kushima, R. ARID1A expression in gastric adenocarcinoma: Clinicopathological significance and correlation with DNA mismatch repair status. World J. Gastroenterol. 2015, 21, 2159–2168. [Google Scholar] [CrossRef] [PubMed]
- Tober, J.M.; Halske, C.; Behrens, H.-M.; Krueger, S.; Roecken, C. Intratumoral heterogeneity and loss of ARID1A expression in gastric cancer correlates with increased PD-L1 expression in Western patients. Hum. Pathol. 2019, 94, 98–109. [Google Scholar] [CrossRef] [PubMed]
- Trizzino, M.; Barbieri, E.; Petracovici, A.; Wu, S.; Welsh, S.A.; Owens, T.A.; Licciulli, S.; Zhang, R.; Gardini, A. The Tumor Suppressor ARID1A Controls Global Transcription via Pausing of RNA Polymerase II. Cell Rep. 2018, 23, 3933–3945. [Google Scholar] [CrossRef]
- Li, J.; Wang, W.; Zhang, Y.; Cieślik, M.; Guo, J.; Tan, M.; Green, M.D.; Wang, W.; Lin, H.; Li, W.; et al. Epigenetic Driver Mutations in ARID1A Shape Cancer Immune Phenotype and Immunotherapy. Available online: https://www.jci.org/articles/view/134402/pdf (accessed on 21 March 2020).
- Kim, Y.-B.; Ahn, J.M.; Bae, W.J.; Sung, C.O.; Lee, D. Functional loss of ARID1A is tightly associated with high PD-L1 expression in gastric cancer. Int. J. Cancer 2019, 145, 916–926. [Google Scholar] [CrossRef]
- Türeci, O.; Sahin, U.; Schulze-Bergkamen, H.; Zvirbule, Z.; Lordick, F.; Koeberle, D.; Thuss-Patience, P.; Ettrich, T.; Arnold, D.; Bassermann, F.; et al. A multicentre, phase IIa study of zolbetuximab as a single agent in patients with recurrent or refractory advanced adenocarcinoma of the stomach or lower oesophagus: The MONO study. Ann. Oncol. 2019, 30, 1487–1495. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, C.S.; Doi, T.; Jang, R.W.; Muro, K.; Satoh, T.; Machado, M.; Sun, W.; Jalal, S.I.; Shah, M.A.; Metges, J.-P.; et al. Safety and Efficacy of Pembrolizumab Monotherapy in Patients With Previously Treated Advanced Gastric and Gastroesophageal Junction Cancer: Phase 2 Clinical KEYNOTE-059 Trial. JAMA Oncol. 2018, 4, e180013. [Google Scholar] [CrossRef]
- UCSF Stomach Cancer Trial: A Phase 3 Efficacy, Safety and Tolerability Study of Zolbetuximab (Experimental Drug) Plus mFOLFOX6 Chemotherapy Compared to Placebo Plus mFOLFOX6 as Treatment for Gastric and Gastroesophageal Junction (GEJ) Cancer. Available online: https://clinicaltrials.ucsf.edu/trial/NCT03504397 (accessed on 21 June 2020).
- Choi, S.J.; Jung, S.W.; Huh, S.; Chung, Y.-S.; Cho, H.; Kang, H. Alteration of DNA Methylation in Gastric Cancer with Chemotherapy. J. Microbiol. Biotechnol. 2017, 27, 1367–1378. [Google Scholar] [CrossRef]
- Cheng, Y.; He, C.; Wang, M.; Ma, X.; Mo, F.; Yang, S.; Han, J.; Wei, X. Targeting epigenetic regulators for cancer therapy: Mechanisms and advances in clinical trials. Sig. Transduct. Target. Ther. 2019, 4, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stojanovic, J.; Tognetto, A.; Tiziano, D.F.; Leoncini, E.; Posteraro, B.; Pastorino, R.; Boccia, S. MicroRNAs expression profiles as diagnostic biomarkers of gastric cancer: A systematic literature review. Biomarkers 2019, 24, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, C.; Sharma, A.R.; Sharma, G.; Doss, C.G.P.; Lee, S.-S. Therapeutic miRNA and siRNA: Moving from Bench to Clinic as Next Generation Medicine. Mol. Ther. Nucl. Acids 2017, 8, 132–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quemener, A.M.; Bachelot, L.; Forestier, A.; Donnou-Fournet, E.; Gilot, D.; Galibert, M.-D. The powerful world of antisense oligonucleotides: From bench to bedside. Wiley Interdiscip. Rev. RNA 2020, e1594. [Google Scholar] [CrossRef] [PubMed]
- Lundin, K.E.; Gissberg, O.; Smith, C.I.E. Oligonucleotide Therapies: The Past and the Present. Hum. Gene Ther. 2015, 26, 475–485. [Google Scholar] [CrossRef] [Green Version]
- Paik, J.; Duggan, S. Volanesorsen: First Global Approval. Drugs 2019, 79, 1349–1354. [Google Scholar] [CrossRef]
- Keam, S.J. Inotersen: First Global Approval. Drugs 2018, 78, 1371–1376. [Google Scholar] [CrossRef]
- Wong, E.; Goldberg, T. Mipomersen (kynamro): A novel antisense oligonucleotide inhibitor for the management of homozygous familial hypercholesterolemia. Pharm. Ther. 2014, 39, 119–122. [Google Scholar]
- Le, B.T.; Raguraman, P.; Kosbar, T.R.; Fletcher, S.; Wilton, S.D.; Veedu, R.N. Antisense Oligonucleotides Targeting Angiogenic Factors as Potential Cancer Therapeutics. Mol. Ther. Nucleic. Acids 2019, 14, 142–157. [Google Scholar] [CrossRef] [Green Version]
- Kamiyama, M.; Ichikawa, Y.; Ishikawa, T.; Chishima, T.; Hasegawa, S.; Hamaguchi, Y.; Nagashima, Y.; Miyagi, Y.; Mitsuhashi, M.; Hyndman, D.; et al. VEGF receptor antisense therapy inhibits angiogenesis and peritoneal dissemination of human gastric cancer in nude mice. Cancer Gene Ther. 2002, 9, 197–201. [Google Scholar] [CrossRef] [Green Version]
- Corcoran, R.B.; Chabner, B.A. Application of Cell-free DNA Analysis to Cancer Treatment. N. Engl. J. Med. 2018, 379, 1754–1765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maron, S.B.; Chase, L.M.; Lomnicki, S.; Kochanny, S.; Moore, K.L.; Joshi, S.S.; Landron, S.; Johnson, J.; Kiedrowski, L.A.; Nagy, R.J.; et al. Circulating Tumor DNA Sequencing Analysis of Gastroesophageal Adenocarcinoma. Clin. Cancer Res. 2019, 25, 7098–7112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pectasides, E.; Stachler, M.D.; Derks, S.; Liu, Y.; Maron, S.; Islam, M.; Alpert, L.; Kwak, H.; Kindler, H.; Polite, B.; et al. Genomic Heterogeneity as a Barrier to Precision Medicine in Gastroesophageal Adenocarcinoma. Cancer Discov. 2018, 8, 37–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.-S.; Liu, Z.-X.; Lu, Y.-X.; Bao, H.; Wu, X.; Zeng, Z.-L.; Liu, Z.; Zhao, Q.; He, C.-Y.; Lu, J.-H.; et al. Liquid biopsies to track trastuzumab resistance in metastatic HER2-positive gastric cancer. Gut 2019, 68, 1152–1161. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Nam, S.K.; Seo, S.H.; Park, K.U.; Ahn, S.-H.; Park, D.J.; Kim, H.-H.; Kim, W.H.; Lee, H.S. Comparative analysis of HER2 copy number between plasma and tissue samples in gastric cancer using droplet digital PCR. Sci. Rep. 2020, 10, 4177. [Google Scholar] [CrossRef]
- Shoda, K.; Masuda, K.; Ichikawa, D.; Arita, T.; Miyakami, Y.; Watanabe, M.; Konishi, H.; Imoto, I.; Otsuji, E. HER2 amplification detected in the circulating DNA of patients with gastric cancer: A retrospective pilot study. Gastric. Cancer 2015, 18, 698–710. [Google Scholar] [CrossRef]
- Leal, A.; van Grieken, N.C.T.; Palsgrove, D.N.; Phallen, J.; Medina, J.E.; Hruban, C.; Broeckaert, M.A.M.; Anagnostou, V.; Adleff, V.; Bruhm, D.C.; et al. White blood cell and cell-free DNA analyses for detection of residual disease in gastric cancer. Nat. Commun. 2020, 11, 525. [Google Scholar] [CrossRef] [Green Version]
- Böger, C.; Krüger, S.; Behrens, H.M.; Bock, S.; Haag, J.; Kalthoff, H.; Röcken, C. Epstein-Barr virus-associated gastric cancer reveals intratumoral heterogeneity of PIK3CA mutations. Ann. Oncol. 2017, 28, 1005–1014. [Google Scholar] [CrossRef]
- Shoda, K.; Ichikawa, D.; Fujita, Y.; Masuda, K.; Hiramoto, H.; Hamada, J.; Arita, T.; Konishi, H.; Kosuga, T.; Komatsu, S.; et al. Clinical utility of circulating cell-free Epstein-Barr virus DNA in patients with gastric cancer. Oncotarget 2017, 8, 28796–28804. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| EBV Protein | Function | Cellular Receptor(s) | Reference |
|---|---|---|---|
| BMRF2 | A lytic protein of EBV; attachment to epithelial cells at the basolateral surface | β1 or α5β1 integrins | [6] |
| gp350/220 | Attachment to cells | CR2/CD21 the major receptor. | [3] |
| gp42 * | Binds B cells, the entry-receptor-binding protein in epithelial cell block gH/gL interaction with integrins | MHC class II | [7] |
| gH/gL- gB | The heterodimer is necessary for membrane fusion. Regulates fusion with epithelial and B cells | αvβ6 and αvβ8 integrins | [8] |
| gH/gL | Receptor for entry into B cells and epithelial cells | EphA2, an erythropoietin-producing hepatocellular (Eph) family member of receptor tyrosine kinases | [5] |
| Cell Type | Latency | BHRF1/miR | BART/miR | BARF1 | EBNA | LMP | EBER | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3A | 3B | 3C | LP | 1 | 2A | 2B | ||||||
| B cell | I | + | + | − | + | − | − | − | − | − | − | − | − | + |
| II | + | + | − | + | − | − | − | − | + | + | + | + | + | |
| III | + | + | − | + | + | + | + | + | + | + | + | + | + | |
| GC cell | I (II) | − | + | + | + | − | − | − | − | − | − | +/− | − | + |
| EBV Latent Genes | Mechanism of Tumorigenesis | References |
|---|---|---|
| BARF1 | Induces anti-apoptotic Bcl-2 and cyclin D genes Damages macrophage colony-stimulating factor | [68,71] |
| EBNA 1 | Induces ROS accumulation and impairs response to DNA damage Suppression actives EBV reactivation | [91,92] |
| LMP2A | Activates NF-κb, Notch, and PI3K/Akt pathways Downregulates HLA Upregulates miR-155-5p | [93,94,95] |
| EBV RNAs | ||
| EBER | Induces IGF-1, an insulin-like growth factor | [76] |
| miR-BARTs | ||
| BART1-5p | Targets glucosaminyl(N-acetyl) transferase 3 (GCNT3) pathway | [96] |
| BART9 | Decreases E-cadherin expression | [82] |
| BART5-3p | Increases the degradation of P53 and inhibits the tumor suppressor DICE1 gene | [80,82,97] |
| BART5 | Targets PUMA | [98] |
| BART9, -11, -12 | Downregulates Bim expression | [99] |
| BART4-5p | Suppresses Bid | [100] |
| BART20-5p | Interacts with the 3′ UTR region of the Bcl-2-associated agonist of cell death (BAD) gene | [101] |
| BART16 | Suppress type I interferon (IFN) signaling | [102,103] |
| BART15-3p BART6-3p | Oncosuppressor action | [17,83] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Re, V.; Brisotto, G.; Repetto, O.; De Zorzi, M.; Caggiari, L.; Zanussi, S.; Alessandrini, L.; Canzonieri, V.; Miolo, G.; Puglisi, F.; et al. Overview of Epstein–Barr-Virus-Associated Gastric Cancer Correlated with Prognostic Classification and Development of Therapeutic Options. Int. J. Mol. Sci. 2020, 21, 9400. https://doi.org/10.3390/ijms21249400
De Re V, Brisotto G, Repetto O, De Zorzi M, Caggiari L, Zanussi S, Alessandrini L, Canzonieri V, Miolo G, Puglisi F, et al. Overview of Epstein–Barr-Virus-Associated Gastric Cancer Correlated with Prognostic Classification and Development of Therapeutic Options. International Journal of Molecular Sciences. 2020; 21(24):9400. https://doi.org/10.3390/ijms21249400
Chicago/Turabian StyleDe Re, Valli, Giulia Brisotto, Ombretta Repetto, Mariangela De Zorzi, Laura Caggiari, Stefania Zanussi, Lara Alessandrini, Vincenzo Canzonieri, Gianmaria Miolo, Fabio Puglisi, and et al. 2020. "Overview of Epstein–Barr-Virus-Associated Gastric Cancer Correlated with Prognostic Classification and Development of Therapeutic Options" International Journal of Molecular Sciences 21, no. 24: 9400. https://doi.org/10.3390/ijms21249400