3.2.3. Detailed Procedures
(1) Detailed Procedures for the Preparation of Intermediates 4a–l
The title compound was prepared from 3a (500.0 mg, 2.45 mmol), p-toluenesulfonyl chloride (585.0 mg, 3.07 mmol), and NaH (147.3 mg of a 60% in mineral oil, 3.68 mmol) in dry THF (16 mL) according to general procedure A (reaction time 2 h); 753 mg of a yellow solid was yielded (82% crude yield) and used in the next step without further purification. ESI-MS: (m/z) 380.3 [M + Na]+, 356.3 [M − H]−.
The title compound was prepared from 3b (250.0 mg, 2.05 mmol), p-toluenesulfonyl chloride (268.8 mg, 1.41 mmol), and NaH (67.7 mg of a 60% dispersion in mineral oil, 1.69 mmol) in dry THF (7.5 mL) according to general procedure A (reaction time 1.5 h); 412 mg of a yellow solid was yielded (97% crude yield) and used in the next step without further purification. ESI-MS: (m/z) 398.2 [M + Na]+, 374.1 [M − H]−.
The title compound was prepared from
3c as described previously [
5].
The title compound was prepared from 3d (560.0 mg, 1.98 mmol), p-toluenesulfonyl chloride (472.4 mg, 2.48 mmol), and sodium hydride (118.9 mg of a 60% dispersion in mineral oil, 2.97 mmol) in dry THF (15 mL) according to general procedure A (reaction time 20 min); 834 mg of a yellow solid was yielded (96% crude yield) and used in the next step without further purification. ESI-MS: (m/z) 457.8 [M + Na]+, 433.9 [M − H]−.
The title compound was prepared from 3e (421.0 mg, 1.35 mmol), p-toluenesulfonyl chloride (322.5 mg, 1.69 mmol), and sodium hydride (81.2 mg of a 60% dispersion in mineral oil, 2.03 mmol) in dry THF (10 mL) according to general procedure A (reaction time 1 h); 606 mg of a dark yellow solid was yielded (93% crude yield) and used in the next step without further purification. ESI-MS: (m/z) 506.3 [M + Na]+; 482.4 [M − H]−.
The title compound was prepared from 3f (710.0 mg, 3.03 mmol) and NaH (182.3 mg of a 60% dispersion in mineral oil, 4.56 mmol) in dry THF (10 mL) according to general procedure A (reaction time 30 min). p-Toluenesulfonyl chloride (695.1 mg, 3.65 mmol) was added as solution in THF (2 mL); 1.15 g was yielded (98% yield) and used in the next step without further purification.
NaH (176.7 mg of a 60% dispersion in mineral oil, 4.42 mmol) was added portion-wise to an ice-cooled stirring suspension of 3g (800.0 mg, 2.95 mmol) in dry THF (10 mL). The mixture was stirred under ice-cooling for 30 min. A solution of p-toluenesulfonyl chloride (673.8 mg, 3.53 mmol) in dry THF (2 mL) was drop-added, while the mixture was left to warm to rt and stirring continued at rt for 30 min. The mixture was poured into saturated NH4Cl solution (100 mL). The resulting precipitate was filtered off, washed with demineralized water, and dried over P2O5 in vacuo; 1.2 g was yielded (96% crude yield) and used in the next step without further purification. ESI-MS: (m/z) 448.0 [M + H]+, 424.0 [M − H]−.
The title compound was prepared from 3h (550.0 mg, 2.31 mmol), p-toluenesulfonyl chloride (550.2 mg, 2.89 mmol). and NaH (138.6 mg of a 60% dispersion in mineral oil, 3.47 mmol) in dry THF (18 mL) according to general procedure A (reaction time 40 min); 890 mg of a yellow solid was yielded (98% crude yield) and used in the next step without further purification. ESI-MS: (m/z) 413.9 [M + Na]+, 390.0 [M − H]−.
The title compound was prepared from 3i (740.0 mg, 2.62 mmol), p-toluenesulfonyl chloride (624.2 mg, 3.27 mmol), and NaH (157.2 mg of a 60% dispersion in mineral oil, 3.93 mmol) in dry THF (33 mL) according to general procedure A (reaction time 1.5 h); 1.1 g was yielded (96% crude yield) and used in the next step without further purification. ESI-MS: (m/z) 457.7 [M + Na]+, 433.8 [M − H]−.
The title compound was prepared from 3j (480.0 mg, 2.05 mmol), p-toluenesulfonyl chloride (489.6 mg, 2.57 mmol), and sodium hydride (123.3 mg of a 60% dispersion in mineral oil, 3.08 mmol) in dry THF (16 mL) according to general procedure A in a (reaction time 30 min); 750 mg of a brown solid was yielded (94% crude yield) and used in the next step without further purification. ESI-MS: (m/z) 410.7 [M + Na]+, 386.7 [M − H]−.
3k (600.0 mg, 2.52 mmol) was suspended in dry THF (20 mL), and NaH (151.2 mg of a 60% dispersion in mineral oil, 3.78 mmol) was added. The mixture was stirred at rt and under N2 atmosphere for 15 min. p-Toluenesulfonyl chloride (600.6 mg, 3.15 mmol) was added and the mixture stirred at rt and under N2 atmosphere for 40 min. Saturated NH4Cl solution (50 mL), EtOAc (30 mL), and some MeOH were added, and phases were separated. The aqueous layer was extracted with DCM (4 × 30 mL). Combined organic layers were dried over Na2SO4. Volatiles were removed under reduced pressure to yield 1.1 g of a brown solid (>100% crude yield), which contained excessive p-toluenesulfonyl chloride. The crude product was used in the next step without further purification. ESI-MS: (m/z) 414.7 [M + Na]+, 390.7 [M − H]−.
The title compound was prepared from 3l (470.0 mg, 1.86 mmol), p-toluenesulfonyl chloride (444.3 mg, 2.33 mmol), and NaH (111.9 mg of a 60% dispersion in mineral oil, 2.80 mmol) in dry THF (16 mL) according to general procedure A (reaction time 20 min); 759 mg of a beige solid was yielded (100% crude yield) and used in the next step without further purification.
(2) Detailed Procedures for the Preparation of Enantiopure Intermediates (R)-5c,d and (S)-5c,d
The title compound was prepared from 4c (465.0 mg, 1.19 mmol), (R)-1-Boc-3-aminopiperidine (284.9 mg, 1.42 mmol), and DIPEA (444.3 mg, 3.44 mmol) in dry DMF (11 mL) according to general procedure B (reaction time 16 h). Purification by flash column chromatography (SiO2, hexane–EtOAc–MeOH 60:38:2) gave 462 mg of a pale yellow solid (69% yield). 1H-NMR (300 MHz, CDCl3) δ 8.60 (s, 1H), 8.49 (s, 1H), 8.05 (d, J = 7.9 Hz, 2H), 7.68 (s, 1H), 7.35 (d, J = 7.5 Hz, 1H), 7.25 (d, J = 8.9 Hz, 2H, overlap with CHCl3 signal), 6.44–4.91 (m, 1H), 4.55–4.31 (m, 1H), 4.23–3.66 (m, 2H), 3.54–2.97 (m, 2H), 2.46–2.01 (m, 4H), 1.97–1.79 (m, 1H), 1.75–1.30 (m, 11H); ESI-MS: (m/z) 578.0 [M + Na]+, 553.9 [M − H]−; HPLC method A: tr = 10.118 min.
The title compound was prepared from 4d (465.0 mg, 1.07 mmol), (R)-1-Boc-3-aminopiperidine (277.2 mg, 1.38 mmol), and DIPEA (412.9 mg, 3.19 mmol) in dry DMF (12 mL) according to general procedure B (reaction time 7 h). Purification by flash column chromatography (SiO2, hexane–EtOAc–MeOH 67:31.5:1.5) gave 400 mg of a light yellow solid (62% yield). ESI-MS: (m/z) 599.8 [M + H]+, 621.7 [M + Na]+, 597.7 [M − H]−; HPLC method A: tr = 10.848 min.
The title compound was prepared from 4c (460.0 mg, 1.17 mmol), (S)-1-Boc-3-aminopiperidine (305.3 mg, 1.53 mmol), and DIPEA (444.8 mg, 3.44 mmol) in dry DMF (11 mL) according to general procedure B (reaction time 9 h). Purification by flash column chromatography (SiO2, DCM–MeOH 97.5:2.5) gave 523 mg of a yellow solid (80% yield). ESI-MS: (m/z) 578.8 [M + Na]+, 554.8 [M − H]−; HPLC method A: tr = 10.577 min.
The title compound was prepared from 4d (510.0 mg, 1.17 mmol), (S)-1-Boc-3-aminopiperidine (315.8 mg, 1.58 mmol), and DIPEA (452.9 mg, 3.50 mmol) in dry DMF (14 mL) according to general procedure B (reaction time 6 h). Purification by flash column chromatography (SiO2, hexane–EtOAc–MeOH 67:31.5:1.5) gave 462 mg of a light yellow solid (66% yield). 1H-NMR (300 MHz, CDCl3) δ 8.67 (s, 1H), 8.60 (s, 1H), 8.06 (d, J = 8.4 Hz, 2H), 7.62 (br s, 1H), 7.49 (dd, J = 8.1, 1.3 Hz, 1H), 7.25 (d, 2H, overlap with CHCl3 signal), 6.14–5.03 (m, 1H), 4.45–4.32 (m, 1H), 4.20–3.72 (m, 2H), 3.47–2.98 (m, 2H), 2.42–2.02 (m, 4H), 1.92–1.78 (m, 1H), 1.75–1.32 (m, 11H); ESI-MS: (m/z) 599.8 [M + H]+, 621.8 [M + Na]+, 597.9 [M − H]−; HPLC method A: tr = 10.767 min.
(3) Detailed Procedures for the Preparation of Intermediates 6a,b and 6d–l
The title compound was prepared by a two-step procedure. In the first step, 4a (100.0 mg, 0.28 mmol), 1-Boc-3-(methylamino)piperidine (71.9 mg, 0.34 mmol), and DIPEA (108.4 mg, 0.84 mmol) were reacted in dry DMF (3 mL) according to general procedure B (reaction time 16 h) to afford 145 mg of crude tert-butyl 3-(methyl(9-tosyl-9H-pyrimido[4,5-b]indol-4-yl)amino)piperidine-1-carboxylate (97% crude yield), used in the second step without further purification. ESI-MS: (m/z) 558.1 [M + Na]+, 533.9 [M − H]−; HPLC method B: 10.875 min. The crude material obtained from the first step was reacted with NatBuO (182.1 mg, 1.89 mmol) in dry THF (5 mL) according to general procedure D (reaction time 1 h). Purification by flash column chromatography (SiO2, DCM–EtOH 95:5) gave 60 mg of the title compound (58% yield). 1H-NMR (300 MHz, DMSO-d6) δ 12.10 (s, 1H), 8.42 (s, 1H), 7.76 (br s, 1H), 7.50 (d, J = 7.7 Hz, 1H), 7.43–7.35 (m, 1H), 7.25 (t, J = 7.4 Hz, 1H), 4.30–3.72 (m, 3H), 3.20–3.04 (m, 4H), 2.79–2.60 (m, 1H), 2.09–1.71 (m, 3H), 1.51–0.99 (m, 10H); HPLC method B: tr = 8.335 min.
The title compound was prepared by a two-step procedure. In the first step, 4b (420.0 mg, 1.12 mmol), 1-Boc-3-(methylamino)piperidine (287.4 mg, 1.34 mmol), and DIPEA (433.0 mg, 3.35 mmol) were reacted in dry DMF (15 mL) according to general procedure B (reaction time 16 h) to afford 549 mg of crude tert-butyl 3-((7-fluoro-9-tosyl-9H-pyrimido[4,5-b]indol-4-yl)(methyl)amino)piperidine-1-carboxylate as a yellow solid (89% crude yield), used in the second step without further purification. Purification of a small portion for analytical purposes was performed by flash column chromatography (SiO2, petroleum ether–EtOAc gradient elution from 2:1 to 1:1). 1H-NMR (400 MHz, CDCl3) δ 8.63 (s, 1H), 8.26 (dd, J = 10.2, 2.4 Hz, 1H), 8.10 (d, J = 8.4 Hz, 2H), 7.65 (s, 1H), 7.26 (d, J = 8.2 Hz, 2H, overlap with CHCl3 signal), 7.17 (td, J = 8.7, 2.4 Hz, 1H), 4.49–3.89 (m, 3H), 3.13 (s, 3H), 3.11–3.02 (m, 1H), 2.76–2.65 (m, 1H), 1.99–1.70 (m, 3H), 1.67–1.29 (m, 10H); ESI-MS: (m/z) 554.7 [M + H]+, 576.7 [M + Na]+, 552.7 [M − H]−; HPLC method A: tr = 10.806 min.
The crude material obtained from the first step was reacted with KtBuO (780.0 mg, 6.95 mmol) in HPLC grade THF (28 mL) according to general procedure D. Purification by flash column chromatography (SiO2, DCM–MeOH gradient elution from 97.5:2.5 to 93:7) gave 191 mg of a beige solid (48% yield). 1H-NMR (400 MHz, CDCl3) δ 11.48 (s, 1H), 8.54 (s, 1H), 7.79–7.69 (m, 1H), 7.24 (dd, J = 8.9, 2.3 Hz, 1H), 7.05 (td, J = 9.1, 2.4 Hz, 1H), 4.59–3.98 (m, 3H), 3.27 (s, 3H), 3.12–3.02 (m, 1H), 2.77–2.60 (m, 1H), 2.08–1.77 (m, 3H), 1.69–1.54 (m, 1H), 1.43 (s, 9H); ESI-MS: (m/z) 422.5 [M + Na]+, 398.5 [M − H]−; HPLC method A: tr = 9.140 min.
The title compound was prepared by a two-step procedure. In the first step, 4d (752.0 mg, 1.73 mmol), 1-Boc-3-(methylamino)piperidine (479.4 mg, 2.23 mmol), and DIPEA (667.4 mg, 5.17 mmol) were reacted in dry DMF (22 mL) according to general procedure B (reaction time 6 h) to afford 1.2 g of crude tert-butyl 3-((7-bromo-9-tosyl-9H-pyrimido[4,5-b]indol-4-yl)(methyl)amino)piperidine-1-carboxylate as a yellow solid (>100% crude yield), used in the second step without further purification. A small portion was purified for analytical purposes by flash column purification (SiO2; DCM–EtOH 97.5:2.5). 1H-NMR (300 MHz, CDCl3) δ 8.75–8.55 (m, 2H), 8.17–8.03 (m, 2H), 7.61–7.46 (m, 2H), 7.31–7.21 (m, 2H, overlap with CHCl3 signal), 4.46–3.93 (m, 3H), 3.20–2.95 (m, 4H), 2.75–2.61 (m, 1H), 2.37 (s, 3H), 2.00–1.71 (m, 3H), 1.62–1.32 (m, 10H); 13C NMR (50 MHz, CDCl3) δ 160.5, 157.2, 154.8, 154.3, 145.8, 136.3, 135.4, 129.8, 128.2, 127.3, 123.7, 120.7, 120.5, 117.5, 101.2, 80.0, 55.6, 46.7, 44.0 (br), 33.7 (br), 28.5, 28.0, 24.8, 21.8; ESI-MS: (m/z) 636.1 [M + Na]+, 612.2 [M − H]−; HPLC method A: tr = 11.296 min. The crude material obtained from the first step was reacted with KtBuO (1.4 g, 12.05 mmol) in dry THF (50 mL) according to general procedure D (reaction time 1 h). Purification by flash column chromatography (SiO2, DCM:MeOH gradient elution from 97.5:2.5 to 93:7) gave 488 mg of a light brown solid (62% yield). 1H-NMR (300 MHz, CDCl3) δ 11.46 (br s, 1H), 8.56 (s, 1H), 7.71–7.62 (m, 2H), 7.40 (dd, J = 8.6, 1.8 Hz, 1H), 4.52–4.01 (m, 3H), 3.27 (s, 3H), 3.13–3.01 (m, 1H), 2.77–2.62 (m, 1H), 2.09–1.77 (m, 3H), 1.72–1.56 (m, 1H), 1.43 (s, 9H); ESI-MS: (m/z) 481.9 [M + Na]+, 458.0 [M − H]−; HPLC method A: tr = 9.546 min.
The title compound was prepared by a two-step procedure. In the first step, 4e (590.0 mg, 1.22 mmol), 1-Boc-3-(methylamino)piperidine (339.8 mg, 1.59 mmol), and DIPEA (473.0 mg, 3.66 mmol) were reacted in dry DMF (16 mL) according to general procedure B. Dissident from the general procedure, the mixture was stirred at 60 °C for 2 h; 779 mg of crude tert-butyl 3-((7-iodo-9-tosyl-9H-pyrimido[4,5-b]indol-4-yl)(methyl)amino)piperidine-1-carboxylate as a beige solid (97% crude yield) was used in the second step without further purification. Purification of a small portion for analytical purposes was performed by flash column chromatography (SiO2, petroleum ether–EtOAc gradient elution from 3:2 to 1:1). 1H-NMR (300 MHz, CDCl3) δ 8.86 (d, J = 1.4 Hz, 1H), 8.62 (s, 1H), 8.09 (d, J = 8.4 Hz, 2H), 7.74 (dd, J = 8.4, 1.4 Hz, 1H), 7.41 (d, J = 7.9 Hz, 1H), 7.27 (d, J = 8.1 Hz, 2H; overlap with CHCl3 signal), 4.47–3.90 (m, 3H), 3.11 (s, 3H), 3.08–2.98 (m, 1H), 2.77–2.60 (m, 1H), 2.37 (s, 3H), 2.01–1.71 (m, 3H), 1.64–1.29 (m, 10H); 13C NMR (50 MHz, CDCl3) δ 160.5, 157.0, 154.8, 154.4, 145.8, 136.4, 135.4, 133.2, 129.8, 128.2, 124.0, 123.1, 121.3, 101.2, 91.3, 80.0, 55.6 (br), 46.7, 44.1 (br), 33.7 (br), 28.5, 28.1, 24.8, 21.8; ESI-MS: (m/z) 684.7 [M + Na]+, 660.8 [M − H]−; HPLC method A: tr = 10.993 min.
The crude material obtained from the first step was reacted with KtBuO (807.4 mg, 7.2 mmol) in dry THF (30 mL) according to general procedure D (reaction time 1 h). Purification twice by flash column chromatography (SiO2, DCM–MeOH 95:5 and SiO2, DCM–MeOH gradient elution from 97.5:2.5 to 93:7) gave 292 mg of a beige solid (56% yield). 1H-NMR (300 MHz, CDCl3) δ 11.30 (s, 1H), 8.52 (s, 1H), 7.92 (d, J = 1.1 Hz, 1H), 7.61 (dd, J = 8.5, 1.4 Hz, 1H), 7.53 (d, J = 8.5 Hz, 1H), 4.55–4.04 (m, 3H), 3.29 (s, 3H), 3.11–2.98 (m, 1H), 2.78–2.61 (m, 1H), 2.10–1.76 (m, 3H), 1.74–1.30 (m, 10H); ESI-MS: (m/z) 530.6 [M + Na]+, 506.6 [M − H]−; HPLC method A: tr = 9.541 min.
4f (250.0 mg, 0.64 mmol), 1-Boc-3-(methylamino)piperidine (165.8, 0.77 mmol), and DIPEA (249.5 mg, 1.93 mmol) were stirred in dry DMF (10 mL) at 70 °C overnight. After cooling down to rt, NatBuO (433.6 mg, 4.51 mmol) was added and stirring continued at rt for 1 h. Saturated NH4Cl solution (150 mL) was added. The resulting precipitate was filtered off, washed with water, and dried over P2O5 in vacuo. Purification by flash column chromatography (SiO2, DCM–EtOH 97:3) gave 172 mg (64% yield). 1H-NMR (300 MHz, DMSO-d6) δ 12.00 (s, 1H), 8.37 (s, 1H), 7.64 (br s, 1H), 6.98 (d, J = 2.3 Hz, 1H), 6.85 (dd, J = 8.7, 1.6 Hz, 1H), 4.24–3.80 (m, 6H), 3.19–3.03 (m, 4H), 2.79–2.60 (m, 1H), 2.05–1.70 (m, 3H), 1.47–1.02 (m, 10H); ESI-MS: (m/z) 412.0 [M + H]+, 433.9 [M + Na]+, 410.1 [M − H]−; HPLC method B: tr = 8.342 min.
4g (600.0 mg, 1.41 mmol), 1-Boc-3-(methylamino)piperidine (362.4 mg, 1.69 mmol), and DIPEA (546.7 mg, 4.23 mmol) were stirred in dry DMF (20 mL) at 70 °C overnight. After cooling down to rt, NatBuO (947.9 mg, 9.86 mmol) was added and stirring continued at rt for 1 h. Saturated NH4Cl solution (200 mL) was added. The resulting precipitate was filtered off, washed with water, and dried over P2O5 in vacuo. Purification by flash column chromatography (SiO2, DCM–EtOH 97:3) gave 280 mg (44% yield). 1H-NMR (300 MHz, DMSO-d6) δ 12.48 (s, 1H), 8.47 (s, 1H), 7.97 (br s, 1H), 7.75 (s, 1H), 7.54 (d, J = 8.3 Hz, 1H), 4.39–3.83 (m, 3H), 3.21 (s, 3H), 3.18–3.06 (m, 1H), 2.81–2.61 (m, 1H), 2.12–1.71 (m, 3H), 1.53–1.01 (m, 10H); HPLC method B: tr = 10.213 min.
The title compound was prepared by a two-step procedure. In the first step, 4h (450.0 mg, 1.15 mmol), 1-Boc-3-(methylamino)piperidine (368.8 mg, 1.72 mmol), and DIPEA (444.7 mg, 3.44 mmol) were reacted in dry DMF (17 mL) according to general procedure B (reaction time 14 h) to afford 682 mg of crude tert-butyl 3-((6-chloro-9-tosyl-9H-pyrimido[4,5-b]indol-4-yl)(methyl)amino)piperidine-1-carboxylate as a beige solid (>100% crude yield), used in the second step without further purification. Purification of a small portion for analytical purposes was performed by flash column chromatography (SiO2, petroleum ether–EtOAc gradient elution from 65:35 to 1:1). 1H-NMR (300 MHz, CDCl3) δ 8.61 (s, 1H), 8.43 (d, J = 9.0 Hz, 1H), 8.05 (d, J = 8.4 Hz, 2H), 7.58 (d, J = 2.0 Hz, 1H), 7.46 (dd, J = 9.0, 2.1 Hz, 1H), 7.24 (d, J = 9.2 Hz, 2H; overlapping with CHCl3 signal), 4.30–3.94 (m, 3H), 3.13 (s, 3H), 3.07–2.94 (m, 1H), 2.75–2.61 (m, 1H), 2.35 (s, 3H), 2.16–2.04 (m, 1H), 1.98–1.78 (m, 2H), 1.74–1.55 (m, 1H), 1.38 (s, 9H); ESI-MS: (m/z) 592.0 [M + Na]+, 568.1 [M − H]−; HPLC method A: tr = 10.903 min.
The crude material obtained from the first step was reacted with KtBuO (774.5 mg, 6.91 mmol) in HPLC grade THF (29 mL) according to general procedure D (reaction time 2.5 h). Purification by flash column chromatography (SiO2, DCM–MeOH gradient elution from 97.5:2.5 to 93:7) gave 252 mg of a yellow solid (61% yield). 1H-NMR (300 MHz, CDCl3) δ 11.46 (br s, 1H), 8.54 (s, 1H), 7.73 (d, J = 1.6 Hz, 1H), 7.45 (d, J = 8.5 Hz, 1H), 7.38 (dd, J = 8.6, 1.8 Hz, 1H), 4.54–4.40 (m, 1H), 4.38–4.01 (m, 2H), 3.30 (s, 3H), 3.10–2.97 (m, 1H), 2.77–2.62 (m, 1H), 2.22–2.11 (m, 1H), 2.03–1.84 (m, 2H), 1.81–1.63 (m, 1H), 1.40 (s, 9H); ESI-MS: (m/z) 416.1 [M + H]+, 438.1 [M + Na]+, 414.1 [M − H]−; HPLC method A: tr = 9.385 min.
The title compound was prepared by a two-step procedure. In the first step, 4i (650 mg, 1.49 mmol), 1-Boc-3-(methylamino)piperidine (415.1 mg, 1.94 mmol), and DIPEA (576.5 mg, 4.46 mmol) were reacted in dry DMF (20 mL) according to procedure B (reaction time 5 h) to yield 871 mg of crude tert-butyl 3-((6-bromo-9-tosyl-9H-pyrimido[4,5-b]indol-4-yl)(methyl)amino)piperidine-1-carboxylate as a dark yellow solid (95% crude yield), used in the second step without further purification. Purification of a small portion for analytical purposes was performed by flash column chromatography (SiO2, petroleum ether–EtOAc gradient elution from 2:1 to 1:1). 1H-NMR (300 MHz, CDCl3) δ 8.61 (s, 1H), 8.39 (d, J = 9.0 Hz, 1H), 8.05 (d, J = 8.3 Hz, 2H), 7.73 (d, J = 1.9 Hz, 1H), 7.60 (dd, J = 9.0, 1.9 Hz, 1H), 7.25 (d, J = 8.4 Hz, 2H, overlap with CHCl3 signal), 4.30–3.96 (m, 3H), 3.13 (s, 3H), 3.06–2.93 (m, 1H), 2.75–2.60 (m, 1H), 2.36 (s, 3H), 2.16–2.07 (m, 1H), 1.98–1.80 (m, 2H), 1.75–1.58 (m, 1H), 1.37 (s, 9H); 13C NMR (50 MHz, CDCl3) δ 160.6, 157.5, 155.0, 154.6, 145.7, 135.4, 134.4, 129.8, 129.4, 128.0, 125.3, 123.7, 117.3, 115.8, 100.5, 79.9, 55.2, 46.4 (br), 44.0 (br), 34.3, 28.4, 28.1, 24.8, 21.8; ESI-MS: (m/z) 614.2 [M + H]+, 636.1 [M + Na]+, 612.2 [M − H]−; HPLC method A: tr = 11.194 min.
The crude material obtained from the first step was reacted with KtBuO (976.5 mg, 8.70 mmol) in dry THF (36 mL) according to general procedure D (reaction time 1 h). Purification by flash column chromatography (SiO2, DCM–MeOH gradient elution from 97.5:2.5 to 93:7) gave 364 mg of a beige solid (64% yield). 1H-NMR (300 MHz, CDCl3) δ 11.26 (br s, 1H), 8.53 (s, 1H), 7.87 (d, J = 0.8 Hz, 1H), 7.52 (dd, J = 8.5, 1.6 Hz, 1H), 7.40 (d, J = 8.5 Hz, 1H), 4.54–4.40 (m, 1H), 4.35–4.03 (m, 2H), 3.30 (s, 3H), 3.09–2.96 (m, 1H), 2.77–2.63 (m, 1H), 2.24–2.13 (m, 1H), 2.04–1.67 (m, 3H), 1.40 (s, 9H); 13C NMR (50 MHz, CDCl3) δ 160.2, 157.1, 155.1, 152.8, 135.5, 127.8, 125.4, 121.9, 113.7, 112.8, 98.0, 79.9, 54.8, 46.6 (br), 44.1 (br), 33.8, 28.5, 28.3, 24.9; ESI-MS: (m/z) 482.3 [M + Na]+, 458.3 [M − H]−; HPLC method A: tr = 9.550 min.
The title compound was prepared by a two-step procedure. In the first step, 4j (730.0 mg, 1.88 mmol), 1-Boc-3-(methylamino)piperidine (524.4 mg, 2.45 mmol), and DIPEA (729.8 mg, 5.65 mmol) were reacted in dry DMF (22 mL) according to general procedure B (reaction time 6.5 h) to afford 975 mg of crude tert-butyl 3-((6-methoxy-9-tosyl-9H-pyrimido[4,5-b]indol-4-yl)(methyl)amino)piperidine-1-carboxylate as a beige solid (92% crude yield), used in the next step without further purification. Purification of a small portion for analytical purposes was performed by flash column chromatography (SiO2). 1H-NMR (400 MHz, CDCl3) δ 8.63 (s, 1H), 8.40 (d, J = 9.0 Hz, 1H), 8.03 (d, J = 7.8 Hz, 2H), 7.22 (d, J = 7.9 Hz, 2H), 7.16–7.07 (m, 2H), 4.24–3.98 (m, 3H), 3.89 (s, 3H), 3.14 (s, 3H), 3.08–2.99 (m, 1H), 2.75–2.64 (m, 1H), 2.34 (s, 3H), 2.10–2.02 (m, 1H), 1.95–1.76 (m, 2H), 1.69–1.57 (m, 1H), 1.45–1.22 (m, 9H); ESI-MS: (m/z) 588.5 [M + Na]+, 564.6 [M − H]−; HPLC method A: tr = 10.013 min.
The crude material obtained from the first step was reacted with KtBuO (1180.2 mg, 10.52 mmol) in dry THF (45 mL) according to general procedure D (reaction time 30 min). Purification by flash column chromatography (SiO2, DCM–MeOH gradient elution from 97.5:2.5 to 93:7) gave 369 mg of a beige solid (60% yield). 1H-NMR (400 MHz, DMSO-d6) δ 11.95 (s, 1H), 8.40 (s, 1H), 7.41 (d, J = 8.7 Hz, 1H), 7.19 (s, 1H), 7.06 (dd, J = 8.8, 2.4 Hz, 1H), 4.25–4.15 (m, 1H), 4.06–3.57 (m, 5H), 3.15 (s, 3H), 3.11–3.01 (m, 1H), 2.80–2.61 (m, 1H), 2.20–1.89 (m, 2H), 1.87–1.77 (m, 1H), 1.55–1.43 (m, 1H), 1.39–0.91 (m, 9H); ESI-MS: (m/z) 412.4 [M + H]+, 434.5 [M + Na]+, 410.4 [M − H]−; HPLC method A: tr = 8.549 min.
The title compound was prepared by a two-step procedure. In the first step, 4k (675.0 mg, 1.72 mmol), 1-Boc-3-(methylamino)piperidine (516.3 mg, 2.41 mmol), and DIPEA (667.3 mg, 5.16 mmol) were reacted in dry DMF (25 mL) according to general procedure B. Purification of a small portion for analytical purposes was performed by flash column chromatography (SiO2; DCM–MeOH gradient elution from 96.5:3.5 to 92.5:7.5). 1H-NMR (300 MHz, CDCl3) δ 8.51 (s, 1H), 8.38 (d, J = 7.8 Hz, 1H), 8.08 (d, J = 6.6 Hz, 2H), 7.45–7.30 (m, 2H), 7.25 (d, 2H, overlap with CHCl3 signal), 4.60–3.88 (m, 3H), 3.25–2.55 (m, 5H), 2.36 (s, 3H), 2.19–1.62 (m, 4H), 1.42 (s, 9H); ESI-MS: (m/z) 592.1 [M + Na]+, 568.1 [M − H]−; HPLC method A: 10.604 min. The crude material obtained from the first step was reacted with KtBuO (1.2 g, 10.71 mmol) in HPLC grade THF (45 mL) according to general procedure D (reaction time 2 h). Purification by flash column chromatography (SiO2, DCM–MeOH gradient elution from 96.5:3.5 to 92.5:7.5) gave 379 mg of a beige solid (53% yield over two steps). 1H-NMR (400 MHz, DMSO-d6) δ 12.35 (s, 1H), 8.41 (s, 1H), 7.44 (d, J = 7.5 Hz, 1H), 7.38 (t, J = 7.8 Hz, 1H), 7.28 (d, J = 7.5 Hz, 1H), 4.35–3.65 (m, 3H), 3.09–2.87 (m, 4H), 2.84–2.63 (m, 1H), 2.18–1.66 (m, 3H), 1.50–1.09 (m, 10H); ESI-MS: (m/z) 438.1 [M + Na]+, 414.1 [M − H]−; HPLC method A: tr = 9.317 min.
The title compound was prepared by a two-step procedure. In the first step, 4l (535.0 mg, 1.32 mmol), 1-Boc-3-(methylamino)piperidine (423.3 mg, 1.98 mmol), and DIPEA (510.6 mg, 3.95 mmol) were reacted in dry DMF (20 mL) according to general procedure B (reaction time 6.5 h) to afford 750 mg of crude tert-butyl-3-((7-chloro-2-methyl-9-tosyl-9H-pyrimido[4,5-b]indol-4-yl)(methyl)amino)piperidine-1-carboxylate as a beige solid (98% crude yield), used in the next step without further purification. Purification of a small portion for analytical purposes was performed by flash column chromatography (SiO2, petroleum ether–EtOAc gradient elution from 7:3 to 3:7). 1H-NMR (300 MHz, CDCl3) δ 8.52 (d, J = 1.8 Hz, 1H), 8.12 (d, J = 8.3 Hz, 2H), 7.55 (d, J = 8.2 Hz, 1H), 7.35 (dd, J = 8.5, 1.9 Hz, 1H), 7.26 (d, J = 8.1 Hz, 2H, overlap with CHCl3 signal), 4.45–3.91 (m, 3H), 3.15–2.96 (m, 4H), 2.75–2.59 (m, 4H), 2.37 (s, 3H), 1.98–1.70 (m, 3H), 1.64–1.31 (m, 10H); ESI-MS: (m/z) 606.5 [M + Na]+, 582.5 [M − H]−; HPLC method A: tr = 12.593 min.
The crude material obtained from the first step was reacted with KtBuO (874.0 mg, 7.79 mmol) in dry THF (32 mL) according to general procedure D (reaction time 2.5 h). Purification by flash column chromatography (SiO2, DCM–MeOH gradient elution from 97.5:2.5 to 93:7) gave 358 mg of a light yellow solid (75% yield). 1H-NMR (400 MHz, DMSO-d6) δ 12.03 (s, 1H), 7.78–7.60 (m, 1H), 7.45 (d, J = 1.8 Hz, 1H), 7.26–7.13 (m, 1H), 4.30–3.78 (m, 3H), 3.12 (s, 3H), 3.09–3.01 (m, 1H), 2.80–2.57 (m, 1H), 2.50 (s, 3H, overlap with DMSO-d5 signal), 2.08–1.68 (m, 3H), 1.54–0.99 (m, 10H); 13C NMR (101 MHz, DMSO-d6) δ 162.5, 159.6, 158.4, 153.9, 137.4, 128.8, 123.3, 120.2, 118.6, 110.8, 95.2, 78.7, 54.5, 46.0 (br), 43.3 (br), 32.5, 27.8, 27.4, 25.8, 24.6; ESI-MS: (m/z) 430.5 [M + H]+, 452.5 [M + Na]+; 428.6 [M − H]−; HPLC method A: tr = 10.332 min.
(4) Detailed Procedures for the Preparation of Enantiopure Intermediates (R)-6c,d and (S)-6c,d
The title compound was prepared by a two-step procedure. In the first step, (R)-5c (350.0 mg, 0.63 mmol), methyl iodide (134.0 mg, 0.94 mmol), and NaH (37.7 mg of a 60% dispersion in mineral oil, 0.94 mmol) were reacted in dry DMF (15 mL) according to general procedure C (reaction time of 3 h) to afford 346 mg of crude tert-butyl (R)-3-((7-chloro-9-tosyl-9H-pyrimido[4,5-b]indol-4-yl)(methyl)amino)piperidine-1-carboxylate as an off-white solid (96% crude yield), used in the next step without further purification. Purification for analytical purposes was performed by flash column chromatography (SiO2, DCM–MeOH gradient elution from 98:2 to 97.5:2.5). 1H-NMR (200 MHz, CDCl3) δ 8.61 (s, 1H), 8.52 (d, J = 1.6 Hz, 1H), 8.09 (d, J = 8.2 Hz, 2H), 7.60 (d, J = 8.6 Hz, 1H), 7.38 (dd, J = 8.5, 1.6 Hz, 1H), 7.26 (d, J = 8.1 Hz, 2H, overlap with CHCl3 signal), 4.56–3.88 (m, 3H), 3.18–2.94 (m, 4H), 2.79–2.57 (m, 1H), 2.35 (s, 3H), 2.07–1.68 (m, 3H), 1.66–1.51 (m, 1H), 1.40 (s, 9H).; 13C NMR (50 MHz, CDCl3) δ 160.2, 157.3, 154.8, 154.0, 145.8, 136.2, 135.4, 132.8, 129.8, 128.2, 124.6, 123.4, 120.2, 114.7, 101.2, 80.0, 55.7, 46.7, 44.0 (br), 33.8, 28.5, 28.1, 24.8, 21.8; ESI-MS: (m/z) 591.8 [M + Na]+, 567.7 [M − H]−; HPLC method A: tr = 10.548 min.
The crude material obtained from the first step was reacted with KtBuO (476.7 mg, 4.25 mmol) in dry THF (19 mL) according to general procedure D (reaction time 1 h). Purification by flash column chromatography (DCM–MeOH gradient elution from 96.5:3.5 to 93:7) gave 183 mg of an off-white solid (73% yield). 1H-NMR (200 MHz, CDCl3) δ 11.91 (br s, 1H), 8.53 (s, 1H), 7.65 (d, J = 8.6 Hz, 1H), 7.46 (s, 1H), 7.21 (d, J = 8.3 Hz, 1H, overlap with CHCl3 signal), 4.75–3.89 (m, 3H), 3.24 (s, 3H), 3.15–2.95 (m, 1H), 2.82–2.53 (m, 1H), 2.21–1.59 (m, 4H), 1.51 (s, 9H); 13C NMR (50 MHz, CDCl3) δ 160.1, 156.8, 154.9, 152.1, 137.5, 131.1, 123.6, 121.5, 118.7, 111.7, 98.6, 80.0, 55.1, 46.8 (br), 44.0 (br), 33.4, 28.5, 28.2, 24.9; ESI-MS: (m/z) 416.0 [M + H]+, 437.9 [M + Na]+, 413.8 [M − H]−; HPLC method A: tr = 9.052 min.
The title compound was prepared by a two-step procedure. In the first step, (R)-5d (400.0 mg, 0.67 mmol), methyl iodide (141.8 mg, 1.0 mmol), and NaH (40.0 mg of a 60% dispersion in mineral oil, 1.0 mmol) were reacted in dry DMF (14 mL) according to general procedure C (reaction time 2.5 h) to afford 382 mg of crude tert-butyl (R)-3-((7-bromo-9-tosyl-9H-pyrimido[4,5-b]indol-4-yl)(methyl)amino)piperidine-1-carboxylate as an off-white solid (93% crude yield), used in the next without further purification. Purification of a small portion for analytical purposes was performed by flash column chromatography (SiO2, hexane–EtOAc 3:2). 1H-NMR (300 MHz, CDCl3) δ 8.68 (s, 1H), 8.62 (s, 1H), 8.10 (d, J = 8.4 Hz, 2H), 7.60–7.48 (m, 2H), 7.27 (d, J = 8.0 Hz, 2H, overlap with CHCl3 signal), 4.45–3.97 (m, 3H), 3.11 (s, 3H), 3.09–2.98 (m, 1H), 2.75–2.61 (m, 1H), 2.37 (s, 3H), 1.95–1.72 (m, 3H), 1.59–1.31 (m, 10H); 13C NMR (75 MHz, CDCl3) δ 160.41, 157.10, 154.69, 154.23, 145.69, 136.14, 135.26, 129.70, 128.02, 127.23, 123.66, 120.65, 120.32, 117.28, 101.06, 79.80, 55.48, 46.56, 43.99 (br), 33.71 (br), 28.36, 27.93, 24.75, 21.68. ESI-MS: (m/z) 635.9 [M + Na]+, 612.1 [M − H]−; HPLC method A: tr = 11.342 min.
The crude material obtained from the first step was reacted with KtBuO (488.0 mg, 4.35 mmol) in dry THF (20 mL) according to general procedure D (reaction time 45 min). Purification by flash column chromatography (SiO2, DCM–MeOH 96:4) gave 175 mg of a white solid (61% yield). 1H-NMR (300 MHz, CDCl3) δ 11.59 (br s, 1H), 8.58 (s, 1H), 7.72–7.62 (m, 2H), 7.40 (dd, J = 8.6, 1.8 Hz, 1H), 4.48–4.02 (m, 3H), 3.26 (s, 3H), 3.14–3.00 (m, 1H), 2.77–2.61 (m, 1H), 2.11–1.75 (m, 3H), 1.70–1.54 (m, 1H), 1.43 (s, 9H); ESI-MS: (m/z) 482.0 [M + Na]+, 458.2 [M − H]−; HPLC method A: tr = 9.693 min.
The title compound was prepared by a two-step procedure. In the first step, (S)-5c (465.0 mg, 0.84 mmol), methyl iodide (178.0 mg, 1.25 mmol), and NaH (50.2 mg of a 60% dispersion in mineral oil, 1.25 mmol) were reacted in dry DMF (15 mL) according to general procedure C (reaction time 3.5 h) to afford 440 mg of crude tert-Butyl (S)-3-((7-chloro-9-tosyl-9H-pyrimido[4,5-b]indol-4-yl)(methyl)amino)piperidine-1-carboxylate as a yellow solid (92% crude yield), used in the next step without further purification. ESI-MS: (m/z) 591.8 [M + Na]+, 567.7 [M − H]−; HPLC method A: tr = 11.053 min.
The crude material obtained from the first step was reacted with KtBuO (606.2 mg, 5.40 mmol) in dry THF (24 mL) according to general procedure D (reaction time 1 h). Purification by flash column chromatography (SiO2, DCM–MeOH gradient elution from 96.5:3.5 to 92.5:7.5) gave 203 mg of a white solid (63% yield). 1H-NMR (300 MHz, CDCl3) δ 11.71 (br s, 1H), 8.57 (s, 1H), 7.71 (d, J = 8.6 Hz, 1H), 7.53 (d, J = 1.7 Hz, 1H), 7.26 (dd, J = 8.6, 1.9 Hz, 1H, overlap with CHCl3 signal), 4.59–3.97 (m, 3H), 3.27 (s, 3H), 3.16–2.99 (m, 1H), 2.83–2.58 (m, 1H), 2.11–1.54 (m, 4H), 1.43 (s, 9H); ESI-MS: (m/z) 437.9 [M + Na]+, 413.8 [M − H]−; HPLC method A: tr = 9.451 min.
The title compound was prepared by a two-step procedure. In the first step, (S)-5d (585.0 mg, 0.97 mmol), NaH (58.4 mg of a 60% dispersion in mineral oil, 1.46 mmol), and methyl iodide (207.4 mg, 1.46 mmol) were reacted in dry DMF (10 mL) according to general procedure C (reaction time 3 h) to afford 578 mg of crude tert-butyl (S)-3-((7-bromo-9-tosyl-9H-pyrimido[4,5-b]indol-4-yl)(methyl)amino)piperidine-1-carboxylate as a light yellow solid (96% crude yield), used in the next step without further purification.
The crude material obtained from the first step was reacted with KtBuO (639.1 mg, 5.70 mmol) in dry THF (25 mL) according to general procedure D (reaction time 2 h). Purification by flash column chromatography (SiO2, DCM–MeOH 96:4) gave 214 mg of a white solid (57% yield). 1H-NMR (400 MHz, CDCl3) δ 12.09 (s, 1H), 8.56 (s, 1H), 7.66 (d, J = 1.6 Hz, 1H), 7.64 (d, J = 8.6 Hz, 1H), 7.38 (dd, J = 8.6, 1.8 Hz, 1H), 4.61–3.96 (m, 1H), 3.26 (s, 1H), 3.11–3.01 (m, 1H), 2.78–2.61 (m, 1H), 2.09–1.75 (m, 1H), 1.72–1.55 (m, 1H), 1.44 (s, 1H).
(5) Detailed Procedures for the Preparation of Intermediates 7a–l
The title compound was prepared from 6a (60.0 mg, 0.16 mmol) in dry DCM (1 mL) and TFA (0.2 mL) according to general procedure E (reaction time 1.5 h); 36 mg of a light brown solid was yielded (81% crude yield) and used in the next step without further purification. 1H-NMR (400 MHz, MeOD) δ 8.36 (s, 1H), 7.84 (d, J = 8.0 Hz, 1H), 7.52 (d, J = 8.0 Hz, 1H), 7.41 (td, J = 7.7, 1.0 Hz, 1H), 7.34–7.27 (m, 1H), 3.30–3.23 (m, 4H), 3.14–3.07 (m, 1H), 3.07–3.00 (m, 1H), 2.72–2.63 (m, 1H), 2.20–1.94 (m, 3H), 1.83–1.70 (m, 1H); ESI-MS: (m/z) 282.3 [M + H]+, 280.3 [M − H]−; HPLC method A: tr = 2.232 min.
The title compound was prepared from 6b (170.0 mg, 0.43 mmol) in dry DCM (5 mL) and TFA (1 mL) according to general procedure E (reaction time 1.5 h); 119 mg of a beige solid was yielded (93% crude yield) and used in the next step without further purification. 1H-NMR (200 MHz, MeOD) δ 8.33 (s, 1H), 7.78 (dd, J = 8.8, 5.2 Hz, 1H), 7.22 (dd, J = 9.3, 2.4 Hz, 1H), 7.06 (td, J = 9.3, 2.5 Hz, 1H), 4.54–4.37 (m, 1H), 3.24 (s, 3H), 3.17–2.83 (m, 3H), 2.62–2.45 (m, 1H), 2.16–1.55 (m, 4H); ESI-MS: (m/z) 300.3 [M + H]+, 298.3 [M − H]−; HPLC method A: tr = 3.036 min.
The title compound was prepared as described previously [
5].
6d (425.0 mg, 0.92 mmol) was stirred in dry DCM (9 mL) and TFA (1.5 mL) at rt for 1 h. The mixture was concentrated under reduced pressure. Residual TFA was neutralized by addition of saturated NaHCO3 solution, resulting in a precipitate which was filtered off and washed with saturated NaHCO3 solution and demineralized water and was then dried over P2O5 in vacuo; 285 mg of an off-white solid was yielded (86% crude yield) and used in the next step without further purification. 1H-NMR (300 MHz, MeOD) δ 8.35 (s, 1H), 7.70 (d, J = 8.6 Hz, 1H), 7.65 (d, J = 1.7 Hz, 1H), 7.40 (dd, J = 8.6, 1.9 Hz, 1H), 4.55–4.37 (m, 1H), 3.24 (s, 3H), 3.15–3.06 (m, 1H), 3.02–2.95 (m, 1H), 2.95–2.85 (m, 1H), 2.61–2.48 (m, 1H), 2.15–1.84 (m, 3H), 1.78–1.60 (m, 1H); 13C NMR (75 MHz, DMSO-d6) δ 159.6, 157.3, 153.8, 137.7, 124.1, 123.1, 118.9, 117.2, 113.7, 97.1, 55.9, 48.7, 45.5, 32.7, 28.3, 26.5; ESI-MS: (m/z) 360.1 [M + H]+, 358.1 [M − H]−; HPLC method A: tr = 4.033 min.
6e (240.0 mg, 0.473 mmol) was suspended in dry DCM (7 mL), and TFA was added (1.5 mL). The mixture was stirred at rt for 30 min and then concentrated under reduced pressure. Residual TFA was neutralized by addition of saturated NaHCO3 solution (30 mL), which resulted in a precipitate. The precipitate was filtered off, washed with saturated NaHCO3 solution and demineralized water, and dried over P2O5 in vacuo; 199 mg of a beige solid was yielded (>100% crude yield) and used in the next step without further purification. 1H-NMR (300 MHz, MeOD) δ 8.35 (s, 1H), 7.86 (s, 1H), 7.65–7.52 (m, 2H), 4.55–4.40 (m, 1H), 3.24 (s, 3H), 3.14–3.05 (m, 1H), 3.02–2.84 (m, 2H), 2.60–2.47 (m, 1H), 2.15–1.84 (m, 3H), 1.77–1.59 (m, 1H); ESI-MS: (m/z) 408.3 [M + H]+, 406.3 [M − H]−; HPLC method A: tr = 4.563 min.
TFA (0.4 mL) was added to a solution of 6f (126.0 mg, 0.31 mmol) in dry DCM (10 mL). The mixture was stirred at rt for 1.5 h and then concentrated under reduced pressure. Saturated NaHCO3 solution was added to the residue, and the mixture was extracted with EtOAc (3 × 20 mL). Combined organic layers were dried over Na2SO4 and evaporated to dryness to afford 90 mg (95% crude yield). 1H-NMR (300 MHz, DMSO-d6) δ 11.95 (s, 1H), 8.33 (s, 1H), 7.68 (d, J = 8.8 Hz, 1H), 6.97 (d, J = 2.4 Hz, 1H), 6.88 (dd, J = 8.8, 2.4 Hz, 1H), 4.40–4.26 (m, 1H), 3.84 (s, 3H), 3.13 (s, 3H), 3.06–2.80 (m, 3H), 2.51–2.41 (m, 1H, overlap with DMSO-d5 signal), 2.00–1.71 (m, 3H), 1.60–1.39 (m, 1H); ESI-MS: (m/z) 312.0 [M + H]+, 310.1 [M − H]−; HPLC method B: tr = 2.684 min.
TFA (1 mL) was added to a solution of 6g (237.0 mg, 0.53 mmol) in dry DCM (10 mL). The mixture was stirred at rt for 2 h and then concentrated under reduced pressure. Saturated NaHCO3 solution was added to the residue, and the mixture was then extracted with EtOAc (3 × 20 mL). Combined organic layers were dried over Na2SO4 and evaporated to dryness; 194 mg was yielded (100% crude yield) and used in the next step without further purification. 1H-NMR (300 MHz, DMSO-d6) δ 12.30 (br s, 1H), 8.44 (s, 1H), 8.01 (d, J = 8.5 Hz, 1H), 7.74 (s, 1H), 7.56 (dd, J = 8.5, 1.2 Hz, 1H), 4.54–4.40 (m, 1H), 3.22 (s, 3H), 3.12–3.05 (m, 1H), 3.01–2.86 (m, 2H), 2.58–2.49 (m, 1H, overlap with DMSO-d5 signal), 2.01–1.75 (m, 3H), 1.66–1.49 (m, 1H); HPLC method B: tr = 4.956 min.
The title compound was prepared from 6h (215.0 mg, 0.52 mmol) in dry DCM (5.6 mL) and TFA (1.1 mL) according to general procedure E (reaction time 1 h); 163 mg of a yellow solid (100% crude yield) was yielded, used in the next step without further purification. 1H-NMR (300 MHz, MeOD) δ 8.35 (s, 1H), 7.76 (d, J = 1.8 Hz, 1H), 7.48 (d, J = 8.6 Hz, 1H), 7.38 (dd, J = 8.6, 2.0 Hz, 1H), 4.54–4.39 (m, 1H), 3.25 (s, 3H), 3.11–2.95 (m, 2H), 2.95–2.85 (m, 1H), 2.63–2.49 (m, 1H), 2.23–1.88 (m, 3H), 1.83–1.65 (m, 1H); 13C NMR (101 MHz, DMSO-d6) δ 159.6, 157.5, 153.9, 135.1, 124.6, 124.3, 121.6, 120.9, 112.5, 96.8, 55.8, 48.8, 45.5, 32.5, 28.4, 26.3; ESI-MS: (m/z) 316.1 [M + H]+, 314.0 [M − H]−; HPLC method A: tr = 4.102 min.
The title compound was prepared from 6i in dry DCM (6.3 mL) and TFA (1 mL) according to general procedure E (reaction time 1 h). Purification by flash column chromatography (SiO2, DCM– (2N NH3 in MeOH) 9:1) gave 218 mg of a beige solid (93% yield). 1H-NMR (300 MHz, MeOD) δ 8.35 (s, 1H), 7.91 (d, J = 1.6 Hz, 1H), 7.52 (dd, J = 8.6, 1.8 Hz, 1H), 7.43 (d, J = 8.6 Hz, 1H), 4.51–4.40 (m, 1H), 3.24 (s, 3H), 3.08–2.95 (m, 2H), 2.94–2.85 (m, 1H), 2.61–2.48 (m, 1H), 2.22–1.89 (m, 3H), 1.84–1.66 (m, 1H); 13C NMR (75 MHz, DMSO-d6) δ 159.6, 157.4, 154.0, 135.4, 127.1, 124.6, 121.6, 113.1, 112.5, 96.7, 56.0, 48.9, 45.5, 32.5, 28.6, 26.5; ESI-MS: (m/z) 360.1 [M + H]+, 358.1 [M − H]−; HPLC method A: tr = 4.058 min.
The title compound was prepared from 6j (325.0 mg, 0.78 mmol) in dry DCM (10 mL) and TFA (2 mL) according to general procedure E (reaction time 2 h); 207 mg of a beige solid (84% yield) was yielded. 1H-NMR (200 MHz, MeOD) δ 8.32 (s, 1H), 7.43 (d, J = 8.8 Hz, 1H), 7.29 (d, J = 2.4 Hz, 1H), 7.07 (dd, J = 8.9, 2.3 Hz, 1H), 4.48–4.29 (m, 1H), 3.89 (s, 3H), 3.22 (s, 3H), 3.13–2.81 (m, 3H), 2.60–2.44 (m, 1H), 2.23–1.84 (m, 3H), 1.82–1.58 (m, 1H); ESI-MS: (m/z) 312.2 [M + H]+, 310.2 [M − H]−; HPLC method A: tr = 2.084 min.
The title compound was prepared from 6k (340.0 mg, 0.82 mmol) in dry DCM (9 mL) and TFA (1.8 mL) according to general procedure E (reaction time 2 h); 244 mg of a beige solid (95% crude yield) was yielded, used in the next step without further purification. 1H-NMR (300 MHz, MeOD) δ 8.31 (s, 1H), 7.43 (dd, J = 7.9, 1.2 Hz, 1H), 7.35 (t, J = 7.8 Hz, 1H), 7.26 (dd, J = 7.7, 1.2 Hz, 1H), 4.52–4.33 (m, 1H), 3.19–2.90 (m, 5H), 2.89–2.68 (m, 1H), 2.57–2.41 (m, 1H), 2.20–1.97 (m, 1H), 1.96–1.74 (m, 2H), 1.73–1.56 (m, 1H); ESI-MS: (m/z) 316.1 [M + H]+, 314.1 [M − H]−; HPLC method A: tr = 3.814 min.
The title compound was prepared from 6l (310.0 mg, 0.72 mmol) in TFA (2 mL) and dry DCM (10 mL) according to general procedure E (reaction time 2.5 h); 239 mg of a yellow solid (100% crude yield) was yielded, used in the next step without further purification. 1H-NMR (300 MHz, MeOD) δ 7.70 (d, J = 8.6 Hz, 1H), 7.46 (d, J = 1.9 Hz, 1H), 7.22 (dd, J = 8.6, 2.0 Hz, 1H), 4.57–4.44 (m, 1H), 3.23 (s, 3H), 3.15–3.06 (m, 1H), 3.02–2.93 (m, 1H), 2.93–2.84 (m, 1H), 2.59–2.47 (m, 4H), 2.13–1.84 (m, 3H), 1.77–1.59 (m, 1H); 13C NMR (101 MHz, DMSO-d6) δ 162.5, 159.6, 158.4, 137.3, 128.6, 123.3, 120.2, 118.8, 110.7, 94.9, 55.8, 48.8, 45.5, 32.4, 28.3, 26.4, 25.9; ESI-MS: (m/z) 330.1 [M + H]+, 328.1 [M − H]−; HPLC method A: tr = 4.602 min.
(6) Detailed Procedures for the Preparation of Enantiopure Intermediates (R)-7c,d and (S)-7c,d
The title compound was prepared from (R)-6c (160.0 mg, 0.39 mmol) in dry DCM (5 mL) and TFA (1 mL) according to general procedure E (reaction time 45 min); 120 mg of a yellow solid (99% crude yield) was yielded, used in the next step without further purification. 1H-NMR (400 MHz, MeOD) δ 8.35 (s, 1H), 7.78 (d, J = 8.6 Hz, 1H), 7.50 (d, J = 1.9 Hz, 1H), 7.28 (dd, J = 8.6, 2.0 Hz, 1H), 4.56–4.47 (m, 1H), 3.26 (s, 3H), 3.20–3.13 (m, 1H), 3.06–3.00 (m, 1H), 2.99–2.92 (m, 1H), 2.64–2.55 (m, 1H), 2.16–1.90 (m, 3H), 1.78–1.65 (m, 1H); ESI-MS: (m/z) 316.0 [M + H]+, 338.0 [M + Na]+, 313.9 [M − H]−; HPLC method A: tr = 3.411 min.
The title compound was prepared from (R)-6d (175.0 mg, 0.38 mmol) in dry DCM (5 mL) and TFA (1 mL) according to general procedure E (reaction time 2.5 h); 135 mg of a beige solid (99% crude yield) was yielded, used in the next step without further purification. 1H-NMR (300 MHz, MeOD) δ 8.35 (s, 1H), 7.70 (d, J = 8.6 Hz, 1H), 7.65 (d, J = 1.8 Hz, 1H), 7.40 (dd, J = 8.6, 1.9 Hz, 1H), 4.54–4.41 (m, 1H), 3.24 (s, 3H), 3.15–3.06 (m, 1H), 3.02–2.95 (m, 1H), 2.94–2.84 (m, 1H), 2.61–2.48 (m, 1H), 2.15–1.85 (m, 3H), 1.77–1.60 (m, 1H); ESI-MS: (m/z) 360.0 [M + H]+, 358.1 [M − H]−; HPLC method A: tr = 3.985 min.
The title compound was prepared from (S)-6c (180.0 mg, 0.43 mmol) in dry DCM (5 mL) and TFA (1 mL) according to general procedure E (reaction time 1.5 h); 142 mg of a beige solid (>100% crude yield) was yielded, used in the next step without further purification. 1H-NMR (300 MHz, MeOD) δ 8.34 (s, 1H), 7.76 (d, J = 8.6 Hz, 1H), 7.49 (d, J = 1.9 Hz, 1H), 7.27 (dd, J = 8.6, 2.0 Hz, 1H), 4.58–4.41 (m, 1H), 3.25 (s, 3H), 3.19–3.09 (m, 1H), 3.05–2.97 (m, 1H), 2.97–2.86 (m, 1H), 2.64–2.50 (m, 1H), 2.19–1.83 (m, 3H), 1.80–1.59 (m, 1H); ESI-MS: (m/z) 316.0 [M + H]+, 338.0 [M + Na]+, 313.9 [M − H]−; HPLC method A: tr = 3.897 min.
The title compound was prepared from (S)-6d (190.0 mg, 0.41 mmol) in dry DCM (5 mL) and TFA (1 mL) according to general procedure E (reaction time 1 h); 141 mg of a beige solid (95% crude yield) was yielded, used in the next step without further purification. 1H-NMR (400 MHz, MeOD) δ 8.35 (s, 1H), 7.69 (d, J = 8.6 Hz, 1H), 7.65 (d, J = 1.8 Hz, 1H), 7.40 (dd, J = 8.6, 1.9 Hz, 1H), 4.53–4.43 (m, 1H), 3.24 (s, 3H), 3.14–3.08 (m, 1H), 3.02–2.95 (m, 1H), 2.95–2.87 (m, 1H), 2.59–2.50 (m, 1H), 2.12–1.86 (m, 3H), 1.75–1.62 (m, 1H); ESI-MS: (m/z) 360.2 [M + H]+, 358.2 [M − H]−; HPLC method A: tr = 4.025 min.
(7) Detailed Procedures for the Preparation of Final Compounds 8–41
The title compound was prepared from 7c (64.0 mg, 0.20 mmol), 3-furoic acid (27.3 mg, 0.24 mmol), PyBOP (130.8 mg, 0.24 mmol), and DIPEA (78.6 mg, 0.61 mmol) in dry DCM (total amount 10 mL) according to general procedure F (reaction time 1.5 h). Purification by flash column chromatography (SiO2, DCM–MeOH gradient elution from 96.5:3.5 to 93.5:6.5) gave 50 mg of a white solid (60% yield). 1H-NMR (400 MHz, DMSO-d6) δ 12.25 (s, 1H), 8.38 (s, 1H), 8.26–7.97 (m, 1H), 7.92–7.77 (m, 1H), 7.73 (t, J = 1.7 Hz, 1H), 7.48 (d, J = 2.0 Hz, 1H), 7.24 (dd, J = 8.6, 2.1 Hz, 1H), 6.85–6.58 (m, 1H), 4.81–3.79 (m, 3H), 3.30–2.56 (m, 5H), 2.15–1.94 (m, 2H), 1.94–1.74 (m, 1H), 1.63–1.47 (m, 1H); ESI-MS: (m/z) 410.0 [M + H]+, 432.0 [M + Na]+, 407.9 [M − H]−; HPLC method A: tr = 7.624 min.
The title compound was prepared from 7c (70.0 mg, 0.22 mmol), ethyl potassium malonate (45.3 mg, 0.27 mmol), PyBOP (138.4 mg, 0.27 mmol), and DIPEA (86.0 mg, 0.67 mmol) in dry DCM (total amount 10 mL) according to general procedure F. Additional ethyl potassium malonate (5.7 mg, 0.03 mmol) and PyBOP (17.3 mg, 0.03 mmol) were added after a reaction time of 2.5 h, and then stirring continued for 1 h. Purification twice by flash column chromatography (SiO2, DCM–MeOH gradient elution from 97.5:2.5 to 93.5:6.5 and SiO2, DCM–EtOH gradient elution from 95:5 to 93:7) gave 50 mg of a white solid (53% yield). 1H-NMR shows a 3:2 mixture of amide bond rotamers. 1H-NMR (400 MHz, DMSO-d6) δ 12.32–12.14 (m, 1H), 8.46–8.34 (m, 1H), 7.86–7.77 (m, 1H), 7.52–7.44 (m, 1H), 7.32–7.16 (m, 1H), 4.61–4.50 (m, 0.6H), 4.42–4.34 (m, 0.4H), 4.33–4.16 (m, 1H), 4.10–3.95 (m, 2H), 3.95–3.90 (m, 0.4H), 3.79–3.68 (m, 0.6H), 3.64–3.47 (m, 2H), 3.33–3.28 (m, 0.4H, overlap with water signal), 3.28–3.15 (m, 3H), 3.08–2.95 (m, 1.2H), 2.63–2.54 (m, 0.4H), 2.10–1.74 (m, 3H), 1.58–1.40 (m, 1H), 1.17–1.08 (m, 3H); 13C NMR (101 MHz, DMSO-d6) δ 167.7, 167.6, 164.5, 164.4, 159.43, 159.40, 157.51, 157.48, 153.7, 153.6, 137.44, 137.40, 129.3, 124.03, 123.97, 120.5, 120.4, 118.4, 118.3, 110.83, 110.81, 97.4, 97.2, 60.5, 60.4, 54.7, 54.5, 47.6, 45.7, 44.0, 41.5, 40.9, 40.8, 33.9, 32.6, 27.2, 27.1, 24.8, 24.3, 13.89, 13.86; ESI-MS: (m/z) 430.0 [M + H]+, 451.9 [M + Na]+, 427.8 [M − H]−; HPLC method A: tr = 7.274 min.
The title compound was prepared from 7c (60.0 mg, 0.19 mmol), 4-(dimethylamino) benzoic acid (37.7 mg, 0.22 mmol), PyBOP (118.6 mg, 0.22 mmol), and DIPEA (73.7 mg, 0.57 mmol) in dry DCM (total amount 7 mL) according to general procedure F (reaction time 40 min). Purification by flash column chromatography (SiO2, DCM–MeOH gradient elution from 95.5:4.5 to 93.5:6.5) gave 70 mg of an off-white solid (80% yield). 1H-NMR (400 MHz, DMSO-d6) δ 12.25 (s, 1H), 8.39 (s, 1H), 7.80 (d, J = 8.0 Hz, 1H), 7.50 (d, J = 1.9 Hz, 1H), 7.25 (dd, J = 8.6, 2.0 Hz, 1H), 7.17 (d, J = 7.8 Hz, 2H), 6.60 (d, J = 7.4 Hz, 2H), 4.50–3.85 (m, 3H), 3.28–3.13 (m, 4H), 3.04–2.78 (m, 7H), 2.12–1.99 (m, 2H), 1.93–1.76 (m, 1H), 1.65–1.46 (m, 1H); ESI-MS: (m/z) 463.9 [M + H]+, 485.9 [M + Na]+, 461.8 [M − H]−; HPLC method A: tr = 8.928 min.
The title compound was prepared from 7c (60.0 mg, 0.19 mmol), butyric acid (20.9 mg, 0.24 mmol), TBTU (76.3 mg, 0.24 mmol), and DIPEA (73.7 mg, 0.57 mmol) in dry DCM (total amount 10 mL) according to general procedure F (reaction time 1 h). Purification by flash column chromatography (SiO2, DCM–MeOH 94.5:5.5) gave 54 mg of a white solid (74% yield). 1H-NMR shows a 5:4 mixture of amide bond rotamers. 1H-NMR (400 MHz, CDCl3) δ 12.42 (br s, 1H), 8.58–8.42 (m, 1H), 7.74–7.59 (m, 1H), 7.51–7.38 (m, 1H), 7.25–7.16 (m, 1H), 5.05–4.86 (m, 0.45H), 4.81–4.65 (m, 0.55H), 4.53–4.29 (m, 1H), 4.28–4.16 (m, 0.55H), 3.97–3.81 (m, 0.45H), 3.37–3.19 (m, 3H), 3.19–3.10 (m, 0.55H), 3.05–2.89 (m, 0.9H), 2.57–2.30 (m, 2.55H), 2.22–1.85 (m, 3H), 1.82–1.53 (m, 3H), 0.98 (t, J = 7.3 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 172.4, 171.9, 160.1, 157.7, 157.3, 152.8, 152.6, 137.6, 137.5, 131.2, 131.1, 123.8, 123.7, 121.5, 121.4, 118.7, 111.7, 111.5, 98.8, 98.5, 55.3, 55.2, 47.8, 46.0, 45.0, 42.2, 35.7, 34.4, 33.4, 28.8, 28.0, 25.6, 25.0, 19.0, 14.3, 14.2; ESI-MS: (m/z) 408.3 [M + Na]+, 384.3 [M − H]−; HPLC method A: tr = 8.285 min.
The title compound was prepared from 7c (60.0 mg, 0.19 mmol), isovaleric acid (22.3 mg, 0.22 mmol), TBTU (76.3 mg, 0.24 mmol), and DIPEA (73.7 mg, 0.57 mmol) in dry DCM (total amount 10 mL) according to general procedure F (reaction time 1 h). Purification by flash column chromatography (SiO2, DCM–MeOH 95:5) gave 61 mg of a white solid (80% yield). 1H-NMR shows a 5:4 mixture of amide bond rotamers. 1H-NMR (400 MHz, CDCl3) δ 12.30 (br s, 1H), 8.56–8.47 (m, 1H), 7.73–7.63 (m, 1H), 7.50–7.41 (m, 1H), 7.25–7.19 (m, 1H), 5.04–4.93 (m, 0.45H), 4.80–4.70 (m, 0.55H), 4.52–4.29 (m, 1H), 4.28–4.20 (m, 0.55H), 3.97–3.85 (m, 0.45H), 3.34–3.20 (m, 3H), 3.18–3.10 (m, 0.55H), 3.05–2.91 (m, 0.9H), 2.55–2.46 (m, 0.55H), 2.45–2.24 (m, 2H), 2.23–1.86 (m, 4H), 1.75–1.54 (m, 1H), 1.01–0.93 (m, 6H); 13C NMR (101 MHz, CDCl3) δ 171.9, 171.4, 160.1, 157.7, 157.3, 152.8, 152.6, 137.6, 137.5, 131.22, 131.15, 123.8, 123.7, 121.6, 121.4, 118.7, 111.7, 111.5, 98.9, 98.6, 55.4, 55.3, 48.0, 46.2, 45.1, 42.5, 42.3, 34.4, 33.4, 28.8, 28.0, 26.0, 25.9, 25.7, 25.0, 22.93, 22.89; ESI-MS: (m/z) 422.3 [M + Na]+, 398.3 [M − H]−; HPLC method A: tr = 8.646 min.
The title compound was prepared from 7c (75.0 mg, 0.24 mmol), 3-(dimethylamino) propionic acid hydrochloride (47.4 mg, 0.31 mmol), PyBOP (160.7 mg, 0.31 mmol), and TEA (72.1 mg, 0.71 mmol) in dry DCM (total amount 10 mL) according to general procedure F (reaction time 1.5 h). During the extractive work-up the organic layer was not washed with saturated NH4Cl solution due to the basic amino function of the introduced substituent. Purification twice by flash column chromatography (SiO2, DCM–(2N NH3 in MeOH) gradient elution from 95:5 to 9:1 and SiO2, DCM:(2N NH3 in MeOH) gradient elution from 92.5:7.5 to 9:1) gave 60 mg of a white solid (61% yield). 1H-NMR shows a 3:2 mixture of amide bond rotamers. 1H-NMR (400 MHz, CDCl3) δ 13.05–12.30 (m, 1H), 8.52–8.41 (m, 1H), 7.65–7.56 (m, 1H), 7.44–7.36 (m, 1H), 7.21–7.11 (m, 1H), 4.96–4.83 (m, 0.4H), 4.76–4.62 (m, 0.6H), 4.47–4.20 (m, 1.6H), 3.95–3.84 (m, 0.4H), 3.32–3.17 (m, 3H), 3.17–3.08 (m, 0.6H), 3.03–2.95 (m, 0.4H), 2.93–2.86 (m, 0.4H), 2.85–2.57 (m, 4H), 2.55–2.45 (m, 0.6H), 2.32 (s, 6H), 2.18–1.83 (m, 3H), 1.73–1.52 (m, 1H); 13C NMR (101 MHz, CDCl3) δ 170.7, 170.2, 160.1, 160.0, 157.8, 157.7, 153.0, 137.7, 137.6, 131.0, 130.9, 123.7, 123.6, 121.22, 121.15, 118.7, 118.6, 111.6, 111.4, 98.8, 98.5, 55.3, 55.2, 54.9, 47.6, 46.0, 45.4, 45.3, 44.9, 42.3, 34.4, 33.4, 31.8, 31.5, 28.7, 27.9, 25.5, 24.8; ESI-MS: (m/z) 415.4 [M + H]+, 413.2 [M − H]−; HPLC method A: tr = 4.833 min.
The title compound was prepared from 7c (70.0 mg, 0.22 mmol), acetic acid (20.0 mg, 0.33 mmol), PyBOP (144.4 mg, 0.28 mmol), and DIPEA (86.1 mg, 0.67 mmol) in dry DCM (total amount 12 mL) according to general procedure F (reaction time 1.5 h). Purification twice by flash column chromatography (SiO2, DCM–MeOH gradient elution from 96:4 to 93.5:6.5 and SiO2, EtOAc/MeOH 9:1) gave 44 mg of an off-white solid (55% yield). 1H-NMR shows a 5:4 mixture of amide bond rotamers. 1H-NMR (300 MHz, DMSO-d6) δ 12.30–12.14 (m, 1H), 8.41 (s, 1H), 7.87–7.78 (m, 1H), 7.51–7.44 (m, 1H), 7.30–7.17 (m, 1H), 4.57–4.47 (m, 0.55H), 4.43–4.12 (m, 1.45H), 4.07–3.95 (m, 0.45H), 3.84–3.72 (m, 0.55H), 3.33–3.16 (m, 3.45H), 3.07–2.85 (m, 1.1H), 2.57–2.44 (m, 0.45H, overlap with DMSO-d5 signal), 2.10–1.86 (m, 5H), 1.86–1.73 (m, 1H), 1.58–1.35 (m, 1H); 13C NMR (101 MHz, DMSO-d6) δ 168.3, 168.2, 159.5, 159.4, 157.50, 157.47, 153.7, 153.6, 137.4, 129.2, 124.0, 123.9, 120.4, 120.3, 118.4, 118.3, 110.8, 97.4, 97.1, 54.7, 54.5, 47.6, 45.7, 43.6, 41.0, 33.8, 32.6, 27.3, 27.1, 25.1, 24.4, 21.3; ESI-MS: (m/z) 380.4 [M + Na]+, 356.5 [M − H]−; HPLC method A: tr = 6.993 min.
The title compound was prepared from 7c (50.0 mg, 0.16 mmol), 2-cyclopropylacetic acid (19.8 mg, 0.20 mmol), TBTU (63.6 mg, 0.20 mmol), and DIPEA (61.4 mg, 0.48 mmol) in dry DCM (total amount 10 mL) according to general procedure F (reaction time 1.5 h). Purification by flash column chromatography (SiO2, DCM–MeOH gradient elution from 96:4 to 93.5:6.5) gave 52 mg of a beige solid (83% yield). 1H-NMR shows a 5:4 mixture of amide bond rotamers. 1H-NMR (300 MHz, CDCl3) δ 12.20 (br s, 1H), 8.72–8.27 (m, 1H), 7.79–7.59 (m, 1H), 7.56–7.39 (m, 1H), 7.29–7.17 (m, 1H, overlap with CHCl3 signal), 5.07–4.88 (m, 0.45H), 4.81–4.66 (m, 0.55H), 4.58–4.30 (m, 1H), 4.27–4.12 (m, 0.55H), 3.94–3.79 (m, 0.45H), 3.38–2.91 (m, 4.55H), 2.60–2.29 (m, 2.45H), 2.25–1.83 (m, 3H), 1.81–1.52 (m, 1H), 1.18–0.96 (m, 1H), 0.65–0.40 (m, 2H), 0.34–0.08 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 172.0, 171.5, 160.0, 157.6, 157.0, 152.7, 152.4, 137.6, 131.2, 123.9, 123.6, 121.6, 121.4, 118.7, 111.7, 98.8, 55.3, 55.2, 47.9, 46.2, 45.1, 42.2, 38.9, 38.8, 34.4, 33.5, 28.8, 28.0, 25.6, 24.9, 7.5, 4.70, 4.66; ESI-MS: (m/z) 398.3 [M + H]+, 420.3 [M + Na]+, 396.3 [M − H]−; HPLC method A: tr = 8.031 min.
The title compound was prepared from 7c (60.0 mg, 0.17 mmol), propionic acid (16.9 mg, 0.23 mmol), PyBOP (118.6 mg, 0.23 mmol). and DIPEA (73.7 mg, 0.57 mmol) in dry DCM (total amount 10 mL) according to general procedure F (reaction time 40 min). Purification by flash column chromatography (SiO2, DCM–MeOH gradient elution from 96:4 to 93.5:6.5) gave 38 mg of a white solid (54% yield). 1H-NMR shows a 5:4 mixture of amide bond rotamers. 1H-NMR (400 MHz, CDCl3) δ 12.58 (br s, 1H), 8.59–8.37 (m, 1H), 7.74–7.58 (m, 1H), 7.52–7.40 (m, 1H), 7.24–7.15 (m, 1H), 5.02–4.88 (m, 0.45H), 4.81–4.67 (m, 0.55H), 4.52–4.30 (m, 1H), 4.29–4.18 (m, 0.55H), 3.95–3.81 (m, 0.45H), 3.37–3.19 (m, 3H), 3.19–3.07 (m, 0.55H), 3.07–2.85 (m, 1H), 2.65–2.34 (m, 2.45H), 2.21–1.83 (m, 3H), 1.76–1.54 (m, 1H), 1.26–1.12 (m, 3H); 13C NMR (101 MHz, CDCl3) δ 173.2, 172.7, 159.92, 159.88, 157.2, 156.5, 152.4, 151.8, 137.6, 137.5, 131.1, 123.8, 123.6, 121.5, 121.3, 118.6, 111.72, 111.68, 98.5, 98.4, 55.2, 55.1, 47.5, 45.8, 44.9, 42.2, 34.5, 33.5, 28.7, 28.0, 26.9, 26.8, 25.5, 24.9, 9.8; ESI-MS: (m/z) 393.9 [M + Na]+, 369.8 [M − H]−; HPLC method A: tr = 7.599 min.
1-(3-((7-Chloro-9H-pyrimido[4,5-b]indol-4-yl)(methyl)amino)piperidin-1-yl)-3,3,3-trifluoropropan-1-one (17)
The title compound was prepared from 7c (65.0 mg, 0.21 mmol), trifluoropropionic acid (33.0 mg, 0.26 mmol), TBTU (82.6 mg, 0.26 mmol), and DIPEA (79.8 mg, 0.62 mmol) in dry DCM (total amount 10 mL) according to general procedure F (reaction time 2 h). Purification by flash column chromatography (SiO2, DCM–MeOH 95:5) gave 44 mg of a light yellow solid (50% yield). 1H-NMR shows a 3:2 mixture of amide bond rotamers. 1H-NMR (300 MHz, DMSO-d6) δ 12.23 (s, 1H), 8.44–8.33 (m, 1H), 7.89–7.76 (m, 1H), 7.54–7.44 (m, 1H), 7.30–7.16 (m, 1H), 4.65–4.52 (m, 0.6H), 4.43–4.16 (m, 1.4H), 4.05–3.95 (m, 0.4H), 3.84–3.62 (m, 2.6H), 3.34–3.17 (m, 3.4H), 3.08–2.93 (m, 1.2H), 2.68–2.54 (m, 0.4H), 2.11–1.72 (m, 3H), 1.62–1.40 (m, 1H); ESI-MS: (m/z) 447.8 [M + Na]+, 423.8 [M − H]−; HPLC method A: tr = 8.124 min.
7c (60.0 mg, 0.19 mmol), pivalic acid (24.3 mg, 0.24 mmol), TBTU (70.3 mg, 0.24 mmol), and DIPEA (73.7 mg, 0.57 mmol) were stirred in dry DCM (10 mL) at rt and under N2 atmosphere, but no conversion was observed. Therefore, the stirring mixture was cooled down to 0 °C, and pivaloyl chloride (237 µL of a freshly prepared 1-M solution in dry DCM, 0.24 mmol) was added. The cooling was removed, and the mixture was stirred for 45 min under N2 atmosphere. Saturated NH4Cl solution (20 mL) was added, and the mixture was diluted with DCM. Phases were separated, and the organic layer was washed with saturated NH4Cl solution (20 mL) and saturated NaHCO3 solution (2 × 20 mL), then dried over Na2SO4, and concentrated under reduced pressure. Purification of the residue by flash column chromatography (DCM–MeOH 95:5) gave 36 mg of a white solid (47% yield). 1H-NMR (300 MHz, CDCl3) δ 12.39 (br s, 1H), 8.49 (s, 1H), 7.67 (d, J = 8.5 Hz, 1H), 7.46 (s, 1H), 7.21 (d, J = 8.3 Hz, 1H), 4.85–4.68 (m, 1H), 4.59–4.37 (m, 2H), 3.29 (s, 3H), 3.12–3.00 (m, 1H), 2.82–2.67 (m, 1H), 2.17–1.85 (m, 3H), 1.75–1.57 (m, 1H), 1.31 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 177.0, 160.0, 157.4, 152.5, 137.6, 131.1, 123.7, 121.3, 118.7, 111.6, 98.6, 55.2, 47.8, 45.7, 39.0, 33.9, 28.6, 28.5, 25.2; ESI-MS: (m/z) 400.5 [M + H]+, 422.5 [M + Na]+, 398.5 [M − H]−; HPLC method A: tr = 8.748 min.
Cyclopropanecarbonyl chloride (190 µL of a freshly prepared 1M solution in dry THF, 0.19 mmol) was slowly added to an ice-cooled stirring solution of 7c (50.0 mg, 0.16 mmol) and TEA (32.0 mg, 0.32 mmol) in dry THF (10 mL) under N2 atmosphere. The mixture was left to warm to rt and stirred under N2 atmosphere. Additional cyclopropanecarbonyl chloride solution was added after 1 h (79 µL, 0.08 mmol) and 2 h (158 µL, 0.16 mmol) after cooling down the mixture each time; however, full consumption of the starting material was not achieved. The mixture was evaporated to dryness. Purification of the residue by flash column chromatography (DCM–MeOH 95:5) gave 46 mg of a white solid (76% yield). 1H-NMR shows a 5:4 mixture of amide bond rotamers. 1H-NMR (400 MHz, DMSO-d6) δ 12.23 (s, 1H), 8.41 (s, 1H), 7.86–7.76 (m, 1H), 7.48 (s, 1H), 7.32–7.14 (m, 1H), 4.63–4.48 (m, 0.55H), 4.44–4.14 (m, 2.45H), 3.30–3.14 (m, 3H), 3.12–2.92 (m, 1H), 2.64–2.54 (m, 0.45H), 2.12–1.74 (m, 4H), 1.56–1.38 (m, 1H), 0.78–0.55 (m, 4H), missing 0.45H below water signal; 13C NMR (101 MHz, CDCl3) δ 172.5, 160.1, 157.2 (br), 152.5 (br), 137.6, 131.1, 123.7, 121.4, 118.7, 111.6, 98.7, 55.3, 47.9 (br), 45.9 (br), 42.9 (br), 34.2 (br), 28.7 (br), 24.9 (br), 11.4, 7.6, 7.4; ESI-MS: (m/z) 384.2 [M + H]+, 406.2 [M + Na]+, 382.2 [M − H]−; HPLC method A: tr = 7.837 min.
Acryloyl chloride (228 µL of a freshly prepared 1M solution in dry THF, 0.23 mmol) was slowly added to a stirring solution of 7c (60.0 mg, 0.19 mmol) and TEA (38.5 mg, 0.38 mmol) in dry THF (10 mL) under N2 atmosphere and ice/MeOH cooling. The mixture was left to warm to rt and stirred until reaction control indicated sufficient conversion. Extractive work-up followed by flash column chromatography (DCM–MeOH 95:5) gave 50 mg of a white solid (71% yield). 1H-NMR shows a 5:4 mixture of amide bond rotamers. 1H-NMR (400 MHz, DMSO-d6) δ 12.23 (s, 1H), 8.41 (s, 1H), 7.88–7.75 (m, 1H), 7.48 (d, J = 1.8 Hz, 1H), 7.22 (d, J = 8.3 Hz, 1H), 6.94–6.74 (m, 1H), 6.15–6.03 (m, 1H), 5.74–5.62 (m, 1H), 4.65–4.54 (m, 0.55H), 4.50–4.38 (m, 0.45H), 4.34–4.19 (m, 1.45H), 4.10–3.98 (m, 0.55H), 3.29–3.16 (m, 3H), 3.10–2.97 (m, 1.1H), 2.70–2.58 (m, 0.45H), 2.10–1.78 (m, 3H), 1.53–1.37 (m, 1H), missing 0.45H below water signal; 13C NMR (101 MHz, DMSO-d6) δ 164.49, 159.40, 157.45, 153.58, 137.40, 129.23, 128.45, 126.98, 123.91, 120.33, 118.34, 110.81, 97.33, 55.07, 54.43, 47.22, 45.09, 44.16, 41.65, 33.63, 32.67, 27.22, 25.37, 24.31; ESI-MS: (m/z) 370.1 [M + H]+, 392.2 [M + Na]+, 368.1 [M − H]−; HPLC method A: tr = 7.384 min.
7c (60.0 mg, 0.19 mmol) and 2-(oxetan-3-ylidene)acetonitrile (36.1 mg, 0.38 mmol) were stirred in EtOH at 70 °C for 6 d. The mixture was concentrated under reduced pressure. Purification of the residue by flash column chromatography (SiO2, DCM–MeOH gradient elution from 96:4 to 93.5:6.5) gave 59 mg of a beige solid (76% yield). 1H-NMR (400 MHz, DMSO-d6) δ 12.21 (s, 1H), 8.41 (s, 1H), 7.79 (d, J = 8.6 Hz, 1H), 7.48 (d, J = 1.5 Hz, 1H), 7.27 (dd, J = 8.6, 1.6 Hz, 1H), 4.52–4.31 (m, 5H), 3.17 (s, 3H), 3.01 (s, 2H), 2.82–2.73 (m, 1H), 2.61–2.54 (m, 1H), 2.49–2.44 (m, 1H, overlap with DMSO-d5 signal), 2.18–2.06 (m, 1H), 1.93–1.69 (m, 3H), 1.60–1.47 (m, 1H); 13C NMR (50 MHz, DMSO-d6) δ 159.5, 157.5, 153.8, 137.4, 129.2, 123.8, 120.3, 119.3, 118.5, 110.9, 97.1, 78.1, 61.0, 55.1, 48.0, 45.0, 33.1, 27.3, 24.7, 17.4; ESI-MS: (m/z) 411.0 [M + H]+, 433.0 [M + Na]+, 409.0 [M − H]−; HPLC method A: tr = 7.888 min.
Cyanoacetic acid (36.7 mg, 0.31 mmol) and PyBOP (179.8 mg, 0.34 mmol) were stirred in dry DCM (3 mL) at rt for 20 min. A solution of 7a (80.4 mg, 0.29 mmol) and DIPEA (43.9 mg, 0.34 mmol) in dry DCM (2 mL) was drop-added. The mixture was stirred at rt for 2 h and then concentrated under reduced pressure. Saturated NaHCO3 solution was added to the residue, and the mixture was extracted with EtOAc (3 × 10 mL). Combined organic layers were dried over Na2SO4 and concentrated under reduced pressure. Purification of the residue by flash column chromatography (SiO2, DCM–EtOH gradient elution from 98:2 to 9:1) gave 55 mg (55% yield). 1H-NMR shows a 3:2 mixture of amide bond rotamers: (300 MHz, DMSO-d6) δ 12.19–11.99 (m, 1H), 8.47–8.36 (m, 1H), 7.90–7.78 (m, 1H), 7.54–7.45 (m, 1H), 7.44–7.34 (m, 1H), 7.31–7.19 (m, 1H), 4.55–4.43 (m, 0.6H), 4.39–4.21 (m, 1.4H), 4.18–3.97 (m, 2H), 3.94–3.84 (m, 0.4H), 3.69–3.56 (m, 0.6H), 3.31–3.14 (m, 3.4H), 3.10–2.93 (m, 1.2H), 2.70–2.56 (m, 0.4H), 2.10–1.74 (m, 3H), 1.66–1.42 (m, 1H); 13C NMR (101 MHz, DMSO-d6) δ 161.6, 161.4, 159.6, 159.5, 157.0, 153.3, 136.7, 124.8, 122.7, 122.6, 120.4, 120.3, 119.32, 119.28, 116.1, 116.0, 111.2, 97.9, 97.7, 54.6, 54.2, 47.4, 45.5, 44.2, 42.1, 34.0, 32.7, 27.1, 26.9, 24.8, 24.7, 24.1; ESI-MS: (m/z) 370.9 [M + Na]+, 346.9 [M − H]−; HPLC method B: tr = 3.316 min.
7b (80.0 mg, 0.27 mmol) and DIPEA (51.7 mg, 0.40 mmol) were stirred in dry DCM (3 mL). A suspension of cyanoacetic acid (25.0 mg, 0.29 mmol) and PyBOP (166.9 mg, 0.32 mmol) in dry DCM (3 mL) was drop-added. The mixture was stirred at rt for 2 h and then concentrated under reduced pressure. Saturated NaHCO3 solution (10 mL) was added to the residue, and the mixture was extracted with EtOAc (3 × 10 mL). Combined organic layers were dried over Na2SO4 and concentrated under reduced pressure. Purification of the residue by flash column chromatography (SiO2, DCM–EtOH gradient elution from 98:2 to 90:10 (twice)) gave 28 mg (29% yield); 1H-NMR shows a 5:4 mixture of amide bond rotamers. 1H-NMR (300 MHz, DMSO-d6) δ 12.28–12.15 (m, 1H), 8.45–8.37 (m, 1H), 7.90–7.77 (m, 1H), 7.30–7.21 (m, 1H), 7.16–7.01 (m, 1H), 4.53–4.44 (m, 0.55H), 4.40–4.17 (m, 1.45H), 4.15–3.98 (m, 2H), 3.92–3.84 (m, 0.45H), 3.66–3.58 (m, 0.55H), 3.30–3.16 (m, 3H), 3.09–2.96 (m, 1.1H), 2.69–2.57 (m, 0.45H), 2.09–1.89 (m, 2H), 1.88–1.75 (m, 1H), 1.67–1.41 (m, 1H), missing 0.45H below water signal; ESI-MS: (m/z) 389.2 [M + Na]+, 365.1 [M − H]−; HPLC method B: tr = 4.514 min.
Cyanoacetic acid (31.8 mg, 0.37 mmol) and PyBOP (213.3 mg, 0.4 mmol) were stirred in dry DCM (5 mL) at rt. A suspension of 7d (120.0 mg, 0.33 mmol) and DIPEA (66.0 mg, 0.5 mmol) in dry DCM (5 mL) was added. The mixture was stirred at rt for 2 h and then concentrated under reduced pressure. Saturated NaHCO3 solution was added to the residue, and the mixture was extracted with EtOAc (3 × 10 mL). Combined organic layers were dried over Na2SO4 and concentrated under reduced pressure. Purification of the residue by flash column chromatography (SiO2, DCM–EtOH gradient elution from 98:2 to 9:1) gave 72 mg (51% yield). 1H-NMR shows a 5:4 mixture of amide bond rotamers. 1H-NMR (300 MHz, DMSO-d6) δ 12.29–12.17 (m, 1H), 8.47–8.38 (m, 1H), 7.82–7.73 (m, 1H), 7.65–7.59 (m, 1H), 7.43–7.33 (m, 1H), 4.54–4.45 (m, 0.55H), 4.40–4.18 (m, 1.45H), 4.14–3.98 (m, 2H), 3.92–3.82 (m, 0.45H), 3.68–3.57 (m, 0.55H), 3.29–3.17 (m, 3H), 3.09–2.95 (m, 1.1H), 2.70–2.57 (m, 0.45H), 2.08–1.74 (m, 3H), 1.67–1.41 (m, 1H), missing 0.45H below water signal; ESI-MS: (m/z) 448.9 [M + Na]+, 424.8 [M − H]−; HPLC method B: tr = 6.305 min.
The title compound was prepared from 7e (75 mg, 0.18 mmol), cyanoacetic acid (19.6 mg, 0.23 mmol), TBTU (73.9 mg, 0.23 mmol), and DIPEA (71.4 mg, 0.55 mmol) in dry DCM (total amount 12 mL) according to general procedure F (reaction time 2 h). Purification by flash column chromatography (SiO2, DCM–MeOH 96:4) gave 61 mg of a white solid (70% yield); 1H-NMR shows a 3:2 mixture of amide bond rotamers. 1H-NMR (400 MHz, DMSO-d6) δ 12.24–12.12 (m, 1H), 8.47–8.38 (m, 1H), 7.79 (s, 1H), 7.68–7.59 (m, 1H), 7.58–7.47 (m, 1H), 4.55–4.42 (m, 0.6H), 4.40–4.19 (m, 1.4H), 4.19–3.98 (m, 2H), 3.92–3.82 (m, 0.4H), 3.69–3.56 (m, 0.6H), 3.32–3.13 (m, 3H), 3.09–2.95 (m, 1.2H), 2.68–2.57 (m, 0.4H), 2.09–1.89 (m, 2H), 1.89–1.75 (m, 1H), 1.67–1.44 (m, 1H), missing 0.4H below water signal; 13C NMR (101 MHz, DMSO-d6) δ 161.6, 161.5, 159.5, 159.4, 156.9, 153.8, 138.0, 128.8, 128.7, 124.6, 124.5, 119.6, 119.02, 118.96, 116.1, 116.0, 97.4, 97.2, 89.5, 89.4, 54.6, 54.2, 47.3, 45.5, 44.1, 42.0, 34.0, 32.7, 27.0, 26.9, 24.8, 24.6, 24.1. ESI-MS: (m/z) 497.2 [M + Na]+, 473.2 [M − H]−; HPLC method A: tr = 7.258 min.
7f (120.0 mg, 0.39 mmol) and DIPEA (99.6 mg, 0.77 mmol) were stirred in dry DCM (5 mL) at rt. A suspension of cyanoacetic acid (36.1 mg, 0.42 mmol) and PyBOP (240.7 mg, 0.46 mmol) in dry DCM (5 mL) was drop-added. The mixture was stirred at rt for 2 h and then concentrated under reduced pressure. Saturated NaHCO3 solution (10 mL) was added to the residue, and the mixture was extracted with EtOAc (3 × 10 mL). Combined organic layers were dried over Na2SO4 and concentrated under reduced pressure. Purification of the residue by flash column chromatography (SiO2, DCM–EtOH gradient elution from 98:2 to 9:1 (twice)) gave 62 mg (43% yield). 1H-NMR shows a 3:2 mixture of amide bond rotamers. 1H-NMR (300 MHz, DMSO-d6) δ 12.05–11.88 (m, 1H), 8.43–8.28 (m, 1H), 7.79–7.61 (m, 1H), 7.02–6.92 (m, 1H), 6.92–6.79 (m, 1H), 4.52–4.40 (m, 0.6H), 4.38–4.16 (m, 1.4H), 4.15–3.97 (m, 2H), 3.93–3.77 (m, 3.4H), 3.68–3.56 (m, 0.6H), 3.27–3.12 (m, 3H), 3.08–2.93 (m, 1.2H), 2.69–2.58 (m, 0.4H), 2.10–1.74 (m, 3H), 1.65–1.41 (m, 1H), missing 0.4H below water signal; ESI-MS: (m/z) 379.0 [M + H]+, 400.9 [M + Na]+, 378.0 [M − H]−; HPLC method B: tr = 3.635 min.
7g (100.0 mg, 0.29 mmol) and DIPEA (54.3 mg, 0.42 mmol) were stirred in dry DCM (5 mL) at rt. A suspension of cyanoacetic acid (26.8 mg, 0.31 mmol) and PyBOP (178.7 mg, 0.36 mmol) in dry DCM (5 mL) was drop-added. The mixture was stirred at rt for 2 h and then concentrated under reduced pressure. Saturated NaHCO3 solution (10 mL) was added to the residue, and the mixture was extracted with EtOAc (3 × 10 mL). Combined organic layers were dried over Na2SO4 and concentrated under reduced pressure. Purification of the residue twice by flash column chromatography (SiO2, DCM–EtOH gradient elution from 97:3 to 4:1 and SiO2, DCM–(2N NH3 in MeOH) gradient elution from 99:1 to 92:8) gave 28 mg (23% yield); 1H-NMR shows a 3:2 mixture of amide bond rotamers. 1H-NMR (300 MHz, DMSO-d6) δ 12.45 (s, 1H), 8.50–8.43 (m, 1H), 8.09–7.99 (m, 1H), 7.74 (s, 1H), 7.59–7.48 (m, 1H), 4.58–4.49 (m, 0.6H), 4.41–4.27 (m, 1.4H), 4.16–4.00 (m, 2H), 3.93–3.84 (m, 0.4H), 3.71–3.60 (m, 0.6H), 3.33–3.21 (m, 3.4H), 3.10–2.95 (m, 1.2H), 2.69–2.58 (m, 0.4H), 2.11–1.76 (m, 3H), 1.70–1.45 (m, 1H); ESI-MS: (m/z) 438.9 [M + Na]+, 415.0 [M − H]−; HPLC method B: tr = 6.640 min.
The title compound was prepared from 7l (65.0 mg, 0.20 mmol), cyanoacetic acid (21.0 mg, 0.25 mmol), TBTU (79.1 mg, 0.25 mmol), and DIPEA (76.4 mg, 0.59 mmol) in dry DCM (total amount 10 mL) according to general procedure F (reaction time 2 h). Purification by flash column chromatography (SiO2, DCM–MeOH gradient elution from 95.5:4.5 to 93:7) gave 48 mg of a grey-yellow solid (61% yield). 1H-NMR shows a 1:1 mixture of amide bond rotamers. 1H-NMR (300 MHz, DMSO-d6) δ 12.02 (s, 1H), 7.84–7.71 (m, 1H), 7.45 (d, J = 2.0 Hz, 1H), 7.28–7.15 (m, 1H), 4.57–4.48 (m, 0.5H), 4.39–4.30 (m, 0.5H), 4.29–3.97 (m, 3H), 3.95–3.86 (m, 0.5H), 3.67–3.58 (m, 0.5H), 3.34–3.27 (m, 0.5H), 3.26–3.14 (m, 3H), 3.08–2.92 (m, 1H), 2.70–2.58 (m, 0.5H), 2.53–2.47 (m, 3H, overlap with DMSO-d5 signal), 2.13–1.72 (m, 3H), 1.67–1.43 (m, 1H); 13C NMR (101 MHz, DMSO-d6) δ 162.6, 162.5, 161.7, 161.5, 159.5, 159.4, 158.5, 158.4, 137.5, 137.4, 128.8, 123.7, 123.5, 120.3, 118.6, 118.5, 116.2, 116.0, 110.8, 95.2, 95.0, 54.5, 54.3, 47.2, 45.6, 44.1, 42.3, 34.2, 32.7, 27.4, 27.0, 25.8, 25.7, 25.0, 24.9, 24.7, 24.3; ESI-MS: (m/z) 396.9 [M + H]+, 418.9 [M + Na]+, 394.9 [M − H]−; HPLC method A: tr = 7.427 min.
The title compound was prepared from 7a (60.0 mg, 0.21 mmol), propionic acid (19.8 mg, 0.27 mmol), TBTU (85.6 mg, 0.27 mmol), and DIPEA (82.7 mg, 0.64 mmol) in dry DCM (total amount 10 mL) according to general procedure F (reaction time 1 h). Purification by flash column chromatography (SiO2; DCM–MeOH 94:6) gave 45 mg of a beige solid (63% yield). 1H-NMR shows a 5:4 mixture of amide bond rotamers. 1H-NMR (400 MHz, DMSO-d6) δ 12.08 (s, 1H), 8.43–8.36 (m, 1H), 7.88–7.78 (m, 1H), 7.52–7.45 (m, 1H), 7.43–7.35 (m, 1H), 7.30–7.17 (m, 1H), 4.62–4.49 (m, 0.55H), 4.45–4.37 (m, 0.45H), 4.35–4.27 (m, 0.45H), 4.26–4.16 (m, 0.55H), 4.08–4.00 (m, 0.45H), 3.88–3.75 (m, 0.55H), 3.29–3.15 (m, 3.45H), 3.02–2.87 (m, 1.1H), 2.56–2.46 (m, 0.45H, overlap with DMSO-d6 signal), 2.46–2.26 (m, 2H), 2.07–1.87 (m, 2H), 1.85–1.75 (m, 1H), 1.52–1.37 (m, 1H), 1.02–0.92 (m, 3H); ESI-MS: (m/z) 338.7 [M + H]+, 360.7 [M + Na]+, 336.7 [M − H]−; HPLC method A: tr = 6.107 min.
The title compound was prepared from 7b (50.0 mg, 0.17 mmol), propionic acid (15.5 mg, 0.21 mmol), TBTU (67.5 mg, 0.21 mmol), and DIPEA (64.8 mg, 0.50 mmol) in dry DCM (total amount 15 mL) according to general procedure F (reaction time 45 min). Purification by flash column chromatography (SiO2; DCM–MeOH 95:5) gave 50 mg of a white solid (84% yield). 1H-NMR (400 MHz, DMSO-d6) δ 12.28–12.14 (m, 1H), 8.44–8.34 (m, 1H), 7.86–7.76 (m, 1H), 7.30–7.20 (m, 1H), 7.13–6.99 (m, 1H), 4.60–4.51 (m, 0.55H), 4.45–4.36 (m, 0.45H), 4.32–4.23 (m, 0.45H), 4.21–4.11 (m, 0.55H), 4.08–3.98 (m, 0.45H), 3.87–3.74 (m, 0.55H), 3.28–3.14 (m, 3.45H), 3.00–2.85 (m, 1.1H), 2.54–2.46 (m, 0.45H, overlap with DMSO-d5 signal), 2.44–2.26 (m, 2H), 2.05–1.86 (m, 2H), 1.83–1.75 (m, 1H), 1.52–1.37 (m, 1H), 1.03–0.92 (m, 3H); 13C NMR (101 MHz, DMSO-d6) δ 171.54, 171.37, 160.25 (d, J = 240.1 Hz), 159.33, 159.21, 157.72, 153.12, 153.03, 137.47 (d, J = 12.5 Hz), 124.14–123.87 (m), 116.17, 108.07 (d, J = 23.5 Hz), 97.80 (d, J = 26.1 Hz), 54.80, 54.55, 46.74, 44.79, 43.87, 41.22, 33.75, 32.55, 27.52, 27.24, 25.65, 25.15, 24.46, 9.43. ESI-MS: (m/z) 378.3 [M + Na]+, 354.4 [M − H]−; HPLC method B: tr = 7.075 min.
The title compound was prepared from 7l (28.0 mg, 0.09 mmol), propionic acid (7.9 mg, 0.11 mmol), TBTU (34.1 mg, 0.11 mmol), and DIPEA (32.9 mg, 0.26 mmol) in dry DCM (total amount 5 mL) according to general procedure F (reaction time 2 h). Purification by flash column chromatography (SiO2, DCM–MeOH gradient elution from 96:4 to 94:6) gave 20 mg of a beige solid (61% yield). 1H-NMR shows a 3:2 mixture of amide bond rotamers. 1H-NMR (400 MHz, DMSO-d6) δ 12.02 (s, 1H), 7.85–7.71 (m, 1H), 7.48–7.40 (m, 1H), 7.29–7.13 (m, 1H), 4.64–4.53 (m, 0.4H), 4.49–4.37 (m, 0.6H), 4.34–4.23 (m, 0.6H), 4.20–4.06 (m, 1H), 3.89–3.77 (m, 0.4H), 3.29–3.14 (m, 3.6H), 3.02–2.86 (m, 0.8H), 2.63–2.54 (m, 0.6H), 2.47–2.29 (m, 2H), 2.10–1.75 (m, 3H), 1.54–1.39 (m, 1H), 1.08–0.92 (m, 3H); ESI-MS: (m/z) 408.2 [M + Na]+, 384.2 [M − H]−; HPLC method A: tr = 7.761 min.
The title compound was prepared from 7d (115.0 mg, 0.32 mmol), propionic acid (29.6 mg, 0.40 mmol), TBTU (128.1 mg, 0.40 mmol), and DIPEA (123.8 mg, 0.96 mmol) in dry DCM (total amount 15 mL) according to general procedure F (reaction time 30 min). Purification by flash column chromatography (SiO2, DCM–MeOH 94:6) gave 48 mg of an off-white solid (36% yield). 1H-NMR shows a 5:4 mixture of amide bond rotamers. 1H-NMR (400 MHz, CDCl3) δ 12.75–12.17 (m, 1H), 8.58–8.47 (m, 1H), 7.67–7.53 (m, 2H), 7.40–7.30 (m, 1H), 5.01–4.90 (m, 0.45H), 4.80–4.68 (m, 0.55H), 4.54–4.40 (m, 0.55H), 4.38–4.29 (m, 0.45H), 4.28–4.20 (m, 0.55H), 3.95–3.82 (m, 0.45H), 3.36–3.20 (m, 3H), 3.19–3.09 (m, 0.55H), 3.05–2.88 (m, 0.9H), 2.67–2.36 (m, 2.55H), 2.21–1.86 (m, 3H), 1.76–1.56 (m, 1H), 1.28–1.13 (m, 3H); 13C NMR (101 MHz, CDCl3) δ 173.2, 172.6, 160.1, 160.0, 157.54, 157.46, 153.0, 137.9, 124.0, 123.92, 123.88, 119.1, 119.0, 118.73, 118.68, 114.6, 114.4, 98.4, 55.2, 55.0, 47.5, 45.8, 45.0, 42.2, 34.4, 33.4, 28.8, 28.0, 26.9, 26.8, 25.6, 24.9, 9.8; ESI-MS: (m/z) 438.0 [M + Na]+, 414.0 [M − H]−; HPLC method A: tr = 8.212 min.
The title compound was prepared from 7e (43.0 mg, 0.11 mmol), propionic acid (9.8 mg, 0.13 mmol), TBTU (42.4 mg, 0.13 mmol), and DIPEA (40.9 mg, 0.32 mmol) in dry DCM (total amount 8 mL) according to general procedure F (reaction time 2 h). Purification by flash column chromatography (SiO2, DCM–MeOH 95:5) gave 25 mg of an off-white solid (51% yield). 1H-NMR shows a 1:1 mixture of amide bond rotamers. 1H-NMR (400 MHz, DMSO-d6) δ 12.20–12.11 (m, 1H), 8.43–8.37 (m, 1H), 7.82–7.76 (m, 1H), 7.67–7.60 (m, 1H), 7.56–7.47 (m, 1H), 4.59–4.52 (m, 0.5H), 4.44–4.37 (m, 0.5H), 4.35–4.26 (m, 0.5H), 4.23–4.14 (m, 0.5H), 4.07–3.98 (m, 0.5H), 3.85–3.79 (m, 0.5H), 3.26–3.15 (m, 3.5H), 3.01–2.87 (m, 1H), 2.54–2.46 (m, 0.5H, overlap with DMSO-d5 signal), 2.45–2.25 (m, 2H), 2.05–1.75 (m, 3H), 1.53–1.37 (m, 1H), 1.02–0.93 (m, 3H); ESI-MS: (m/z) 464.9 [M + H]+, 486.9 [M + Na]+, 462.8 [M − H]−; HPLC method A: tr = 8.443 min.
Cyanoacetic acid (16.8 mg, 0.20 mmol) and TBTU (63.5 mg, 0.20 mmol) were stirred in dry DCM (5 mL) at rt and under N2 atmosphere for 15 min. A suspension of 7h (50.0 mg, 0.16 mmol) and DIPEA (61.4 mg, 0.48 mmol) in dry DCM (5 mL) was added to the activated acid, and the mixture was stirred at rt and under N2 atmosphere for 2.5 h. A precipitate formed. The mixture was diluted with DCM, and MeOH was added to dissolve the precipitate. The solution was washed with saturated NaHCO3 solution (2 × 20 mL) and saturated NH4Cl solution (2 × 20 mL). The organic layer was dried over Na2SO4 and concentrated under reduced pressure. Purification of the residue by flash column chromatography (SiO2, DCM–MeOH gradient elution from 95:5 to 92:8) gave 34 mg of a white solid (56% yield); 1H-NMR shows a 5:4 mixture of amide rotamers. 1H-NMR (400 MHz, DMSO-d6) δ 12.32–12.18 (m, 1H), 8.46–8.36 (m, 1H), 7.83–7.73 (m, 1H), 7.53–7.45 (m, 1H), 7.44–7.36 (m, 1H), 4.51–4.42 (m, 0.55H), 4.40–4.25 (m, 1.45H), 4.17–3.99 (m, 2H), 3.93–3.85 (m, 0.45H), 3.69–3.60 (m, 0.55H), 3.29–3.19 (m, 3H), 3.07–2.91 (m, 1.1H), 2.68–2.57 (m, 0.45H), 2.10–1.94 (m, 2H), 1.89–1.79 (m, 1H), 1.71–1.42 (m, 1H), missing 0.45H below water signal; 13C NMR (101 MHz, DMSO-d6) δ 161.6, 161.5, 159.5, 157.5, 153.8, 135.20, 135.18, 124.7, 124.6, 121.8, 121.7, 120.72, 120.71, 116.02, 115.97, 112.6, 97.0, 96.9, 54.6, 53.8, 47.4, 45.4, 43.9, 42.0, 34.1, 33.1, 27.0, 26.9, 24.9, 24.6, 24.1; ESI-MS: (m/z) 405.4 [M + Na]+, 381.4 [M − H]−; HPLC method A: tr = 6.666 min.
A mixture of cyanoacetic acid (21.3 mg, 0.25 mmol) and EDCI·HCl (47.9 mg, 0.25 mmol) was stirred in dry DCM (6 mL) at rt and under N2 atmosphere for 20 min. A suspension of 7i (60.0 mg, 0.17 mmol) and DIPEA (64.6 mg, 0.51 mmol) in dry DCM (4 mL) was added, and the mixture was stirred at rt and under N2 atmosphere. Due to slow conversion, reactants were added repeatedly: EDCI·HCl (47.9 mg, 0.25 mmol) after 3 h of stirring, cyanoacetic acid (21.3 mg, 0.25 mmol) after 5 h of stirring, and again EDCI·HCl (63.9 mg, 0.33 mmol) after 20 h of stirring. Sufficient conversion was achieved after a reaction time of 2 days. The mixture was diluted with DCM, washed with saturated NH4Cl solution and saturated NaHCO3 solution, dried over Na2SO4, and concentrated under reduced pressure. Purification of the residue by flash column chromatography (SiO2, DCM–MeOH 95:5) gave 16 mg of an off-white solid (22% yield). 1H-NMR shows a 5:4 mixture of amide bond rotamers. 1H-NMR (400 MHz, DMSO-d6) δ 12.34–12.22 (m, 1H), 8.44 (s, 0.45H), 8.40 (s, 0.55H), 7.96–7.87 (m, 1H), 7.57–7.50 (m, 1H), 7.48–7.41 (m, 1H), 4.49–4.41 (m, 0.55H), 4.40–4.24 (m, 1.45H), 4.15–3.99 (m, 2H), 3.92–3.84 (m, 0.45H), 3.70–3.60 (m, 0.55H), 3.29–3.19 (m, 3H), 3.07–2.92 (m, 1.1H), 2.69–2.57 (m, 0.45H), 2.11–1.95 (m, 2H), 1.90–1.80 (m, 1H), 1.72–1.43 (m, 1H), missing 0.45H below water signal; 13C NMR (101 MHz, DMSO-d6) δ 161.6, 161.5, 159.5, 159.4, 157.4, 153.9, 135.5, 127.3, 127.2, 124.7, 124.6, 121.4, 121.3, 116.1, 116.0, 113.1, 112.5, 112.4, 96.8, 96.8, 54.7, 53.9, 47.4, 45.4, 43.9, 42.0, 34.0, 33.1, 27.0, 26.9, 24.9, 24.6, 24.1. ESI-MS: (m/z) 449.4 [M + Na]+, 425.5 [M − H]−; HPLC method A: tr = 6.749 min.
The title compound was prepared from 7j (60.0 mg, 0.19 mmol), cyanoacetic acid (20.5 mg, 0.24 mmol), TBTU (77.3 mg, 0.24 mmol), and DIPEA (74.7 mg, 0.58 mmol) in dry DCM (total amount 10 mL) according to general procedure F (reaction time 0.5 h). Purification by flash column chromatography (SiO2; DCM–MeOH 95:5) gave 38 mg of a beige solid (52% yield). 1H-NMR shows a 5:4 mixture of amide bond rotamers. 1H-NMR (400 MHz, DMSO-d6) δ 12.00–11.87 (m, 1H), 8.43–8.36 (m, 1H), 7.44–7.37 (m, 1H), 7.34–7.20 (m, 1H), 7.09–7.01 (m, 1H), 4.40–3.97 (m, 4H), 3.92–3.80 (m, 3.45H), 3.65–3.57 (m, 0.55H), 3.30–3.17 (m, 3H), 3.06–2.92 (m, 1.1H), 2.69–2.57 (m, 0.4H), 2.15–1.98 (m, 2H), 1.90–1.79 (m, 1H), 1.71–1.44 (m, 1H), missing 0.45H below water signal; 13C NMR (101 MHz, DMSO-d6) δ 161.6, 161.4, 159.9, 159.6, 157.3, 157.2, 154.0, 153.9, 153.1, 131.4, 131.3, 119.9, 119.7, 116.00, 115.96, 113.6, 112.9, 111.9, 111.8, 106.8, 106.0, 98.3, 97.9, 55.6, 55.5, 54.7, 54.3, 47.4, 45.4, 44.0, 42.0, 34.0, 32.7, 27.2, 27.1, 24.9, 24.8, 24.6, 24.1; ESI-MS: (m/z) 379.1 [M + H]+, 401.2 [M + Na]+, 377.2 [M − H]−; HPLC method A: tr = 4.053 min.
The title compound was prepared from 7k (55.0 mg, 0.17 mmol), cyanoacetic acid (18.5 mg, 0.22 mmol), TBTU (69.9 mg, 0.22 mmol), and DIPEA (67.5 mg, 0.52 mmol) in dry DCM (total amount 10 mL) according to general procedure F (reaction time 1 h). A precipitate formed during the reaction and was dissolved by adding DCM and MeOH prior to the extractive work-up. Repeated purification by flash column chromatography (SiO2, DCM–MeOH 94:6; SiO2, DCM–MeOH gradient elution from 96:4 to 92:8; SiO2, DCM–MeOH gradient elution from 96:4 to 92:8; and SiO2, DCM–EtOH 95:5) gave 35 mg of a beige solid (52% yield). 1H-NMR shows a 1:1 mixture of amide bond rotamers. 1H-NMR (300 MHz, DMSO-d6) δ 12.35 (s, 1H), 8.50–8.32 (m, 1H), 7.50–7.34 (m, 2H), 7.33–7.25 (m, 1H), 4.42–3.95 (m, 4H), 3.92–3.80 (m, 0.5H), 3.66–3.55 (m, 0.5H), 3.09–2.89 (m, 4H), 2.71–2.58 (m, 0.5H), 2.07–1.73 (m, 3H), 1.71–1.42 (m, 1H), missing 0.5H below water signal; ESI-MS: (m/z) 405.4 [M + Na]+, 381.3 [M − H]−; HPLC method A: tr = 6.396 min.
The title compound was prepared from 7h (50.0 mg, 0.16 mmol), propionic acid (14.7 mg, 0.20 mmol), TBTU (63.5 mg, 0.20 mmol), and DIPEA (61.4 mg, 0.48 mmol) in dry DCM (10 mL) according to general procedure F (reaction time 45 min). Purification by flash column chromatography (SiO2, DCM–MeOH gradient elution from 96.5:3.5 to 92.5: 7.5) gave 42 mg of an off-white solid (71% yield); 1H-NMR shows a 1:1 mixture of amide bond rotamers. 1H-NMR (400 MHz, DMSO-d6) δ 12.25 (s, 1H), 8.39 (s, 1H), 7.84–7.69 (m, 1H), 7.52–7.45 (m, 1H), 7.43–7.37 (m, 1H), 4.55–4.46 (m, 0.5H), 4.46–4.38 (m, 0.5H), 4.35–4.19 (m, 1H), 4.12–4.00 (m, 0.5H), 3.89–3.77 (m, 0.5H), 3.31–3.18 (m, 3.5H), 3.02–2.92 (m, 0.5H), 2.92–2.83 (m, 0.5H), 2.55–2.26 (m, 2.5H, overlap with DMSO-d5 signal), 2.08–1.93 (m, 2H), 1.90–1.75 (m, 1H), 1.60–1.35 (m, 1H), 1.04–0.91 (m, 3H); 13C NMR (101 MHz, DMSO-d6) δ 171.5, 171.4, 159.5, 157.5, 153.8, 153.8, 135.2, 124.7, 124.6, 124.5, 121.8, 121.6, 120.7, 112.6, 96.9, 96.8, 54.8, 54.1, 46.8, 44.7, 43.6, 41.1, 33.8, 32.9, 27.4, 27.2, 25.6, 25.0, 24.4, 9.3; ESI-MS: (m/z) 394.4 [M + Na]+, 370.4 [M − H]−; HPLC method A: tr = 7.789 min.
Propionic acid (16.7 mg, 0.23 mmol) and TBTU (72.4 mg, 0.23 mmol) were stirred in dry DCM (4 mL) at rt and under N2 atmosphere for 15 min. A suspension of 7i (65.0 mg, 0.18 mmol) in dry DCM (8 mL) was added to the activated acid, followed by addition of DIPEA (70.0 mg, 0.54 mmol). The mixture was stirred at rt and under N2 atmosphere for 50 min; then diluted with DCM; and washed with saturated NaHCO3 solution (2 × 25 mL), saturated NH4Cl solution (2 × 25 mL), and saturated NaCl solution (25 mL). The organic layer was dried over Na2SO4 and concentrated under reduced pressure. Purification of the residue by flash column chromatography (DCM–MeOH 95:5) gave 24 mg of an off-white solid (32% yield); 1H-NMR shows a 1:1 mixture of amide bond rotamers. 1H-NMR (400 MHz, DMSO-d6) δ 12.26 (s, 1H), 8.39 (s, 1H), 7.96–7.81 (m, 1H), 7.56–7.48 (m, 1H), 7.48–7.40 (m, 1H), 4.50–4.36 (m, 1H), 4.34–4.16 (m, 1H), 4.08–4.01 (m, 0.5H), 3.87–3.77 (m, 0.5H), 3.31–3.18 (m, 3.5H), 3.02–2.92 (m, 0.5H), 2.92–2.82 (m, 0.5H), 2.54–2.26 (m, 2.5H, overlap with DMSO-d5 signal), 2.09–1.96 (m, 2H), 1.92–1.76 (m, 1H), 1.60–1.36 (m, 1H), 1.02–0.90 (m, 3H); 13C NMR (101 MHz, DMSO-d6) δ 171.6, 171.5, 159.5, 157.4, 153.9, 153.8, 135.5, 127.3, 124.7, 124.5, 121.4, 113.2, 112.5, 96.8, 96.7, 54.9, 54.1, 46.9, 44.7, 43.7, 41.2, 33.8, 32.9, 27.4, 27.3, 25.7, 25.1, 24.4, 9.4; ESI-MS: (m/z) 438.0 [M + Na]+, 414.0 [M − H]−; HPLC method A: tr = 8.360 min.
The title compound was prepared from 7j (60.0 mg, 0.19 mmol), cyanoacetic acid (17.9 mg, 0.24 mmol), TBTU (77.3 mg, 0.24 mmol), and DIPEA (74.7 mg, 0.58 mmol) in dry DCM (total amount 10 mL) according to general procedure F (reaction time 2 h). Purification by flash column chromatography (SiO2; DCM–MeOH 95:5) gave 43 mg of a beige solid (61% yield). 1H-NMR (300 MHz, DMSO-d6) δ 12.03–11.84 (m, 1H), 8.45–8.31 (m, 1H), 7.46–7.36 (m, 1H), 7.33–7.19 (m, 1H), 7.12–6.99 (m, 1H), 4.50–4.34 (m, 1H), 4.33–4.09 (m, 1H), 4.06–3.96 (m, 0.45H), 3.90–3.74 (m, 3.55H), 3.30–3.14 (m, 3.45H), 3.03–2.81 (m, 1.1H), 2.57–2.44 (m, 0.45H, overlap with DMSO-d5 signal), 2.41–2.23 (m, 2H), 2.12–1.77 (m, 3H), 1.62–1.37 (m, 1H), 1.01–0.86 (m, 3H); 13C NMR (101 MHz, DMSO-d6) δ 171.42, 171.38, 159.9, 159.6, 157.3, 157.2, 154.0, 153.9, 153.1, 131.3, 119.8, 119.8, 113.5, 113.0, 111.9, 111.8, 106.7, 105.9, 98.3, 97.8, 55.6, 55.5, 54.9, 54.5, 46.8, 44.6, 43.6, 41.2, 33.6, 32.6, 27.6, 27.3, 25.6, 25.1, 24.4, 9.3; ESI-MS: (m/z) 390.2 [M + Na]+, 366.2 [M − H]−; HPLC method A: tr = 5.463 min.
The title compound was prepared from 7k (55.0 mg, 0.17 mmol), propionic acid (16.2 mg, 0.22 mmol), TBTU (69.9 mg, 0.22 mmol), and DIPEA (67.5 mg, 0.52 mmol) in dry DCM (total amount 10 mL) according to general procedure F (reaction time 2 h). Purification by flash column chromatography (SiO2, DCM–MeOH 94:6) gave 46 mg of a light beige solid (71% yield). NMR shows a 5:4 mixture of amide rotamers. 1H-NMR (300 MHz, Pyr-d5) δ 14.06–13.63 (m, 1H), 8.88–8.80 (m, 1H), 7.70–7.61 (m, 1H), 7.50–7.36 (m, 2H), 4.87–4.77 (m, 0.55H), 4.69–4.55 (m, 0.45H), 4.55–4.42 (m, 0.55H), 4.27–4.15 (m, 0.55H), 3.73–3.58 (m, 0.45H), 3.23–2.95 (m, 4H), 2.95–2.78 (m, 0.45H), 2.72–2.22 (m, 2.55H), 2.17–1.96 (m, 1H), 1.95–1.42 (m, 3H), 1.31–1.11 (m, 3H), missing 0.45H below water signal; ESI-MS: (m/z) 372.3 [M + H]+, 394.4 [M + Na]+, 370.3 [M − H]−; HPLC method A: tr = 7.646 min.
(8) Detailed Procedures for the Preparation of Enantiopure Final Compounds (R)-2, (R)-20 and (R)-28
The title compound was prepared from (R)-7c (50.0 mg, 0.16 mmol), cyanoacetic acid (16.2 mg, 0.19 mmol), PyBOP (98.9 mg, 0.19 mmol), and DIPEA (61.4 mg, 0.48 mmol) in dry DCM (total amount 10 mL) according to general procedure F (reaction time 2 h). Purification twice by flash column chromatography (SiO2, DCM–MeOH gradient elution from 1:0 to 92:8 and SiO2, DCM–EtOH gradient elution from 96.5:3.5 to 92:8) gave 33 mg of a white solid (54% yield). 1H-NMR shows a 3:2 mixture of amide bond rotamers. 1H-NMR (400 MHz, DMSO-d6) δ 12.33–12.17 (m, 1H), 8.48–8.36 (m, 1H), 7.90–7.77 (m, 1H), 7.53–7.45 (m, 1H), 7.31–7.18 (m, 1H), 4.55–4.46 (m, 0.6H), 4.39–4.20 (m, 1.4H), 4.18–4.00 (m, 2H), 3.94–3.83 (m, 0.4H), 3.69–3.58 (m, 0.6H), 3.29–3.17 (m, 3H), 3.09–2.97 (m, 1.2H), 2.70–2.58 (m, 0.4H), 2.12–1.74 (m, 3H), 1.68–1.43 (m, 1H), missing 0.4H below water signal; 13C NMR (101 MHz, DMSO-d6) δ 161.6, 161.5, 159.4, 159.3, 157.5, 153.7, 137.4, 129.3, 129.2, 124.1, 124.0, 120.4, 118.4, 118.3, 116.14, 116.08, 110.8, 97.3, 97.2, 54.6, 54.2, 47.3, 45.5, 44.1, 42.0, 34.1, 32.8, 27.0, 26.9, 24.9, 24.7, 24.2; ESI-MS: (m/z) 404.9 [M + Na]+, 380.8 [M − H]−; HPLC method A: tr = 6.644 min.
The title compound was prepared from (R)-7d (70.0 mg, 0.19 mmol), cyanoacetic acid (20.7 mg, 0.24 mmol), TBTU (78.0 mg, 0.24 mmol), and DIPEA (75.3 mg, 0.58 mmol) in dry DCM (total amount 12 mL) according to general procedure F (reaction time 30 min). Purification by flash column chromatography (SiO2, DCM–MeOH 94.5:5.5) gave 63 mg of a white solid (76% yield); 1H-NMR shows a 3:2 mixture of amide bond rotamers. 1H-NMR (300 MHz, DMSO-d6) δ 12.30–12.14 (m, 1H), 8.49–8.37 (m, 1H), 7.82–7.72 (m, 1H), 7.66–7.58 (m, 1H), 7.44–7.31 (m, 1H), 4.55–4.44 (m, 0.6H), 4.40–4.19 (m, 1.4H), 4.16–3.98 (m, 2H), 3.93–3.82 (m, 0.4H), 3.68–3.57 (m, 0.6H), 3.28–3.14 (m, 3.4H), 3.09–2.94 (m, 1.2H), 2.70–2.56 (m, 0.4H), 2.09–1.74 (m, 3H), 1.67–1.41 (m, 1H); 13C NMR (101 MHz, DMSO-d6) δ 161.6, 161.4, 159.44, 159.36, 157.3, 153.7, 137.7, 124.4, 124.2, 123.1, 123.0, 118.7, 118.6, 117.38, 117.35, 116.1, 116.0, 113.7, 97.3, 97.2, 54.6, 54.2, 47.3, 45.5, 44.1, 42.0, 34.0, 32.8, 27.0, 26.9, 24.8, 24.6, 24.1; ESI-MS: (m/z) 449.4 [M + Na]+, 425.5 [M − H]−; HPLC method A: tr = 6.672 min.
The title compound was prepared from (R)-7d (45.0 mg, 0.13 mmol), propionic acid (11.6 mg, 0.16 mmol), TBTU (50.1 mg, 0.16 mmol), and DIPEA (48.4 mg, 0.38 mmol) in dry DCM (total amount 10 mL) according to general procedure F (reaction time 40 min). Purification by flash column chromatography (SiO2, DCM–MeOH gradient elution from 96:4 to 92:8) gave 28 mg (54% yield). 1H-NMR shows a 5:4 mixture of amide bond rotamers. 1H-NMR (400 MHz, CDCl3) δ 12.89–12.09 (m, 1H), 8.52 (s, 1H), 7.73–7.55 (m, 2H), 7.43–7.29 (m, 1H), 5.01–4.88 (m, 0.45H), 4.80–4.67 (m, 0.55H), 4.55–4.41 (m, 0.55H), 4.39–4.29 (m, 0.45H), 4.28–4.18 (m, 0.55H), 3.94–3.80 (m, 0.45H), 3.35–3.19 (m, 3H), 3.18–3.09 (m, 0.55H), 3.04–2.87 (m, 0.9H), 2.64–2.35 (m, 2.55H), 2.20–1.84 (m, 3H), 1.75–1.54 (m, 1H), 1.26–1.17 (m, 3H); 13C NMR (101 MHz, CDCl3) δ 173.2, 172.6, 160.1, 157.6, 157.4, 153.0, 137.84, 137.76, 124.1, 124.03, 123.96, 119.2, 119.1, 118.84, 118.79, 114.6, 114.4, 98.8, 98.5, 55.2, 55.1, 47.5, 45.8, 45.0, 42.3, 34.5, 33.4, 29.8, 28.8, 28.0, 26.9, 25.6, 24.9, 9.8; ESI-MS: (m/z) 416.0 [M + H]+, 438.0 [M + Na]+, 414.0 [M − H]−; HPLC method A: tr = 8.046 min.
(9) Detailed Procedures for the Preparation of Enantiopure Final Compounds (S)-2 and (S)-20
The title compound was prepared from (S)-7c (64.0 mg, 0.20 mmol), cyanoacetic acid (20.7 mg, 0.24 mmol), PyBOP (126.6 mg, 0.24 mmol), and DIPEA (78.6 mg, 0.61 mmol) in dry DCM (total amount 10 mL) according to general procedure F (reaction time 3 h). Purification by flash column chromatography (SiO2, DCM–MeOH gradient elution from 95.5:4.5 to 92:8) gave 54 mg of a white solid (70% yield). 1H-NMR shows a 3:2 mixture of amide bond rotamers: (300 MHz, DMSO-d6) δ 12.31–12.16 (m, 1H), 8.48–8.37 (m, 1H), 7.89–7.77 (m, 1H), 7.53–7.43 (m, 1H), 7.32–7.18 (m, 1H), 4.58–4.44 (m, 0.6H), 4.41–4.19 (m, 1.4H), 4.16–3.98 (m, 2H), 3.93–3.81 (m, 0.4H), 3.71–3.56 (m, 0.6H), 3.29–3.17 (m, 3H), 3.10–2.94 (m, 1.2H), 2.70–2.56 (m, 0.4H), 2.10–1.73 (m, 3H), 1.68–1.42 (m, 1H), missing 0.4H below water signal; ESI-MS: (m/z) 405.1 [M + Na]+, 380.9 [M − H]−; HPLC method A: tr = 6.679 min.
The title compound was prepared from (S)-7d (65.0 mg, 0.18 mmol), cyanoacetic acid (19.2 mg, 0.23 mmol), TBTU (72.4 mg, 0.23 mmol), and DIPEA (70.0 mg, 0.54 mmol) in dry DCM (total amount 12 mL) according to general procedure F (reaction time 30 min). Purification by flash column chromatography (SiO2, DCM–MeOH 94.5:5.5) gave 48 mg of an off-white solid (62% yield). 1H-NMR shows a 3:2 mixture of amide bond rotamers. 1H-NMR (300 MHz, DMSO-d6) δ 12.32–12.12 (m, 1H), 8.49–8.37 (m, 1H), 7.83–7.72 (m, 1H), 7.67–7.57 (m, 1H), 7.44–7.30 (m, 1H), 4.56–4.44 (m, 0.6H), 4.41–4.19 (m, 1.4H), 4.17–3.99 (m, 2H), 3.93–3.81 (m, 0.4H), 3.69–3.58 (m, 0.6H), 3.32–3.14 (m, 3.4H), 3.11–2.95 (m, 1.2H), 2.70–2.56 (m, 0.4H), 2.10–1.73 (m, 3H), 1.67–1.41 (m, 1H); ESI-MS: (m/z) 449.3 [M + Na]+, 425.3 [M − H]−; HPLC method A: tr = 6.649 min.
(10) Detailed Procedures for the Preparation of Intermediates 43 and 44·HCl
tert-Butyl piperidin-3-yl-carbamate (42) (1.0 g, 4.99 mmol) and cyanoacetic acid (470.0 mg, 5.49 mmol) were stirred in dry DCM (15 mL) at 0°C and under N2 atmosphere. A solution of DCC (1.1 g, 5.49 mmol) in dry DCM (11 mL) was drop-added. The mixture was stirred at rt overnight and then filtered rinsing the residue with fresh DCM. The filtrate was concentrated under reduced pressure. Purification of the residue by flash column chromatography (SiO2, DCM–EtOAc 7:3) gave 942 mg (71% yield); 1H-NMR shows a 3:2 mixture of amide bond rotamers. 1H-NMR (300 MHz, DMSO-d6) δ 7.08–6.49 (m, 1H), 4.10–3.88 (m, 2.6H), 3.81–3.67 (m, 0.4H), 3.53–3.19 (m, 2H, overlap with water signal), 3.06–2.91 (m, 1.4H), 2.69–2.58 (m, 0.6H), 1.85–1.36 (m, 13H).
4N HCl in dioxane (2.1 mL) was added to a solution of 43 (200.0 mg, 0.75 mmol) in dry THF (2 mL). The mixture was stirred at rt overnight. The resulting precipitate was filtered off, washed with Et2O, and dried under reduced pressure. The yield was 130 mg (96% crude yield), used in the next step without further purification. 1H-NMR shows a 3:2 mixture of amide bond rotamers. 1H-NMR (400 MHz, DMSO-d6) δ 8.61–8.25 (m, 3H), 4.26–3.95 (m, 2.4H), 3.70–3.64 (m, 0.6H), 3.55–3.50 (m, 0.4H), 3.45–3.36 (m, 1H), 3.31–3.04 (m, 2.6H), 2.03–1.89 (m, 1H), 1.77–1.62 (m, 2H), 1.54–1.36 (m, 1H); 13C NMR (101 MHz, DMSO-d6) δ 162.1, 161.7, 116.2, 116.1, 47.7, 46.1, 46.0, 45.3, 43.9, 41.6, 27.4, 27.1, 25.3, 25.2, 25.1, 22.0, 21.1.
(11) Detailed Procedures for the Preparation of Final Compounds 45–50
The title compound was prepared from 4a (100.0 mg, 0.27 mmol), 44·HCl (113.8 mg, 0.56 mmol), DIPEA (181.0 mg, 1.40 mmol), and NatBuO (188.0 mg, 1.96 mmol) in dry DMF (5 mL) according to general procedure G. Purification by flash column chromatography (SiO2, DCM–EtOH gradient elution from 97:3 to 85:15) gave 34 mg (36% yield). 1H-NMR shows a 5:4 mixture of amide bond rotamers. 1H-NMR (300 MHz, DMSO-d6) δ 11.98–11.84 (m, 1H), 8.42–8.34 (m, 1H), 8.33–8.26 (m, 2H), 7.50–7.43 (m, 1H), 7.42–7.33 (m, 1H), 7.29–7.20 (m, 1H), 6.86–6.63 (m, 1H), 4.51–4.23 (m, 2H), 4.16–3.98 (m, 2H), 3.93–3.83 (m, 0.45H), 3.69–3.57 (m, 0.55H), 3.21–3.11 (m, 0.45H), 3.08–2.97 (m, 0.55H), 2.94–2.84 (m, 0.55H), 2.73–2.60 (m, 0.45H), 2.13–1.97 (m, 1H), 1.92–1.72 (m, 2H), 1.69–1.42 (m, 1H); ESI-MS: (m/z) 335.1 [M + H]+, 357.0 [M + Na]+, 332.9 [M − H]−; HPLC method B: tr = 2.649 min.
The title compound was prepared from 4b (100.0 mg, 0.27 mmol), 44·HCl (81.3 mg, 0.40 mmol), DIPEA (171.9 mg, 1.33 mmol), and NatBuO (179.0 mg, 1.86 mmol) in dry DMF (5 mL) according to general procedure G. The precipitate formed upon addition of saturated NH4Cl solution was not extracted with EtOAc but instead filtered off, washed with water, and dried over P2O5 in vacuo. Purification by flash column chromatography (SiO2, DCM–EtOH gradient elution from 94:6 to 9:1) gave 29 mg (31% yield). 1H-NMR shows a 5:4 mixture of amide bond rotamers. 1H-NMR (300 MHz, DMSO-d6) δ 12.14–11.96 (m, 1H), 8.41–8.27 (m, 2H), 7.26–7.18 (m, 1H), 7.17–7.06 (m, 1H), 6.92–6.73 (m, 1H), 4.49–4.23 (m, 2H), 4.16–3.97 (m, 2H), 3.93–3.83 (m, 0.45H), 3.70–3.59 (m, 0.55H), 3.20–3.08 (m, 0.45H), 3.08–2.95 (m, 0.55H), 2.90–2.80 (m, 0.55H), 2.72–2.61 (m, 0.45H), 2.13–1.95 (m, 1H), 1.88–1.74 (m, 2H), 1.68–1.42 (m, 1H); 13C NMR (101 MHz, DMSO-d6) δ 161.59, 161.56, 161.5, 161.4, 159.1, 159.1, 156.1, 155.6, 155.6, 154.4, 154.4, 137.1, 137.0, 122.9, 122.83, 122.80, 122.7, 116.2, 116.1, 116.02, 115.99, 107.9, 107.6, 97.9, 97.6, 95.7, 95.5, 50.0, 47.5, 46.4, 46.3, 45.8, 42.2, 29.83, 29.75, 24.94, 24.86, 24.3, 23.7; ESI-MS: (m/z) 375.3 [M + Na]+, 351.1 [M − H]−; HPLC method B: tr = 3.525 min.
The title compound was prepared by a two-step procedure.
In the first step 4c (200.0 mg, 0.51 mmol), 44·HCl (135.0 mg, 0.66 mmol), and DIPEA (197.7 mg, 1.53 mmol) were reacted in dry DMF (3.5 mL) at 70 °C for 19 h. Additional 44·HCl (26.0 mg, 0.128 mmol) was added, and stirring at 70 °C continued for 6 h. After cooling down to rt, the mixture was poured into ice-cold water and saturated NH4Cl solution was added (30 mL). The resulting precipitate was filtered off, washed with water, and dried over P2O5 in vacuo. Purification by flash column chromatography (SiO2, DCM–MeOH 96.5:3.5) gave 104 mg of 3-(3-((7-Chloro-9-tosyl-9H-pyrimido[4,5-b]indol-4-yl)amino)piperidin-1-yl)-3-oxopropanenitrile as a pale yellow solid (39% yield). 1H-NMR shows a 5:4 mixture of amide bond rotamers. 1H-NMR (300 MHz, DMSO-d6) δ 8.55–8.44 (m, 1H), 8.42–8.30 (m, 2H), 8.00 (d, J = 8.4 Hz, 2H), 7.64–7.52 (m, 1H), 7.39 (d, J = 8.3 Hz, 2H), 7.22–7.05 (m, 1H), 4.44–4.19 (m, 2H), 4.15–3.92 (m, 2H), 3.84–3.74 (m, 0.45H), 3.67–3.57 (m, 0.55H), 3.16–3.06 (m, 0.45H), 3.05–2.92 (m, 0.55H), 2.87–2.75 (m, 0.55H), 2.70–2.55 (m, 0.45H), 2.32 (s, 3H), 2.07–1.90 (m, 1H), 1.90–1.68 (m, 2H), 1.68–1.35 (m, 1H); ESI-MS: (m/z) 544.8 [M + Na]+, 520.7 [M − H]−; HPLC method A: tr = 8.439 min.
The purified material obtained from the first step (91.0 mg, 0.17 mmol) was reacted with KtBuO (136.7 mg, 1.22 mmol) in dry THF (10 mL) according to general procedure D (reaction time 2 h). Purification by flash column chromatography (SiO2, DCM–MeOH gradient elution from 95:5 to 92:8) gave 41 mg of a white solid (64% yield). 1H-NMR shows a 5:4 mixture of amide bond rotamers. 1H-NMR (300 MHz, DMSO-d6) δ 12.07 (s, 1H), 8.49–8.26 (m, 2H), 7.54–7.41 (m, 1H), 7.36–7.21 (m, 1H), 6.99–6.78 (m, 1H), 4.51–4.24 (m, 2H), 4.17–3.97 (m, 2H), 3.93–3.82 (m, 045H), 3.72–3.57 (m, 0.55H), 3.20–3.09 (m, 0.45H), 3.08–2.95 (m, 0.55H), 2.90–2.79 (m, 0.55H), 2.74–2.59 (m, 0.45H), 2.15–1.96 (m, 1H), 1.91–1.72 (m, 2H), 1.71–1.38 (m, 1H); 13C NMR (101 MHz, DMSO-d6) δ 161.6, 161.4, 155.90, 155.85, 155.8, 155.03, 154.99, 137.0, 129.1, 129.0, 122.9, 122.8, 120.0, 118.22, 118.18, 116.2, 116.1, 110.7, 95.6, 95.4, 49.9, 47.5, 46.5, 46.2, 45.8, 42.2, 29.8, 29.7, 24.94, 24.86, 24.3, 23.7; ESI-MS: (m/z) 391.0 [M + Na]+, 366.9 [M − H]−; HPLC method A: tr = 6.023 min.
4d (50.0 mg, 0.11 mmol), 44·HCl (35.0 mg, 0.17 mmol), and DIPEA (73.7 mg, 0.57 mmol) were stirred in a solvent mixture of dry dioxane (1 mL) and dry DMF (0.1 mL) at 70 °C overnight. Additional 44·HCl (35.0 mg, 0.17 mmol) and DIPEA (73.7 mg, 0.57 mmol) were added, and stirring at 70 °C continued overnight. The mixture was concentrated under reduced pressure, the residue diluted with dry THF (4 mL). NatBuO (77.0 mg, 0.80 mmol) was added, and the mixture was stirred at rt for 1 h. Saturated NH4Cl solution (30 mL) was added, and the mixture was extracted with EtOAc (3 × 20 mL). Combined organic layers were dried over Na2SO4 and concentrated under reduced pressure. Purification of the residue by flash column chromatography (SiO2, DCM–EtOH gradient elution from 97:3 to 4:1) gave 25 mg (53% yield). 1H-NMR shows a 5:4 mixture of amide bond rotamers. 1H-NMR (400 MHz, DMSO-d6) δ 12.11–12.03 (m, 1H), 8.43–8.34 (m, 1H), 8.33–8.26 (m, 1H), 7.60 (s, 1H), 7.45–7.38 (m, 1H), 6.99–6.80 (m, 1H), 4.49–4.41 (m, 0.55H), 4.39–4.26 (m, 1.45H), 4.15–3.99 (m, 2H), 3.90–3.83 (m, 0.45H), 3.68–3.61 (m, 0.55H), 3.19–3.10 (m, 0.45H), 3.05–2.96 (m, 0.55H), 2.87–2.79 (m, 0.55H), 2.70–2.61 (m, 0.45H), 2.12–1.97 (m, 1H), 1.87–1.75 (m, 2H), 1.67–1.40 (m, 1H); 13C NMR (101 MHz, DMSO-d6) δ 161.5, 161.4, 155.9, 155.8, 155.7, 155.13, 155.09, 137.3, 123.2, 123.1, 122.7, 118.51, 118.47, 117.2, 117.1, 116.2, 116.1, 113.6, 95.6, 95.4, 49.9, 47.5, 46.5, 46.2, 45.8, 42.2, 29.8, 29.7, 24.9, 24.9, 24.3, 23.7; ESI-MS: (m/z) 434.8 [M + Na]+, 410.7 [M − H]−; HPLC method B: tr = 5.144 min.
The title compound was prepared from 4f (496.0 mg, 1.28 mmol), 44·HCl (392.2 mg, 1.92 mmol), DIPEA (828.5 mg, 6.41 mmol), and NatBuO (863.7 mg, 8.97 mmol) in dry DMF (20 mL) according to general procedure G but was stirred at rt for 3 d after addition of NatBuO. Purification by flash column chromatography (SiO2, DCM–EtOH gradient elution from 97:3 to 4:1) gave 70 mg (15% yield). 1H-NMR shows a 5:4 mixture of amide bond rotamers. 1H-NMR (400 MHz, DMSO-d6) δ 11.88–11.77 (m, 1H), 8.37–8.27 (m, 1H), 8.22–8.16 (m, 1H), 6.98–6.94 (m, 1H), 6.90–6.84 (m, 1H), 6.74–6.57 (m, 1H), 4.48–4.24 (m, 2H), 4.15–4.00 (m, 2H), 3.91–3.81 (m, 3.45H), 3.67–3.59 (m, 0.55H), 3.18–3.09 (m, 0.45H), 3.06–2.96 (m, 0.55H), 2.89–2.82 (m, 0.55H), 2.71–2.61 (m, 0.45H), 2.11–1.98 (m, 1H), 1.88–1.74 (m, 2H), 1.66–1.40 (m, 1H); 13C NMR (101 MHz, DMSO-d6) δ 161.5, 161.4, 157.7, 157.6, 155.4, 155.2, 155.1, 153.5, 153.4, 137.7, 122.4, 122.2, 116.2, 116.1, 112.9, 112.8, 108.8, 96.1, 95.9, 95.0, 55.3, 50.1, 47.5, 46.3, 45.8, 42.2, 29.84, 29.76, 24.9, 24.8, 24.2, 23.7; ESI-MS: (m/z) 387.0 [M + Na]+, 363.1 [M − H]−; HPLC method B: tr = 2.768 min.
The title compound was prepared from 4g (180.0 mg, 0.42 mmol), 44·HCl (129.1 mg, 0.63 mmol), DIPEA (272.7 mg, 2.11 mmol), and NatBuO (284.4 mg, 2.96 mmol) in dry DMF (10 mL) according to general procedure G. Purification by flash column chromatography (SiO2, 1.DCM–EtOH gradient elution from 97:3 to 4:1, 2.DCM–(2N NH3 in MeOH) gradient elution from 99:1 to 92:8) gave 12 mg (7% yield). 1H-NMR shows a 5:4 mixture of amide bond rotamers. 1H-NMR (300 MHz, DMSO-d6) δ 12.31 (s, 1H), 8.60–8.51 (m, 1H), 8.50–8.39 (m, 1H), 7.73 (s, 1H), 7.62–7.53 (m, 1H), 7.17–6.98 (m, 1H), 4.52–4.24 (m, 2H), 4.16–3.97 (m, 2H), 3.93–3.83 (m, 1H), 3.71–3.61 (m, 1H), 3.22–3.12 (m, 1H), 3.08–2.96 (m, 1H), 2.92–2.82 (m, 1H), 2.73–2.62 (m, 1H), 2.15–1.97 (m, 1H), 1.92–1.74 (m, 2H), 1.70–1.40 (m, 1H); ESI-MS: (m/z) 424.9 [M + Na]+, 401.0 [M − H]−; HPLC method B: tr = 5.880 min.

 ,
, 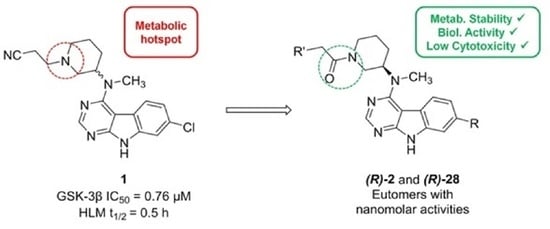

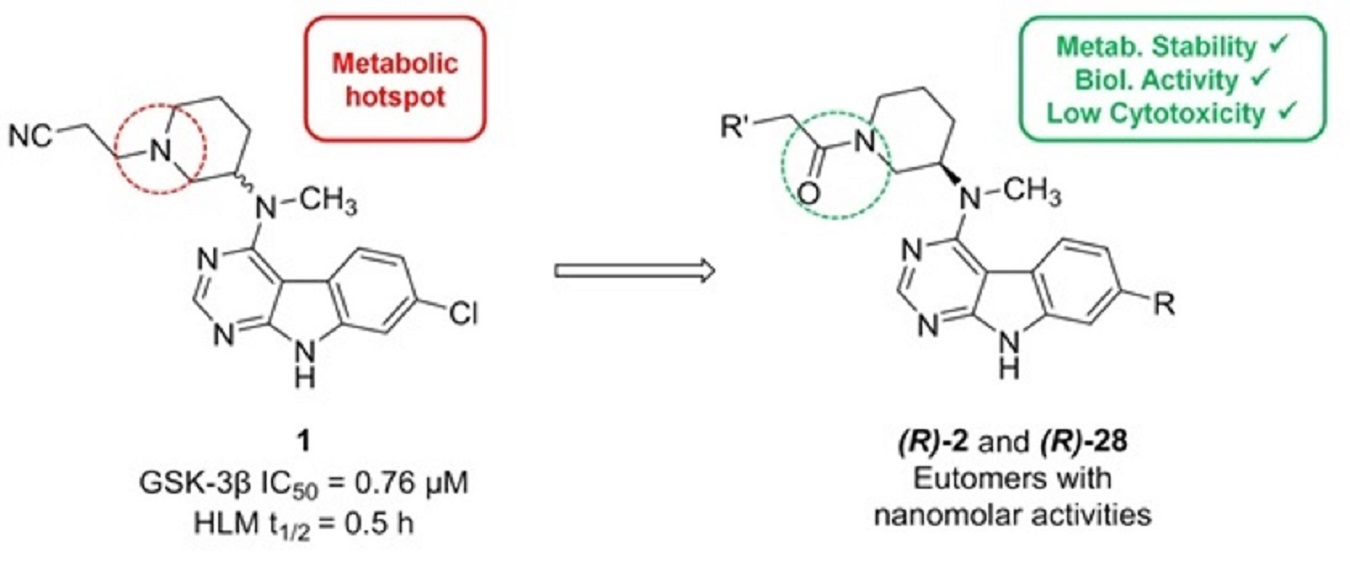{kind=link}
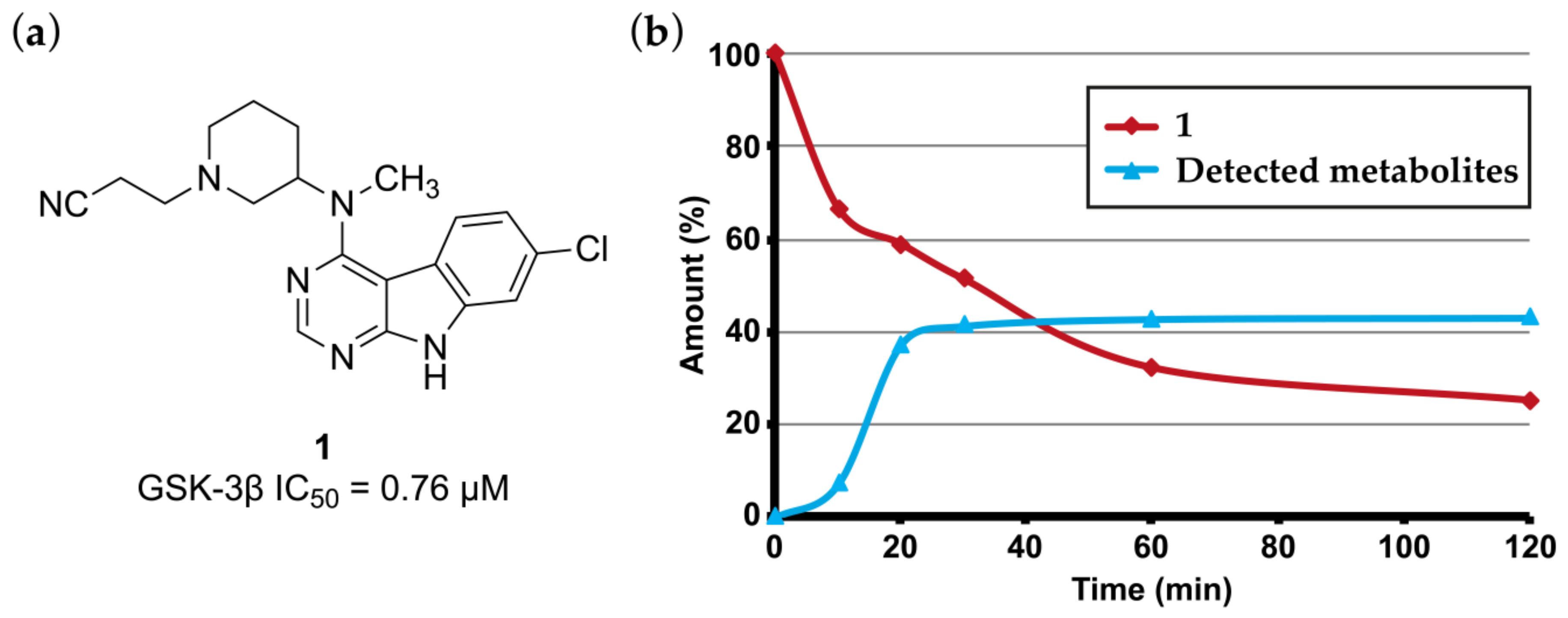{kind=link}
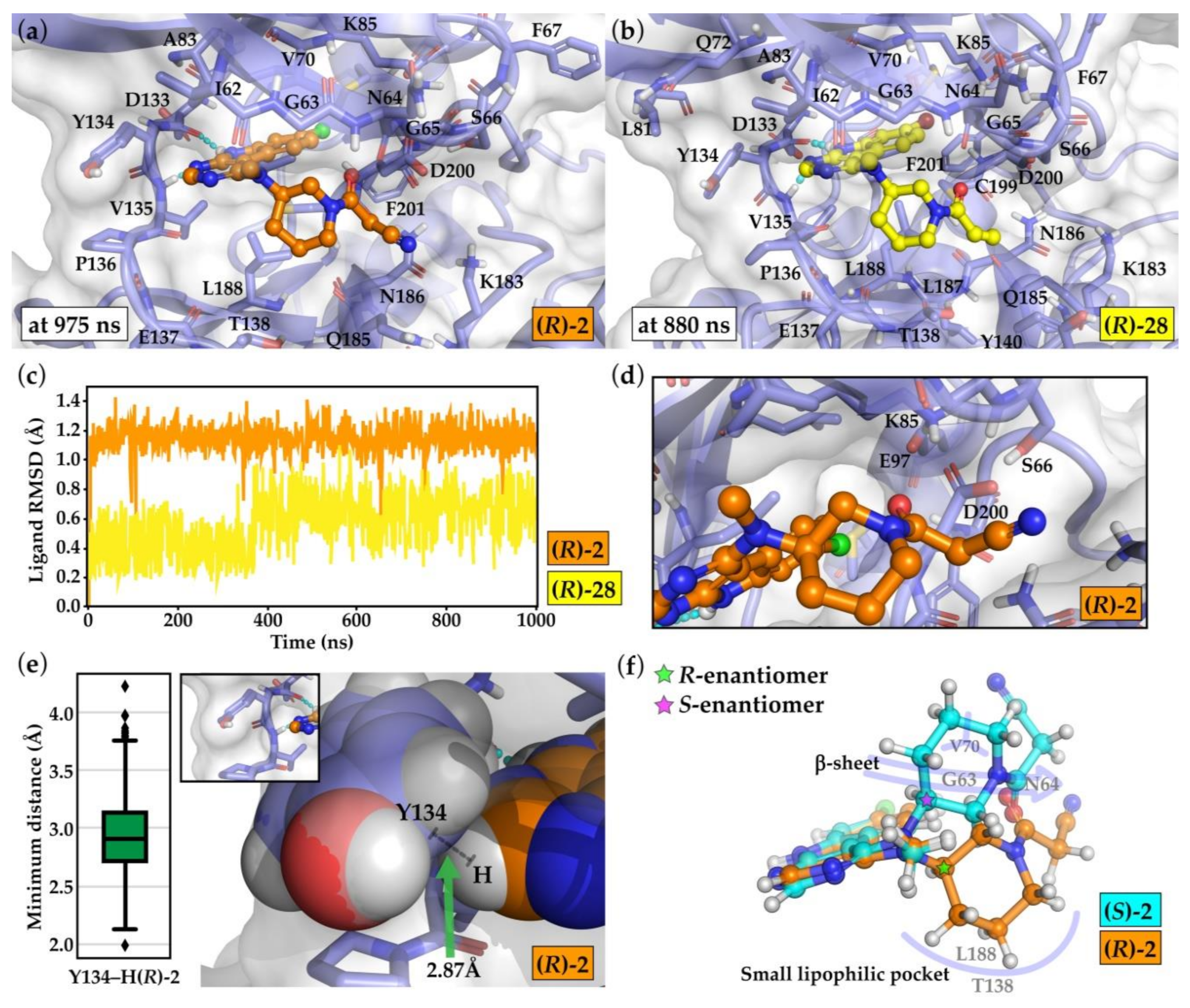{kind=link}
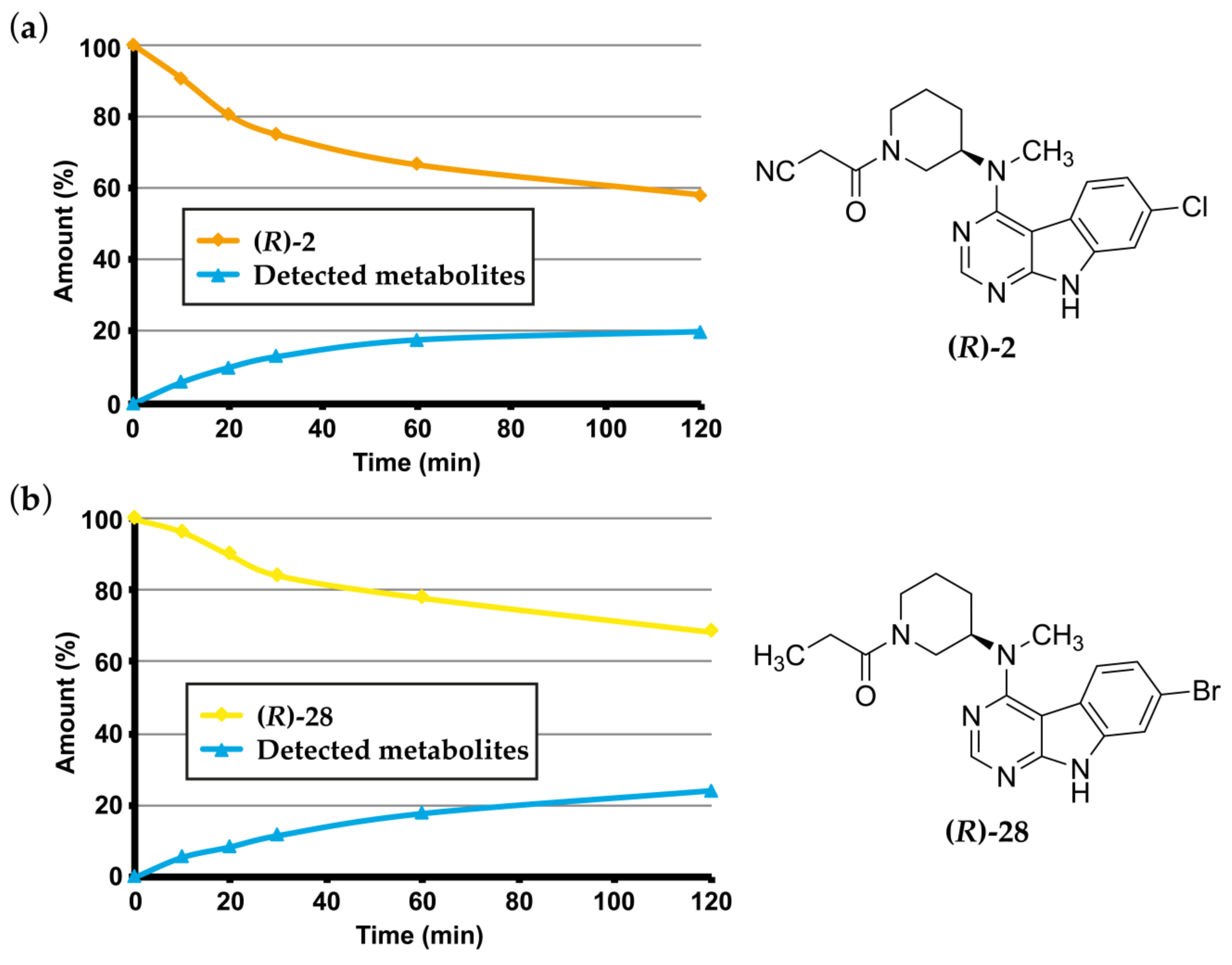{kind=link}
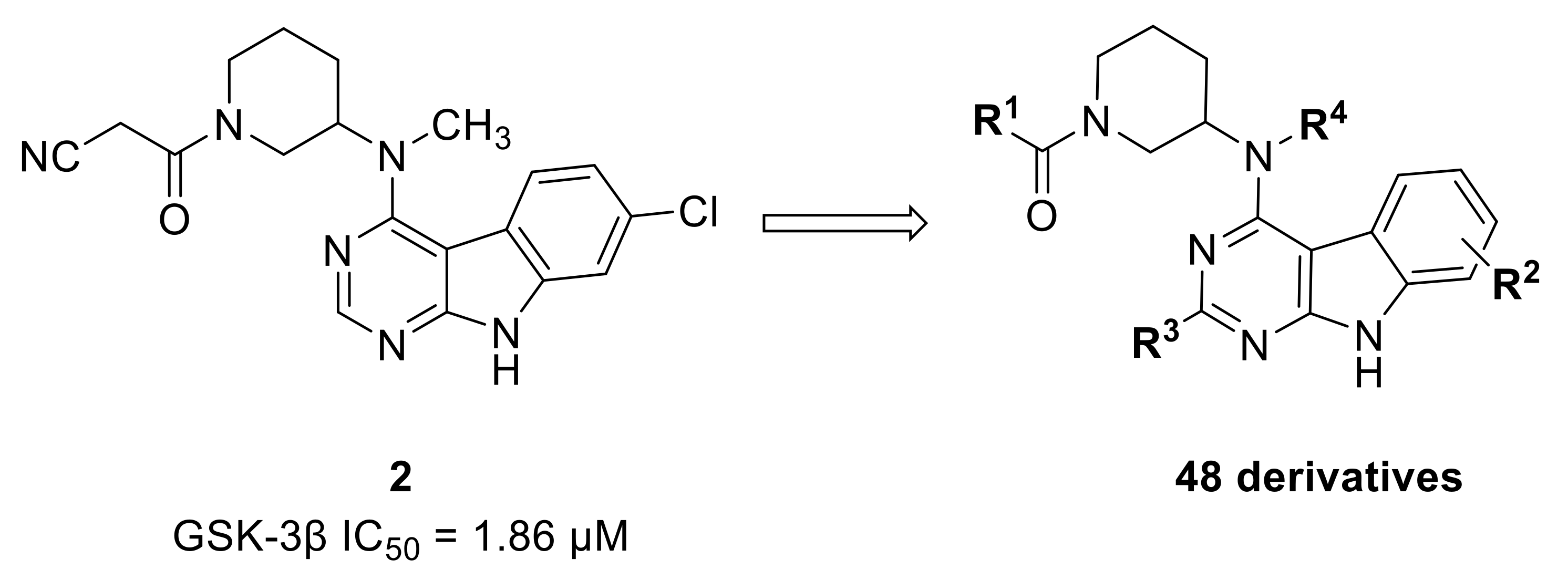{kind=link}
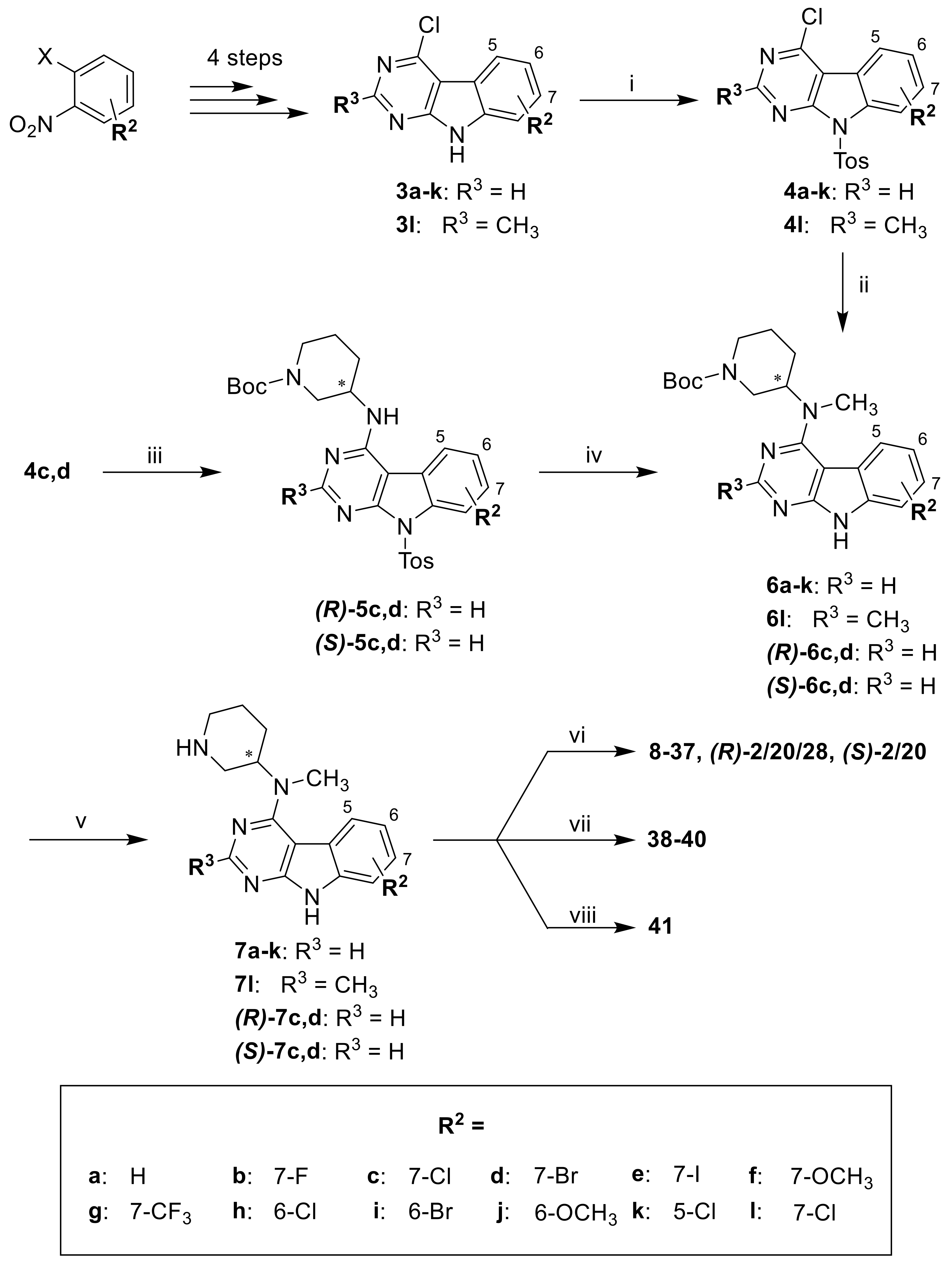{kind=link}
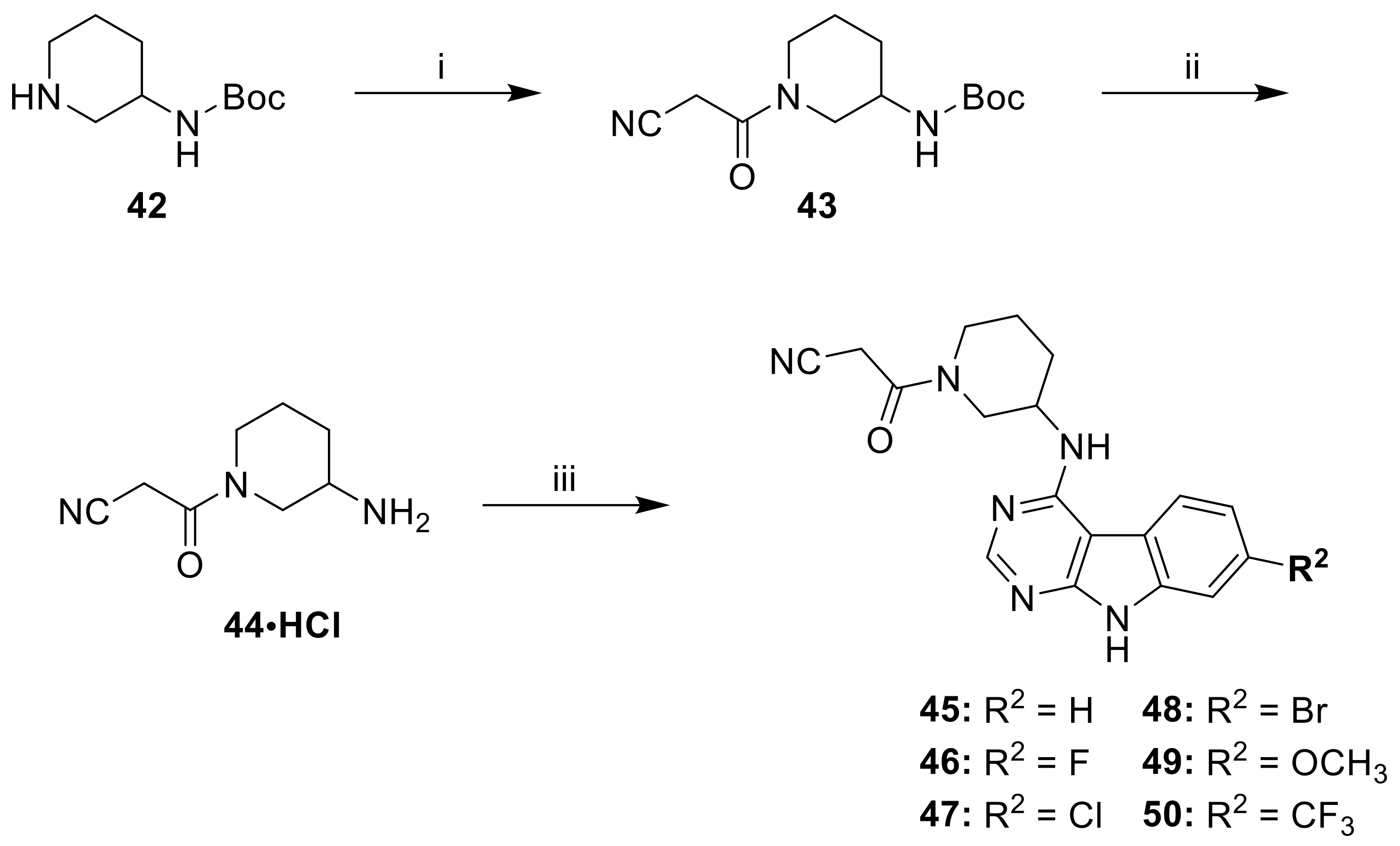{kind=link}


























