Damage-Associated Molecular Patterns and Their Signaling Pathways in Primary Blast Lung Injury: New Research Progress and Future Directions


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
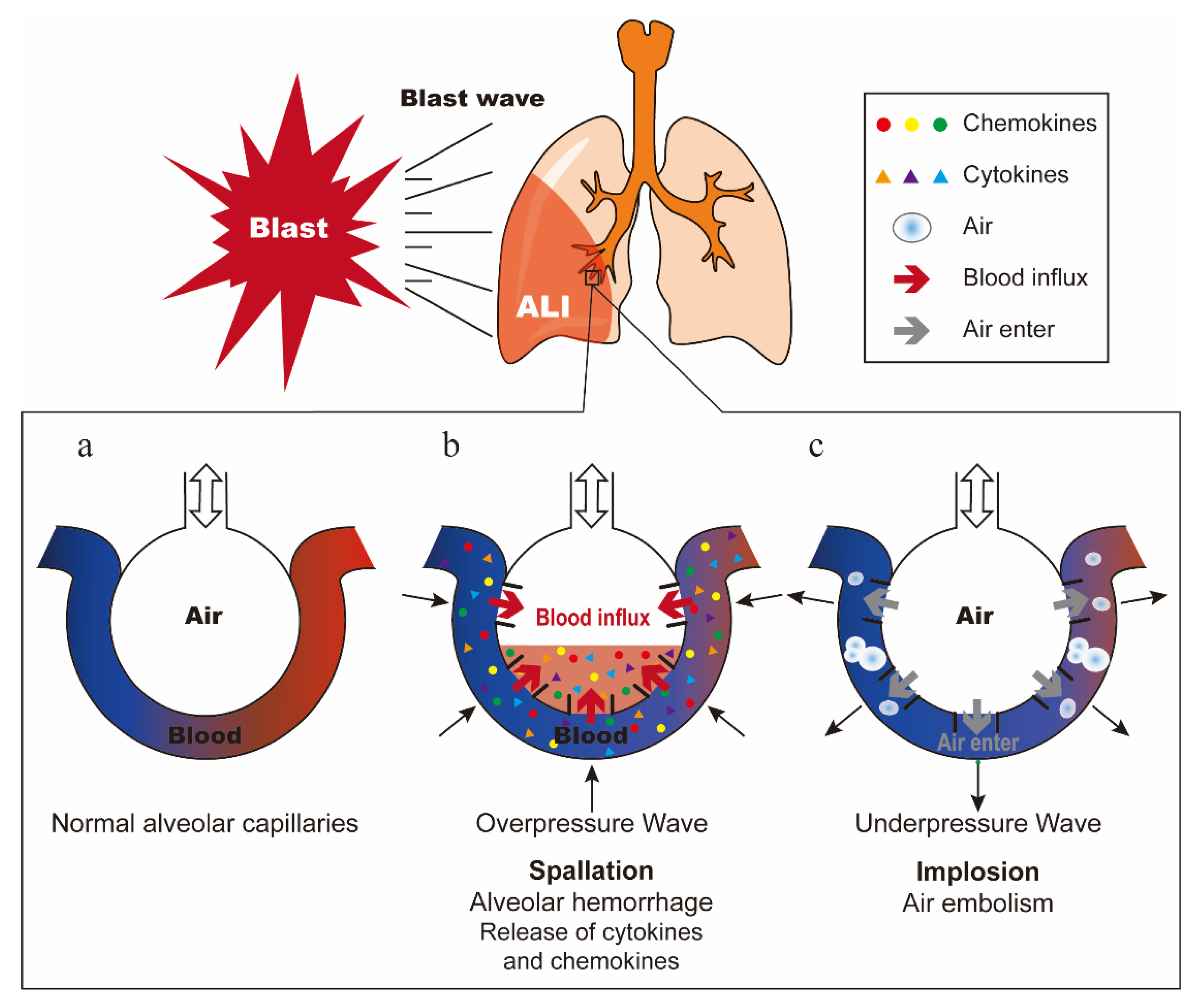
2. Overview of Primary Blast Lung Injury
2.1. The Mechanism of Primary Blast Lung Injury
2.2. Pathophysiological Characteristics and Clinical Diagnosis of Primary Blast Lung Injury
2.3. The Medical Treatment of Primary Blast Lung Injury
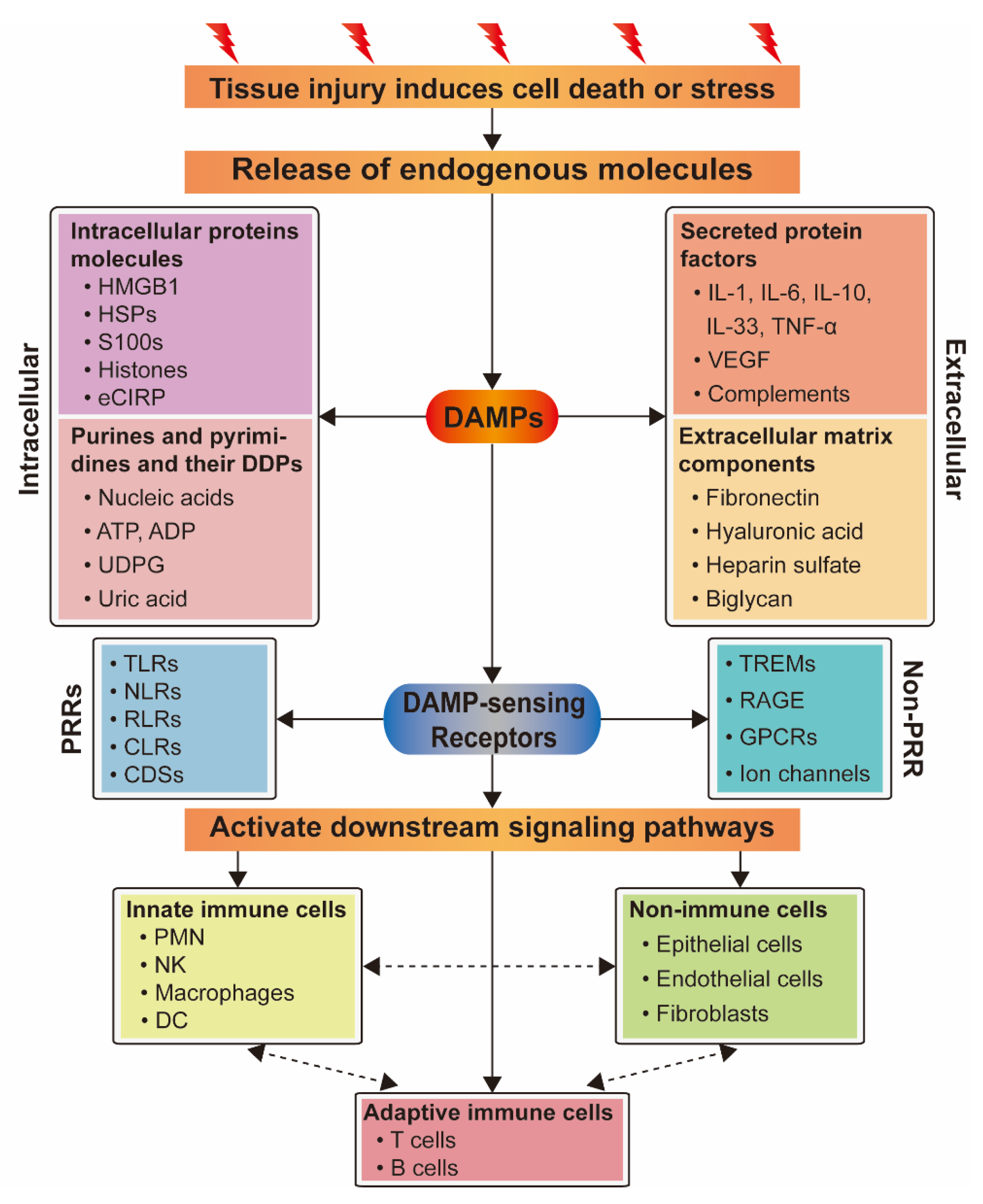
3. DAMPs and Their Sensing Receptors
3.1. Damage-Associated Molecular Pattern
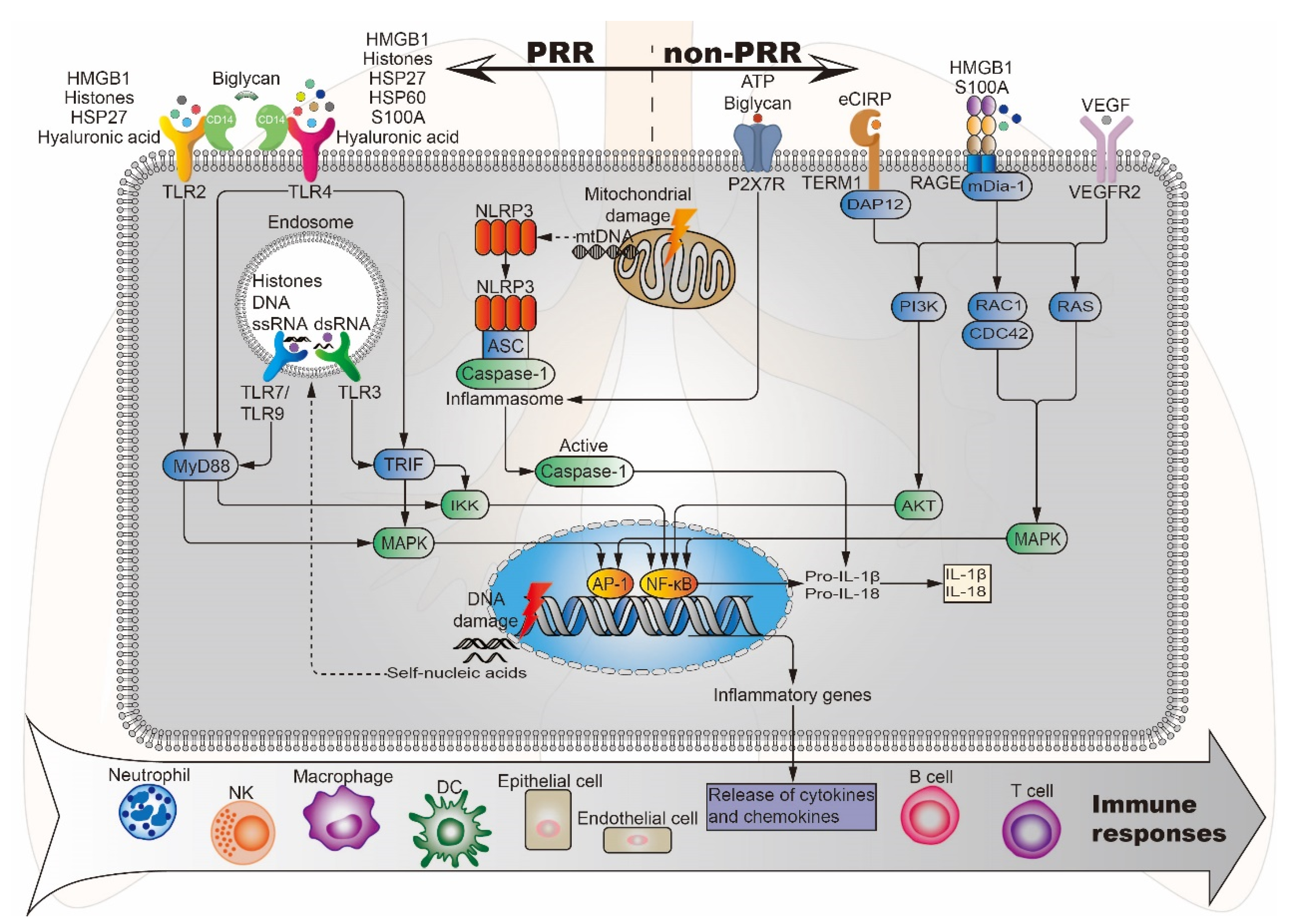
3.2. DAMP-Sensing Receptors
3.2.1. Pattern Recognition Receptors Bound by Damage-Associated Molecular Patterns
3.2.2. Other Receptors/Channels Bound by DAMPs
3.3. Crosstalk Between DAMP and Their Sensing Receptors
4. DAMPs and Their Signaling Pathways Related to PBLI
4.1. Intracellular Protein Molecules
4.1.1. Nuclear Protein HMGB1
4.1.2. Histones
4.1.3. Calcium Binding Protein S100A
4.1.4. Heat Shock Proteins
4.1.5. Cold-Inducible RNA-Binding Protein
4.2. Secretory Protein Factors
4.2.1. Cytokines IL-1, IL-6, IL-10, IL-33, and TNF-α
4.2.2. Vascular Endothelial Growth Factor
4.2.3. Complements
4.3. Purines and Pyrimidines and Their Derived Degradation Products
Nucleic Acids
4.4. Extracellular Matrix Components
4.4.1. Hyaluronic Acid
4.4.2. Biglycan
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AECs | Alveolar Epithelial Cells |
| AGEs | Advanced Glycation End Products |
| AIM2 | Absent in Melanoma 2 |
| ALI | Acute Lung Injuries |
| ARDS | Acute Respiratory Distress Syndrome |
| ASC | Apoptosis-Associated Speck-Like Protein |
| ASK1 | Apoptosis Signal-Regulating Kinase-1 |
| ATP | Adenosine Triphosphate |
| BALF | Bronchoalveolar Lavage Fluid |
| BGN | Biglycan |
| BOP | Blast Overpressure |
| bTBI | Blast-Induced Traumatic Brain Injury |
| CaSRs | Calcium-Sensing Receptors |
| CDSs | Cytoplasmic DNA Sensors |
| cGAMP | Cyclic GMP-AMP |
| cGAS | GMP–AMP Synthase |
| CLRs | C-Type Lectin Receptors |
| ComC | Complement Cascade |
| COVID-19 | Coronavirus Disease 2019 |
| CRS | Cytokine Release Syndrome |
| CSIF | Cytokine Synthesis Inhibitory Factor |
| DAF | Decay-Accelerating Factor |
| DAMP | Damage-Associated Molecular Pattern |
| DCs | Dendritic Cells |
| DDPs | Derived Degradation Products |
| DNGR1 | Dendritic Cell Natural Killer Lectin Group Receptor 1 |
| eCIRP | Extracellular Cold-Inducible RNA-Binding Protein |
| ECM | Extracellular Matrix |
| FE | Fat Embolism |
| FPRs | N-Formyl Peptide Receptors |
| GGO | Ground-Glass Opacity |
| GPCRs | G-Protein-Coupled Receptors |
| GPRC6A | G-Protein-Coupled Receptor Family C Group 6 Member A |
| HA | Hyaluronic Acid |
| Hb | Hemoglobin |
| HCVECs | Human Coronary Vascular Endothelial Cells |
| HMGB1 | High Mobility Group Box 1 |
| HMW-HA | High Molecular Weight Hyaluronic Acid |
| HSPs | Heat Shock Proteins |
| ICAM | Intercellular Adhesion Molecule |
| IFN-I | Type I Interferon |
| IFN-α | Interferon-Alpha |
| IgV | Immunoglobulin Variable |
| IL-10R1 | IL-10 Receptor 1 |
| IL-1ra | IL-1 receptor antagonist |
| IL-6 | Interleukin 6 |
| IL-6R | Interleukin-6 Receptor |
| iNOS | Inducible Nitric Oxide Synthase |
| IRAK | IL-1 Receptor-Associated Kinase |
| IRI | Ischemia/Reperfusion Injury |
| JNK | C-Jun N-Terminal Kinase |
| LATS1/2 | Large Tumor Suppressor Kinase 1/2 |
| LDL | Low-Density Lipoprotein |
| LMW-HA | Low Molecular Weight Hyaluronic Acid |
| LPS | Lipopolysaccharide |
| MBL | Mannan-Binding Lectin |
| MCP | Monocyte Chemoattractant Protein |
| MINCLE | Macrophage-Inducible C-type Lectin |
| MOF | Multiple Organ Failure |
| MSCs | Mesenchymal Stem Cells |
| mtDNA | Mitochondrial DNA |
| MyD88 | Myeloid Differentiation Protein 88 |
| NET | Neutrophil Extracellular Trap |
| NF-kB | Nuclear Factor-κB |
| NKs | Natural Killer Cells |
| NLRs | NOD-Like Receptors |
| ox-mtDNA | Oxidized Mitochondrial DNA |
| P2X7R | P2X7 Receptor |
| P2YRs | P2Y Receptors |
| PBI | Primary Blast Injury |
| PBLI | Primary Blast Lung Injury |
| PFC | Perfluorocarbon |
| PKR | Double-Stranded RNA-Dependent Protein Kinase |
| PLGF | Placental Growth Factor |
| PMNs | Polymorphonuclear Neutrophils |
| PRRs | Pattern Recognition Receptors |
| RAGE | Receptor for Advanced Glycation End Products |
| RIG-I | Retinoic Acid-Inducible Gene-I |
| RLRs | Retinoic Acid-Inducible Gene-I Like Receptors |
| ROS | Reactive Oxygen Species |
| S100s | S100 Proteins |
| SOFA | Sequential Organ Failure Assessment |
| STING | Stimulator of Interferon Genes |
| TAZ | Transcriptional Coactivator with PDZ-Binding Motif |
| TBK1 | TANK-Binding Kinase 1 |
| TLRs | Toll-Like Receptors |
| TNF-α | Tumor Necrosis Factor-α |
| TREMs | Triggering Receptors Expressed on Myeloid Cells |
| TRP | Transient Receptor Potential |
| UDPG | Uridine Diphosphate Glucose |
| VEGF | Vascular Endothelial Growth Factor |
| VEGFR | Vascular Endothelial Growth Factor Receptor |
References
- Wild, H.; Stewart, B.T.; LeBoa, C.; Stave, C.D.; Wren, S.M. Epidemiology of Injuries Sustained by Civilians and Local Combatants in Contemporary Armed Conflict: An Appeal for a Shared Trauma Registry Among Humanitarian Actors. World J. Surg. 2020, 44, 1863–1873. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Du, J.; Zhuang, Z.; Wang, Z.G.; Jiang, J.X.; Yang, C. Incidence, casualties and risk characteristics of civilian explosion blast injury in China: 2000–2017 data from the state Administration of Work Safety. Mil. Med. Res. 2020, 7, 29. [Google Scholar] [CrossRef] [PubMed]
- Wolf, S.J.; Bebarta, V.S.; Bonnett, C.J.; Pons, P.T.; Cantrill, S.V. Blast injuries. Lancet 2009, 374, 405–415. [Google Scholar] [CrossRef]
- Mathews, Z.R.; Koyfman, A. Blast Injuries. J. Emerg. Med. 2015, 49, 573–587. [Google Scholar] [CrossRef]
- Scott, T.E.; Kirkman, E.; Haque, M.; Gibb, I.E.; Mahoney, P.; Hardman, J.G. Primary blast lung injury—A review. Br. J. Anaesth. 2017, 118, 311–316. [Google Scholar] [CrossRef] [Green Version]
- Mackenzie, I.M.; Tunnicliffe, B. Blast injuries to the lung: Epidemiology and management. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2011, 366, 295–299. [Google Scholar] [CrossRef]
- Sziklavari, Z.; Molnar, T.F. Blast injures to the thorax. J. Thorac. Dis. 2019, 11, S167–S171. [Google Scholar] [CrossRef]
- Westrol, M.S.; Donovan, C.M.; Kapitanyan, R. Blast Physics and Pathophysiology of Explosive Injuries. Ann. Emerg. Med. 2017, 69, S4–S9. [Google Scholar] [CrossRef]
- Jani, N.; Falvo, M.J.; Sotolongo, A.; Osinubi, O.Y.; Tseng, C.L.; Rowneki, M.; Montopoli, M.; Morley, S.W.; Mitchell, V.; Helmer, D.A. Blast Injury and Cardiopulmonary Symptoms in U.S. Veterans: Analysis of a National Registry. Ann. Intern. Med. 2017, 167, 753–755. [Google Scholar] [CrossRef]
- Fievisohn, E.; Bailey, Z.; Guettler, A.; VandeVord, P. Primary Blast Brain Injury Mechanisms: Current Knowledge, Limitations, and Future Directions. J. Biomech. Eng. 2018, 140. [Google Scholar] [CrossRef]
- Butt, Y.; Kurdowska, A.; Allen, T.C. Acute Lung Injury: A Clinical and Molecular Review. Arch. Pathol. Lab. Med. 2016, 140, 345–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Land, W. Allograft injury mediated by reactive oxygen species: From conserved proteins of Drosophila to acute and chronic rejection of human transplants. Part III: Interaction of (oxidative) stress-induced heat shock proteins with toll-like receptor-bearing cells of innate immunity and its consequences for the development of acute and chronic allograft rejection. Transplant. Rev. 2003, 17, 67–86. [Google Scholar] [CrossRef]
- Zedler, S.; Faist, E. The impact of endogenous triggers on trauma-associated inflammation. Curr. Opin. Crit. Care 2006, 12, 595–601. [Google Scholar] [CrossRef] [PubMed]
- Relja, B.; Mors, K.; Marzi, I. Danger signals in trauma. Eur. J. Trauma Emerg. Surg. Off. Publ. Eur. Trauma Soc. 2018, 44, 301–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandolfi, F.; Altamura, S.; Frosali, S.; Conti, P. Key Role of DAMP in Inflammation, Cancer, and Tissue Repair. Clin. Ther. 2016, 38, 1017–1028. [Google Scholar] [CrossRef] [Green Version]
- Schardin, H. The Physical Principles of the Effects of a Detonation; German Aviation Medicine, World War II: Department of the US Air Force, Office of the Surgeon General: Washington, DC, USA, 1950; pp. 1207–1224. [Google Scholar]
- Ho, A.M. A simple conceptual model of primary pulmonary blast injury. Med. Hypotheses 2002, 59, 611–613. [Google Scholar] [CrossRef]
- Yeh, D.D.; Schecter, W.P. Primary blast injuries--an updated concise review. World J. Surg. 2012, 36, 966–972. [Google Scholar] [CrossRef]
- Guzzi, L.M.; Argyros, G. The management of blast injury. Eur. J. Emerg. Med. Off. J. Eur. Soc. Emerg. Med. 1996, 3, 252–255. [Google Scholar] [CrossRef]
- Ho, A.M.; Ling, E. Systemic air embolism after lung trauma. Anesthesiology 1999, 90, 564–575. [Google Scholar] [CrossRef]
- Gorbunov, N.V.; Asher, L.V.; Ayyagari, V.; Atkins, J.L. Inflammatory leukocytes and iron turnover in experimental hemorrhagic lung trauma. Exp. Mol. Pathol. 2006, 80, 11–25. [Google Scholar] [CrossRef]
- Kirkman, E.; Watts, S. Characterization of the response to primary blast injury. Philos. Trans. R. Soci. Lond. Ser. B Biol. Sci. 2011, 366, 286–290. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.E.; Garner, J. Pathophysiology of primary blast injury. J. R. Army Med. Corps 2019, 165, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.H.; Guo, G.H. Advances in the research of blast lung injury. Chin. J. Burn. 2016, 32, 156–159. [Google Scholar] [CrossRef]
- Oikonomou, A.; Thiessen, R.; Prassopoulos, P. CT findings in blast lung injury. BMJ Case Rep. 2012, 2012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avidan, V.; Hersch, M.; Armon, Y.; Spira, R.; Aharoni, D.; Reissman, P.; Schecter, W.P. Blast lung injury: Clinical manifestations, treatment, and outcome. Am. J. Surg. 2005, 190, 927–931. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.F.; Zhu, F.; Fang, H.; Xu, D.Y.; Xu, L.; Chen, T.S.; Zheng, Y.J.; Xiao, S.C.; Xia, Z.F. Management of combined massive burn and blast injury: A 20-year experience. Burns. J. Int. Soc. Burn. Inj 2020, 46, 75–82. [Google Scholar] [CrossRef]
- Scott, T.E.; Das, A.; Haque, M.; Bates, D.G.; Hardman, J.G. Management of primary blast lung injury: A comparison of airway pressure release versus low tidal volume ventilation. Intensive Care Med. Exp. 2020, 8, 26. [Google Scholar] [CrossRef]
- McDonald Johnston, A.; Alderman, J.E. Thoracic Injury in Patients Injured by Explosions on the Battlefield and in Terrorist Incidents. Chest 2020, 157, 888–897. [Google Scholar] [CrossRef]
- Singh, S.K.; Kumar, A.; Katyal, S. A terrorist bomb blast, a real challenge for any tertiary care health provider. Anesth. Essays Res. 2014, 8, 229–232. [Google Scholar] [CrossRef]
- Byrnes, D.; Masterson, C.H.; Artigas, A.; Laffey, J.G. Mesenchymal Stem/Stromal Cells Therapy for Sepsis and Acute Respiratory Distress Syndrome. Semin. Respir. Crit. Care Med. 2020. [Google Scholar] [CrossRef]
- Sadeghian Chaleshtori, S.; Mokhber Dezfouli, M.R.; Jabbari Fakhr, M. Mesenchymal stem/stromal cells: The therapeutic effects in animal models of acute pulmonary diseases. Respir. Res. 2020, 21, 110. [Google Scholar] [CrossRef] [PubMed]
- Relja, B.; Land, W.G. Damage-associated molecular patterns in trauma. Eur. J. Trauma Emerg. Surg. Off. Publ. Eur. Trauma Soc. 2019, 46, 751–775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, S. Danger-Associated Molecular Patterns (DAMPs): The Derivatives and Triggers of Inflammation. Curr. Allergy Asthma Rep. 2018, 18, 63. [Google Scholar] [CrossRef] [PubMed]
- Krysko, D.V.; Agostinis, P.; Krysko, O.; Garg, A.D.; Bachert, C.; Lambrecht, B.N.; Vandenabeele, P. Emerging role of damage-associated molecular patterns derived from mitochondria in inflammation. Trends Immunol. 2011, 32, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Frevert, C.W.; Felgenhauer, J.; Wygrecka, M.; Nastase, M.V.; Schaefer, L. Danger-Associated Molecular Patterns Derived from the Extracellular Matrix Provide Temporal Control of Innate Immunity. J. Histochem. Cytochem. Off. J. Histochem. Soc. 2018, 66, 213–227. [Google Scholar] [CrossRef] [PubMed]
- Vénéreau, E.; Ceriotti, C.; Bianchi, M.E. DAMPs from Cell Death to New Life. Front. Immunol. 2015, 6, 422. [Google Scholar] [CrossRef] [Green Version]
- Bianchi, M.E. DAMPs, PAMPs and alarmins: All we need to know about danger. J. Leukoc. Biol. 2007, 81, 1–5. [Google Scholar] [CrossRef]
- Chen, G.Y.; Nuñez, G. Sterile inflammation: Sensing and reacting to damage. Nature reviews. Immunology 2010, 10, 826–837. [Google Scholar] [CrossRef] [Green Version]
- Vourc’h, M.; Roquilly, A.; Asehnoune, K. Trauma-Induced Damage-Associated Molecular Patterns-Mediated Remote Organ Injury and Immunosuppression in the Acutely Ill Patient. Front. Immunol. 2018, 9, 1330. [Google Scholar] [CrossRef] [Green Version]
- Gong, T.; Liu, L.; Jiang, W.; Zhou, R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nature reviews. Immunology 2020, 20, 95–112. [Google Scholar] [CrossRef]
- Yang, M.; Wang, H.Y.; Chen, J.C.; Zhao, J. Regulation of airway inflammation and remodeling in asthmatic mice by TLR3/TRIF signal pathway. Mol. Immunol. 2017, 85, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Lin, G.; Han, Z.; Chai, J. Structural Biology of NOD-Like Receptors. Adv. Exp. Med. Biol. 2019, 1172, 119–141. [Google Scholar] [CrossRef]
- Brubaker, S.W.; Bonham, K.S.; Zanoni, I.; Kagan, J.C. Innate immune pattern recognition: A cell biological perspective. Annu. Rev. Immunol. 2015, 33, 257–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Rivero Vaccari, J.C.; Brand, F.J., 3rd; Berti, A.F.; Alonso, O.F.; Bullock, M.R.; De Rivero Vaccari, J.P. Mincle signaling in the innate immune response after traumatic brain injury. J. Neurotrauma 2015, 32, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Komada, T.; Chung, H.; Lau, A.; Platnich, J.M.; Beck, P.L.; Benediktsson, H.; Duff, H.J.; Jenne, C.N.; Muruve, D.A. Macrophage Uptake of Necrotic Cell DNA Activates the AIM2 Inflammasome to Regulate a Proinflammatory Phenotype in CKD. J. Am. Soc. Nephrol. 2018, 29, 1165–1181. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 2011, 34, 637–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, T.; Jiang, W.; Zhou, R. Control of Inflammasome Activation by Phosphorylation. Trends Biochem. Sci. 2018, 43, 685–699. [Google Scholar] [CrossRef] [PubMed]
- Ablasser, A.; Hur, S. Regulation of cGAS- and RLR-mediated immunity to nucleic acids. Nat. Immunol. 2020, 21, 17–29. [Google Scholar] [CrossRef]
- Liddicoat, B.J.; Piskol, R.; Chalk, A.M.; Ramaswami, G.; Higuchi, M.; Hartner, J.C.; Li, J.B.; Seeburg, P.H.; Walkley, C.R. RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science 2015, 349, 1115–1120. [Google Scholar] [CrossRef] [Green Version]
- Haddad, Y.; Lahoute, C.; Clément, M.; Laurans, L.; Metghalchi, S.; Zeboudj, L.; Giraud, A.; Loyer, X.; Vandestienne, M.; Wain-Hobson, J.; et al. The Dendritic Cell Receptor DNGR-1 Promotes the Development of Atherosclerosis in Mice. Circ. Res. 2017, 121, 234–243. [Google Scholar] [CrossRef]
- Li, X.D.; Wu, J.; Gao, D.; Wang, H.; Sun, L.; Chen, Z.J. Pivotal roles of cGAS-cGAMP signaling in antiviral defense and immune adjuvant effects. Science 2013, 341, 1390–1394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rathinam, V.A.; Jiang, Z.; Waggoner, S.N.; Sharma, S.; Cole, L.E.; Waggoner, L.; Vanaja, S.K.; Monks, B.G.; Ganesan, S.; Latz, E.; et al. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat. Immunol. 2010, 11, 395–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bierhaus, A.; Schiekofer, S.; Schwaninger, M.; Andrassy, M.; Humpert, P.M.; Chen, J.; Hong, M.; Luther, T.; Henle, T.; Klöting, I.; et al. Diabetes-associated sustained activation of the transcription factor nuclear factor-kappaB. Diabetes 2001, 50, 2792–2808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasparotto, J.; Girardi, C.S.; Somensi, N.; Ribeiro, C.T.; Moreira, J.C.F.; Michels, M.; Sonai, B.; Rocha, M.; Steckert, A.V.; Barichello, T.; et al. Receptor for advanced glycation end products mediates sepsis-triggered amyloid-β accumulation, Tau phosphorylation, and cognitive impairment. J. Biol. Chem. 2018, 293, 226–244. [Google Scholar] [CrossRef] [Green Version]
- Gilman, A.G. G proteins: Transducers of receptor-generated signals. Annu. Rev. Biochem. 1987, 56, 615–649. [Google Scholar] [CrossRef]
- Moran, M.M.; McAlexander, M.A.; Bíró, T.; Szallasi, A. Transient receptor potential channels as therapeutic targets. Nature reviews. Drug Discov. 2011, 10, 601–620. [Google Scholar] [CrossRef]
- Ford, J.W.; McVicar, D.W. TREM and TREM-like receptors in inflammation and disease. Curr. Opin. Immunol. 2009, 21, 38–46. [Google Scholar] [CrossRef] [Green Version]
- Tammaro, A.; Derive, M.; Gibot, S.; Leemans, J.C.; Florquin, S.; Dessing, M.C. TREM-1 and its potential ligands in non-infectious diseases: From biology to clinical perspectives. Pharmacol. Ther. 2017, 177, 81–95. [Google Scholar] [CrossRef]
- Ho, C.C.; Liao, W.Y.; Wang, C.Y.; Lu, Y.H.; Huang, H.Y.; Chen, H.Y.; Chan, W.K.; Chen, H.W.; Yang, P.C. TREM-1 expression in tumor-associated macrophages and clinical outcome in lung cancer. Am. J. Respir. Crit. Care Med. 2008, 177, 763–770. [Google Scholar] [CrossRef]
- Stefano, L.; Racchetti, G.; Bianco, F.; Passini, N.; Gupta, R.S.; Panina Bordignon, P.; Meldolesi, J. The surface-exposed chaperone, Hsp60, is an agonist of the microglial TREM2 receptor. J. Neurochem. 2009, 110, 284–294. [Google Scholar] [CrossRef]
- Yeh, F.L.; Wang, Y.; Tom, I.; Gonzalez, L.C.; Sheng, M. TREM2 Binds to Apolipoproteins, Including APOE and CLU/APOJ, and Thereby Facilitates Uptake of Amyloid-Beta by Microglia. Neuron 2016, 91, 328–340. [Google Scholar] [CrossRef] [PubMed]
- Hudson, B.I.; Lippman, M.E. Targeting RAGE Signaling in Inflammatory Disease. Annu. Rev. Med. 2018, 69, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Weiß, E.; Kretschmer, D. Formyl-Peptide Receptors in Infection, Inflammation, and Cancer. Trends Immunol. 2018, 39, 815–829. [Google Scholar] [CrossRef] [PubMed]
- Idzko, M.; Ferrari, D.; Eltzschig, H.K. Nucleotide signalling during inflammation. Nature 2014, 509, 310–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossol, M.; Pierer, M.; Raulien, N.; Quandt, D.; Meusch, U.; Rothe, K.; Schubert, K.; Schöneberg, T.; Schaefer, M.; Krügel, U.; et al. Extracellular Ca2+ is a danger signal activating the NLRP3 inflammasome through G protein-coupled calcium sensing receptors. Nat. Commun. 2012, 3, 1329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Virgilio, F.; Dal Ben, D.; Sarti, A.C.; Giuliani, A.L.; Falzoni, S. The P2X7 Receptor in Infection and Inflammation. Immunity 2017, 47, 15–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, L.; Chen, C.; Chen, Y.; Li, Y.; Tang, F.; Huang, H.; He, W.; Zhang, R.; Shen, L. High-Mobility Group Box 1 (HMGB1) and Autophagy in Acute Lung Injury (ALI): A Review. Medical science monitor. Int. Med. J. Exp. Clin. Res 2019, 25, 1828–1837. [Google Scholar] [CrossRef]
- Li, R.; Shang, Y.; Yu, Y.; Zhou, T.; Xiong, W.; Zou, X. High-mobility group box 1 protein participates in acute lung injury by activating protein kinase R and inducing M1 polarization. Life Sci. 2020, 246, 117415. [Google Scholar] [CrossRef]
- Cohen, M.J.; Brohi, K.; Calfee, C.S.; Rahn, P.; Chesebro, B.B.; Christiaans, S.C.; Carles, M.; Howard, M.; Pittet, J.F. Early release of high mobility group box nuclear protein 1 after severe trauma in humans: Role of injury severity and tissue hypoperfusion. Critical Care 2009, 13, R174. [Google Scholar] [CrossRef] [Green Version]
- Silk, E.; Zhao, H.; Weng, H.; Ma, D. The role of extracellular histone in organ injury. Cell Death Dis. 2017, 8, e2812. [Google Scholar] [CrossRef] [Green Version]
- Abrams, S.T.; Zhang, N.; Manson, J.; Liu, T.; Dart, C.; Baluwa, F.; Wang, S.S.; Brohi, K.; Kipar, A.; Yu, W.; et al. Circulating histones are mediators of trauma-associated lung injury. Am. J. Respir. Crit. Care Med. 2013, 187, 160–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hua, X.; Zhang, H.; Jia, J.; Chen, S.; Sun, Y.; Zhu, X. Roles of S100 family members in drug resistance in tumors: Status and prospects. Biomed. Pharmacother. Biomed. Pharmacother. 2020, 127, 110156. [Google Scholar] [CrossRef] [PubMed]
- Donato, R.; Cannon, B.R.; Sorci, G.; Riuzzi, F.; Hsu, K.; Weber, D.J.; Geczy, C.L. Functions of S100 proteins. Curr. Mol. Med. 2013, 13, 24–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertheloot, D.; Latz, E. HMGB1, IL-1α, IL-33 and S100 proteins: Dual-function alarmins. Cell. Mol. Immunol. 2017, 14, 43–64. [Google Scholar] [CrossRef] [Green Version]
- Lorenz, E.; Muhlebach, M.S.; Tessier, P.A.; Alexis, N.E.; Duncan Hite, R.; Seeds, M.C.; Peden, D.B.; Meredith, W. Different expression ratio of S100A8/A9 and S100A12 in acute and chronic lung diseases. Respir. Med. 2008, 102, 567–573. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, D.; Zenker, S.; Rossaint, J.; Hölscher, A.; Pohlen, M.; Zarbock, A.; Roth, J.; Vogl, T. Alarmin S100A8 Activates Alveolar Epithelial Cells in the Context of Acute Lung Injury in a TLR4-Dependent Manner. Front. Immunol. 2017, 8, 1493. [Google Scholar] [CrossRef] [Green Version]
- Tolle, L.B.; Standiford, T.J. Danger-associated molecular patterns (DAMPs) in acute lung injury. J. Pathol. 2013, 229, 145–156. [Google Scholar] [CrossRef] [Green Version]
- Jin, C.; Cleveland, J.C.; Ao, L.; Li, J.; Zeng, Q.; Fullerton, D.A.; Meng, X. Human myocardium releases heat shock protein 27 (HSP27) after global ischemia: The proinflammatory effect of extracellular HSP27 through toll-like receptor (TLR)-2 and TLR4. Mol. Med. 2014, 20, 280–289. [Google Scholar] [CrossRef]
- Cheng, W.; Li, Y.; Hou, X.; Zhang, N.; Ma, J.; Ding, F.; Li, F.; Miao, Z.; Zhang, Y.; Qi, Q.; et al. HSP60 is involved in the neuroprotective effects of naloxone. Mol. Med. Rep. 2014, 10, 2172–2176. [Google Scholar] [CrossRef] [Green Version]
- Wong, H.R.; Menendez, I.Y.; Ryan, M.A.; Denenberg, A.G.; Wispé, J.R. Increased expression of heat shock protein-70 protects A549 cells against hyperoxia. Am. J. Physiol. 1998, 275, L836–L841. [Google Scholar] [CrossRef]
- Tanaka, K.; Tanaka, Y.; Namba, T.; Azuma, A.; Mizushima, T. Heat shock protein 70 protects against bleomycin-induced pulmonary fibrosis in mice. Biochem. Pharmacol. 2010, 80, 920–931. [Google Scholar] [CrossRef]
- Zhong, P.; Huang, H. Recent progress in the research of cold-inducible RNA-binding protein. Future Sci. Oa 2017, 3, 246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aziz, M.; Brenner, M.; Wang, P. Extracellular CIRP (eCIRP) and inflammation. J. Leukoc. Biol. 2019, 106, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Gurien, S.D.; Aziz, M.; Cagliani, J.; Denning, N.L.; Last, J.; Royster, W.; Coppa, G.F.; Wang, P. An eCIRP-derived small peptide targeting TREM-1 attenuates hemorrhagic shock. J. Trauma Acute Care Surg. 2020. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, H.; Zhou, J.; Zhong, Y.; Ali, M.M.; McGuire, F.; Nagarkatti, P.S.; Nagarkatti, M. Role of cytokines as a double-edged sword in sepsis. In Vivo 2013, 27, 669–684. [Google Scholar] [PubMed]
- Pedersen, B.K.; Febbraio, M.A. Muscle as an endocrine organ: Focus on muscle-derived interleukin-6. Physiol. Rev. 2008, 88, 1379–1406. [Google Scholar] [CrossRef] [Green Version]
- Scheller, J.; Chalaris, A.; Schmidt-Arras, D.; Rose-John, S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochimica Biophysica Acta 2011, 1813, 878–888. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, B.K. Muscle as a secretory organ. Comprehensive Physiology 2013, 3, 1337–1362. [Google Scholar] [CrossRef]
- Mosser, D.M.; Zhang, X. Interleukin-10: New perspectives on an old cytokine. Immunol. Rev. 2008, 226, 205–218. [Google Scholar] [CrossRef]
- Soderholm, A.T.; Barnett, T.C.; Sweet, M.J.; Walker, M.J. Group A streptococcal pharyngitis: Immune responses involved in bacterial clearance and GAS-associated immunopathologies. J. Leukoc. Biol. 2018, 103, 193–213. [Google Scholar] [CrossRef]
- Shurety, W.; Pagan, J.K.; Prins, J.B.; Stow, J.L. Endocytosis of uncleaved tumor necrosis factor-alpha in macrophages. Lab. Investig. J. Tech. Methods Pathol. 2001, 81, 107–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, C.; Liu, Y.; Zhang, Y.; Cong, P.; Shi, X.; Liu, Y.; Shi Hongxu Jin, L.; Hou, M. Shock waves increase pulmonary vascular leakage, inflammation, oxidative stress, and apoptosis in a mouse model. Exp. Biol. Med. 2018, 243, 934–944. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Liang, Z.; Li, H.; Li, C.; Yang, Z.; Li, Y.; She, D.; Cao, L.; Wang, W.; Liu, C.; et al. Perfluorocarbon reduces cell damage from blast injury by inhibiting signal paths of NF-kappaB, MAPK and Bcl-2/Bax signaling pathway in A549 cells. PLoS ONE 2017, 12, e0173884. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Li, M.; Zhou, Z.; Guan, X.; Xiang, Y. Can we use interleukin-6 (IL-6) blockade for coronavirus disease 2019 (COVID-19)-induced cytokine release syndrome (CRS)? J. Autoimmun. 2020. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Wu, Z.; Li, J.W.; Zhao, H.; Wang, G.Q. The cytokine release syndrome (CRS) of severe COVID-19 and Interleukin-6 receptor (IL-6R) antagonist Tocilizumab may be the key to reduce the mortality. Int. J. Antimicrob. Agents 2020, 4, 105954. [Google Scholar] [CrossRef]
- Ferrara, N. VEGF and the quest for tumour angiogenesis factors. Nature reviews. Cancer 2002, 2, 795–803. [Google Scholar] [CrossRef]
- Lin, C.K.; Lin, Y.H.; Huang, T.C.; Shi, C.S.; Yang, C.T. VEGF mediates fat embolism-induced acute lung injury via VEGF receptor 2 and the MAPK cascade. Sci. Rep. 2019, 9, 11713. [Google Scholar] [CrossRef] [Green Version]
- Ferrara, N. Vascular endothelial growth factor: Basic science and clinical progress. Endocr. Rev. 2004, 25, 581–611. [Google Scholar] [CrossRef]
- Medford, A.R.; Ibrahim, N.B.; Millar, A.B. Vascular endothelial growth factor receptor and coreceptor expression in human acute respiratory distress syndrome. J. Crit. Care 2009, 24, 236–242. [Google Scholar] [CrossRef] [Green Version]
- Kaner, R.J.; Crystal, R.G. Compartmentalization of vascular endothelial growth factor to the epithelial surface of the human lung. Mol. Med. 2001, 7, 240–246. [Google Scholar] [CrossRef] [Green Version]
- Medford, A.R.; Millar, A.B. Vascular endothelial growth factor (VEGF) in acute lung injury (ALI) and acute respiratory distress syndrome (ARDS): Paradox or paradigm? Thorax 2006, 61, 621–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, M.; Qiu, Z. Changes in TNF-alpha, IL-6, IL-10 and VEGF in rats with ARDS and the effects of dexamethasone. Exp. Ther. Med. 2019, 17, 383–387. [Google Scholar] [CrossRef] [PubMed]
- Standiford, T.J.; Ward, P.A. Therapeutic targeting of acute lung injury and acute respiratory distress syndrome. Trans. Res. J. Lab. Clin. Med. 2016, 167, 183–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Aderemi, O.A.; Zhao, Q.; Edsall, P.R.; Simovic, M.O.; Lund, B.J.; Espinoza, M.D.; Woodson, A.M.; Li, Y.; Cancio, L.C. Early Complement and Fibrinolytic Activation in a Rat Model of Blast-Induced Multi-Organ Damage. Mil. Med. 2019, 184, 282–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Yang, Z.; Chavko, M.; Liu, B.; Aderemi, O.A.; Simovic, M.O.; Dubick, M.A.; Cancio, L.C. Complement inhibition ameliorates blast-induced acute lung injury in rats: Potential role of complement in intracellular HMGB1-mediated inflammation. PLoS ONE 2018, 13, e0202594. [Google Scholar] [CrossRef]
- Wu, G.; Zhu, Q.; Zeng, J.; Gu, X.; Miao, Y.; Xu, W.; Lv, T.; Song, Y. Extracellular mitochondrial DNA promote NLRP3 inflammasome activation and induce acute lung injury through TLR9 and NF-κB. J. Thorac. Dis. 2019, 11, 4816–4828. [Google Scholar] [CrossRef]
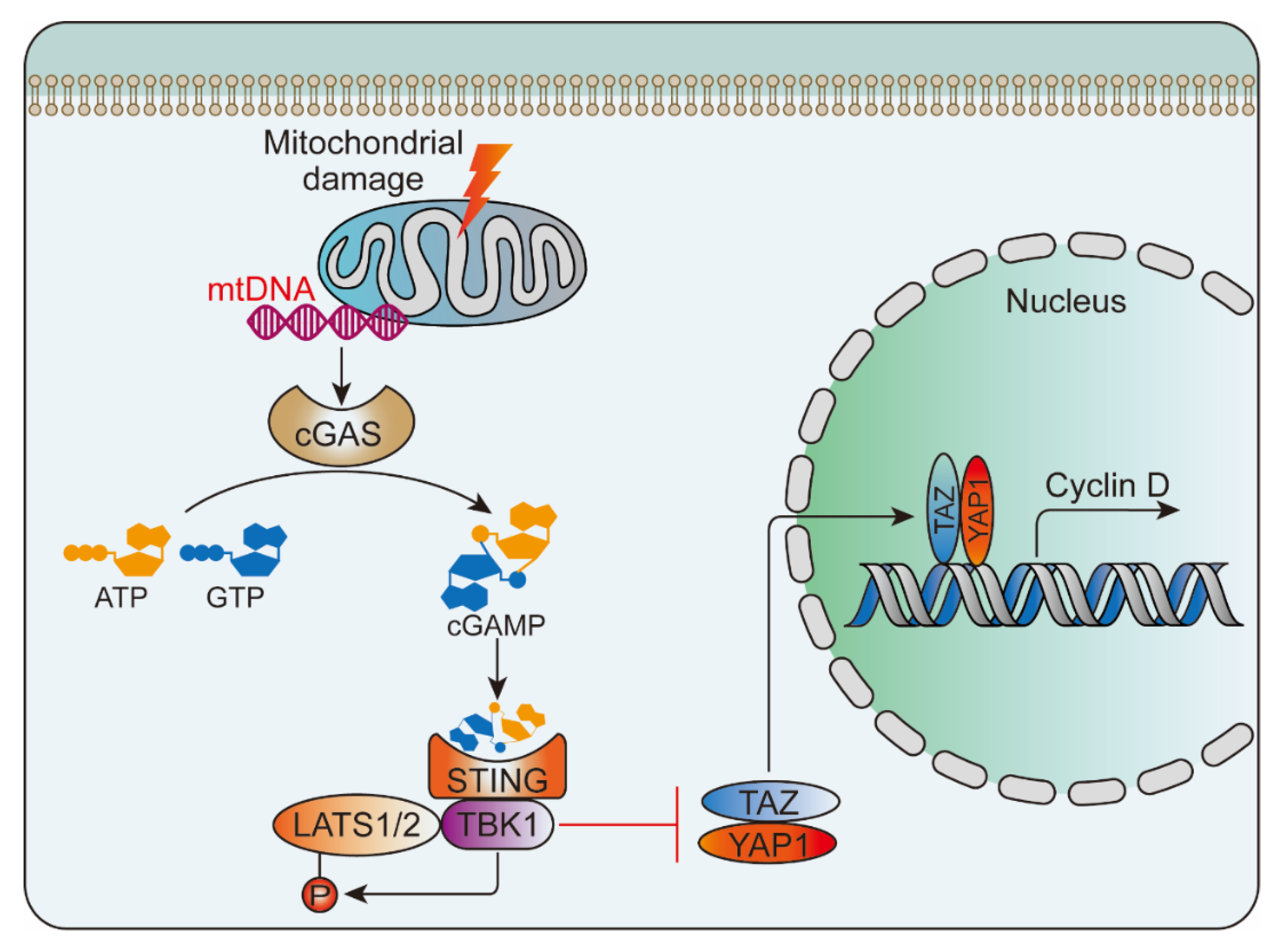
- Huang, L.S.; Hong, Z.; Wu, W.; Xiong, S.; Zhong, M.; Gao, X.; Rehman, J.; Malik, A.B. mtDNA Activates cGAS Signaling and Suppresses the YAP-Mediated Endothelial Cell Proliferation Program to Promote Inflammatory Injury. Immunity 2020, 52, 475–486.e475. [Google Scholar] [CrossRef]
- Lv, Y.; Kim, K.; Sheng, Y.; Cho, J.; Qian, Z.; Zhao, Y.Y.; Hu, G.; Pan, D.; Malik, A.B.; Hu, G. YAP Controls Endothelial Activation and Vascular Inflammation Through TRAF6. Circ. Res. 2018, 123, 43–56. [Google Scholar] [CrossRef]
- He, J.; Bao, Q.; Zhang, Y.; Liu, M.; Lv, H.; Liu, Y.; Yao, L.; Li, B.; Zhang, C.; He, S.; et al. Yes-Associated Protein Promotes Angiogenesis via Signal Transducer and Activator of Transcription 3 in Endothelial Cells. Circ. Res. 2018, 122, 591–605. [Google Scholar] [CrossRef]
- Ratajczak, M.Z.; Adamiak, M.; Thapa, A.; Bujko, K.; Brzezniakiewicz-Janus, K.; Lenkiewicz, A.M. NLRP3 inflammasome couples purinergic signaling with activation of the complement cascade for the optimal release of cells from bone marrow. Leukemia 2019, 33, 815–825. [Google Scholar] [CrossRef] [Green Version]
- Cicko, S.; Köhler, T.C.; Ayata, C.K.; Müller, T.; Ehrat, N.; Meyer, A.; Hossfeld, M.; Zech, A.; Di Virgilio, F.; Idzko, M. Extracellular ATP is a danger signal activating P2X7 receptor in a LPS mediated inflammation (ARDS/ALI). Oncotarget 2018, 9, 30635–30648. [Google Scholar] [CrossRef] [PubMed]
- Gaudet, A.D.; Popovich, P.G. Extracellular matrix regulation of inflammation in the healthy and injured spinal cord. Exp. Neurol. 2014, 258, 24–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaharudin, A.; Aziz, Z. Effectiveness of hyaluronic acid and its derivatives on chronic wounds: A systematic review. J. Wound Care 2016, 25, 585–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, T.; Bianco, P.; Fisher, L.W.; Longenecker, G.; Smith, E.; Goldstein, S.; Bonadio, J.; Boskey, A.; Heegaard, A.M.; Sommer, B.; et al. Targeted disruption of the biglycan gene leads to an osteoporosis-like phenotype in mice. Nat. Genet. 1998, 20, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Roedig, H.; Nastase, M.V.; Wygrecka, M.; Schaefer, L. Breaking down chronic inflammatory diseases: The role of biglycan in promoting a switch between inflammation and autophagy. FEBS J. 2019, 286, 2965–2979. [Google Scholar] [CrossRef] [Green Version]
- Tighe, R.M.; Garantziotis, S. Hyaluronan interactions with innate immunity in lung biology. Matrix Biol. J. Int. Soc. Matrix Biol. 2019, 78–79, 84–99. [Google Scholar] [CrossRef]
- Jiang, D.; Liang, J.; Fan, J.; Yu, S.; Chen, S.; Luo, Y.; Prestwich, G.D.; Mascarenhas, M.M.; Garg, H.G.; Quinn, D.A.; et al. Regulation of lung injury and repair by Toll-like receptors and hyaluronan. Nat. Med. 2005, 11, 1173–1179. [Google Scholar] [CrossRef]
- Xu, C.; Shi, Q.; Zhang, L.; Zhao, H. High molecular weight hyaluronan attenuates fine particulate matter-induced acute lung injury through inhibition of ROS-ASK1-p38/JNK-mediated epithelial apoptosis. Environ. Toxicol. Pharmacol. 2018, 59, 190–198. [Google Scholar] [CrossRef]
- Koslowski, R.; Pfeil, U.; Fehrenbach, H.; Kasper, M.; Skutelsky, E.; Wenzel, K.W. Changes in xylosyltransferase activity and in proteoglycan deposition in bleomycin-induced lung injury in rat. Eur. Respir. J. 2001, 18, 347–356. [Google Scholar] [CrossRef]
- Babelova, A.; Moreth, K.; Tsalastra-Greul, W.; Zeng-Brouwers, J.; Eickelberg, O.; Young, M.F.; Bruckner, P.; Pfeilschifter, J.; Schaefer, R.M.; Gröne, H.J.; et al. Biglycan, a danger signal that activates the NLRP3 inflammasome via toll-like and P2X receptors. J. Biol. Chem. 2009, 284, 24035–24048. [Google Scholar] [CrossRef] [Green Version]
- Roedig, H.; Nastase, M.V.; Frey, H.; Moreth, K.; Zeng-Brouwers, J.; Poluzzi, C.; Hsieh, L.T.; Brandts, C.; Fulda, S.; Wygrecka, M.; et al. Biglycan is a new high-affinity ligand for CD14 in macrophages. Matrix Biol. J. Int. Soc. Matrix Biol. 2019, 77, 4–22. [Google Scholar] [CrossRef] [PubMed]
- Poluzzi, C.; Nastase, M.V.; Zeng-Brouwers, J.; Roedig, H.; Hsieh, L.T.; Michaelis, J.B.; Buhl, E.M.; Rezende, F.; Manavski, Y.; Bleich, A.; et al. Biglycan evokes autophagy in macrophages via a novel CD44/Toll-like receptor 4 signaling axis in ischemia/reperfusion injury. Kidney Int. 2019, 95, 540–562. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, N.; Geng, C.; Hou, S.; Fan, H.; Gong, Y. Damage-Associated Molecular Patterns and Their Signaling Pathways in Primary Blast Lung Injury: New Research Progress and Future Directions. Int. J. Mol. Sci. 2020, 21, 6303. https://doi.org/10.3390/ijms21176303
Li N, Geng C, Hou S, Fan H, Gong Y. Damage-Associated Molecular Patterns and Their Signaling Pathways in Primary Blast Lung Injury: New Research Progress and Future Directions. International Journal of Molecular Sciences. 2020; 21(17):6303. https://doi.org/10.3390/ijms21176303
Chicago/Turabian StyleLi, Ning, Chenhao Geng, Shike Hou, Haojun Fan, and Yanhua Gong. 2020. "Damage-Associated Molecular Patterns and Their Signaling Pathways in Primary Blast Lung Injury: New Research Progress and Future Directions" International Journal of Molecular Sciences 21, no. 17: 6303. https://doi.org/10.3390/ijms21176303