Multiple Sulfatase Deficiency: A Disease Comprising Mucopolysaccharidosis, Sphingolipidosis, and More Caused by a Defect in Posttranslational Modification

 , , and
, , and
Abstract
:1. Introduction
2. From a Variant Form of MLD to a Unique Posttranslational Modification and the Discovery of the SUMF1 Gene
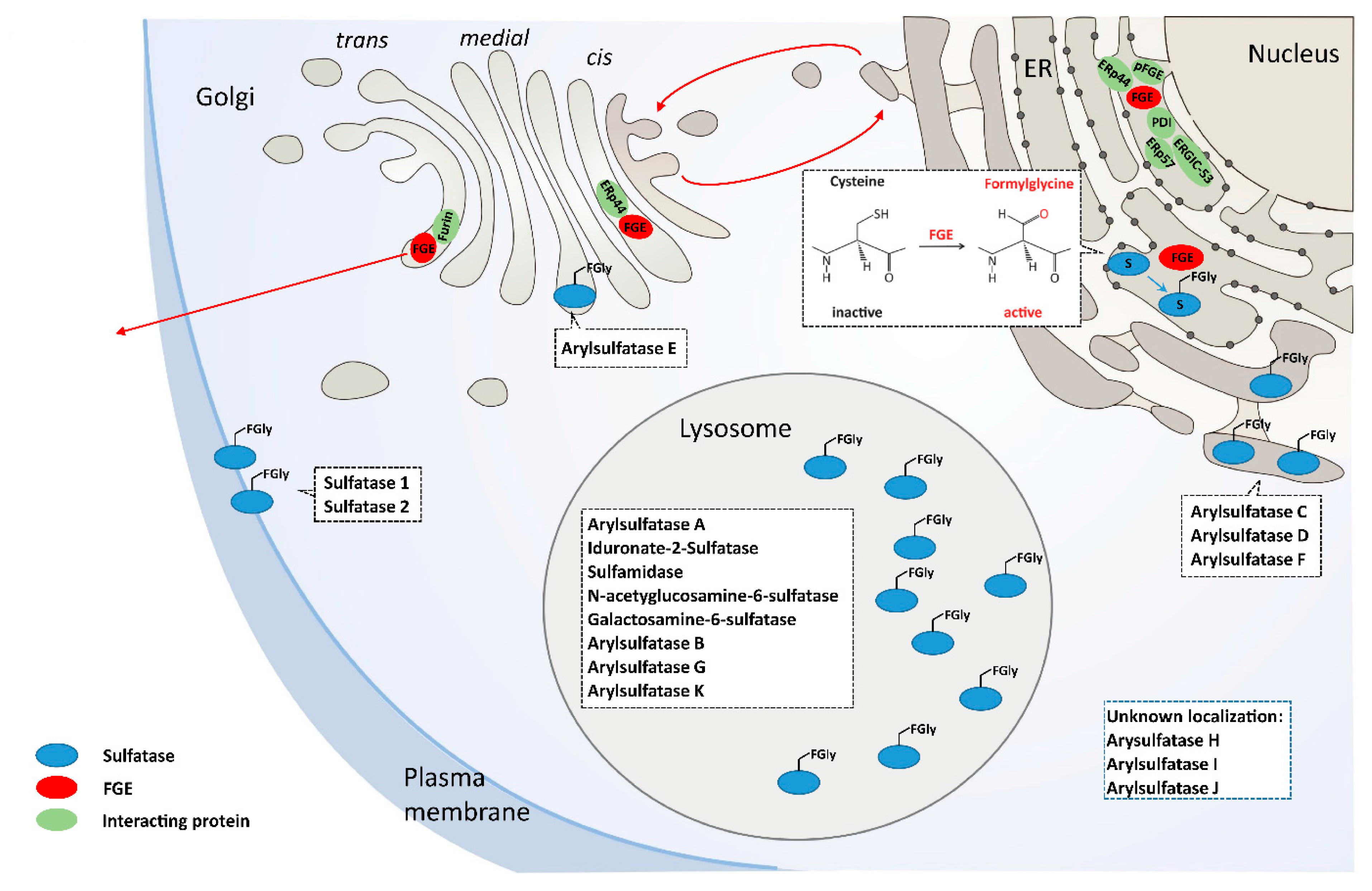
3. FGE the MSD Protein
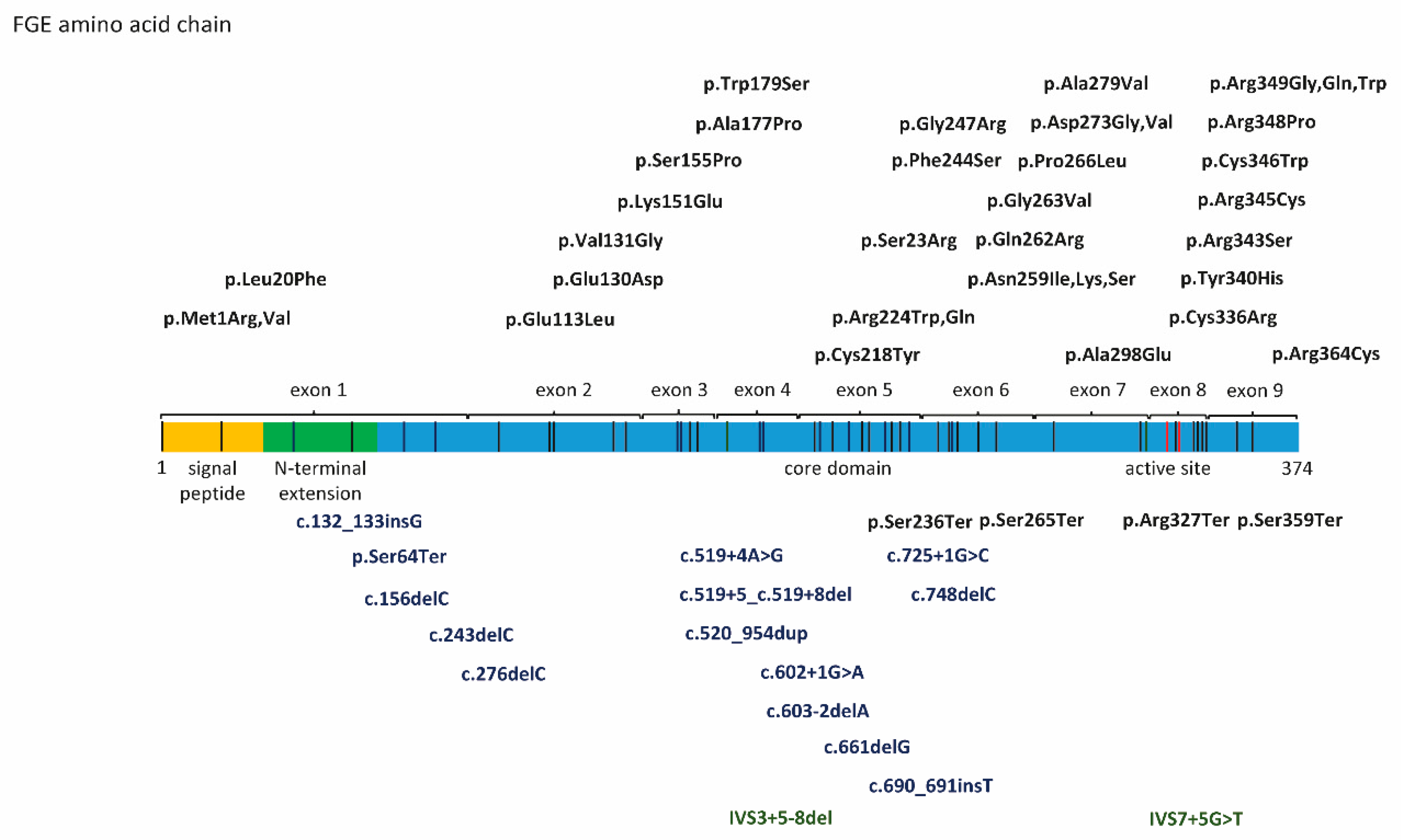
4. SUMF1 Mutations and Functional Consequences
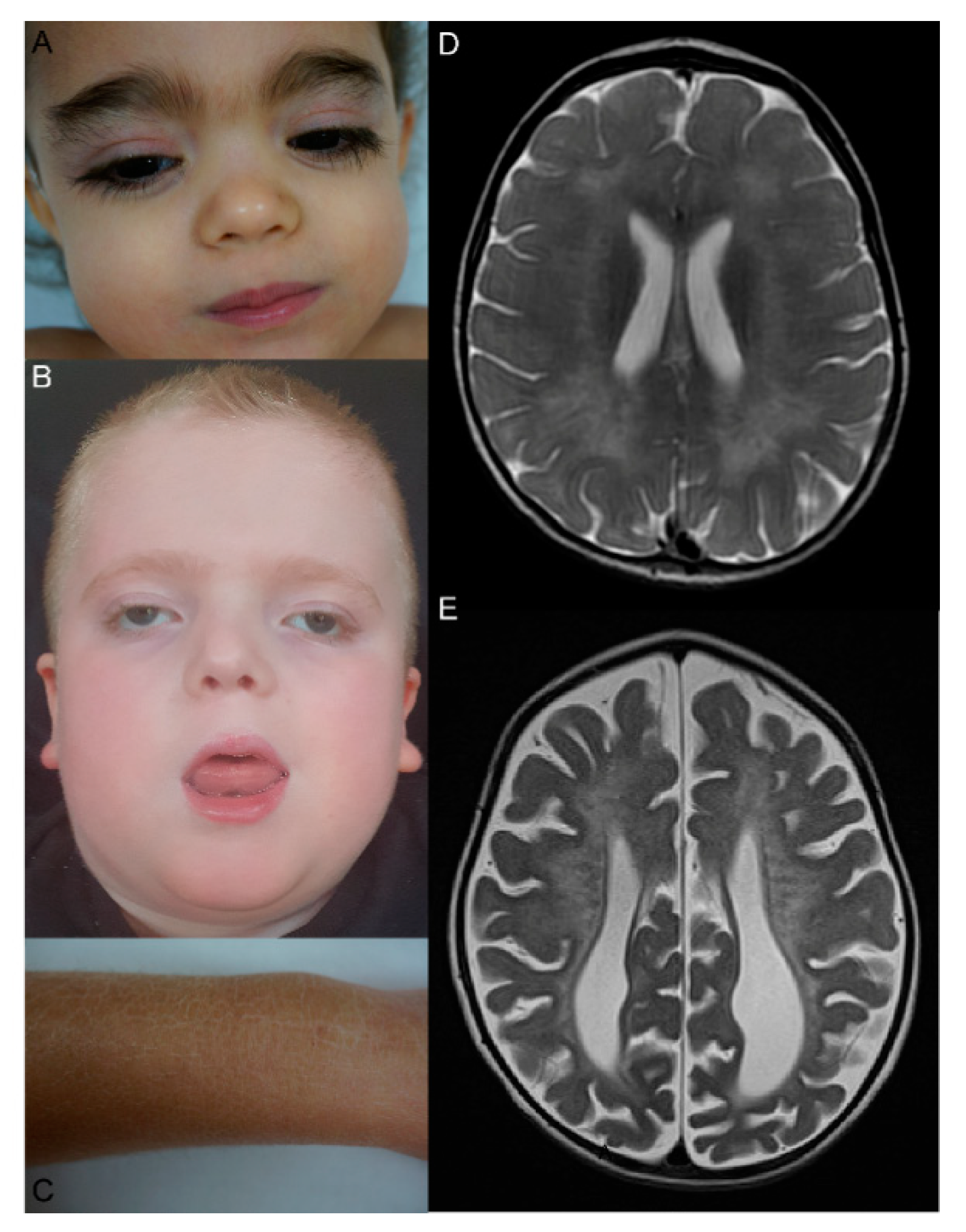
5. Clinical, MRI, and Ultrastructural Features, Disease Classifications, Genotype-Phenotype Correlation
6. Biomarkers and Diagnosis
7. Care of MSD Patients
8. MSD Animal Disease Models
9. Patient Organizations and Research Towards a Therapy for MSD
10. Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cosma, M.P.; Pepe, S.; Annunziata, I.; Newbold, R.F.; Grompe, M.; Parenti, G.; Ballabio, A. The Multiple Sulfatase Deficiency Gene Encodes an Essential and Limiting Factor for the Activity of Sulfatases. Cell 2003, 113, 445–456. [Google Scholar] [CrossRef]
- Dierks, T.; Schlotawa, L.; Frese, M.-A.; Radhakrishnan, K.; Von Figura, K.; Schmidt, B. Molecular basis of multiple sulfatase deficiency, mucolipidosis II/III and Niemann–Pick C1 disease—Lysosomal storage disorders caused by defects of non-lysosomal proteins. Biochim. Biophys. Acta (BBA) Bioenerg. 2009, 1793, 710–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dierks, T.; Schmidt, B.; Borissenko, L.V.; Peng, J.; Preusser, A.; Mariappan, M.; Von Figura, K. Multiple Sulfatase Deficiency Is Caused by Mutations in the Gene Encoding the Human Cα-Formylglycine Generating Enzyme. Cell 2003, 113, 435–444. [Google Scholar] [CrossRef] [Green Version]
- Meikle, P.J.; Hopwood, J.J.; Clague, A.E.; Carey, W.F. Prevalence of lysosomal storage disorders. JAMA 1999, 281, 249–254. [Google Scholar] [CrossRef]
- Austin, J.; Armstrong, D.; Shearer, L. Metachromatic form of diffuse cerebral sclerosis. V. The nature and significance of low sulfatase activity: A controlled study of brain, liver and kidney in four patients with metachromatic leukodystrophy (MLD). Arch. Neurol. 1965, 13, 593–614. [Google Scholar] [CrossRef]
- Austin, J.H. Studies in Metachromatic Leukodystrophy. Arch. Neurol. 1973, 28, 258. [Google Scholar] [CrossRef]
- Eto, Y.; Wiesmann, U.N.; Carson, J.H.; Herschkowitz, N.N. Multiple sulfatase deficiencies in cultured skin fibroblasts. Occurrence in patients with a variant form of metachromatic leukodystrophy. Arch. Neurol. 1974, 30, 153–156. [Google Scholar] [CrossRef]
- Horwitz, A.L. Genetic complementation studies of multiple sulfatase deficiency. Proc. Natl. Acad. Sci. USA 1979, 76, 6496–6499. [Google Scholar] [CrossRef] [Green Version]
- Chang, P.L.; Davidson, R.G. Complementation of arylsulfatase A in somatic hybrids of metachromatic leukodystrophy and multiple sulfatase deficiency disorder fibroblasts. Proc. Natl. Acad. Sci. USA 1980, 77, 6166–6170. [Google Scholar] [CrossRef] [Green Version]
- Fedde, K.; Horwitz, A.L. Complementation of multiple sulfatase deficiency in somatic cell hybrids. Am. J. Hum. Genet. 1984, 36, 623–633. [Google Scholar]
- Ballabio, A.; Parenti, G.; Napolitano, E.; Di Natale, P.; Andria, G. Genetic complementation of steroid sulphatase after somatic cell hybridization of X-linked ichthyosis and multiple sulphatase deficiency. Qual. Life Res. 1985, 70, 315–317. [Google Scholar] [CrossRef] [PubMed]
- Rommerskirch, W.; Von Figura, K. Multiple sulfatase deficiency: Catalytically inactive sulfatases are expressed from retrovirally introduced sulfatase cDNAs. Proc. Natl. Acad. Sci. USA 1992, 89, 2561–2565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, B.; Selmer, T.; Ingendoh, A.; Von Figurat, K. A novel amino acid modification in sulfatases that is defective in multiple sulfatase deficiency. Cell 1995, 82, 271–278. [Google Scholar] [CrossRef] [Green Version]
- Dierks, T.; Dickmanns, A.; Preusser-Kunze, A.; Schmidt, B.; Mariappan, M.; Von Figura, K.; Ficner, R.; Rudolph, M.G. Molecular Basis for Multiple Sulfatase Deficiency and Mechanism for Formylglycine Generation of the Human Formylglycine-Generating Enzyme. Cell 2005, 121, 541–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mariappan, M.; Gande, S.L.; Radhakrishnan, K.; Schmidt, B.; Dierks, T.; Von Figura, K. The Non-catalytic N-terminal Extension of Formylglycine-generating Enzyme Is Required for Its Biological Activity and Retention in the Endoplasmic Reticulum. J. Boil. Chem. 2008, 283, 11556–11564. [Google Scholar] [CrossRef] [Green Version]
- Fraldi, A.; Zito, E.; Annunziata, F.; Lombardi, A.; Cozzolino, M.; Monti, M.; Spampanato, C.; Ballabio, A.; Pucci, P.; Sitia, R.; et al. Multistep, sequential control of the trafficking and function of the multiple sulfatase deficiency gene product, SUMF1 by PDI, ERGIC-53 and ERp44. Hum. Mol. Genet. 2008, 17, 2610–2621. [Google Scholar] [CrossRef]
- Schlotawa, L.; Wachs, M.; Bernhard, O.; Mayer, F.J.; Dierks, T.; Schmidt, B.; Radhakrishnan, K. Recognition and ER Quality Control of Misfolded Formylglycine-Generating Enzyme by Protein Disulfide Isomerase. Cell Rep. 2018, 24, 27–37. [Google Scholar] [CrossRef] [Green Version]
- Mariappan, M.; Preusser-Kunze, A.; Balleininger, M.; Eiselt, N.; Schmidt, B.; Gande, S.L.; Wenzel, D.; Dierks, T.; von Figura, K. Expression, localization, structural, and functional characterization of pFGE, the paralog of the Calpha-formylglycine-generating enzyme. J. Biol. Chem. 2005, 280, 15173–15179. [Google Scholar] [CrossRef] [Green Version]
- Gande, S.L.; Mariappan, M.; Schmidt, B.; Pringle, T.H.; Von Figura, K.; Dierks, T. Paralog of the formylglycine-generating enzyme—Retention in the endoplasmic reticulum by canonical and noncanonical signals. FEBS J. 2008, 275, 1118–1130. [Google Scholar] [CrossRef]
- Zito, E.; Fraldi, A.; Pepe, S.; Annunziata, I.; Kobinger, G.; Di Natale, P.; Ballabio, A.; Cosma, M.P. Sulphatase activities are regulated by the interaction of sulphatase-modifying factor 1 with SUMF2. EMBO Rep. 2005, 6, 655–660. [Google Scholar] [CrossRef] [Green Version]
- Roeser, D.; Preusser-Kunze, A.; Schmidt, B.; Gasow, K.; Wittmann, J.G.; Dierks, T.; Von Figura, K.; Rudolph, M.G. A general binding mechanism for all human sulfatases by the formylglycine-generating enzyme. Proc. Natl. Acad. Sci. USA 2005, 103, 81–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dierks, T.; Lecca, M.; Schlotterhose, P.; Schmidt, B.; Von Figura, K. Sequence determinants directing conversion of cysteine to formylglycine in eukaryotic sulfatases. EMBO J. 1999, 18, 2084–2091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Recksiek, M.; Selmer, T.; Dierks, T.; Schmidt, B.; Von Figura, K. Sulfatases, Trapping of the Sulfated Enzyme Intermediate by Substituting the Active Site Formylglycine. J. Boil. Chem. 1998, 273, 6096–6103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrico, I.S.; Carlson, B.L.; Bertozzi, C.R. Introducing genetically encoded aldehydes into proteins. Nat. Methods 2007, 3, 321–322. [Google Scholar] [CrossRef] [PubMed]
- Rupniewski, I.; Rabuka, D. Site-Specific Labeling of Proteins Using the Formylglycine-Generating Enzyme (FGE). Breast Cancer 2019, 2012, 63–81. [Google Scholar] [CrossRef]
- Diez-Roux, G.; Ballabio, A. Sulfatases and Human Disease. Annu. Rev. Genom. Hum. Genet. 2005, 6, 355–379. [Google Scholar] [CrossRef] [PubMed]
- Sardiello, M.; Annunziata, I.; Roma, G.; Ballabio, A. Sulfatases and sulfatase modifying factors: An exclusive and promiscuous relationship. Hum. Mol. Genet. 2005, 14, 3203–3217. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Ai, X.; Freeman, S.D.; Pownall, M.E.; Lu, Q.; Kessler, D.S.; Emerson, C.P. QSulf1, a heparan sulfate 6-O-endosulfatase, inhibits fibroblast growth factor signaling in mesoderm induction and angiogenesis. Proc. Natl. Acad. Sci. USA 2004, 101, 4833–4838. [Google Scholar] [CrossRef] [Green Version]
- Dhoot, G.K. Regulation of Wnt Signaling and Embryo Patterning by an Extracellular Sulfatase. Science 2001, 293, 1663–1666. [Google Scholar] [CrossRef]
- Muenzer, J.; Wraith, J.E.; Beck, M.; Giugliani, R.; Harmatz, P.; Eng, C.M.; Vellodi, A.; Martin, R.; Ramaswami, U.; Gucsavas-Calikoglu, M.; et al. A phase II/III clinical study of enzyme replacement therapy with idursulfase in mucopolysaccharidosis II (Hunter syndrome). Genet. Med. 2006, 8, 465–473. [Google Scholar] [CrossRef] [Green Version]
- Takakusaki, Y.; Hisayasu, S.; Hirai, Y.; Shimada, T. Coexpression of formylglycine-generating enzyme is essential for synthesis and secretion of functional arylsulfatase A in a mouse model of metachromatic leukodystrophy. Hum. Gene Ther. 2005, 16, 929–936. [Google Scholar] [CrossRef] [PubMed]
- Fraldi, A.; Hemsley, K.M.; Crawley, A.; Lombardi, A.; Lau, A.A.; Sutherland, L.; Auricchio, A.; Ballabio, A.; Hopwood, J.J. Functional correction of CNS lesions in an MPS-IIIA mouse model by intracerebral AAV-mediated delivery of sulfamidase and SUMF1 genes. Hum. Mol. Genet. 2007, 16, 2693–2702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sevin, C.; Verot, L.; Benraiss, A.; Van Dam, D.; Bonnin, D.; Nagels, G.; Fouquet, F.; Gieselmann, V.; Vanier, M.T.; De Deyn, P.P.; et al. Partial cure of established disease in an animal model of metachromatic leukodystrophy after intracerebral adeno-associated virus-mediated gene transfer. Gene Ther. 2006, 14, 405–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomatsu, S.; Montaño, A.M.; Ohashi, A.; Gutierrez, M.A.; Oikawa, H.; Oguma, T.; Dung, V.C.; Nishioka, T.; Orii, T.; Sly, W.S. Enzyme replacement therapy in a murine model of Morquio A syndrome. Hum. Mol. Genet. 2007, 17, 815–824. [Google Scholar] [CrossRef] [Green Version]
- Preusser-Kunze, A.; Mariappan, M.; Schmidt, B.; Gande, S.L.; Mutenda, K.; Wenzel, D.; Von Figura, K.; Dierks, T. Molecular Characterization of the Human Cα-formylglycine-generating Enzyme. J. Boil. Chem. 2005, 280, 14900–14910. [Google Scholar] [CrossRef] [Green Version]
- Zito, E.; Buono, M.; Pepe, S.; Settembre, C.; Annunziata, I.; Surace, E.M.; Dierks, T.; Monti, M.; Cozzolino, M.; Pucci, P.; et al. Sulfatase modifying factor 1 trafficking through the cells: From endoplasmic reticulum to the endoplasmic reticulum. EMBO J. 2007, 26, 2443–2453. [Google Scholar] [CrossRef] [Green Version]
- Spooner, R.A.; Smith, D.C.; Easton, A.J.; Roberts, L.M.; Lord, J.M. Retrograde transport pathways utilised by viruses and protein toxins. Virol. J. 2006, 3, 26. [Google Scholar] [CrossRef] [Green Version]
- Frankel, L.B.; Di Malta, C.; Wen, J.; Eskelinen, E.-L.; Ballabio, A.; Lund, A.H. A non-conserved miRNA regulates lysosomal function and impacts on a human lysosomal storage disorder. Nat. Commun. 2014, 5, 5840. [Google Scholar] [CrossRef] [Green Version]
- Paul, D.M.; Chadah, T.; Senthilkumar, B.; Sethumadhavan, R.; Rajasekaran, R. Structural distortions due to missense mutations in human formylglycine-generating enzyme leading to multiple sulfatase deficiency. J. Biomol. Struct. Dyn. 2017, 36, 3575–3585. [Google Scholar] [CrossRef]
- Holder, P.G.; Jones, L.C.; Drake, P.M.; Barfield, R.M.; Bañas, S.; De Hart, G.W.; Baker, J.; Rabuka, D. Reconstitution of Formylglycine-generating Enzyme with Copper(II) for Aldehyde Tag Conversion. J. Boil. Chem. 2015, 290, 15730–15745. [Google Scholar] [CrossRef] [Green Version]
- Knop, M.; Lemnaru, R.; Seebeck, F.P. Mutation of Conserved Residues Increases in Vitro Activity of the Formylglycine-Generating Enzyme. ChemBioChem 2017, 18, 1755–1761. [Google Scholar] [CrossRef]
- Meury, M.; Knop, M.; Seebeck, F.P. Structural Basis for Copper-Oxygen Mediated C−H Bond Activation by the Formylglycine-Generating Enzyme. Angew. Chem. 2017, 56, 8115–8119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Appel, M.; Meier, K.K.; Lafrance-Vanasse, J.; Lim, H.; Tsai, C.-L.; Hedman, B.; Hodgson, K.O.; Tainer, J.; Solomon, E.I.; Bertozzi, C.R. Formylglycine-generating enzyme binds substrate directly at a mononuclear Cu(I) center to initiate O2activation. Proc. Natl. Acad. Sci. USA 2019, 116, 5370–5375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miarzlou, D.A.; Leisinger, F.; Joss, D.; Häussinger, D.; Seebeck, F.P. Structure of formylglycine-generating enzyme in complex with copper and a substrate reveals an acidic pocket for binding and activation of molecular oxygen. Chem. Sci. 2019, 10, 7049–7058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlotawa, L.; Ennemann, E.C.; Radhakrishnan, K.; Schmidt, B.; Chakrapani, A.; Christen, H.-J.; Moser, H.; Steinmann, B.; Dierks, T.; Gärtner, J. SUMF1 mutations affecting stability and activity of formylglycine generating enzyme predict clinical outcome in multiple sulfatase deficiency. Eur. J. Hum. Genet. 2011, 19, 253–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Annunziata, I.; Bouché, V.; Lombardi, A.; Settembre, C.; Ballabio, A. Multiple sulfatase deficiency is due to hypomorphic mutations of theSUMF1 gene. Hum. Mutat. 2007, 28, 928. [Google Scholar] [CrossRef]
- Cosma, M.P.; Pepe, S.; Parenti, G.; Settembre, C.; Annunziata, I.; Wade-Martins, R.; Di Domenico, C.; Di Natale, P.; Mankad, A.; Cox, B.; et al. Molecular and functional analysis ofSUMF1 mutations in multiple sulfatase deficiency. Hum. Mutat. 2004, 23, 576–581. [Google Scholar] [CrossRef]
- Sabourdy, F.; Mourey, L.; Le Trionnaire, E.; Bednarek, N.; Caillaud, C.; Chaix, Y.; Delrue, M.-A.; Dusser, A.; Froissart, R.; Garnotel, R.; et al. Natural disease history and characterisation of SUMF1 molecular defects in ten unrelated patients with multiple sulfatase deficiency. Orphanet J. Rare Dis. 2015, 10, 31. [Google Scholar] [CrossRef] [Green Version]
- Schlotawa, L.; Radhakrishnan, K.; Baumgartner, M.R.; Schmid, R.; Schmidt, B.; Dierks, T.; Gärtner, J. Rapid degradation of an active formylglycine generating enzyme variant leads to a late infantile severe form of multiple sulfatase deficiency. Eur. J. Hum. Genet. 2013, 21, 1020–1023. [Google Scholar] [CrossRef] [Green Version]
- Schlotawa, L.; Steinfeld, R.; Von Figura, K.; Dierks, T.; Gärtner, J. Molecular analysis ofSUMF1 mutations: Stability and residual activity of mutant formylglycine-generating enzyme determine disease severity in multiple sulfatase deficiency. Hum. Mutat. 2007, 29, 205. [Google Scholar] [CrossRef]
- Jaszczuk, L.; Schlotawa, L.; Dierks, T.; Ohlenbusch, A.; Koppenhöfer, D.; Babicz, M.; Lejman, M.; Radhakrishnan, K.; Ługowska, A. Expanding the genetic cause of multiple sulfatase deficiency: A novel SUMF1 variant in a patient displaying a severe late infantile form of the disease. Mol. Genet. Metab. 2017, 121, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Schlotawa, L.; Dierks, T.; Christoph, S.; Cloppenburg, E.; Ohlenbusch, A.; Korenke, G.C.; Gärtner, J. Severe neonatal multiple sulfatase deficiency presenting with hydrops fetalis in a preterm birth patient. JIMD Rep. 2019, 49, 48–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Settembre, C.; Fraldi, A.; Jahreiss, L.; Spampanato, C.; Venturi, C.; Medina, D.L.; De Pablo, R.; Tacchetti, C.; Rubinsztein, D.C.; Ballabio, A. A block of autophagy in lysosomal storage disorders. Hum. Mol. Genet. 2007, 17, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Platt, F.M.; d’Azzo, A.; Davidson, B.L.; Neufeld, E.F.; Tifft, C.J. Lysosomal storage diseases. Nat. Rev. Dis. Primers 2018, 4, 27. [Google Scholar] [CrossRef] [PubMed]
- Ahrens-Nicklas, R.; Schlotawa, L.; Ballabio, A.; Brunetti-Pierri, N.; De Castro, M.; Dierks, T.; Eichler, F.; Ficicioglu, C.; Finglas, A.; Gaertner, J.; et al. Complex care of individuals with multiple sulfatase deficiency: Clinical cases and consensus statement. Mol. Genet. Metab. 2018, 123, 337–346. [Google Scholar] [CrossRef]
- Guerra, W.F.; Verity, M.A.; Philippart, M.; Fluharty, A.L.; Nguyen, H.T. Multiple Sulfatase Deficiency: Clinical, Neuropathological, Ultrastructural and Biochemical Studies. J. Neuropathol. Exp. Neurol. 1990, 49, 406–423. [Google Scholar] [CrossRef]
- Zilberman, U.; Bibi, H.; Baumgartner, M.R.; Patterson, M.; Rahman, S.; Peters, V.; Morava, E.; Zschocke, J. The Effect of Multiple Sulfatase Deficiency (MSD) on Dental Development: Can We Use the Teeth as an Early Diagnostic Tool? JIMD Rep. 2016, 30, 95–101. [Google Scholar] [CrossRef] [Green Version]
- Prasad, C.; Rupar, C.A.; Campbell, C.; Napier, M.; Ramsay, D.; Tay, K.; Sharan, S.; Prasad, A.N. Case of Multiple Sulfatase Deficiency and Ocular Albinism: A Diagnostic Odyssey. Can. J. Neurol. Sci. J. Can. Sci. Neurol. 2014, 41, 626–631. [Google Scholar] [CrossRef] [Green Version]
- Eto, Y.; Gomibuchi, I.; Umezawa, F.; Tsuda, T. Pathochemistry, Pathogenesis and Enzyme Replacement in Multiple-Sulfatase Deficiency. Enzyme 1987, 38, 273–279. [Google Scholar] [CrossRef]
- Schlotawa, L.; Adang, L.; De Castro, M.; Ahrens-Nicklas, R. Multiple Sulfatase Deficiency. In GeneReviews((R)); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; NCBI Bookshelf: Seattle, WA, USA, 1993. [Google Scholar]
- Burch, M.; Fensom, A.H.; Jackson, M.; Pitts-Tucker, T.; Congdon, P.J. Multiple sulphatase deficiency presenting at birth. Clin. Genet. 1986, 30, 409–415. [Google Scholar] [CrossRef]
- Steckel, F.; Hasilik, A.; Figura, K. Synthesis and stability of arylsulfatase A and B in fibroblasts from multiple sulfatase deficiency. JBIC J. Boil. Inorg. Chem. 1985, 151, 141–145. [Google Scholar] [CrossRef] [PubMed]
- Yutaka, T.; Okada, S.; Kato, T.; Inui, K.; Yabuuchi, H. Properties of sulfatases in cultured skin fibroblasts of multiple sulfatase deficient patients. Clin. Genet. 1981, 20, 296–303. [Google Scholar] [CrossRef] [PubMed]
- Fluharty, A.L.; Stevens, R.L.; De La Flor, S.D.; Shapiro, L.J.; Kihara, H. Arysulfatase A modulation with pH in multiple sulfatase deficiency disorder fibroblasts. Am. J. Hum. Genet. 1979, 31, 574–580. [Google Scholar] [PubMed]
- Chang, P.L.; Rosa, N.E.; Ballantyne, S.R.; Davidson, R.G. Biochemical variability of arylsulphatases-A,-B and-C in cultured fibroblasts from patients with multiple sulphatase deficiency. J. Inherit. Metab. Dis. 1983, 6, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Settembre, C.; Annunziata, I.; Spampanato, C.; Zarcone, D.; Cobellis, G.; Nusco, E.; Zito, E.; Tacchetti, C.; Cosma, M.P.; Ballabio, A. Systemic inflammation and neurodegeneration in a mouse model of multiple sulfatase deficiency. Proc. Natl. Acad. Sci. USA 2007, 104, 4506–4511. [Google Scholar] [CrossRef] [Green Version]
- Buono, M.; Visigalli, I.; Biffi, A.; Bergamasco, R.; Cosma, M.P. Sulfatase modifying factor 1-mediated fibroblast growth factor signaling primes hematopoietic multilineage development. J. Cell Boil. 2010, 190, 1647–1660. [Google Scholar] [CrossRef]
- Di Malta, C.; Fryer, J.; Settembre, C.; Andrea, B. Astrocyte dysfunction triggers neurodegeneration in a lysosomal storage disorder. Mol. Genet. Metab. 2013, 108, S35. [Google Scholar] [CrossRef] [Green Version]
- Spampanato, C.; De Leonibus, E.; Dama, P.; Gargiulo, A.; Fraldi, A.; Sorrentino, N.C.; Russo, F.; Nusco, E.; Auricchio, A.; Surace, E.M.; et al. Efficacy of a Combined Intracerebral and Systemic Gene Delivery Approach for the Treatment of a Severe Lysosomal Storage Disorder. Mol. Ther. 2011, 19, 860–869. [Google Scholar] [CrossRef]
- Raffai, F.; Timmis, O. Building the patient community. Gene Ther. 2017, 24, 547–550. [Google Scholar] [CrossRef]
- DeWard, S.J.; Wilson, A.; Bausell, H.; Volz, A.S.; Mooney, K. Practical Aspects of Recruitment and Retention in Clinical Trials of Rare Genetic Diseases: The Phenylketonuria (PKU) Experience. J. Genet. Couns. 2013, 23, 20–28. [Google Scholar] [CrossRef]
- Milne, C.-P.; Ni, W. The Use of Social Media in Orphan Drug Development. Clin. Ther. 2017, 39, 2173–2180. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Sulfatase | Alias | Chromosomal Region | Gene | Localization | Substrate | Disease or Syndrome | Abbreviation | MIM No. |
|---|---|---|---|---|---|---|---|---|
| Arylsulfatase A | Cerebroside-3-sulfatase | 22q13.33 | ARSA | Lysosome | Cerebroside-3-sulfate | Metachromatic Leukodystrophy | MLD | 250,100 |
| Iduronate-2-Sulfatase | Xq28 | IDS | Lysosome | HS, DS, H | Hunter | MPS II | 309,900 | |
| Sulfamidase | N-Sulfoglucosamine-sulfohydrolase | 17q25.3 | SGSH | Lysosome | HS, H | Sanfilippo IIIa | MPS IIIa | 252,900 |
| N-acetyglucosamine-6-sulfatase | 12q14.3 | GNS | Lysosome | HS, H | Sanfilippo IIId | MPS IIId | 252,940 | |
| Galactosamine-6-sulfatase | 16q24.3 | GALNS | Lysosome | CS, KS | Morquio A | MPS IVa | 253,000 | |
| Arylsulfatase B | N-acetylgalactosamine-4-sulfatase | 5q14.1 | ARSB | Lysosome | CS, DS | Maroteaux-Lamy | MPS VI | 253,200 |
| Arylsulfatase G | N-sulfoglucosamine-3-sulfatase | 17q24.2 | ARSG | Lysosome | HS | Usher syndrome type 4 | USH4 | 618,144 |
| Arylsulfatase K | Glucuronate-2-sulfatase | 5q15 | ARSK | Lysosome | HS, DS | unknown | ||
| Arylsulfatase C | Steroidsulfatase | Xp22.31 | STS | ER | Steroid sulfates | X-linked ichthyosis | XLI | 308,100 |
| Arylsulfatase D | Xp22.33 | ARSD | ER | unknown | ||||
| Arylsulfatase F | Xp22.33 | ARSF | ER | unknown | ||||
| Arylsulfatase E | Xp22.33 | ARSE | Golgi | Chondrodysplasia punctata type I | CDPXI | 302,950 | ||
| Sulfatase 1 | Sulf1 | 8q13.2-q.13.3 | SULF1 | Cell surface | HS | unknown | ||
| Sulfatase 2 | Sulf2 | 20q13.12 | SULF2 | Cell surface | HS | unknown | ||
| Arylsulfatase H | Xp22.33 | ARSH | unknown | unknown | ||||
| Arylsulfatase I | Sulf5 | 5q32 | ARSI | unknown | unknown | |||
| Arylsulfatase J | Sulf4 | 4q26 | ARSJ | unknown | unknown |
| System | Clinical Concerns |
|---|---|
| Cardiac and vascular | Arrythmias |
| Cardiac hypertrophy | |
| Cardiac valve issues | |
| Hypertension | |
| Dermatologic | Hirsutism |
| Ichthyosis | |
| Musculoskeletal | Cord compression |
| Dysostosis multiplex | |
| Poor bone health | |
| Tone abnormalities | |
| Neurologic | Peripheral neuropathy |
| Hydrocephalus | |
| Intracranial pressure | |
| Seizures | |
| Nutrition and gastroenterologic | Feeding intolerance |
| Constipation | |
| Hepatosplenomegaly | |
| Gastroesophageal reflux | |
| Gallbladder issues | |
| Ophthalmic | Cataracts |
| Corneal clouding | |
| Glaucoma | |
| Retinopathy | |
| Retinitis pigmentosa | |
| Optic nerve abnormalities | |
| Strabismus | |
| Oral | Dental complications |
| Hyperplastic gums | |
| Poor oral- motor coordination | |
| Tooth enamel abnormalities | |
| Otolaryngologic | Airway obstruction |
| Airway narrowing | |
| Oral and pharyngeal obstruction | |
| Hearing disorders | |
| Recurrent otitis media | |
| Respiratory | Obstructive and recessive lung disease |
| Sleep issues | |
| Apnea (central and peripheral) | |
| Recurrent pneumonia |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schlotawa, L.; Adang, L.A.; Radhakrishnan, K.; Ahrens-Nicklas, R.C. Multiple Sulfatase Deficiency: A Disease Comprising Mucopolysaccharidosis, Sphingolipidosis, and More Caused by a Defect in Posttranslational Modification. Int. J. Mol. Sci. 2020, 21, 3448. https://doi.org/10.3390/ijms21103448
Schlotawa L, Adang LA, Radhakrishnan K, Ahrens-Nicklas RC. Multiple Sulfatase Deficiency: A Disease Comprising Mucopolysaccharidosis, Sphingolipidosis, and More Caused by a Defect in Posttranslational Modification. International Journal of Molecular Sciences. 2020; 21(10):3448. https://doi.org/10.3390/ijms21103448
Chicago/Turabian StyleSchlotawa, Lars, Laura A. Adang, Karthikeyan Radhakrishnan, and Rebecca C. Ahrens-Nicklas. 2020. "Multiple Sulfatase Deficiency: A Disease Comprising Mucopolysaccharidosis, Sphingolipidosis, and More Caused by a Defect in Posttranslational Modification" International Journal of Molecular Sciences 21, no. 10: 3448. https://doi.org/10.3390/ijms21103448