MicroRNA-24-3p Targets Notch and Other Vascular Morphogens to Regulate Post-ischemic Microvascular Responses in Limb Muscles

 , ,
, ,
Abstract
:1. Introduction
2. Results
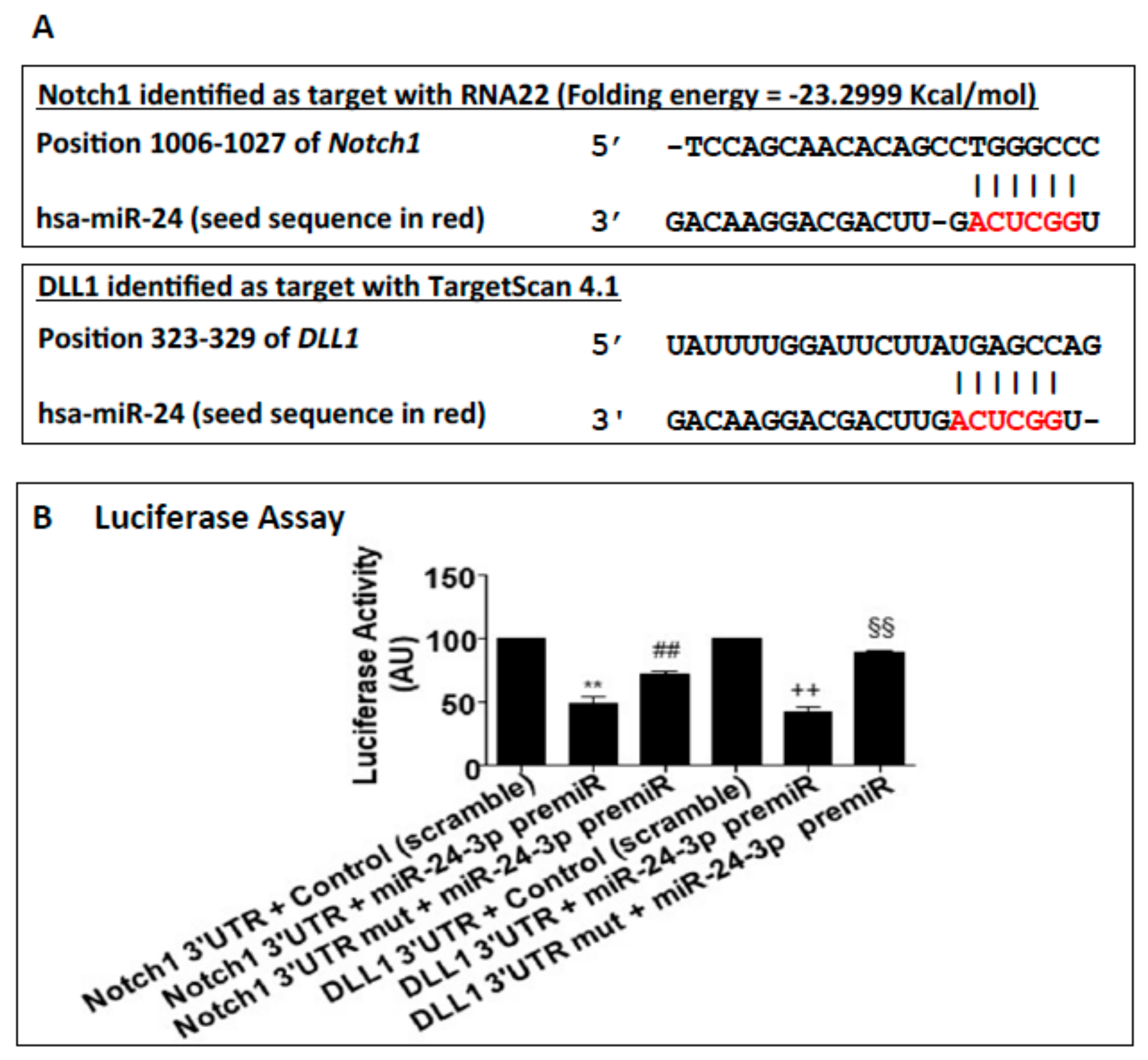
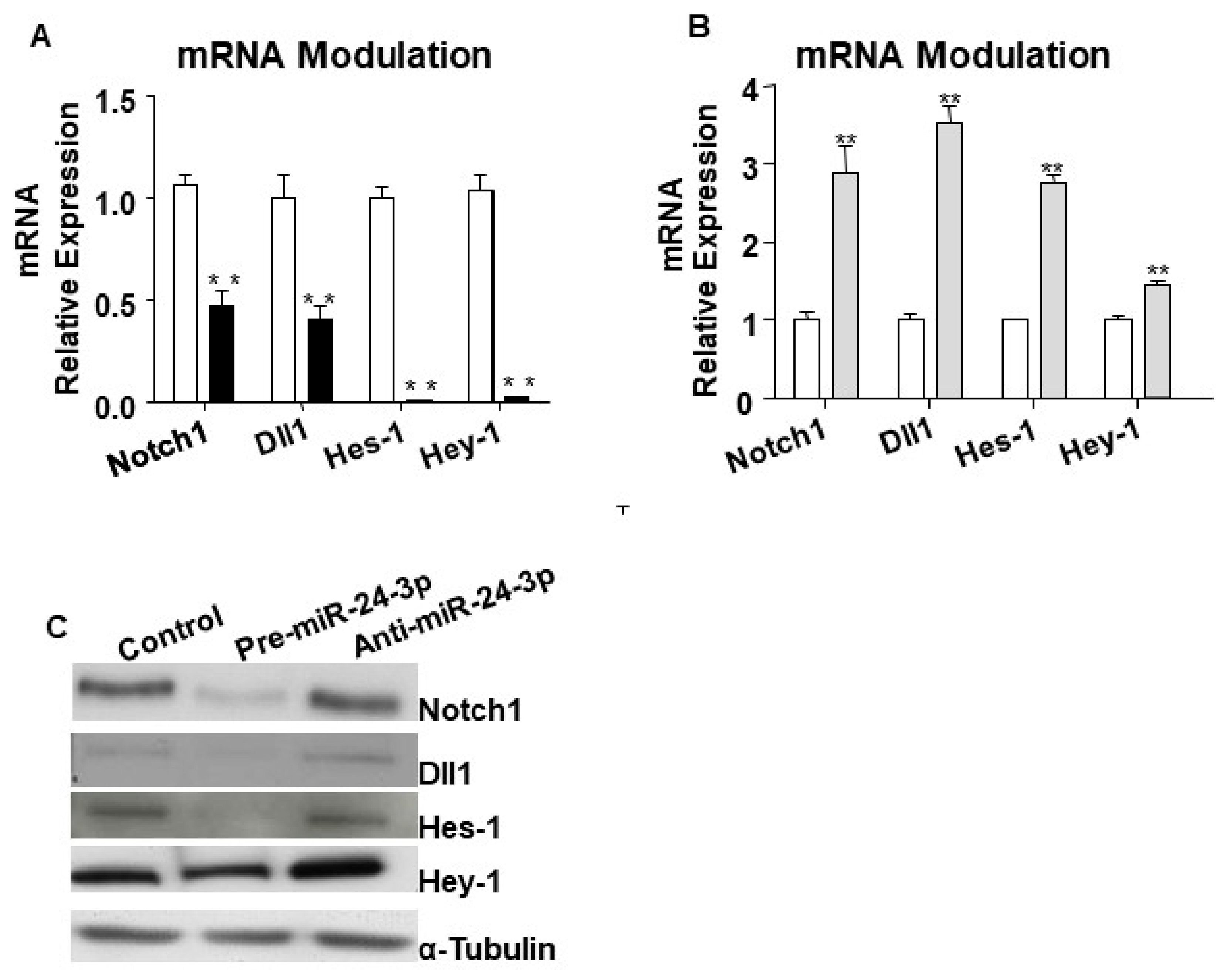
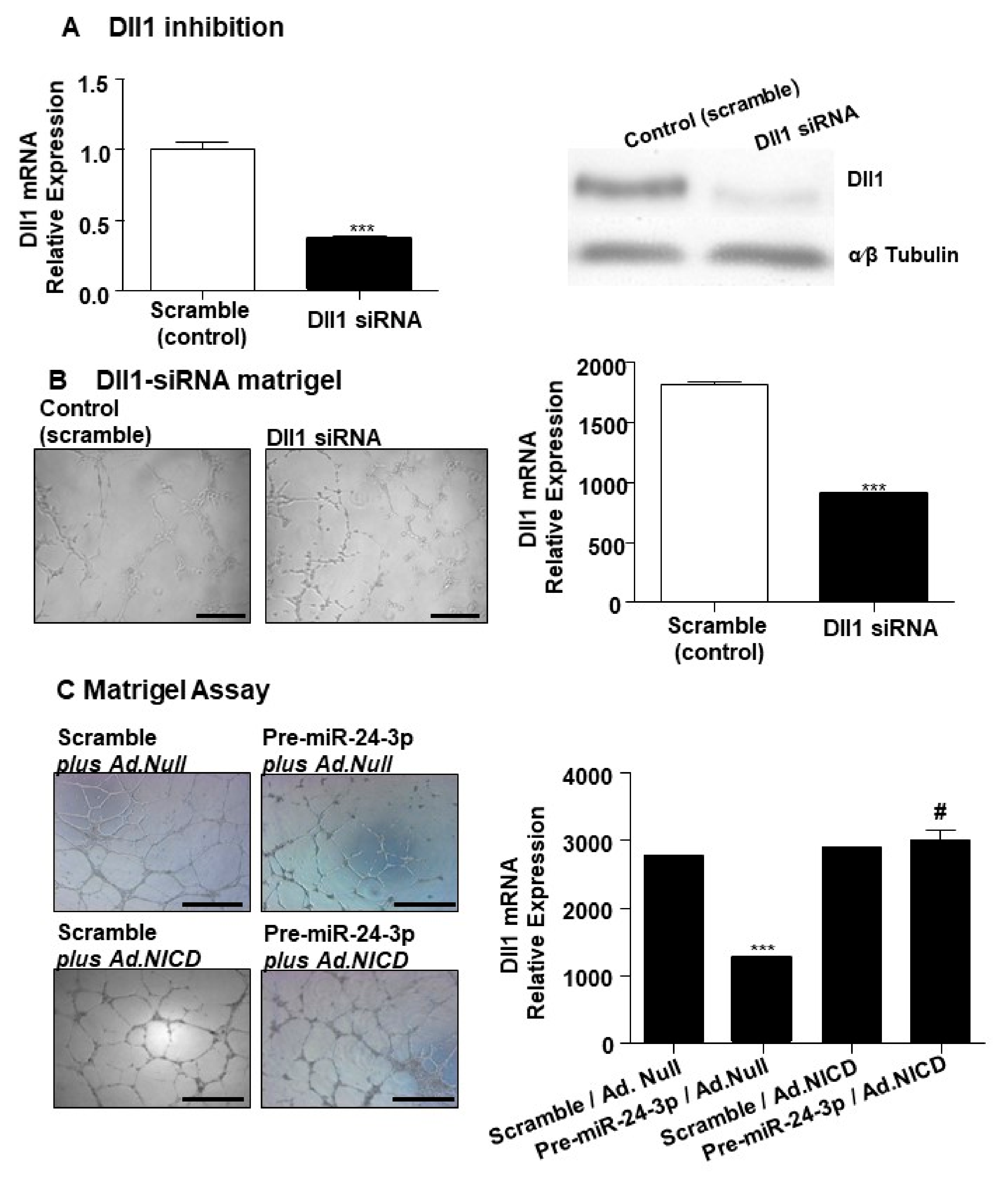
2.1. miR-24-3p Targets Dll1/Notch Signalling and Regulates Vascular Cell Behaviour
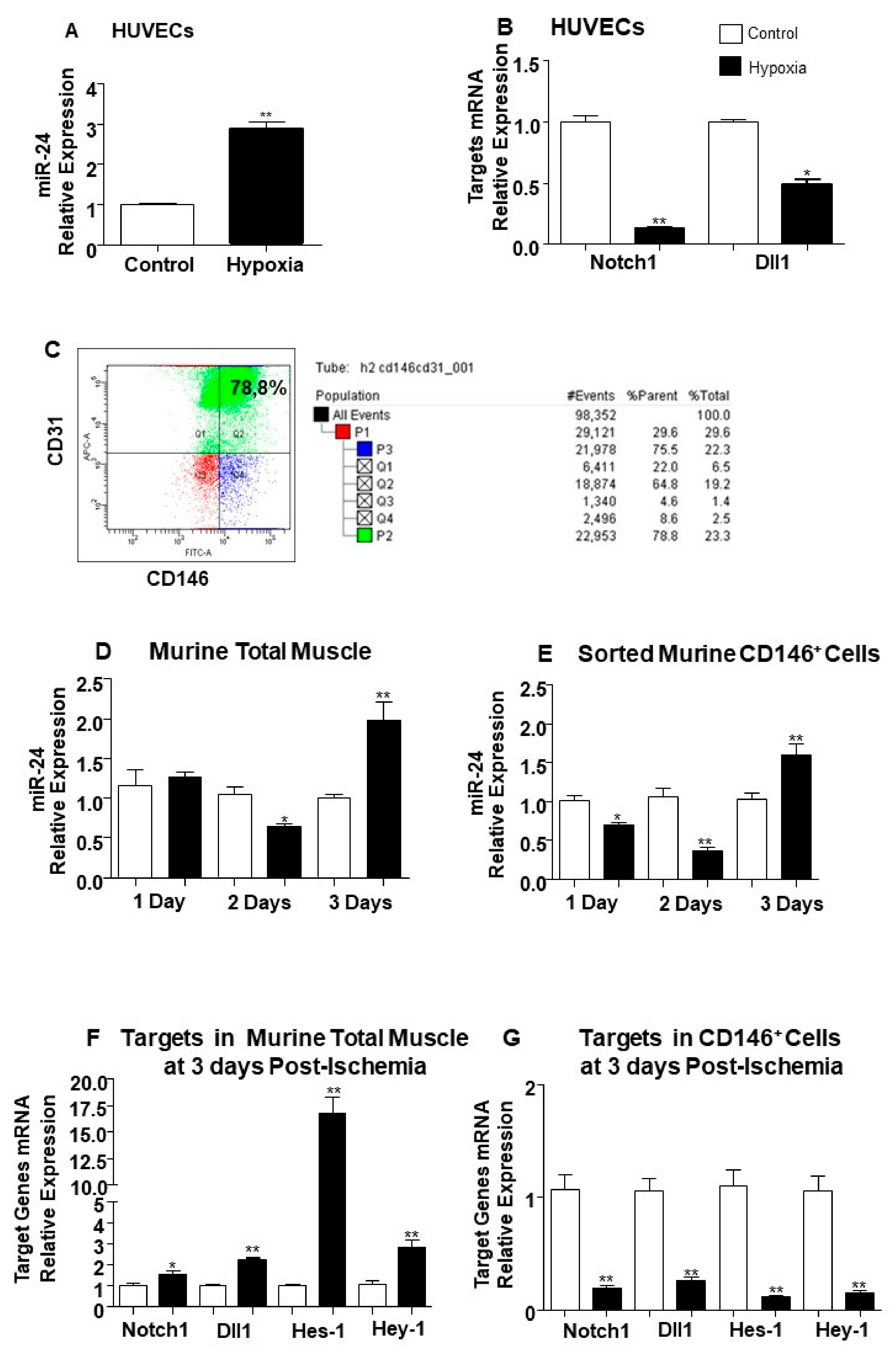
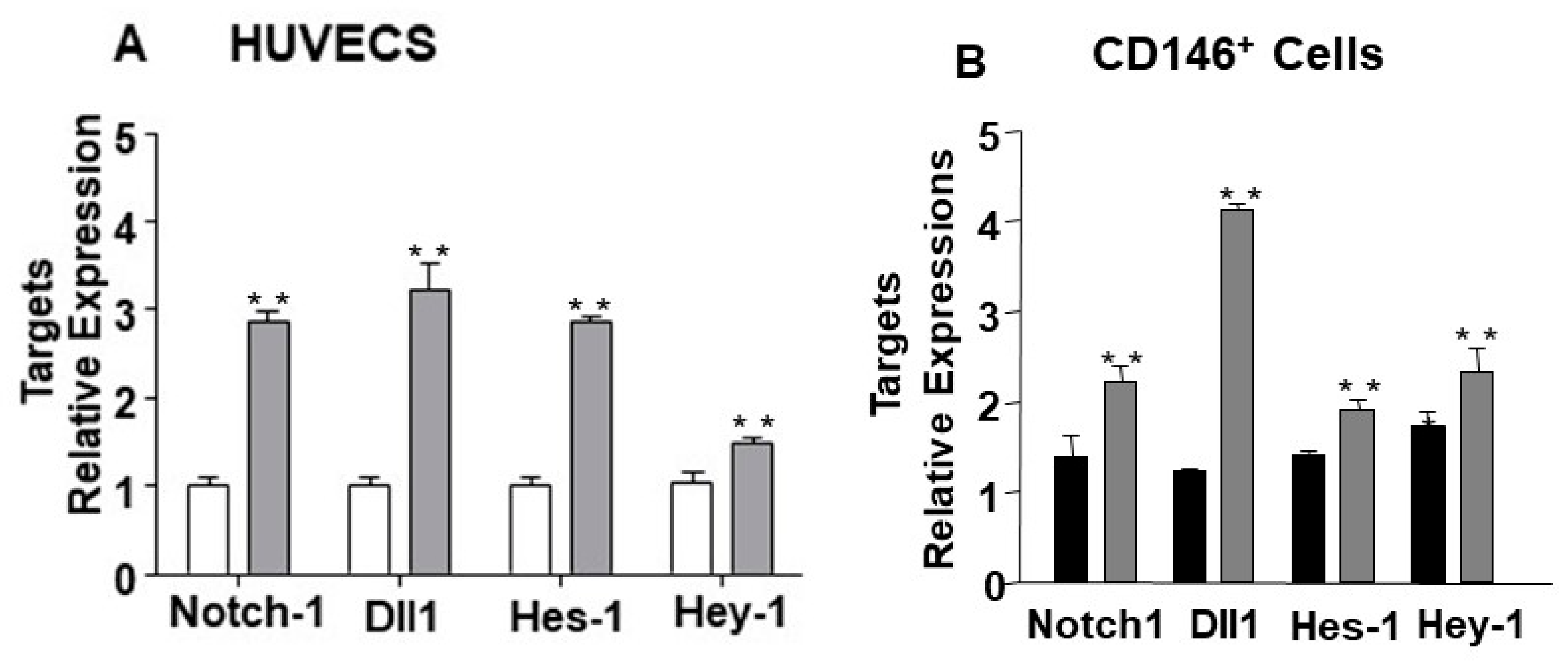
2.2. The Endothelial Expression of miR-24-3p and its Dll1 and Notch-1 Targets is Regulated by Hypoxia and Limb Ischemia
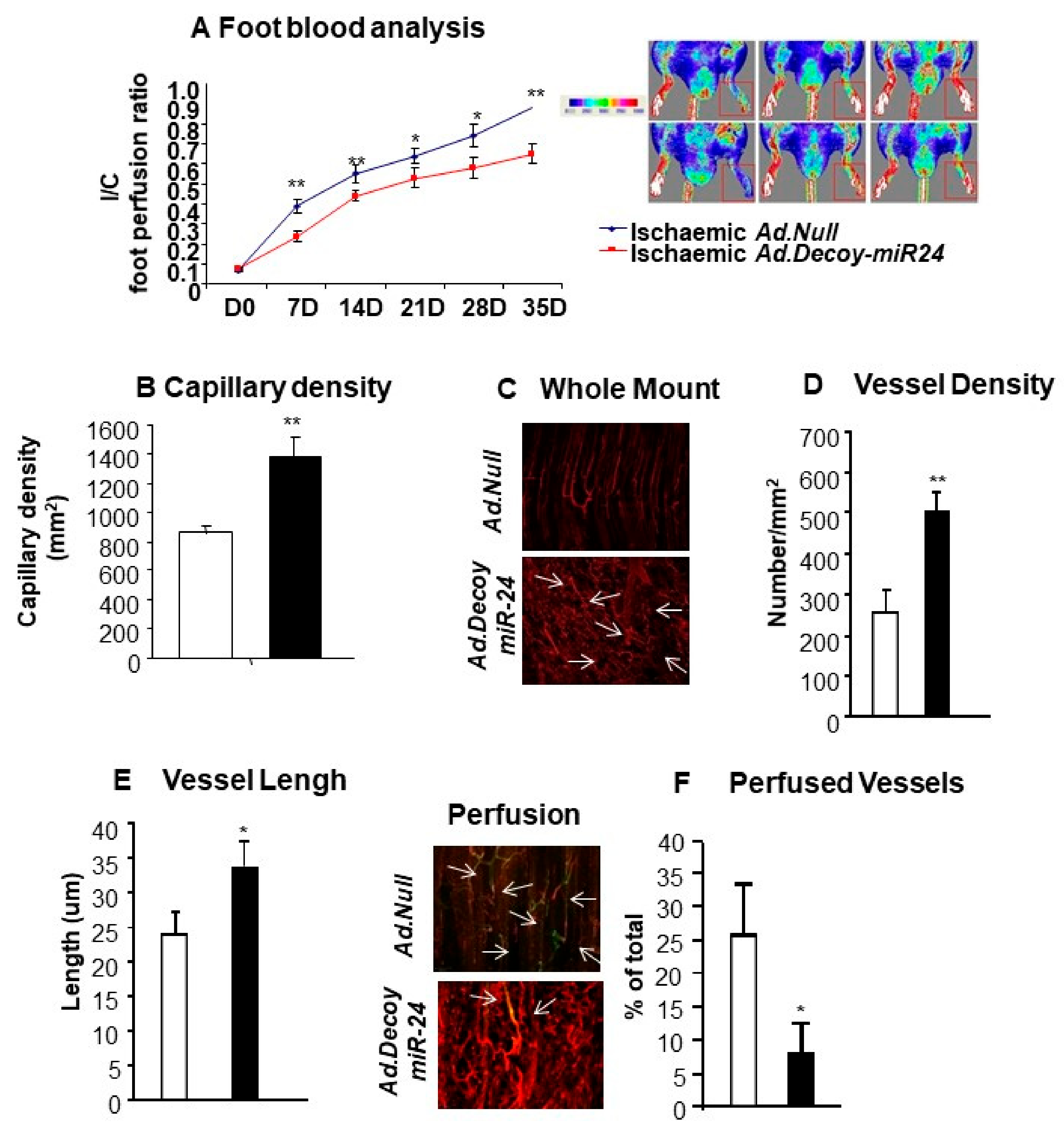
2.3. miR-24-3p Inhibition Induces the Development of Dysfunctional Microvessels and Worsens Blood Flow Recovery in the Limb Ischemia Model
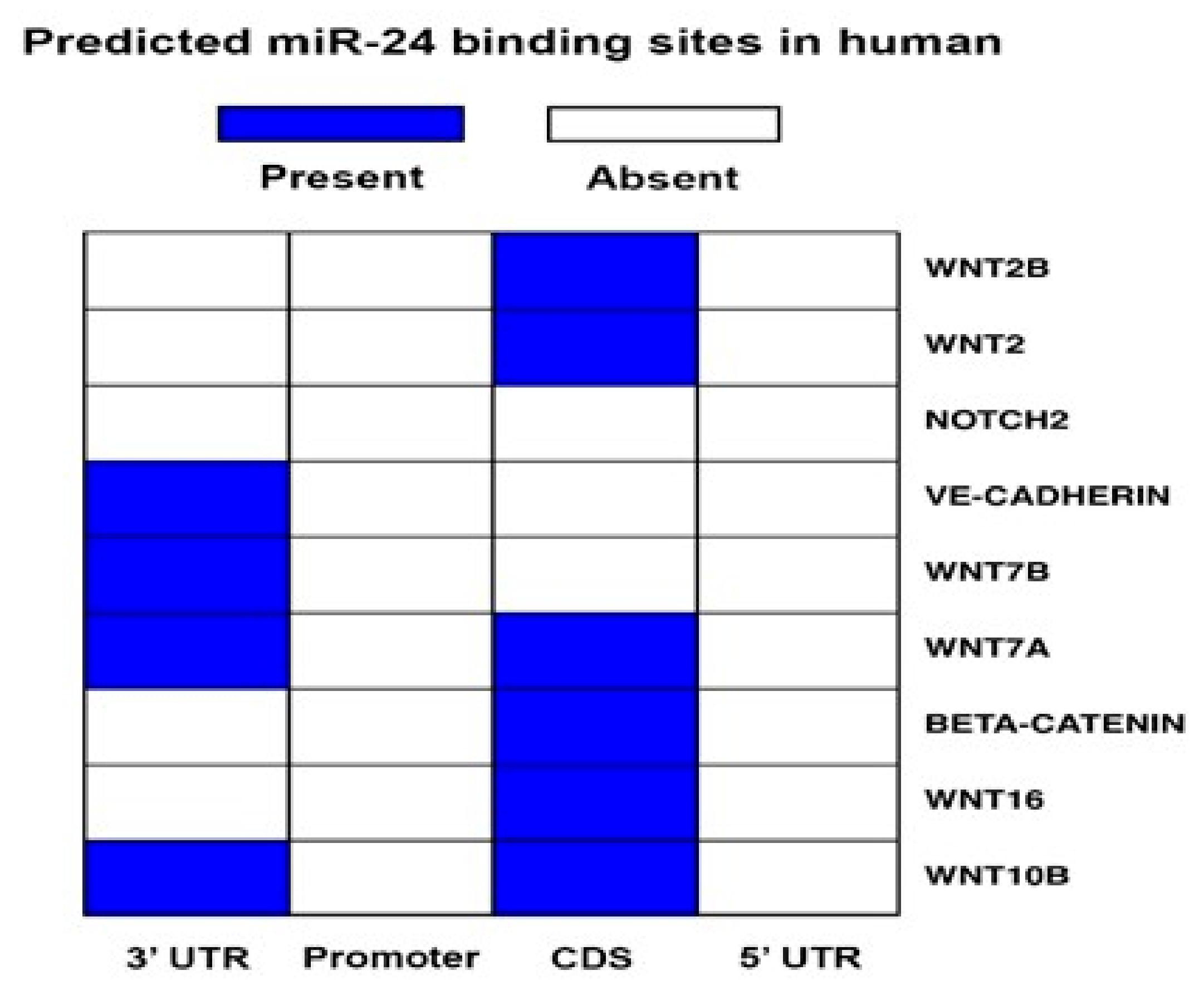
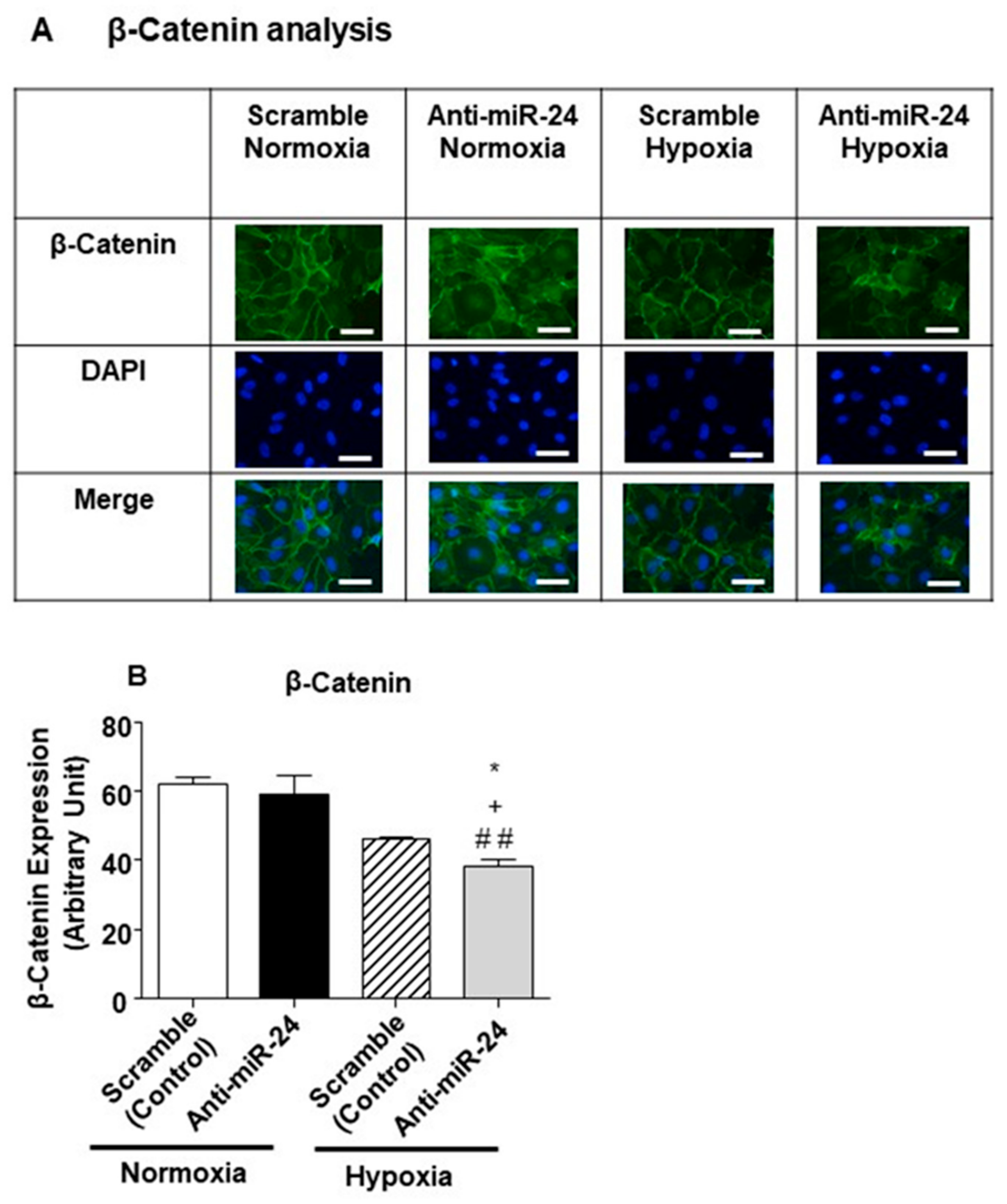
2.4. miR-24-3p Targets the Expression of β-Catenin and Other Vascular Morphogens
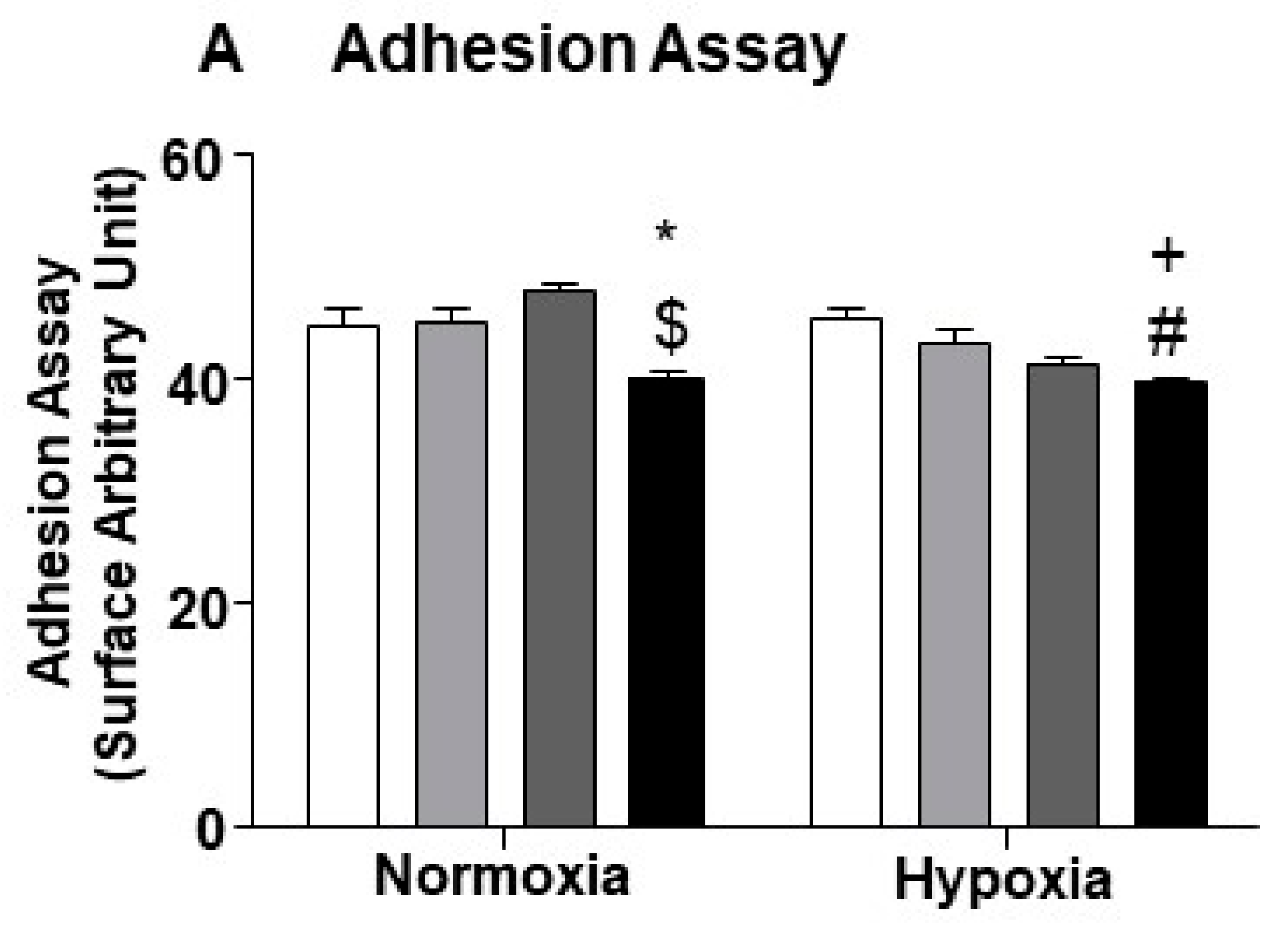
2.5. miR-24-3p Inhibition in ECs Impairs Their Capacity to Adhere with Pericytes
3. Discussion
4. Material and Methods
4.1. Cell Culture
4.2. miR-24-3p Target Prediction and Target Genes Validation
4.3. RNA Extraction and TaqMan Quantitative Real-Time PCR Analysis
4.4. Western Blot Analyses
4.5. miR-24-3p Mimic and Anti-miR-24-3p Transfection
4.6. Dll1 siRNA Transfection
4.7. Adenoviral Vectors
4.8. HUVEC Infections with Adenoviruses
4.9. Biology Assays with HUVECs
4.10. Pericyte Adhesion to HUVECs
4.11. β-catenin Quantification
4.12. In Vivo Studies
4.13. Mouse Model of Unilateral Limb Ischemia and in Vivo Gene Transfer
4.14. Expressional Analyses on Murine Tissues and Cells
4.15. Histology and Immunohistochemistry
4.16. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Ørom, U.A.; Lund, A.H. Experimental identification of microRNA targets. Gene 2010, 451, 1–5. [Google Scholar] [CrossRef] [PubMed]
- El Azzouzi, H.; Leptidis, S.; Doevendans, P.A.; Leon, J.; De Windt, L.J. HypoxamiRs: Regulators of cardiac hypoxia and energy metabolism. Trends Endocrinol. Metab. 2008, 12, 1426–1431. [Google Scholar] [CrossRef] [PubMed]
- Ivan, M.; Adrian, L.; Harris, A.L.; Martelli, F.; Kulshreshtha, R. Hypoxia response and microRNAs: No longer two separate worlds. J. Cell. Mol. Med. 2008, 12, 1426–1431. [Google Scholar] [CrossRef] [PubMed]
- Lorenzen, J.M.; Kaucsar, T.; Schauerte, C.; Schmitt, R.; Rong, S.; Hübner, A.; Scherf, K.; Fiedler, J.; Martino, F.; Kumarswamy, R.; et al. MicroRNA-24 antagonism prevents renal ischemia reperfusion injury. J. Am. Soc. Nephrol. 2014, 25, 2717–2729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiedler, J.; Jazbutyte, V.; Kirchmaier, B.C.; Gupta, S.K.; Lorenzen, J.; Hartmann, D.; Galuppo, P.; Kneitz, S.; Pena, J.T.G.; Sohn-Lee, C.; et al. MicroRNA-24 regulates vascularity after myocardial infarction. Circulation 2011, 124, 720–730. [Google Scholar] [CrossRef] [Green Version]
- Meloni, M.; Marchetti, M.; Garner, K.; Littlejohns, B.; Sala-Newby, G.; Xenophontos, N.; Floris, I.; Suleiman, M.-S.; Madeddu, P.; Caporali, A.; et al. Local inhibition of microRNA-24 improves reparative angiogenesis and left ventricle remodeling and function in mice with myocardial infarction. Mol. Ther. 2013, 21, 1140–1390. [Google Scholar] [CrossRef] [Green Version]
- Zhoua, O.; Gallaghera, R.; Ufret-Vincentya, R.; Lia, X.; Olsonb, E.N.; Wanga, S. Regulation of angiogenesis and choroidal neovascularization by members of microRNA-23∼27∼24 clusters. Proc. Nat. Acad. Sci. USA 2011, 108, 8287–8292. [Google Scholar] [CrossRef] [Green Version]
- Qian, L.; van Laake, L.W.; Huang, Y.; Liu, S.; Wendland, M.F.; Srivastava, D. miR-24 inhibits apoptosis and represses Bim in mouse cardiomyocytes. J. Exp. Med. 2011, 208, 549–560. [Google Scholar] [CrossRef] [Green Version]
- Maegdefessel, L.; Spin, J.; Raaz, U.; Eken, S.M.; Toh, R.; Azuma, J.; Adam, M.; Nagakami, F.; Heymann, H.M.; Chernugobova, E.; et al. miR-24 limits aortic vascular inflammation and murine abdominal aneurysm development. Nat. Commun. 2014, 5, 5214. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. Vasculogenesis, Angiogenesis, and Arteriogenesis: Mechanisms of Blood Vessel Formation and Remodeling. J. Cell. Biochem. 2007, 102, 840–8474. [Google Scholar] [CrossRef] [PubMed]
- Kofler, N.M.; Simons, M. Angiogenesis versus arteriogenesis: Neuropilin 1 modulation of VEGF signaling. F1000Prime Rep. 2015, 7, 26. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Proweller, A. Vascular smooth muscle Notch signals regulate endothelial cell sensitivity to angiogenic stimulation. J. Biol. Chem. 2011, 286, 13741–13753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korn, C.; Scholz, B.; Hu, J.; Srivastava, K.; Wojtarowicz, J.; Arnsperger, T.; Adams, R.H.; Boutros, M.; Augustin, H.G.; Augustin, I. Endothelial cell-derived non-canonical Wnt ligands control vascular pruning in angiogenesis. Development 2014, 141, 1757–1766. [Google Scholar] [CrossRef] [Green Version]
- Herbert, S.P.; Stainier, D.Y.R. Molecular control of endothelial cell behaviour during blood vessel morphogenesis. Nat. Rev. Mol. Cell Biol. 2011, 12, 551–564. [Google Scholar] [CrossRef] [Green Version]
- Napp, L.C.; Augustynik, M.; Paesler, F.; Krishnasamy, K.; Woiterski, J.; Limbourg, A.; Bauersachs, J.; Drexler, H.; Le Noble, F.; Limbourg, F.P. Extrinsic Notch Ligand Delta-Like 1 Regulates Tip Cell Selection and Vascular Branching Morphogenesis. Circ. Res. 2012, 110, 530–535. [Google Scholar] [CrossRef] [Green Version]
- Hellström, M.; Phng, L.-K.; Hofmann, J.J.; Wallgard, E.; Coultas, L.; Lindblom, P.; Alva, J.; Nilsson, A.-K.; Karlsson, L.; Gaiano, N.; et al. Dll4 signalling through Notch1 regulates formation of tip cells during angiogenesis. Nature 2007, 445, 776–780. [Google Scholar] [CrossRef]
- Trinidade, A.; Kumar, S.R.; Scehnet, J.S.; Lopes-da-Costa, L.; Becker, J.; Jiang, W.; Liu, R.; Gill, P.S.; Duarte, A. Overexpression of delta-like 4 induces arterialization and attenuates vessel formation in developing mouse embryos. Blood 2008, 112, 1720–1729. [Google Scholar] [CrossRef] [Green Version]
- Gerhardt, H.; Golding, M.; Fruttiger, M.; Ruhrberg, C.; Lundkvist, A.; Abramsson, A.; Jeltsch, M.; Mitchell, C.; Alitalo, K.; Shima, D.; et al. VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J. Cell Biol. 2003, 161, 1163–1177. [Google Scholar] [CrossRef]
- Shay-Salit, A.; Shushy, M.; Wolfovitz, E.; Yahav, H.; Breviario, F.E.; Nitzan Resnick, N. VEGF receptor 2 and the adherens junction as a mechanical transducer in vascular endothelial cells. Proc. Natl. Acad. Sci. USA 2002, 9, 9462–9467. [Google Scholar] [CrossRef] [Green Version]
- Stewart, D.J.; Kutryk, M.J.; Fitchett, D.; Freeman, M.; Camack, N.; Su, Y.; Siega, A.D.; Bilodeau, L.; Burton, J.R.; Proulx, G.; et al. Trial Investigators. VEGF gene therapy fails to improve perfusion of ischemic myocardium in patients with advanced coronary disease: Results of the NORTHERN trial. Mol. Ther. 2009, 17, 1109–1115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conway, E.M.; Collen, D.; Carmeliet, P. Molecular mechanisms of blood vessel growth. Cardiovasc. Res. 2001, 49, 507–521. [Google Scholar] [CrossRef] [Green Version]
- Slusarski, D.; Corces, V.; Moon, R. Interaction of Wnt and a Frizzled homologue triggers G-protein-linked phosphatidylinositol signalling. Nature 1997, 390, 410–413. [Google Scholar] [CrossRef] [PubMed]
- Phng, L.K.; Potente, M.; Leslie, J.D.; Babbage, J.; Nyqvist, D.; Lobov, I.; Ondr, J.K.; Rao, S.; Lang, R.A.; Thurston, G.; et al. Nrarp Coordinates Endothelial Notch and Wnt Signaling to Control Vessel Density in Angiogenesis. Dev. Cell. 2009, 1, 70–82. [Google Scholar] [CrossRef]
- Potente, M.; Gerhard, H.; Carmeliet, P. Basic and Therapeutic Aspects of Angiogenesis. Cell 2011, 6, 873–887. [Google Scholar] [CrossRef] [Green Version]
- Olsen, J.J.; Öther-Gee Pohl, S.; Deshmukh, A.; Visweswaran, M.; Ward, N.C.; Arfuso, F.; Agostino, M.; Dharmarajan, A. The Role of Wnt Signalling in Angiogenesis. Clin. Biochem Rev. 2017, 38, 131–142. [Google Scholar]
- Wu, C.; Chen, J.; Chen, C.; Wang, W.; Wen, L.; Gao, K.; Chen, X.; Xiong, S.; Zhao, H.; Li, S. Wnt/β-catenin coupled with HIF-1α/VEGF signaling pathways involved in galangin neurovascular unit protection from focal cerebral ischemia. Sci. Rep. 2015, 5, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Blaschuk, O.W.; Rowlands, T.M. Cadherins as modulators of angiogenesis and the structural integrity of blood vessels. Cancer Metastasis Rev. 2000, 19, 1–5. [Google Scholar] [CrossRef]
- Li, J.; Zhao, Y.; Lu, Y.; Ritchie, W.; Grau, G.; Vadas, M.A.; Gamble, J.R. The Poly-cistronic miR-23-27-24 Complexes Target Endothelial Cell Junctions: Differential Functional and Molecular Effects of miR-23a and miR-23b. Mol. Ther. Nucleic Acids 2016, 5, e354. [Google Scholar] [CrossRef]
- Kourtidis, A.; Ngok, S.P.; Pulimeno, P.; Feathers, R.W.; Carpio, L.R.; Baker, T.R.; Carr, J.M.; Yan, I.K.; Borges, S.; Perez, E.A.; et al. Distinct E-cadherin-based complexes regulate cell behaviour through miRNA processing or Src and p120 catenin activity. Nat. Cell Biol. 2015, 17, 1145–1157. [Google Scholar] [CrossRef] [Green Version]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/beta-catenin signaling: Components, mechanisms, and diseases Dev. Cell. 2009, 17, 9–26. [Google Scholar]
- Peifer, M.; Rauskolb, C.; Williams, M.; Riggleman, B.; Wieschaus, E. The segment polari-ty gene armadillo interacts with the wingless signaling pathway in both embryonic and adult pat-tern formation. Development 1991, 111, 1029–1043. [Google Scholar] [PubMed]
- Hausser, J.; Syed, A.P.; Bilen, B.; Zavolan, M. Analysis of CDS-located miRNA target sites suggests that they can effectively inhibit translation. Genome Res. 2013, 23, 604–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camps, C.; Saini, H.K.; Mole, D.R.; Choudhry, H.; Reczko, M.; Guerra-Assunção, J.A.; Tian, Y.M.; Buffa, F.M.; Harris, A.L.; Hatzigeorgiou, A.G.; et al. Integrated analysis of microRNA and mRNA expression and association with HIF binding reveals the complexity of microRNA expression regulation under hypoxia. Mol. Cancer 2014, 13, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, F.; Yang, R.; Wei, Y.; Wang, D.; Zhu, Y.; Wang, X.; Lu, Y.; Wang, Y.; Zen, K.; Li, L. HIF-1α-induced miR-23a∼27a∼24 cluster promotes colorectal cancer progression via reprogramming metabolism. Cancer Lett. 2019, 440–441, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Yamamizu, K.; Matsunaga, T.; Uosaki, H.; Fukushima, H.; Katayama, S.; Hiraoka-Kanie, M.; Mitani, K.; Yamashita, J.K. Convergence of Notch and beta-catenin signaling induces arterial fate in vascular progenitors. J. Cell Biol. 2010, 189, 325–338. [Google Scholar] [CrossRef] [Green Version]
- Hayward, P.; Brennan, K.; Sanders, P.; Balayo, T.; DasGupta, R.; Perrimon, N.; Arias, A.M. Notch modulates Wnt signalling by associating with Armadillo/beta-catenin and regulating its transcriptional activity. Development 2005, 132, 1819–1830. [Google Scholar] [CrossRef] [Green Version]
- Kwon, C.; Cheng, P.; King, I.N.; Andersen, P.; Shenje, L.; Nigam, V.; Srivastava, D. Notch Post-Translationally Regulates β-Catenin Protein in Stem and Progenitor Cells. Nat. Cell Biol. 2011, 13, 1244–1251. [Google Scholar] [CrossRef] [Green Version]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative CT method. Nat Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]
- Jamieson, J.; Macklin, B.; Gerecht, S. Pericytes Derived from Human Pluripotent Stem Cells. Adv. Exp. Med. Biol. 2018, 1109, 111–124. [Google Scholar]
- .Machado, M.J.C.; Boardman, R.; Riu, F.; Emanueli, C.; Benest, A.V.; Bates, D.O. Enhanced notch signaling modulates unproductive revascularization in response to nitric oxide-angiopoietin signaling in a mouse model of peripheral ischemia. Microcirculation 2019, 26, e12549. [Google Scholar] [CrossRef] [PubMed]
- Caporali, A.; Pani, E.; Horrevoets, A.J.G.; Kraenkel, N.; Oikawa, A.; Sala-Newby, G.B.; Meloni, M.; Cristofaro, B.; Graiani, G.; Leroyer, A.; et al. The neurotrophin receptor p75NTR triggers endothelial cell apoptosis and inhibits angiogenesis: Implications for diabetes-induced impairment of reparative neovascularization. Circ. Res. 2008, 103, e15–e26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borggrefea, T.; Lauthb, M.; Zwijsenc, A.; Huylebroeckd, D.; Oswalde, F.; Giaimoa, B.D. The Notch intracellular domain integrates signals from Wnt, Hedgehog, TGFβ/BMP and hypoxia pathways. Biochim. Biophys Acta. 2016, 1863, 303–313. [Google Scholar] [CrossRef]
- Kasper, D.M.; Moro, A.; Ristori, E.; Narayanan, A.; Hill-Teran, G.; Fleming, E.; Moreno-Mateos, M.; Vejnar, C.E.; Zhang, J.; Lee, D.; et al. MicroRNAs Establish Uniform Traits during the Architecture of Vertebrate Embryos. Dev. Cell. 2017, 27, 552–565. [Google Scholar] [CrossRef] [Green Version]
- Wansheng, L.; Xiaosheng, C.; Yu, Z. Effects of microRNA-21 and microRNA-24 inhibitors on neuronal apoptosis in ischemic stroke. Am. J. Transl. Res. 2016, 8, 3179–3187. [Google Scholar]
- Villa, J.C.; Chiu, D.; Brandes, A.H.; Escorcia, F.E.; Villa, C.H.; Maguire, W.F.; Hu, C.J.; de Stanchina, E.; Simon, M.C.; Sisodia, S.S.; et al. Nontranscriptional role of Hif-1α in activation of γ-secretase and notch signaling in breast cancer. Cell Rep. 2014, 21, 1077–1092. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.H.; Kim, H.; Ki, H.; Narae, K.Y.; Yang, I.; Yang, N.; Lee, K.Y.; Kim, N.; Park, H.-S.; Kim, K. Beta-catenin modulates the level and transcriptional activity of Notch1/NICD through its direct interaction. Biochim. Biophys. Acta. 2009, 1793, 290–299. [Google Scholar] [CrossRef] [Green Version]
- Phng, L.K.; Gerhardt, H. Angiogenesis: A team effort coordinated by Notch. Dev. Cell 2009, 16, 196–208. [Google Scholar] [CrossRef]
- Krebs, L.T.; Xue, Y.; Norton, C.R.; Shutter, J.R.; Maguire, M.; Sundberg, J.P.; Gallahan, D.; Closson, V.; Kitajewski, J.; Callahan, R.; et al. Notch signaling is essential for vascular morphogenesis in mice. Genes Dev. 2000, 14, 1343–1352. [Google Scholar]
- Li, B.; Jia, Z.; Wang, T.; Wang, W.; Zhang, C.; Chen, P.; Ma, K.; Zhou, C. Interaction of Wnt/beta-catenin and notch signaling in the early stage of cardiac differentiation of P19CL6 cells. J. Cell. Biochem. 2012, 113, 629–639. [Google Scholar] [CrossRef]
- Gustafsson, M.V.; Zheng, X.; Pereira, T.; Gradin, K.; Jin, S.; Lundkvist, J.; Ruas, J.L.; Poellinger, L.; Lendahl, U.; Bondesson, M. Hypoxia requires notch signaling to maintain the undifferentiated cell state. Dev. Cell. 2005, 9, 617–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Funahashi, Y.; Shawber, C.J.; Vorontchikhina, M.; Sharma, A.; Outtz, H.H.; Kitajewski, J. Notch regulates the angiogenic response via induction of VEGFR-1. J. Angiogenes. Res. 2010, 2, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darden, J.; Payne, L.B.; Zhao, H.; Chappell, J.C. Excess vascular endothelial growth factor-A disrupts pericyte recruitment during blood vessel formation. [CrossRef] [PubMed]
- Avnit-Sagi, T.; Kantorovich, L.; Kredo-Russo, S.; Hornstein, E.; Walker, M.D. The Promoter of the pri-miR-375 Gene Directs Expression Selectively to the Endocrine Pancreas. PLoS One 2009, 4, e5033. [Google Scholar] [CrossRef] [PubMed]
- Benedito, R.; Rocha, S.F.; Woeste, M.; Zamykal, M.; Radtke, F.; Casanovas, O.; Duarte, A.; Pytowski, B.; Adams, R.H. Notch-dependent VEGFR3 upregulation allows angiogenesis without VEGF–VEGFR2 signalling. Nature 2012, 484, 110–114. [Google Scholar] [CrossRef]
- Katare, R.; Riu, F.; Mitchell, K.; Gubernator, M.; Campagnolo, P.; Cui, Y.; Fortunato, O.; Avolio, E.; Cesselli, D.; Beltrami, A.P.; et al. Transplantation of human pericyte progenitor cells improves the repair of infarcted heart through activation of an angiogenic program involving micro-RNA-132. Circ. Res. 2011, 30, 894–906. [Google Scholar] [CrossRef] [Green Version]
- Slovut, D.P.; Sullivan, T.M. Critical limb ischemia: Medical and surgical management. Vasc. Med. 2008, 13, 281–291. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Murine Primers |
|---|---|
| 18S | F 5′-TAGAGGGACAAGTGGCGTTC-3′ R 5′-TGTACAAAGGGCAGGGACTT-3′ |
| Murine Notch1 | F 5′-CCTTGCTCTGCCTAACGC-3′ R 5′-GGAGTCCTGGATCGTTGG-3′ |
| Murine Dll1 | F 5′-GCAGGACCTTCTTTCGCGTAT-3′ R 5′-AAGGGGAATCGGATGGGGTT-3′ |
| Murine Hes1 | F 5′-TCAACACGACACCGGACAAAC-3′ R 5′-ATGCCGGGAGCTATCTTTTCTT-3′ |
| Murine Hey1 | F 5′-CCGACGAGACCGAATCAATAAC-3′ R 5′-TCAGGTGATCCACAGTCATCTG-3′ |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marchetti, M.; Meloni, M.; Anwar, M.; Al-Haj-Zen, A.; Sala-Newby, G.; Slater, S.; Ford, K.; Caporali, A.; Emanueli, C. MicroRNA-24-3p Targets Notch and Other Vascular Morphogens to Regulate Post-ischemic Microvascular Responses in Limb Muscles. Int. J. Mol. Sci. 2020, 21, 1733. https://doi.org/10.3390/ijms21051733
Marchetti M, Meloni M, Anwar M, Al-Haj-Zen A, Sala-Newby G, Slater S, Ford K, Caporali A, Emanueli C. MicroRNA-24-3p Targets Notch and Other Vascular Morphogens to Regulate Post-ischemic Microvascular Responses in Limb Muscles. International Journal of Molecular Sciences. 2020; 21(5):1733. https://doi.org/10.3390/ijms21051733
Chicago/Turabian StyleMarchetti, Micol, Marco Meloni, Maryam Anwar, Ayman Al-Haj-Zen, Graciela Sala-Newby, Sadie Slater, Kerrie Ford, Andrea Caporali, and Costanza Emanueli. 2020. "MicroRNA-24-3p Targets Notch and Other Vascular Morphogens to Regulate Post-ischemic Microvascular Responses in Limb Muscles" International Journal of Molecular Sciences 21, no. 5: 1733. https://doi.org/10.3390/ijms21051733