The Evolving Role of CD8+CD28− Immunosenescent T Cells in Cancer Immunology

1
Department of Neurosurgery, Indiana University School of Medicine, Indianapolis, IN 46202, USA
2
Department of Neurology, University of Illinois at Chicago School of Medicine, Chicago, IL 60612, USA
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2019, 20(11), 2810; https://doi.org/10.3390/ijms20112810
Submission received: 1 May 2019
/
Revised: 5 June 2019
/
Accepted: 6 June 2019
/
Published: 8 June 2019
(This article belongs to the Special Issue Immunophenotyping in Autoimmune Diseases and Cancer)
Abstract
:Functional, tumor-specific CD8+ cytotoxic T lymphocytes drive the adaptive immune response to cancer. Thus, induction of their activity is the ultimate aim of all immunotherapies. Success of anti-tumor immunotherapy is precluded by marked immunosuppression in the tumor microenvironment (TME) leading to CD8+ effector T cell dysfunction. Among the many facets of CD8+ T cell dysfunction that have been recognized—tolerance, anergy, exhaustion, and senescence—CD8+ T cell senescence is incompletely understood. Naïve CD8+ T cells require three essential signals for activation, differentiation, and survival through T-cell receptor, costimulatory receptors, and cytokine receptors. Downregulation of costimulatory molecule CD28 is a hallmark of senescent T cells and increased CD8+CD28− senescent populations with heterogeneous roles have been observed in multiple solid and hematogenous tumors. T cell senescence can be induced by several factors including aging, telomere damage, tumor-associated stress, and regulatory T (Treg) cells. Tumor-induced T cell senescence is yet another mechanism that enables tumor cell resistance to immunotherapy. In this paper, we provide a comprehensive overview of CD8+CD28− senescent T cell population, their origin, their function in immunology and pathologic conditions, including TME and their implication for immunotherapy. Further characterization and investigation into this subset of CD8+ T cells could improve the efficacy of future anti-tumor immunotherapy.
1. Introduction
The conflict between cancer and the immune system has long been established [1]. Immunotherapies are being investigated to augment the anti-tumor effects of the immune system and promote long-term cancer control [2]. CD8+ cytotoxic T cells (CTLs) are the main players driving the adaptive immune response against cancer and execute tumor-specific immune responses, rendering them the primary endpoint to most immunotherapies [3,4]. Establishment of effective antigen-specific CD8+ T cells enabled preliminary clinical success of cancer vaccines, oncolytic viruses, adoptive cellular therapy, and checkpoint inhibitors in several cancers including melanoma, lung cancer, renal cell cancer, Hodgkin’s lymphoma, etc. [5,6,7]. Unfortunately, despite their promise, the efficacy of these treatments varies depending on the type and location of the tumor, and has been ineffective in poorly immunogenic cancers such as glioblastoma (GBM) [7,8,9,10,11,12,13].
A variety of T cell deficiencies have been identified in immunosuppressive tumors that contribute to the ultimate ineffectiveness of CD8+ CTL-mediated tumor killing [14]. Immune tolerance, anergy, and exhaustion of CD8+ T cells have been studied extensively in the past [14,15,16,17]. While the concept of immune senescence, defined by terminally differentiated cells in cell cycle arrest after extensive replication or in response to cellular damage or stress, has been well established with aging and chronic infections [18,19,20,21], our knowledge of its role in cancer is still in its early stages. CD28 is an indispensable costimulatory molecule needed for the activation of T cells and its role is critical to the proper activation of CD8+ CTLs [22]. Current evidence shows that the downregulation of CD28 is a hallmark of senescent CD8+ T cells and CD28− senescent T cells display immunosuppressive functions in cancer [23,24,25,26].
In this review, we will focus on the recent advances in our understanding of CD8+CD28− T cells. We first will discuss cellular senescence and the evolution of the CD28− T cells. Next, we will review the significance of CD8+CD28− T cells in multiple disease processes, including transplant, autoimmune disease, chronic viral infection, and cancer, including CNS tumors. Finally, we will discuss the functional implications of CD28− T cells in onco-immunology and the important areas of future investigation on novel immunotherapeutic strategies.
2. Role of CD8+ T Cells in Cancer Immunology
CD8+ T cells are a subset of lymphocytes committed to detecting peptide antigens presented by major histocompatibility complex (MHC) class I molecules (Figure 1) [27,28]. CD8+ T cells arise from common lymphoid progenitors that migrate from the bone marrow to the thymus where they pass through a series of distinct phases of maturation [29,30]. The naïve CD8+ T cell pool is comprised of polyclonal T cells that express CD28, CCR7, and CD62L, the latter two allowing them to recirculate between blood and secondary lymphoid organs [31,32]. Initial priming of CD8+ T cells involves T cell receptor (TCR) recognition of peptide/MHC complexes presented by professional antigen presenting cells (APCs), such as dendritic cells (DCs). DCs also express surface markers CD70 and CD80/CD86 for binding to CD27 and CD28 receptors expressed on CD8+T cells. This provides a critical secondary signal for T cell activation. Host cells, including cancer cells, can serve as targets for previously activated CD8+ T cells by processing and presenting antigenic tumor peptides by MHC class I.
In addition, CD4+ T helper cells interact with CD8+ T cells and modulate CD8+ T cell activation [34,35,36]. Activated CD4+ T helper cells can secrete a variety of cytokines, such as interferon-gamma (IFN-γ) and IL-2, and facilitate CD8+ T cells’ optimal proliferation and activation [28,37]. CD4+ T cells could also help with DC maturation for expression of costimulatory molecules and secretion of cytokines that contribute to CD8+ T cell priming [38]. A similar mechanism is also carried out by natural killer (NK) cells, showing that there is collaboration between CD4+ T cells with NK and DCs for induction of CD8+ T cell priming [28,38].
Upon activation, effector CD8+ CTLs destroy antigen-expressing cancer cells primarily utilizing two main pathways: granule exocytosis (such as perforin and granzyme) and death ligand/receptor-mediated apoptosis (such as Fas ligand and TRAIL) [39]. Additionally, activated CD8+ T cells release IFN-γ and tumor necrosis factor alpha (TNF-α) to induce cytotoxicity in the target cells and stimulate M1 macrophage-mediated anti-tumor response [28]. In multiple solid tumors, tumor-infiltrating CD8+ CTLs can be used as a prognostic factor [40,41,42,43,44,45,46,47,48]. For example, in breast cancer, significantly increased CD8+ T cells at tumor sites have been shown to have an inverse correlation with advanced tumor stages and a positive correlation with clinical outcomes [41,49,50]. Similar findings of a favorable prognosis associated with the accumulation of tumor-infiltrating CD8+ T cells were reported in colorectal, oral squamous cell, pancreatic, and ovarian carcinomas [43,44,45,47,48,51].
3. CD28 Costimulatory Receptor
The CD28 costimulatory receptor, a 44-kDa membrane glycoprotein, is expressed on nearly all human T lymphocytes at birth [52]. Binding of the CD28 receptor on T cells provides an essential second signal alongside TCR ligation for naïve T cell activation. CD28 signaling has diverse effects on T cell function, including orchestrating membrane raft trapping at the immunological synapse, transcriptional changes, downstream post-translational modifications, and actin cytoskeletal remodeling [52,53,54]. This leads to intracellular biochemical events such as survival and proliferation signals, induction of IL-2, activation of telomerase, stabilization of mRNA for several cytokines, increased glucose metabolism, and enhanced T cell migration and homing [52,55,56].
CD28 family of costimulatory molecules also includes ICOS, CTLA-4, PD-1, PD1H, TIGIT, and BTLA [41]. This family of receptors and ligands has considerable complexity in both binding pattern and biological effects. For instance, CD28 (activating) and CTLA-4 (inhibitory) are highly homologous and compete for the same ligands (CD80 and CD86) and regulate immune response by providing opposing effects [51,52].
The critical role of CD28 in induction of immune response was demonstrated in mice treated with CD28 antagonist, which induced antigen specific tolerance and prevented the progression of autoimmune diseases and organ graft rejection [57]. This has led to the development of abatacept (Orencia® Bristol-Myers Squibb, New York, NY, USA) [58] and belatacept (Nulojix® Bristol-Myers Squibb New York, NY, USA) [59], a modified antibody composed of Fc region of IgG1 fused to the extracellular domain of CTLA-4, which bind to CD80/86 on APCs and block the costimulatory signaling by CD28. Abatacept and belatacept are used clinically to treat rheumatoid arthritis and organ transplant rejection, respectively [56,60].
On the other hand, the use CD28 agonists to awaken T cells from the tolerant state could lead to new therapies to re-activate the immune system for the treatment of infectious disease [61] and cancer [62]. Although, in a phase I trial of systemic administration of CD28 superagonist monoclonal antibodies (mAb) (TGN1412), uncontrolled CD28 signaling led to a potent induction of downstream immune activation independent of TCR-CD3 complex resulting in catastrophic systemic inflammatory syndromes in six volunteer subjects [63]. Investigation of these unexpected serious adverse events have led to better design of clinical trials and appreciation of differences in CD28 expression and regulation between species (critical for transitioning preclinical testing to clinical investigations) [64,65]. An updated CD28 superagonist TAB08 is under clinical testing for rheumatoid arthritis [64]. In addition, localized or targeted use of anti-CD28 mAbs has much potential such as incorporating the intracellular costimulatory domain of CD28 into CAR (chimeric antigen receptor) T cells for adoptive transfer immunotherapy and the use of CD28 agonist aptamer with tumor vaccine [66,67,68].
Importantly, the use of these therapeutics are in clinical trials for a variety of disease states including solid neoplasms and inflammatory diseases (Table 1). Although previous experience with CD28 agonist mAbs has been disappointing, headway is being made in their use in solid tumors and rheumatoid arthritis. Perhaps out of an abundance of caution, current clinical trials for the use of CD28 agonists are testing their safety, efficacy, and tolerability in patients and are undergoing dose escalation studies. Fortunately, significant progress has been made into CAR-T cell therapy incorporating costimulatory domains and has led to the FDA-approval of the CAR-T cell therapy tisagenlecleucel (KYMRIAH® Novartis Pharmaceuticals, Basel, Switzerland) for relapse or refractory acute lymphoblastic leukemia patients [69,70].
Furthermore, recent progress in the manipulation of other costimulatory molecule such as ICOS, CD137 (4-1BB), OX40, and GITR has also demonstrated tremendous therapeutic potential [74]. Activation of ICOS, CD137, and OX40 via mAbs and aptamers improved T cell proliferation, function, and overall antitumor response [74,75,76,77]. Targeting of GITR exhibited effects on both effector and regulatory T cells. Ligation of constitutively expressed GITR on regulatory T cells caused depletion in their number, loss lineage stability and immunosuppressive function [78,79], while GITR agonists work synergistically with PD-1 blockage to promote CD4 and CD8 accumulation in murine ovarian cancer [80]. Blockade of inhibitory receptor CTLA-4 have been shown to be effective in enhancing CD28 signaling and augmenting ICOS stimulation [74,81]. Combination of checkpoint inhibitors and costimulatory agonists presents an exciting avenue of cancer treatment and several clinical studies are currently investigating their benefit [74].
4. Cellular Senescence in the Immune System
Cellular senescence is a state of cell cycle arrest in respond to cellular damage or stress to prevent neoplastic transformation [82]. Cellular senescence have been identified in areas of physiological homeostasis, such as development [82], wound healing [83], and placental natural killer lymphocytes [84]. However, cellular senescence also contributes to the loss of function associated with aging and age-related disease as well as chronic viral infection, neurodegenerative disease, and cancer [18,85,86]. Two categories of cellular senescence have been described in literature: the first is aging associated, telomere-dependent replicative senescence and the second is stress-induced premature senescence, also known as telomere-independent senescence [82,87]. Oncogene-induced senescence is one of the well-described mechanisms for premature senescence [87,88]. Regardless of the initiating mechanism, cells that undergo senescence survive by exhibiting a variety of phenotypical and molecular features (Figure 2), including morphological changes, cell division blockage, change of sensitivity against apoptosis, metabolic dysfunction, and a specialized secretory activity termed senescence associated secretory phenotype (SASP) [20]. Additional characteristics include nuclear p16 and p21 expression [89,90,91], DNA damage [92], senescence associated heterochromatin foci (SAHF) [93], and increased lysosomal senescence-associated β-galactosidase (SA-β-gal) activity [91]. Recently, lipofuscin accumulation was also established as a hallmark of senescent cells [94].
Cellular senescence also occurs in the human immune system [18,95]. The effectiveness of the immune response declines with age particularly in the elderly population [96,97]. Immune deficiencies start to appear in DCs, NK cells, and monocytes/macrophages with aging, and it was proposed that myeloid-derived suppressor cells (MDSC) could also induce senescence in T and B cells compartment in diverse inflammatory conditions [96,97]. Importantly, lymphocytes, especially T cells, show the most considerable changes with aging [95,98]. Among the various complex features that contribute to the aging-associated changes in T cell immunity, the accumulation of CD28− T cells is one of the hallmark phenomena in T cell immunosenescence [26,99,100]. TME can also induce senescence in tumor-infiltrating T cells [14].
5. Role of CD8+CD28− T Cells
Although CD28 is expressed on the majority of the CD8+ T cells at birth [52], normal aging process and activation of CD8+ T cells invariably leads to CD28 downregulation [101]. CD8+CD28− cells represent a distinct population distinguishable from the general population of CD8+CD28+ T cells [99], which are known for their crucial role in the clearance of cancer and intracellularly infected cells, in terms of their phenotype and function [102]. In vitro studies showed purified CD28+ T cells progressively lose CD28 during each successful stimulation, with the CD8+ T cells losing their CD28 more rapidly than the CD4+ T cells [26,103,104]. The differential rate of CD28 loss is associated with the rapid inactivation of telomerase and CD8+ T cells reach replicative senescence faster than CD4+ T cells, at which stage T cells are no longer able to enter mitosis but still remain viable [105]. Thus, these CD8+CD28− T cells are defined as senescent T cells. Less than 50% of the CD8+ T cell compartment of elderly or chronically infected individuals are CD28+ while up to 80% of CD4+ T cells maintain their CD28 expression even in the centenarians [26,103]. Interestingly, a large proportion of CD8+CD28− T cells of elderly persons also have lower levels of CD8 expression [106,107]. Although the significance of this observation is unknown, downregulation of the expression of CD8 and CD4 molecules is characteristic for activated T cells, suggesting that those CD8lowCD28− T cells subset represent senescent lymphocytes that are chronically activated from either common persistent antigens (in the setting of aging) or persistent infection or inflammation (in the setting of cancer) [25,108].
6. Characteristics of CD8+CD28− Senescent T cells
CD8+CD28− T cells are highly oligoclonal and terminally differentiated effector lymphocytes that have lost their capacity to undergo cell division [23,108]. They are functionally heterogeneous and their characteristics vary depending on the context where they are found (Figure 3) [23,108]. They also express a variety of other NK cell-related receptors including KIR, NKG2D, CD56, CD57, CD94, and Fc-γ receptor IIIa and have features crossing the border between innate and adaptive immunity [109,110]. Alterations in the costimulatory receptor NKG2D signaling and expression levels in CD8+ T cells can lead to autoimmune conditions that are either TCR dependent or TCR-independent [111,112,113]. Gained expression of CD57, also known as HNK-1 (human natural killer-1), is a common feature associated with circulating senescent T cells, and increased CD8+CD28−CD57+ senescent T cells were identified in multiple pathological conditions, including HIV infection, multiple myeloma, lung cancer, and chronic inflammation conditions such as diabetes and obesity [99,114,115]. Although expression of CD57 is linked to antigen-induced apoptosis of CD8+ T cells [116], the acquisition of CD94 has been reported to confer resistance to apoptosis in CD8+CD28− T cells. [117] Similarly, CD8+CD28− T cells are often associated with the lack of perforin, rendering them ineffective Ag-specific killers in chronic viral infections [21,118,119,120]. On the other hand, in certain disease processes such as chronic obstructive pulmonary disease (COPD) and rheumatoid arthritis, they have been reported to express increased levels of cytotoxic mediators, perforin and granzyme B, and pro-inflammatory cytokines, IFN-γ and TNFα, where CD8+CD28− T cells can cause significant damages to normal surrounding tissue in an antigen-nonspecific manner [121].
CD8+CD28− T cells are also shown to be immunosuppressive and function as regulatory T cells [122,123,124,125]. For example, CD8+CD28− T cells directly inhibit Ag-presenting function of DCs by inducing inhibitory receptors, such as immunoglobulin like transcript 3 (ILT3) and ILT4, which leads DCs to be immune tolerant than immunogenic [122,126]. Such tolerogenic DCs anergize alloreactive CD4+CD25+ T cells and convert them into regulatory T cells, which in turn, continue the immunosuppressive cascade by tolerizing other DCs and amplify T cell immunosenescence [126,127]. In vivo, CD8+CD28− T cells have been directly correlated with the suppression of antigen-specific T cell responses in patients with plasma cell dyscrasia [123]. Therefore, their characteristics and functions in immunity range from reduced antigen-specific killing to enhanced cytotoxic abilities and from crossing innate immunity function to promoting immune regulation.
7. CD8+CD28− T cells in Pathologic Conditions
CD8+CD28− T cells play a significant role in pathological conditions [18,99,100,121]. High populations of these cells have been associated with chronic viral infections including human immunodeficiency virus (HIV), hepatitis C virus, cytomegalovirus (CMV), and human parvovirus B19 [99]. Shortened telomeres, reduced IL-2 production, and increased IL-6 production were observed in these cells [19]. The loss of CD28 also serves as a prognostic indicator for viral infection. For instance, increased frequency of CD8+CD28− T cells in the early stage of HIV infection correlates with faster progression to AIDS [99]. Additionally, higher levels of CD8+CD28− T cells are associated with subclinical carotid artery disease in HIV-infected women [99]. Older CMV seropositive individuals, who had higher number of CD8+CD28− cells, responded poorly to vaccines and had early mortality compared to aged-matched CMV seronegative counterparts [19].
A heterogeneous role was reported for CD8+CD28− T cells in autoimmune diseases [99,108]. Senescent T cell population is increased in patients with Grave’s disease and ankylosing spondylitis and these cells’ cytotoxicity contributes to autoimmune response [128,129]. In rheumatoid arthritis patients, clinical response to abatacept, a CD80/86-CD28 T cell co-stimulation modulator, is associated with a concomitant decrease in CD8+CD28− T cells, suggesting prognostic value for this phenotype [56]. In contrast, patients with systemic lupus erythematosus were found to have reduced CD8+CD28− T cells [130]. Similarly, in a mice model for multiple sclerosis, adoptive transfer of CD8+CD28− regulatory T cells have been shown to prevent autoimmune encephalomyelitis [131]. Though treated as a single phenotype, CD8+CD28− T cells represent a heterogeneous group that has differential activities in different pathologic conditions. In solid organ transplant recipients, CD8+CD28− T cells have been found to undergo oligoclonal expansion and play a suppressive role and promote allograft tolerance [108,132]. Elevated CD8+CD28− T cell population in liver transplant patients are associated with better graft function and reduced rejection rates [133] and contribute to reducing immunosuppressant dosage [124]. In addition, the presence of CD8+CD28− T cells is associated with decreased CD80/86 expression and increased inhibitory receptor (ILT3, ILT4) in circulating APCs, implying an immunosuppressive role of this subset [126,133].
8. CD8+CD28− T cells and Cancer
The cycle of anti-tumor immunity starts with the presentation of cancer antigens released from cancer cell turnover. Resident tissue DCs or lymph nodes residing DCs capture cancer antigens and present the antigens in the form of peptide-MHC I complex to activate naïve CD8+ T cells. Activated effector CD8+ T cells travel through blood and lymphatic to reach tumor beds where they execute cancer-specific killing. This leads to further endogenous antigen release and DC activation, thereby closing the cycle for anti-tumor immune response [1,28].
The presence of lymphoid aggregates is linked with improved responses to cancer therapies such as standard cytotoxic therapies, vaccine-based treatments, or immune checkpoint blockades. [5,134] Immunologically ‘hot’ tumors, such as melanomas and lung cancers, are thus more amenable to control than ‘cold’ tumors, i.e., tumors with diminished T cell infiltrates, such as GBM. [135,136] This drives modern cancer therapy to investigate how to redirect the TME to attract the right types of immune infiltrates.
Effector arm insufficiency, especially CD8+ T cell dysfunction, is a hallmark of inadequate anti-tumor immune response [16]. Four forms of T cell dysfunction—tolerance, anergy, exhaustion, and senescence—have all been reported in cancer microenvironment [17,35,137]. Immune tolerance is a physiological process where the body eliminates self-reacting T cells. Tumor cells, such as GBM, can mimic peripheral tolerance and facilities FasL-mediated deletion of T cells [17]. Tolerance can be enforced by TGF-β and IL-10 secreted by Tregs that are recruited in the TME as well as cancer cells. [17,138,139]. T cell anergy is a T cell hypo-responsive state with low IL-2 production and poor proliferative capacity [17]. It results from the lack of co-stimulation of TCRs through CD28. This is due to the competitive binding of CTLA-4 expressed on Tregs to CD80 and CD86 on the APCs [14,17,139]. Anergic T cells display very little to no effector function, but expression on inhibitory markers is unclear [15]. T cell exhaustion occurs after excessive and continuous stimulation of the TCR and inflammatory cytokines like IFNα/β. Exhausted T cells have diminished proliferative capacity and have poor cytokine production and effector function. However, they express high levels of inhibitory receptors, or immune checkpoints, such as PD-1, CTLA-4, TIM3, LAG3, etc. [15,140]. The degree of T cell exhaustion can vary with tumor types as well. It is more severe in GBM compared to other cancers such as breast, lung, and melanoma [137]. T cell senescence can be distinguished from anergy and exhaustion in their origins and their surface receptors. For example, senescent T cells express fewer CD28 but more NK receptors, whereas exhausted or anergic T cells express more inhibitory receptors such as PD-1 and CTLA-1 (Figure 4). While anergic and exhausted T cells are hyporesponsive and hypofunctional, senescent T cells were considered metabolically active in their physiological or pathological environment despite being in cell cycle arrest (Figure 3). Though both T cell anergy and T cell exhaustion in natural occurrence are considered reversible, T cell senescence was considered irreversible until recently [14].
Targeting dysfunctional effector T cells has revolutionized the paradigm of tumor immunotherapy and immune checkpoint inhibitors set a paramount example [141]. Utilizing tumor dysregulation of immune checkpoint expression in exhausted dysfunctional T cells, immune checkpoint inhibitors were developed to promote protumor immune landscape [141]. Ipilimumab, CTLA-4 inhibitor, the first immune checkpoint inhibitor approved by FDA, was used to treat patients with advanced melanoma and has demonstrated improved survival when given with gp100 melanoma vaccine [142]. PD-1 inhibitors, pembrolizumab and nivolumab, and PD-L1 inhibitors, atezolizumab, durvalumab, and avelumab soon followed showing promising results. Pembrolizumab and nivolumab demonstrated 40–45% objective response rate in melanoma and non-small cell lung cancer [143]. In urothelial bladder cancer, use of PD-1/PD-L1 inhibitors showed overall response rate between 13% and 24% [144]. With metastatic brain disease, the combination of ipilimumab and nivolumab showed 56% intracranial response with 19% complete response from metastatic melanoma, and pembrolizumab have been shown to demonstrate intracranial activity against melanoma and NSCLC metastasis [145]. Continued investigation of the safety and efficacy of these novel immunomodulating drugs are ongoing in various malignancies [146]. However, it has been reported that the presence of functionally aberrant senescent T cells with loss of CD27 and CD28 and gained expression of CD57 cells was associated with resistance to checkpoint inhibitor blockade [8]. Therefore, senescent T cell phenotypes are possible predictive biomarkers for clinical response to checkpoint inhibitor therapy and potential targets to improve checkpoint inhibitor efficacy.
Increased CD8+CD28− senescent populations displaying heterogeneous roles have been observed in multiple solid and hematogenous tumors [24]. This immunosuppressive phenotype was initially observed in patients with plasma cell dyscrasia, where increased number of CD8+CD28− T cells was present in the bone marrow (i.e., TME) and the amount directly correlated with the suppression of antigen-specific T cell response [123]. Similarly, in patients with lung cancer, the CD8+CD28− T cells express elevated Foxp3 and have been shown to play an immunoregulatory role [147]. High levels of CD8+CD28− T cells were found in patients with advanced stages of non-small-cell lung cancer. Their numbers declined with resection of the tumor, and the decreased level of CD8+CD28− T cells correlates with favorable prognosis in tumor management [148]. In contrast, the CD8+CD28− T cell populations in melanoma patients express perforin, where they contribute to anti-tumor immune response [149].
CD8+CD28- T cell senescence is triggered by a variety of biological processes including telomere damage, Treg cells and tumor-associated stresses [150]. Naturally occurring CD4+CD25hiFoxp3+ Treg (nTreg) and tumor-derived γδ Treg cells can induce responder T cell senescence as an immunosuppressive mechanism [127,151]. Senescent T cells induced by Treg cells have phenotypic changes, including expression of SA-β-Gal, downregulation of co-stimulatory molecules CD27 and CD28, and promotion of cell cycle and growth arrest in G0/G1 phase [127,151]. Importantly, CD8+CD28− senescent T cells induced by Treg cells have potent regulatory activities [150] and deepen immunosuppression in TME [152]. Therefore, CD8+CD28− senescent T cells are important mediators and amplifiers of immunosuppression mediated by Treg cells. The blockage of Treg-induced senescence in responder immune cells is critical in controlling tumor immunosuppression and restoring effector T cell function.
One of the mechanisms for Treg-induced CD8+ T cell immunosenescence is mediated by nuclear kinase ataxia-telangiectasia mutated protein (ATM)-associated DNA damage in responder T cells triggered by glucose competition [150]. MAP ERK1/2 and p38 signaling functionally cooperate with transcription factors STAT1/STAT3 to control Treg-induced senescence in responder T cells [150]. Utilizing these mechanisms, Treg-induced T cell senescence was successfully prevented by inhibiting the DNA damage response and/or STAT signaling in a recent in vivo mice study [150]. Another study has shown that senescent T cells are in fact able to regain function by inhibiting the p38 MAPK pathway [153]. Furthermore, human Toll-like receptor 8 (TLR8) signaling can directly target multiple types of tumors and prevent tumor-induced cell senescence through modulation of levels of endogenous secondary messenger cAMP in tumor cells [154].
9. CD8+CD28− T cells and Glioblastoma
Despite being isolated in the intracranial compartment by the blood brain barrier, GBM, the most common and aggressive primary brain tumor in adults, demonstrates a remarkable level of immunosuppression [155]. Current standard of care for patients with GBM includes surgery, temozolomide chemotherapy, radiotherapy, and corticosteroids, all of which have potent immunosuppressive effects. Tumor cells express surface ligands such as PD-L1 and CD95 (Fas/apoptosis antigen 1) that can lead to T cell suppression via apoptosis and immunosuppressive cytokines like TGF-β, IL-10, and other tolerance factors [139]. Tumor-associate macrophages, modified neutrophils, and Foxp3+ Tregs, are also recruited by the tumor to promote its progression [156,157,158].
T cell dysfunctions including tolerance, anergy, and exhaustion have also been well documented in GBM [14,17]. However, despite of promise of checkpoint inhibitors in the treatment of several solid tumors, their therapeutic efficacy in GBM remains to be validated. Phase III clinical trial Checkmate 143 reported that PD-1 monoclonal antibody (nivolumab) monotherapy failed to demonstrate survival benefits compared to bevacizumab in recurrent GBM patients who were previously treated with chemotherapy and radiotherapy [17,69,155]. Ongoing clinical trials are investigating the tolerability and drug toxicity in combination treatment and in patients with newly diagnosed GBM patients as well as recurrent GBM patients. The muted response to immune activators seen thus far highlights to the need for novel strategies to boost immunity to GBM.
The role of T cell senescence in GBM has been reported but is yet to be fully elucidated. The presence of circulating senescent CD4+CD28−CD57+ T cells was correlated with poor prognosis in GBM patients [159]. CD8+CD28−Foxp3+ T cells, which have been found in other cancers to cause APC dysfunction [160], were also identified in TME from patients with GBM [161]. The APCs isolated from these patients displayed dysfunctional phenotype associated with high levels of ILT2, ILT3, and ILT4 and low levels of CD40, CD80, and CD86 [162]. It is speculated that CD8+CD28− T cells help sculpt an immunosuppressive environment in a similar fashion in GBM.
The potential pro-tumoral effect of CD8+CD28− cells can also be inferred from worse prognosis observed in older GBM patients [163]. Since CD8+CD28− cells are derived from the general population of immature CD8+CD28+ T cells originating from the thymus [99], the production of immature CD8+CD28+ decreases as thymic involution occurs through aging, but also with cancer [164]. This decrease in immature CD8+CD28+ cells due to thymic senescence has also been associated with poor outcome in GMB patients [17].
10. Implications of CD8+CD28− T Cells for the Future of Immunotherapy
Since success of immunotherapy largely relies on addressing effector arm dysfunction, as evident from the success of checkpoint inhibitors, future investigations into new treatment methods should explore strategies to deplete or inhibit regulatory CD8+CD28− T cells and reverse T cell senescence as an adjuvant for more effective immunotherapy. There are four main approaches to rejuvenate T cell pools (1) replacement of senescent cells, (2) reprogramming of the senescent cells to be functional, (3) adoptive transfer of proficient T cells, and (4) restoration of naïve T cell pool [165].
Replacement strategies include selectively removing senescent cells from the circulation and then subsequent expansion of memory and effector T cells. Removal of senescent cells is of particular importance, not only due to their own dysfunction but also due to their ability to and spreads senescence in bystander cells [166]. A possible approach for their removal is to promote selective apoptosis in senescent T cells. In a recent study, an engineered peptide that interferes with FOXO4/p53 interaction induced p53-mediated intrinsic apoptosis in senescent fibroblasts and neutralized doxorubicin-induced chemotoxicity in vivo [167]. Whether this also can be used in inducing apoptosis of senescent T cell remains to be investigated. Targeting commonly known senescent cell anti-apoptotic pathways such as Bcl-2 and ephrins in senescent T cells is also warranted [168]. Homeostatic expansion in the form of autologous stem cell transplantation has been used to reconstitute functional naïve, memory, and affect T cell pools in both autoimmune diseases and hematologic malignancies [169,170]. More recently, senolytic treatment of amyloid-beta (Aβ) peptide -associated senescent oligodendrocyte progenitor cell in mice with Alzheimer’s disease showed successful selective removal of senescent cells from the plaque, reduced neuroinflammation, lessened Aβ load, and ameliorated cognitive deficits [171]. This is of particular interest as a successful application of senolytic therapy in the CNS pathology, such as GBM. CD8+CD28− cells replacement strategies are still in early phases of development, however their successful implementation has the potential to complement the current paradigm of immunotherapies.
Re-programming involves differentiating T cells away from dysfunctional states by enhancing telomerase activity to extend cellular lifespan and preclude replicative senescence [172]. For example, restoring CD28 expression slows replicative senescence in human T cells through increased telomerase activity to increase proliferative potential [173]. Additionally, pharmacological inhibition of SRC homology 2 domain-containing phosphatase-1 (SHP-1), a key regulator of T cell signal transduction machinery, improved TCR/CD28 signaling and successfully improve T cell functions in elderly donors [174]. Aptamers, short, single-stranded DNA or RNA molecules, have been engineered to target immune costimulatory receptors (CD28, OX40 and 4-1BB) and have shown to improve T cell activation and induce antitumor response. Aptamers have the benefits of being chemically synthesized, versatility of targeting motifs, high penetration rate, and ease of neutralization [75]. Re-programming of T cells present the most promising avenue for anti-CD8+CD28− therapy with wide selection of potential targets and treatment mechanisms.
Adoptive transfer is to bypass the co-stimulation requirement to re-differentiate pluripotent stem cells into naïve and cytotoxic T cells to fight malignancy [175]. The development of T cell adoptive immunotherapy using the third generation of CAR technology by incorporating the intracellular costimulatory domains to bypass the requirement for CD28 activation is underway. The third generation CARs T cells were investigated in hematological malignancies and xenograft model of solid tumors and have shown preclinical success [176,177,178,179,180]. While other types of immunotherapy can cause systemic side effects, antigen specificity of CAR therapy limits the adverse effect of immunotherapy to its targets, and they are reversible when the target cell is eliminated, or the engraftment of the CAR T cells is terminated [181]. However, its high specificity can be a weakness in heterogeneous tumors with high mutational profiles since CAR therapy can select for cells negative for the targeted antigen [155]. Such was the case for IL-13Rα2 CAR therapy for GBM where recurrence occurred 7.5 months after treatment despite shrinking all lesions by 77–100% [155].
Finally, restoring and maintaining the thymic environment reverse effects of thymic involution. Using bioengineered thymus organoids with the help of growth-promoting factors and cytokines such as IL-21, it has been shown that significant immune restorative function and rejuvenation of the peripheral T cell pools were achieved in murine models [182]. Unfortunately, current understanding of thymic restoration is not complete enough for clinical implementation, and there are still unanswered questions regarding its feasibility in establishing functional naïve T cell production [183,184]. The safe removal of senescent CD8+ T cells and restoration or differential induction of functional CD8+ cytotoxic T cells would add a promising mechanism to defend host against cancer invasion and fight immunotolerance of malignancy.
There is theoretical concern that reversing the growth arrest by selective blockage of senescent T cells carries a risk of malignancy, which is less of a concern for targeting functionally exhausted T cells [185]. Nevertheless, one may argue that increased theoretical lifetime risk of malignancy is outweighed by the potential immediate benefit of extending the life expectancy, even by a few months to years, in the battle against aggressive cancers, such as GBM with a median overall survival of only 20.6 months. Furthermore, the benefits of targeting both immunosenescence and exhaustion may be more evident with reduced dose requirement for each, thereby reducing risks of drug-associated adverse events. Potential synergistic efficacy to boost immunity against cancer may also be implemented as already seen with GITR stimulation/PD-1 blockade and CTLA-4/ICOS stimulation [74,80,81].
11. Conclusions
In summary, functional, tumor specific CD8+ cytotoxic T cells drive the adaptive immune response to cancer and are the primary endpoint to most immunotherapies. However, the promise of current cancer immunotherapy has been limited by marked immunosuppression in the TME defined by CD8+ T cell dysfunction, especially in immune ‘cold’ cancers, such as GBM. Among the many facets of CD8+ T cell dysfunction, including tolerance, anergy, and exhaustion, CD8+ T cell senescence, as represented by the CD8+CD28− population, is an emerging field and their presence has been described in many cancers. CD8+CD28− T cells contribute to tumor immunosuppression and resistance to immunotherapy. Further characterization and investigation into this subset of CD8+ T cells will provide novel targets for effective immunotherapy and successful cancer control.
Author Contributions
Conceptualization, Supervision, and Funding Acquisition: M.D.; Writing-Original Draft Preparation and Writing-Review & Editing: W.X.H., J.H.W., M.H., K.F., and M.D.
Funding
This work was supported by the NIH K08NS092895 grant (M.D.).
Acknowledgments
Authors would like to thank Christopher Brown MS for his help with the figure illustrations.
Conflicts of Interest
The authors declare that they have no competing interests.
Abbreviations
| APC | Antigen presenting cells |
| CAR | Chimeric antigen receptor |
| CMV | Cytomegalovirus |
| CTL | Cytotoxic T-lymphocytes |
| DC | Dendritic cells |
| GBM | Glioblastoma |
| NK | Natural killer |
| SA-β-Gal | Senescence-associated-β-galactosidase |
| TCR | T cell receptor |
| Th | T helper cells |
| TME | Tumor microenvironment |
| Tregs | T- regulatory cells |
References
- Palucka, A.K.; Coussens, L.M. The basis of oncoimmunology. Cell 2016, 164, 1233–1247. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Lee, H.H.; Lee, H.J.; Choi, W.S.; Lee, J.; Kim, H.S. Natural killer cells as a promising therapeutic target for cancer immunotherapy. Arch. Pharm. Res. 2019, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Spitzer, M.H.; Carmi, Y.; Reticker-Flynn, N.E.; Kwek, S.S.; Madhireddy, D.; Martins, M.M.; Gherardini, P.F.; Prestwood, T.R.; Chabon, J.; Bendall, S.C.; et al. Systemic immunity is required for effective cancer immunotherapy. Cell 2017, 168, 487–502.e15. [Google Scholar] [CrossRef] [PubMed]
- Ostroumov, D.; Fekete-Drimusz, N.; Saborowski, M.; Kuhnel, F.; Woller, N. CD4 and CD8 T lymphocyte interplay in controlling tumor growth. Cell. Mol. Life Sci. 2018, 75, 689–713. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farkona, S.; Diamandis, E.P.; Blasutig, I.M. Cancer immunotherapy: The beginning of the end of cancer? Bmc Med. 2016, 14, 73. [Google Scholar] [CrossRef]
- Moreira, A.; Gross, S.; Kirchberger, M.C.; Erdmann, M.; Schuler, G.; Heinzerling, L. Senescence markers: Predictive for response to checkpoint inhibitors. Int. J. Cancer 2019, 144, 1147–1150. [Google Scholar] [CrossRef]
- Omuro, A.; Vlahovic, G.; Lim, M.; Sahebjam, S.; Baehring, J.; Cloughesy, T.; Voloschin, A.; Ramkissoon, S.H.; Ligon, K.L.; Latek, R.; et al. Nivolumab with or without ipilimumab in patients with recurrent glioblastoma: Results from exploratory phase i cohorts of checkmate 143. Neuro Oncol. 2018, 20, 674–686. [Google Scholar] [CrossRef]
- Reardon, D.A.; Omuro, A.; Brandes, A.A.; Rieger, J.; Wick, A.; Sepulveda, J.; Phuphanich, S.; de Souza, P.; Ahluwalia, M.S.; Lim, M.; et al. OS10.3 randomized phase 3 study evaluating the efficacy and safety of nivolumab vs bevacizumab in patients with recurrent glioblastoma: Checkmate 143. Neuro Oncol. 2017, 19, iii21. [Google Scholar] [CrossRef]
- Powles, T.; Duran, I.; van der Heijden, M.S.; Loriot, Y.; Vogelzang, N.J.; De Giorgi, U.; Oudard, S.; Retz, M.M.; Castellano, D.; Bamias, A.; et al. Atezolizumab versus chemotherapy in patients with platinum-treated locally advanced or metastatic urothelial carcinoma (imvigor211): A multicentre, open-label, phase 3 randomised controlled trial. Lancet 2018, 391, 748–757. [Google Scholar] [CrossRef]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and activity of anti-PD-l1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef]
- Crespo, J.; Sun, H.; Welling, T.H.; Tian, Z.; Zou, W. T cell anergy, exhaustion, senescence, and stemness in the tumor microenvironment. Curr. Opin. Immunol. 2013, 25, 214–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef]
- Thommen, D.S.; Schumacher, T.N. T cell dysfunction in cancer. Cancer Cell 2018, 33, 547–562. [Google Scholar] [CrossRef]
- Woroniecka, K.I.; Rhodin, K.E.; Chongsathidkiet, P.; Keith, K.A.; Fecci, P.E. T-cell dysfunction in glioblastoma: Applying a new framework. Clin. Cancer Res. 2018, 24, 3792–3802. [Google Scholar] [CrossRef]
- Aguilera, M.O.; Delgui, L.R.; Romano, P.S.; Colombo, M.I. Chronic infections: A possible scenario for autophagy and senescence cross-talk. Cells 2018, 7, 162. [Google Scholar] [CrossRef]
- Aberg, J.A. Aging, inflammation, and hiv infection. Top. Antivir. Med. 2012, 20, 101–105. [Google Scholar]
- Childs, B.G.; Gluscevic, M.; Baker, D.J.; Laberge, R.M.; Marquess, D.; Dananberg, J.; van Deursen, J.M. Senescent cells: An emerging target for diseases of ageing. Nat. Rev. Drug Discov. 2017, 16, 718–735. [Google Scholar] [CrossRef]
- Warren, J.A.; Clutton, G.; Goonetilleke, N. Harnessing CD8(+) T cells under hiv antiretroviral therapy. Front. Immunol. 2019, 10, 291. [Google Scholar] [CrossRef]
- Schwartz, R.H. Costimulation of T lymphocytes: The role of CD28, CTLA-4, and B7/BB1 in interleukin-2 production and immunotherapy. Cell 1992, 71, 1065–1068. [Google Scholar] [CrossRef]
- Chen, X.; Liu, Q.; Xiang, A.P. Cd8+CD28- T cells: Not only age-related cells but a subset of regulatory T cells. Cell. Mol. Immunol. 2018, 15, 734–736. [Google Scholar] [CrossRef]
- Filaci, G.; Fenoglio, D.; Fravega, M.; Ansaldo, G.; Borgonovo, G.; Traverso, P.; Villaggio, B.; Ferrera, A.; Kunkl, A.; Rizzi, M.; et al. CD8+ CD28- T regulatory lymphocytes inhibiting T cell proliferative and cytotoxic functions infiltrate human cancers. J. Immunol. 2007, 179, 4323–4334. [Google Scholar] [CrossRef]
- Borthwick, N.J.; Lowdell, M.; Salmon, M.; Akbar, A.N. Loss of CD28 expression on CD8(+) T cells is induced by IL-2 receptor gamma chain signalling cytokines and type i ifn, and increases susceptibility to activation-induced apoptosis. Int. Immunol. 2000, 12, 1005–1013. [Google Scholar] [CrossRef]
- Vallejo, A.N. CD28 extinction in human T cells: Altered functions and the program of T-cell senescence. Immunol. Rev. 2005, 205, 158–169. [Google Scholar] [CrossRef]
- Borst, J.; Ahrends, T.; Babala, N.; Melief, C.J.M.; Kastenmuller, W. CD4(+) T cell help in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2018, 18, 635–647. [Google Scholar] [CrossRef]
- Farhood, B.; Najafi, M.; Mortezaee, K. CD8(+) cytotoxic t lymphocytes in cancer immunotherapy: A review. J. Cell. Physiol. 2019, 234, 8509–8521. [Google Scholar] [CrossRef]
- Singer, A.; Adoro, S.; Park, J.H. Lineage fate and intense debate: Myths, models and mechanisms of CD4- versus CD8-lineage choice. Nat. Rev. Immunol. 2008, 8, 788–801. [Google Scholar] [CrossRef]
- Germain, R.N. T-cell development and the CD4-CD8 lineage decision. Nat. Rev. Immunol. 2002, 2, 309–322. [Google Scholar] [CrossRef]
- Arosa, F.A.; Esgalhado, A.J.; Padrao, C.A.; Cardoso, E.M. Divide, conquer, and sense: Cd8(+)CD28(-) T cells in perspective. Front. Immunol. 2016, 7, 665. [Google Scholar] [CrossRef]
- Fearon, D.T.; Carr, J.M.; Telaranta, A.; Carrasco, M.J.; Thaventhiran, J.E. The rationale for the il-2-independent generation of the self-renewing central memory CD8+ T cells. Immunol. Rev. 2006, 211, 104–118. [Google Scholar] [CrossRef]
- Martinez-Lostao, L.; Anel, A.; Pardo, J. How do cytotoxic lymphocytes kill cancer cells? Clin. Cancer Res. 2015, 21, 5047–5056. [Google Scholar] [CrossRef]
- Janssen, E.M.; Lemmens, E.E.; Wolfe, T.; Christen, U.; von Herrath, M.G.; Schoenberger, S.P. CD4+ T cells are required for secondary expansion and memory in CD8+ T lymphocytes. Nature 2003, 421, 852–856. [Google Scholar] [CrossRef]
- Reading, J.L.; Galvez-Cancino, F.; Swanton, C.; Lladser, A.; Peggs, K.S.; Quezada, S.A. The function and dysfunction of memory CD8(+) T cells in tumor immunity. Immunol. Rev. 2018, 283, 194–212. [Google Scholar] [CrossRef]
- Ara, A.; Ahmed, K.A.; Xiang, J. Multiple effects of CD40-CD40l axis in immunity against infection and cancer. Immuno Targets Ther. 2018, 7, 55–61. [Google Scholar] [CrossRef]
- Kershaw, M.H.; Westwood, J.A.; Darcy, P.K. Gene-engineered T cells for cancer therapy. Nat. Rev. Cancer 2013, 13, 525–541. [Google Scholar] [CrossRef]
- Gottschalk, C.; Mettke, E.; Kurts, C. The role of invariant natural killer T cells in dendritic cell licensing, cross-priming, and memory CD8(+) T cell generation. Front. Immunol. 2015, 6, 379. [Google Scholar] [CrossRef]
- Wu, R.C.; Hwu, P.; Radvanyi, L.G. New insights on the role of CD8(+)CD57(+) T-cells in cancer. Oncoimmunology 2012, 1, 954–956. [Google Scholar] [CrossRef]
- Lohr, J.; Ratliff, T.; Huppertz, A.; Ge, Y.; Dictus, C.; Ahmadi, R.; Grau, S.; Hiraoka, N.; Eckstein, V.; Ecker, R.C.; et al. Effector T-cell infiltration positively impacts survival of glioblastoma patients and is impaired by tumor-derived TGF-β. Clin. Cancer Res. 2011, 17, 4296–4308. [Google Scholar] [CrossRef]
- Mahmoud, S.M.; Paish, E.C.; Powe, D.G.; Macmillan, R.D.; Grainge, M.J.; Lee, A.H.; Ellis, I.O.; Green, A.R. Tumor-infiltrating CD8+ lymphocytes predict clinical outcome in breast cancer. J. Clin. Oncol. 2011, 29, 1949–1955. [Google Scholar] [CrossRef]
- Yang, Z.Q.; Yang, Z.Y.; Zhang, L.D.; Ping, B.; Wang, S.G.; Ma, K.S.; Li, X.W.; Dong, J.H. Increased liver-infiltrating CD8+FOXP3+ regulatory T cells are associated with tumor stage in hepatocellular carcinoma patients. Hum. Immunol. 2010, 71, 1180–1186. [Google Scholar] [CrossRef]
- Sato, E.; Olson, S.H.; Ahn, J.; Bundy, B.; Nishikawa, H.; Qian, F.; Jungbluth, A.A.; Frosina, D.; Gnjatic, S.; Ambrosone, C.; et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 18538–18543. [Google Scholar] [CrossRef]
- Ling, A.; Edin, S.; Wikberg, M.L.; Oberg, A.; Palmqvist, R. The intratumoural subsite and relation of CD8(+) and FOXP3(+) T lymphocytes in colorectal cancer provide important prognostic clues. Br. J. Cancer 2014, 110, 2551–2559. [Google Scholar] [CrossRef]
- Liu, L.; Zhao, G.; Wu, W.; Rong, Y.; Jin, D.; Wang, D.; Lou, W.; Qin, X. Low intratumoral regulatory T cells and high peritumoral CD8(+) T cells relate to long-term survival in patients with pancreatic ductal adenocarcinoma after pancreatectomy. Cancer Immunol. Immunother. 2016, 65, 73–82. [Google Scholar] [CrossRef]
- Coca, S.; Perez-Piqueras, J.; Martinez, D.; Colmenarejo, A.; Saez, M.A.; Vallejo, C.; Martos, J.A.; Moreno, M. The prognostic significance of intratumoral natural killer cells in patients with colorectal carcinoma. Cancer 1997, 79, 2320–2328. [Google Scholar] [CrossRef]
- Preston, C.C.; Maurer, M.J.; Oberg, A.L.; Visscher, D.W.; Kalli, K.R.; Hartmann, L.C.; Goode, E.L.; Knutson, K.L. The ratios of CD8+ T cells to CD4+CD25+ FOXP3+ and FOXP3- T cells correlate with poor clinical outcome in human serous ovarian cancer. PLoS ONE 2013, 8, e80063. [Google Scholar] [CrossRef]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pages, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef]
- Huang, Y.; Ma, C.; Zhang, Q.; Ye, J.; Wang, F.; Zhang, Y.; Hunborg, P.; Varvares, M.A.; Hoft, D.F.; Hsueh, E.C.; et al. CD4+ and CD8+ T cells have opposing roles in breast cancer progression and outcome. Oncotarget 2015, 6, 17462–17478. [Google Scholar] [CrossRef] [Green Version]
- Zhu, S.; Lin, J.; Qiao, G.; Xu, Y.; Zou, H. Differential regulation and function of tumor-infiltrating T cells in different stages of breast cancer patients. Tumour Biol. 2015, 36, 7907–7913. [Google Scholar] [CrossRef]
- Watanabe, Y.; Katou, F.; Ohtani, H.; Nakayama, T.; Yoshie, O.; Hashimoto, K. Tumor-infiltrating lymphocytes, particularly the balance between CD8(+) T cells and CCR4(+) regulatory T cells, affect the survival of patients with oral squamous cell carcinoma. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endodontol. 2010, 109, 744–752. [Google Scholar] [CrossRef]
- Esensten, J.H.; Helou, Y.A.; Chopra, G.; Weiss, A.; Bluestone, J.A. CD28 costimulation: From mechanism to therapy. Immunity 2016, 44, 973–988. [Google Scholar] [CrossRef]
- Zumerle, S.; Molon, B.; Viola, A. Membrane rafts in T cell activation: A spotlight on CD28 costimulation. Front. Immunol. 2017, 8, 1467. [Google Scholar] [CrossRef]
- Porciello, N.; Grazioli, P.; Campese, A.F.; Kunkl, M.; Caristi, S.; Mastrogiovanni, M.; Muscolini, M.; Spadaro, F.; Favre, C.; Nunès, J.A.; et al. A non-conserved amino acid variant regulates differential signalling between human and mouse CD28. Nat. Commun. 2018, 9, 1080. [Google Scholar] [CrossRef] [Green Version]
- Bour-Jordan, H.; Esensten, J.H.; Martinez-Llordella, M.; Penaranda, C.; Stumpf, M.; Bluestone, J.A. Intrinsic and extrinsic control of peripheral T-cell tolerance by costimulatory molecules of the CD28/B7 family. Immunol. Rev. 2011, 241, 180–205. [Google Scholar] [CrossRef]
- Zhang, Q.; Vignali, D.A. Co-stimulatory and co-inhibitory pathways in autoimmunity. Immunity 2016, 44, 1034–1051. [Google Scholar] [CrossRef]
- Lenschow, D.J.; Zeng, Y.; Thistlethwaite, J.R.; Montag, A.; Brady, W.; Gibson, M.G.; Linsley, P.S.; Bluestone, J.A. Long-term survival of xenogeneic pancreatic islet grafts induced by CTLA4IG. Science 1992, 257, 789–792. [Google Scholar] [CrossRef]
- Blair, H.A.; Deeks, E.D. Abatacept: A review in rheumatoid arthritis. Drugs 2017, 77, 1221–1233. [Google Scholar] [CrossRef]
- Vincenti, F.; Rostaing, L.; Grinyo, J.; Rice, K.; Steinberg, S.; Gaite, L.; Moal, M.C.; Mondragon-Ramirez, G.A.; Kothari, J.; Polinsky, M.S.; et al. Belatacept and long-term outcomes in kidney transplantation. N. Engl. J. Med. 2016, 374, 333–343. [Google Scholar] [CrossRef]
- Ford, M.L. T cell cosignaling molecules in transplantation. Immunity 2016, 44, 1020–1033. [Google Scholar] [CrossRef]
- Attanasio, J.; Wherry, E.J. Costimulatory and coinhibitory receptor pathways in infectious disease. Immunity 2016, 44, 1052–1068. [Google Scholar] [CrossRef]
- Callahan, M.K.; Postow, M.A.; Wolchok, J.D. Targeting T cell co-receptors for cancer therapy. Immunity 2016, 44, 1069–1078. [Google Scholar] [CrossRef]
- Suntharalingam, G.; Perry, M.R.; Ward, S.; Brett, S.J.; Castello-Cortes, A.; Brunner, M.D.; Panoskaltsis, N. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N. Engl. J. Med. 2006, 355, 1018–1028. [Google Scholar] [CrossRef]
- Tyrsin, D.; Chuvpilo, S.; Matskevich, A.; Nemenov, D.; Romer, P.S.; Tabares, P.; Hunig, T. From TGN1412 to TAB08: The return of CD28 superagonist therapy to clinical development for the treatment of rheumatoid arthritis. Clin. Exp. Rheumatol. 2016, 34, 45–48. [Google Scholar]
- Hunig, T. The storm has cleared: Lessons from the CD28 superagonist TGN1412 trial. Nat. Rev. Immunol. 2012, 12, 317–318. [Google Scholar] [CrossRef]
- Zhou, J.; Rossi, J. Aptamers as targeted therapeutics: Current potential and challenges. Nat. Rev. Drug Discov. 2017, 16, 440. [Google Scholar] [CrossRef]
- Lozano, T.; Soldevilla, M.M.; Casares, N.; Villanueva, H.; Bendandi, M.; Lasarte, J.J.; Pastor, F. Targeting inhibition of FOXP3 by a CD28 2’-fluro oligonucleotide aptamer conjugated to p60-peptide enhances active cancer immunotherapy. Biomaterials 2016, 91, 73–80. [Google Scholar] [CrossRef]
- Soldevilla, M.M.; Villanueva, H.; Casares, N.; Lasarte, J.J.; Bendandi, M.; Inoges, S.; Lopez-Diaz de Cerio, A.; Pastor, F. MRP1-CD28 bi-specific oligonucleotide aptamers: Target costimulation to drug-resistant melanoma cancer stem cells. Oncotarget 2016, 7, 23182–23196. [Google Scholar] [CrossRef]
- Filley, A.C.; Henriquez, M.; Dey, M. Recurrent glioma clinical trial, checkmate-143: The game is not over yet. Oncotarget 2017, 8, 91779–91794. [Google Scholar] [CrossRef]
- Porter, D.L.; Hwang, W.T.; Frey, N.V.; Lacey, S.F.; Shaw, P.A.; Loren, A.W.; Bagg, A.; Marcucci, K.T.; Shen, A.; Gonzalez, V.; et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci. Transl. Med. 2015, 7, 303ra139. [Google Scholar] [CrossRef]
- Tang, X.Y.; Sun, Y.; Zhang, A.; Hu, G.L.; Cao, W.; Wang, D.H.; Zhang, B.; Chen, H. Third-generation CD28/4-1BB chimeric antigen receptor T cells for chemotherapy relapsed or refractory acute lymphoblastic leukaemia: A non-randomised, open-label phase I trial protocol. BMJ Open 2016, 6, e013904. [Google Scholar] [CrossRef]
- Ahmed, N.; Brawley, V.; Hegde, M.; Bielamowicz, K.; Wakefield, A.; Ghazi, A.; Ashoori, A.; Diouf, O.; Gerken, C.; Landi, D.; et al. Autologous HER2 cmv bispecific CAR T cells are safe and demonstrate clinical benefit for glioblastoma in a phase I trial. J. Immunother. Cancer 2015, 3, O11. [Google Scholar] [CrossRef]
- Cabo, M.; Offringa, R.; Zitvogel, L.; Kroemer, G.; Muntasell, A.; Galluzzi, L. Trial watch: Immunostimulatory monoclonal antibodies for oncological indications. Oncoimmunology 2017, 6, e1371896. [Google Scholar] [CrossRef]
- Emerson, D.A.; Redmond, W.L. Overcoming tumor-induced immune suppression: From relieving inhibition to providing costimulation with T cell agonists. BioDrugs 2018, 32, 221–231. [Google Scholar] [CrossRef]
- Sanmamed, M.F.; Pastor, F.; Rodriguez, A.; Perez-Gracia, J.L.; Rodriguez-Ruiz, M.E.; Jure-Kunkel, M.; Melero, I. Agonists of co-stimulation in cancer immunotherapy directed against CD137, OX40, GITR, CD27, CD28, and icos. Semin. Oncol. 2015, 42, 640–655. [Google Scholar] [CrossRef]
- Pratico, E.D.; Sullenger, B.A.; Nair, S.K. Identification and characterization of an agonistic aptamer against the T cell costimulatory receptor, OX40. Nucleic Acid Ther. 2013, 23, 35–43. [Google Scholar] [CrossRef]
- Weigelin, B.; Bolanos, E.; Teijeira, A.; Martinez-Forero, I.; Labiano, S.; Azpilikueta, A.; Morales-Kastresana, A.; Quetglas, J.I.; Wagena, E.; Sanchez-Paulete, A.R.; et al. Focusing and sustaining the antitumor CTL effector killer response by agonist anti-CD137 mAb. Proc. Natl. Acad. Sci. USA 2015, 112, 7551–7556. [Google Scholar] [CrossRef] [Green Version]
- Coe, D.; Begom, S.; Addey, C.; White, M.; Dyson, J.; Chai, J.G. Depletion of regulatory T cells by anti-gitr mab as a novel mechanism for cancer immunotherapy. Cancer Immunol. Immunother. 2010, 59, 1367–1377. [Google Scholar] [CrossRef]
- Schaer, D.A.; Budhu, S.; Liu, C.; Bryson, C.; Malandro, N.; Cohen, A.; Zhong, H.; Yang, X.; Houghton, A.N.; Merghoub, T.; et al. Gitr pathway activation abrogates tumor immune suppression through loss of regulatory T cell lineage stability. Cancer Immunol. Res. 2013, 1, 320–331. [Google Scholar] [CrossRef]
- Lu, L.; Xu, X.; Zhang, B.; Zhang, R.; Ji, H.; Wang, X. Combined PD-1 blockade and GITR triggering induce a potent antitumor immunity in murine cancer models and synergizes with chemotherapeutic drugs. J. Transl. Med. 2014, 12, 36. [Google Scholar] [CrossRef]
- Fan, X.; Quezada, S.A.; Sepulveda, M.A.; Sharma, P.; Allison, J.P. Engagement of the icos pathway markedly enhances efficacy of CTLA-4 blockade in cancer immunotherapy. J. Exp. Med. 2014, 211, 715–725. [Google Scholar] [CrossRef]
- Munoz-Espin, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef]
- Demaria, M.; Ohtani, N.; Youssef, S.A.; Rodier, F.; Toussaint, W.; Mitchell, J.R.; Laberge, R.M.; Vijg, J.; Van Steeg, H.; Dolle, M.E.; et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev. Cell 2014, 31, 722–733. [Google Scholar] [CrossRef]
- Rajagopalan, S.; Long, E.O. Cellular senescence induced by CD158D reprograms natural killer cells to promote vascular remodeling. Proc. Natl. Acad. Sci. USA 2012, 109, 20596–20601. [Google Scholar] [CrossRef]
- Howcroft, T.K.; Campisi, J.; Louis, G.B.; Smith, M.T.; Wise, B.; Wyss-Coray, T.; Augustine, A.D.; McElhaney, J.E.; Kohanski, R.; Sierra, F. The role of inflammation in age-related disease. Aging 2013, 5, 84–93. [Google Scholar] [CrossRef] [Green Version]
- Myrianthopoulos, V.; Evangelou, K.; Vasileiou, P.V.S.; Cooks, T.; Vassilakopoulos, T.P.; Pangalis, G.A.; Kouloukoussa, M.; Kittas, C.; Georgakilas, A.G.; Gorgoulis, V.G. Senescence and senotherapeutics: A new field in cancer therapy. Pharmacol. Ther. 2019, 193, 31–49. [Google Scholar] [CrossRef]
- Petrakis, T.G.; Komseli, E.S.; Papaioannou, M.; Vougas, K.; Polyzos, A.; Myrianthopoulos, V.; Mikros, E.; Trougakos, I.P.; Thanos, D.; Branzei, D.; et al. Exploring and exploiting the systemic effects of deregulated replication licensing. Semin. Cancer Biol. 2016, 37–38, 3–15. [Google Scholar] [CrossRef]
- Bartkova, J.; Rezaei, N.; Liontos, M.; Karakaidos, P.; Kletsas, D.; Issaeva, N.; Vassiliou, L.V.; Kolettas, E.; Niforou, K.; Zoumpourlis, V.C.; et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 2006, 444, 633–637. [Google Scholar] [CrossRef]
- Rayess, H.; Wang, M.B.; Srivatsan, E.S. Cellular senescence and tumor suppressor gene p16. Int. J. Cancer 2012, 130, 1715–1725. [Google Scholar] [CrossRef]
- Alcorta, D.A.; Xiong, Y.; Phelps, D.; Hannon, G.; Beach, D.; Barrett, J.C. Involvement of the cyclin-dependent kinase inhibitor p16 (INK4A) in replicative senescence of normal human fibroblasts. Proc. Natl. Acad. Sci. USA 1996, 93, 13742–13747. [Google Scholar] [CrossRef]
- Jurk, D.; Wang, C.; Miwa, S.; Maddick, M.; Korolchuk, V.; Tsolou, A.; Gonos, E.S.; Thrasivoulou, C.; Saffrey, M.J.; Cameron, K.; et al. Postmitotic neurons develop a p21-dependent senescence-like phenotype driven by a DNA damage response. Aging Cell 2012, 11, 996–1004. [Google Scholar] [CrossRef]
- Ou, H.L.; Schumacher, B. DNA damage responses and p53 in the aging process. Blood 2018, 131, 488–495. [Google Scholar] [CrossRef] [Green Version]
- Aird, K.M.; Zhang, R. Detection of senescence-associated heterochromatin foci (SAHF). Methods Mol. Biol. 2013, 965, 185–196. [Google Scholar]
- Evangelou, K.; Lougiakis, N.; Rizou, S.V.; Kotsinas, A.; Kletsas, D.; Munoz-Espin, D.; Kastrinakis, N.G.; Pouli, N.; Marakos, P.; Townsend, P.; et al. Robust, universal biomarker assay to detect senescent cells in biological specimens. Aging Cell 2017, 16, 192–197. [Google Scholar] [CrossRef]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. Immunosenescence: The potential role of myeloid-derived suppressor cells (MDSC) in age-related immune deficiency. Cell. Mol. Life Sci. 2019, 76, 1901–1918. [Google Scholar] [CrossRef]
- Akbar, A.N.; Fletcher, J.M. Memory T cell homeostasis and senescence during aging. Curr. Opin. Immunol. 2005, 17, 480–485. [Google Scholar] [CrossRef]
- Swain, S.; Clise-Dwyer, K.; Haynes, L. Homeostasis and the age-associated defect of CD4 T cells. Semin. Immunol. 2005, 17, 370–377. [Google Scholar] [CrossRef] [Green Version]
- Singhal, S.K.; Roder, J.C.; Duwe, A.K. Suppressor cells in immunosenescence. Fed. Proc. 1978, 37, 1245–1252. [Google Scholar]
- Strioga, M.; Pasukoniene, V.; Characiejus, D. CD8+ CD28- and CD8+ CD57+ T cells and their role in health and disease. Immunology 2011, 134, 17–32. [Google Scholar] [CrossRef]
- Vallejo, A.N.; Weyand, C.M.; Goronzy, J.J. T-cell senescence: A culprit of immune abnormalities in chronic inflammation and persistent infection. Trends Mol. Med. 2004, 10, 119–124. [Google Scholar] [CrossRef]
- Eck, S.C.; Chang, D.; Wells, A.D.; Turka, L.A. Differential down-regulation of CD28 by B7-1 and B7-2 engagement. Transplantation 1997, 64, 1497–1499. [Google Scholar] [CrossRef]
- Laidlaw, B.J.; Craft, J.E.; Kaech, S.M. The multifaceted role of CD4(+) T cells in CD8(+) T cell memory. Nat. Rev. Immunol. 2016, 16, 102–111. [Google Scholar] [CrossRef]
- Weng, N.P.; Akbar, A.N.; Goronzy, J. CD28(-) T cells: Their role in the age-associated decline of immune function. Trends Immunol. 2009, 30, 306–312. [Google Scholar] [CrossRef]
- Qin, L.; Jing, X.; Qiu, Z.; Cao, W.; Jiao, Y.; Routy, J.P.; Li, T. Aging of immune system: Immune signature from peripheral blood lymphocyte subsets in 1068 healthy adults. Aging 2016, 8, 848–859. [Google Scholar] [CrossRef]
- Valenzuela, H.F.; Effros, R.B. Divergent telomerase and CD28 expression patterns in human CD4 and CD8 T cells following repeated encounters with the same antigenic stimulus. Clin. Immunol. 2002, 105, 117–125. [Google Scholar] [CrossRef]
- Khan, N.; Shariff, N.; Cobbold, M.; Bruton, R.; Ainsworth, J.A.; Sinclair, A.J.; Nayak, L.; Moss, P.A. Cytomegalovirus seropositivity drives the CD8 T cell repertoire toward greater clonality in healthy elderly individuals. J. Immunol. 2002, 169, 1984–1992. [Google Scholar] [CrossRef]
- Paillard, F.; Sterkers, G.; Vaquero, C. Transcriptional and post-transcriptional regulation of tcr, CD4 and CD8 gene expression during activation of normal human T lymphocytes. EMBO J. 1990, 9, 1867–1872. [Google Scholar] [CrossRef]
- Mou, D.; Espinosa, J.; Lo, D.J.; Kirk, A.D. CD28 negative T cells: Is their loss our gain? Am. J. Transpl. 2014, 14, 2460–2466. [Google Scholar] [CrossRef]
- Tarazona, R.; DelaRosa, O.; Alonso, C.; Ostos, B.; Espejo, J.; Pena, J.; Solana, R. Increased expression of NK cell markers on T lymphocytes in aging and chronic activation of the immune system reflects the accumulation of effector/senescent T cells. Mech. Ageing Dev. 2000, 121, 77–88. [Google Scholar] [CrossRef]
- Seyda, M.; Elkhal, A.; Quante, M.; Falk, C.S.; Tullius, S.G. T cells going innate. Trends Immunol. 2016, 37, 546–556. [Google Scholar] [CrossRef]
- Bauer, S.; Groh, V.; Wu, J.; Steinle, A.; Phillips, J.H.; Lanier, L.L.; Spies, T. Activation of nk cells and T cells by NKG2D, a receptor for stress-inducible mica. Science 1999, 285, 727–729. [Google Scholar] [CrossRef]
- Verneris, M.R.; Karimi, M.; Baker, J.; Jayaswal, A.; Negrin, R.S. Role of NKG2D signaling in the cytotoxicity of activated and expanded CD8+ T cells. Blood 2004, 103, 3065–3072. [Google Scholar] [CrossRef] [Green Version]
- Prajapati, K.; Perez, C.; Rojas, L.B.P.; Burke, B.; Guevara-Patino, J.A. Functions of NKG2D in CD8(+) T cells: An opportunity for immunotherapy. Cell. Mol. Immunol. 2018, 15, 470–479. [Google Scholar] [CrossRef]
- Yi, H.S.; Kim, S.Y.; Kim, J.T.; Lee, Y.S.; Moon, J.S.; Kim, M.; Kang, Y.E.; Joung, K.H.; Lee, J.H.; Kim, H.J.; et al. T-cell senescence contributes to abnormal glucose homeostasis in humans and mice. Cell Death Dis. 2019, 10, 249. [Google Scholar] [CrossRef]
- Onyema, O.O.; Decoster, L.; Njemini, R.; Forti, L.N.; Bautmans, I.; De Waele, M.; Mets, T. Shifts in subsets of CD8+ T-cells as evidence of immunosenescence in patients with cancers affecting the lungs: An observational case-control study. BMC Cancer 2015, 15, 1016. [Google Scholar] [CrossRef]
- Brenchley, J.M.; Karandikar, N.J.; Betts, M.R.; Ambrozak, D.R.; Hill, B.J.; Crotty, L.E.; Casazza, J.P.; Kuruppu, J.; Migueles, S.A.; Connors, M.; et al. Expression of CD57 defines replicative senescence and antigen-induced apoptotic death of CD8+ T cells. Blood 2003, 101, 2711–2720. [Google Scholar] [CrossRef]
- Gunturi, A.; Berg, R.E.; Forman, J. Preferential survival of CD8 T and NK cells expressing high levels of CD94. J. Immunol. 2003, 170, 1737–1745. [Google Scholar] [CrossRef]
- Appay, V.; Nixon, D.F.; Donahoe, S.M.; Gillespie, G.M.; Dong, T.; King, A.; Ogg, G.S.; Spiegel, H.M.; Conlon, C.; Spina, C.A.; et al. Hiv-specific CD8(+) T cells produce antiviral cytokines but are impaired in cytolytic function. J. Exp. Med. 2000, 192, 63–75. [Google Scholar] [CrossRef]
- Kiniry, B.E.; Hunt, P.W.; Hecht, F.M.; Somsouk, M.; Deeks, S.G.; Shacklett, B.L. Differential expression of CD8(+) T cell cytotoxic effector molecules in blood and gastrointestinal mucosa in HIV-1 infection. J. Immunol. 2018, 200, 1876–1888. [Google Scholar] [CrossRef]
- Reuter, M.A.; Del Rio Estrada, P.M.; Buggert, M.; Petrovas, C.; Ferrando-Martinez, S.; Nguyen, S.; Sada Japp, A.; Ablanedo-Terrazas, Y.; Rivero-Arrieta, A.; Kuri-Cervantes, L.; et al. HIV-specific CD8(+) T cells exhibit reduced and differentially regulated cytolytic activity in lymphoid tissue. Cell Rep. 2017, 21, 3458–3470. [Google Scholar] [CrossRef]
- Hodge, G.; Hodge, S. Steroid resistant CD8(+)CD28(null) nkt-like pro-inflammatory cytotoxic cells in chronic obstructive pulmonary disease. Front. Immunol. 2016, 7, 617. [Google Scholar] [CrossRef]
- Cortesini, R.; LeMaoult, J.; Ciubotariu, R.; Cortesini, N.S. Cd8+CD28- t suppressor cells and the induction of antigen-specific, antigen-presenting cell-mediated suppression of th reactivity. Immunol. Rev. 2001, 182, 201–206. [Google Scholar] [CrossRef]
- Plaumann, J.; Engelhardt, M.; Awwad, M.H.S.; Echchannaoui, H.; Amman, E.; Raab, M.S.; Hillengass, J.; Halama, N.; Neuber, B.; Muller-Tidow, C.; et al. IL-10 inducible CD8(+) regulatory T-cells are enriched in patients with multiple myeloma and impact the generation of antigen-specific T-cells. Cancer Immunol. Immunother. 2018, 67, 1695–1707. [Google Scholar] [CrossRef]
- Geng, L.; Liu, J.; Huang, J.; Lin, B.; Yu, S.; Shen, T.; Wang, Z.; Yang, Z.; Zhou, L.; Zheng, S. A high frequency of CD8(+)CD28(-) t-suppressor cells contributes to maintaining stable graft function and reducing immunosuppressant dosage after liver transplantation. Int. J. Med. Sci. 2018, 15, 892–899. [Google Scholar] [CrossRef]
- Vieyra-Lobato, M.R.; Vela-Ojeda, J.; Montiel-Cervantes, L.; Lopez-Santiago, R.; Moreno-Lafont, M.C. Description of CD8(+) regulatory T lymphocytes and their specific intervention in graft-versus-host and infectious diseases, autoimmunity, and cancer. J. Immunol. Res 2018, 2018, 3758713. [Google Scholar] [CrossRef]
- Manavalan, J.S.; Rossi, P.C.; Vlad, G.; Piazza, F.; Yarilina, A.; Cortesini, R.; Mancini, D.; Suciu-Foca, N. High expression of ILT3 and ILT4 is a general feature of tolerogenic dendritic cells. Transpl. Immunol. 2003, 11, 245–258. [Google Scholar] [CrossRef]
- Ye, J.; Huang, X.; Hsueh, E.C.; Zhang, Q.; Ma, C.; Zhang, Y.; Varvares, M.A.; Hoft, D.F.; Peng, G. Human regulatory T cells induce T-lymphocyte senescence. Blood 2012, 120, 2021–2031. [Google Scholar] [CrossRef]
- Sun, Z.; Zhong, W.; Lu, X.; Shi, B.; Zhu, Y.; Chen, L.; Zhang, G.; Zhang, X. Association of graves’ disease and prevalence of circulating IFN-γ-producing CD28(-) T cells. J. Clin. Immunol. 2008, 28, 464–472. [Google Scholar] [CrossRef]
- Maly, K.; Schirmer, M. The story of CD4+ CD28- T cells revisited: Solved or still ongoing? J. Immunol. Res. 2015, 2015, 348746. [Google Scholar]
- Tulunay, A.; Yavuz, S.; Direskeneli, H.; Eksioglu-Demiralp, E. CD8+CD28-, suppressive T cells in systemic lupus erythematosus. Lupus 2008, 17, 630–637. [Google Scholar] [CrossRef]
- Najafian, N.; Chitnis, T.; Salama, A.D.; Zhu, B.; Benou, C.; Yuan, X.; Clarkson, M.R.; Sayegh, M.H.; Khoury, S.J. Regulatory functions of CD8+CD28- T cells in an autoimmune disease model. J. Clin. Investig. 2003, 112, 1037–1048. [Google Scholar] [CrossRef]
- Manavalan, J.S.; Kim-Schulze, S.; Scotto, L.; Naiyer, A.J.; Vlad, G.; Colombo, P.C.; Marboe, C.; Mancini, D.; Cortesini, R.; Suciu-Foca, N. Alloantigen specific CD8+CD28- FOXP3+ T suppressor cells induce ILT3+ ILT4+ tolerogenic endothelial cells, inhibiting alloreactivity. Int. Immunol. 2004, 16, 1055–1068. [Google Scholar] [CrossRef]
- Chang, C.C.; Ciubotariu, R.; Manavalan, J.S.; Yuan, J.; Colovai, A.I.; Piazza, F.; Lederman, S.; Colonna, M.; Cortesini, R.; Dalla-Favera, R.; et al. Tolerization of dendritic cells by T(s) cells: The crucial role of inhibitory receptors ILT3 and ILT4. Nat. Immunol. 2002, 3, 237–243. [Google Scholar] [CrossRef]
- Topalian, S.L.; Taube, J.M.; Anders, R.A.; Pardoll, D.M. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat. Rev. Cancer 2016, 16, 275–287. [Google Scholar] [CrossRef]
- Tomaszewski, W.; Sanchez-Perez, L.; Gajewski, T.F.; Sampson, J.H. Brain tumor microenvironment and host state: Implications for immunotherapy. Clin. Cancer Res. 2019. [Google Scholar] [CrossRef]
- Wargo, J.A.; Reddy, S.M.; Reuben, A.; Sharma, P. Monitoring immune responses in the tumor microenvironment. Curr. Opin. Immunol. 2016, 41, 23–31. [Google Scholar] [CrossRef] [Green Version]
- Woroniecka, K.; Chongsathidkiet, P.; Rhodin, K.; Kemeny, H.; Dechant, C.; Farber, S.H.; Elsamadicy, A.A.; Cui, X.; Koyama, S.; Jackson, C.; et al. T-cell exhaustion signatures vary with tumor type and are severe in glioblastoma. Clin. Cancer Res. 2018, 24, 4175–4186. [Google Scholar] [CrossRef]
- Mirzaei, R.; Sarkar, S.; Yong, V.W. T cell exhaustion in glioblastoma: Intricacies of immune checkpoints. Trends Immunol. 2017, 38, 104–115. [Google Scholar] [CrossRef]
- Nduom, E.K.; Weller, M.; Heimberger, A.B. Immunosuppressive mechanisms in glioblastoma. Neuro Oncol. 2015, 17 (Suppl. 7), vii9–vii14. [Google Scholar] [CrossRef]
- Wherry, E.J. T cell exhaustion. Nat. Immunol. 2011, 12, 492–499. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [Green Version]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Darvin, P.; Toor, S.M.; Sasidharan Nair, V.; Elkord, E. Immune checkpoint inhibitors: Recent progress and potential biomarkers. Exp. Mol. Med. 2018, 50, 165. [Google Scholar] [CrossRef]
- Cheng, W.; Fu, D.; Xu, F.; Zhang, Z. Unwrapping the genomic characteristics of urothelial bladder cancer and successes with immune checkpoint blockade therapy. Oncogenesis 2018, 7, 2. [Google Scholar] [CrossRef]
- Lauko, A.; Thapa, B.; Venur, V.A.; Ahluwalia, M.S. Management of brain metastases in the new era of checkpoint inhibition. Curr. Neurol. Neurosci. Rep. 2018, 18, 70. [Google Scholar] [CrossRef]
- Johnson, D.B.; Sullivan, R.J.; Menzies, A.M. Immune checkpoint inhibitors in challenging populations. Cancer 2017, 123, 1904–1911. [Google Scholar] [CrossRef] [Green Version]
- Meloni, F.; Morosini, M.; Solari, N.; Passadore, I.; Nascimbene, C.; Novo, M.; Ferrari, M.; Cosentino, M.; Marino, F.; Pozzi, E.; et al. Foxp3 expressing CD4+ CD25+ and CD8+CD28- T regulatory cells in the peripheral blood of patients with lung cancer and pleural mesothelioma. Hum. Immunol. 2006, 67, 1–12. [Google Scholar] [CrossRef]
- Chen, C.; Chen, D.; Zhang, Y.; Chen, Z.; Zhu, W.; Zhang, B.; Wang, Z.; Le, H. Changes of CD4+CD25+FOXP3+ and CD8+CD28- regulatory T cells in non-small cell lung cancer patients undergoing surgery. Int. Immunopharmacol. 2014, 18, 255–261. [Google Scholar] [CrossRef]
- Casado, J.G.; Soto, R.; DelaRosa, O.; Peralbo, E.; del Carmen Munoz-Villanueva, M.; Rioja, L.; Pena, J.; Solana, R.; Tarazona, R. CD8 T cells expressing nk associated receptors are increased in melanoma patients and display an effector phenotype. Cancer Immunol. Immunother. 2005, 54, 1162–1171. [Google Scholar] [CrossRef]
- Liu, X.; Mo, W.; Ye, J.; Li, L.; Zhang, Y.; Hsueh, E.C.; Hoft, D.F.; Peng, G. Regulatory T cells trigger effector T cell DNA damage and senescence caused by metabolic competition. Nat. Commun. 2018, 9, 249. [Google Scholar] [CrossRef]
- Ye, J.; Ma, C.; Hsueh, E.C.; Eickhoff, C.S.; Zhang, Y.; Varvares, M.A.; Hoft, D.F.; Peng, G. Tumor-derived gammadelta regulatory T cells suppress innate and adaptive immunity through the induction of immunosenescence. J. Immunol. 2013, 190, 2403–2414. [Google Scholar] [CrossRef]
- Ye, J.; Peng, G. Controlling T cell senescence in the tumor microenvironment for tumor immunotherapy. Oncoimmunology 2015, 4, e994398. [Google Scholar] [CrossRef] [Green Version]
- Lanna, A.; Henson, S.M.; Escors, D.; Akbar, A.N. The kinase p38 activated by the metabolic regulator AMPK and scaffold TAB1 drives the senescence of human T cells. Nat. Immunol. 2014, 15, 965–972. [Google Scholar] [CrossRef] [Green Version]
- Ye, J.; Ma, C.; Hsueh, E.C.; Dou, J.; Mo, W.; Liu, S.; Han, B.; Huang, Y.; Zhang, Y.; Varvares, M.A.; et al. TLR8 signaling enhances tumor immunity by preventing tumor-induced T-cell senescence. EMBO Mol. Med. 2014, 6, 1294–1311. [Google Scholar] [CrossRef]
- Lim, M.; Xia, Y.; Bettegowda, C.; Weller, M. Current state of immunotherapy for glioblastoma. Nat. Rev. Clin. Oncol. 2018, 15, 422–442. [Google Scholar] [CrossRef]
- Broekman, M.L.; Maas, S.L.N.; Abels, E.R.; Mempel, T.R.; Krichevsky, A.M.; Breakefield, X.O. Multidimensional communication in the microenvirons of glioblastoma. Nat. Rev. Neurol. 2018, 14, 482–495. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. The microenvironmental landscape of brain tumors. Cancer Cell 2017, 31, 326–341. [Google Scholar] [CrossRef]
- Sayour, E.J.; McLendon, P.; McLendon, R.; De Leon, G.; Reynolds, R.; Kresak, J.; Sampson, J.H.; Mitchell, D.A. Increased proportion of FoxP3+ regulatory T cells in tumor infiltrating lymphocytes is associated with tumor recurrence and reduced survival in patients with glioblastoma. Cancer Immunol. Immunother. 2015, 64, 419–427. [Google Scholar] [CrossRef]
- Fornara, O.; Odeberg, J.; Wolmer Solberg, N.; Tammik, C.; Skarman, P.; Peredo, I.; Stragliotto, G.; Rahbar, A.; Soderberg-Naucler, C. Poor survival in glioblastoma patients is associated with early signs of immunosenescence in the CD4 T-cell compartment after surgery. Oncoimmunology 2015, 4, e1036211. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.F. CD8+ regulatory T cells, their suppressive mechanisms, and regulation in cancer. Hum. Immunol. 2008, 69, 811–814. [Google Scholar] [CrossRef]
- Kmiecik, J.; Poli, A.; Brons, N.H.; Waha, A.; Eide, G.E.; Enger, P.O.; Zimmer, J.; Chekenya, M. Elevated CD3+ and CD8+ tumor-infiltrating immune cells correlate with prolonged survival in glioblastoma patients despite integrated immunosuppressive mechanisms in the tumor microenvironment and at the systemic level. J. Neuroimmunol. 2013, 264, 71–83. [Google Scholar] [CrossRef] [Green Version]
- Vlad, G.; Cortesini, R.; Suciu-Foca, N. CD8+ T suppressor cells and the ILT3 master switch. Hum. Immunol. 2008, 69, 681–686. [Google Scholar] [CrossRef]
- Ladomersky, E.; Scholtens, D.M.; Kocherginsky, M.; Hibler, E.A.; Bartom, E.T.; Otto-Meyer, S.; Zhai, L.; Lauing, K.L.; Choi, J.; Sosman, J.A.; et al. The coincidence between increasing age, immunosuppression, and the incidence of patients with glioblastoma. Front. Pharmacol. 2019, 10, 200. [Google Scholar] [CrossRef]
- Lamas, A.; Lopez, E.; Carrio, R.; Lopez, D.M. Adipocyte and leptin accumulation in tumor-induced thymic involution. Int. J. Mol. Med. 2016, 37, 133–138. [Google Scholar] [CrossRef]
- Kasakovski, D.; Xu, L.; Li, Y. T cell senescence and CAR-T cell exhaustion in hematological malignancies. J. Hematol. Oncol. 2018, 11, 91. [Google Scholar] [CrossRef]
- Short, S.; Fielder, E.; Miwa, S.; von Zglinicki, T. Senolytics and senostatics as adjuvant tumour therapy. EBioMedicine 2019, 41, 683–692. [Google Scholar] [CrossRef] [Green Version]
- Baar, M.P.; Brandt, R.M.C.; Putavet, D.A.; Klein, J.D.D.; Derks, K.W.J.; Bourgeois, B.R.M.; Stryeck, S.; Rijksen, Y.; van Willigenburg, H.; Feijtel, D.A.; et al. Targeted apoptosis of senescent cells restores tissue homeostasis in response to chemotoxicity and aging. Cell 2017, 169, 132–147.e16. [Google Scholar] [CrossRef]
- Kirkland, J.L.; Tchkonia, T.; Zhu, Y.; Niedernhofer, L.J.; Robbins, P.D. The clinical potential of senolytic drugs. J. Am. Geriatr. Soc. 2017, 65, 2297–2301. [Google Scholar] [CrossRef]
- Rueff, J.; Medinger, M.; Heim, D.; Passweg, J.; Stern, M. Lymphocyte subset recovery and outcome after autologous hematopoietic stem cell transplantation for plasma cell myeloma. Biol. Blood Marrow Transpl. 2014, 20, 896–899. [Google Scholar] [CrossRef]
- Farge, D.; Arruda, L.C.; Brigant, F.; Clave, E.; Douay, C.; Marjanovic, Z.; Deligny, C.; Maki, G.; Gluckman, E.; Toubert, A.; et al. Long-term immune reconstitution and T cell repertoire analysis after autologous hematopoietic stem cell transplantation in systemic sclerosis patients. J. Hematol. Oncol. 2017, 10, 21. [Google Scholar] [CrossRef]
- Zhang, P.; Kishimoto, Y.; Grammatikakis, I.; Gottimukkala, K.; Cutler, R.G.; Zhang, S.; Abdelmohsen, K.; Bohr, V.A.; Misra Sen, J.; Gorospe, M.; et al. Senolytic therapy alleviates abeta-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an alzheimer’s disease model. Nat. Neurosci. 2019, 22, 719–728. [Google Scholar] [CrossRef]
- Allsopp, R. Telomere length and ipsc re-programming: Survival of the longest. Cell Res. 2012, 22, 614–615. [Google Scholar] [CrossRef]
- Parish, S.T.; Wu, J.E.; Effros, R.B. Sustained CD28 expression delays multiple features of replicative senescence in human CD8 T lymphocytes. J. Clin. Immunol. 2010, 30, 798–805. [Google Scholar] [CrossRef]
- Le Page, A.; Fortin, C.; Garneau, H.; Allard, N.; Tsvetkova, K.; Tan, C.T.; Larbi, A.; Dupuis, G.; Fulop, T. Downregulation of inhibitory SRC homology 2 domain-containing phosphatase-1 (SHP-1) leads to recovery of T cell responses in elderly. Cell Commun. Signal. 2014, 12, 2. [Google Scholar] [CrossRef]
- Karagiannis, P.; Iriguchi, S.; Kaneko, S. Reprogramming away from the exhausted T cell state. Semin. Immunol. 2016, 28, 35–44. [Google Scholar] [CrossRef]
- Themeli, M.; Kloss, C.C.; Ciriello, G.; Fedorov, V.D.; Perna, F.; Gonen, M.; Sadelain, M. Generation of tumor-targeted human T lymphocytes from induced pluripotent stem cells for cancer therapy. Nat. Biotechnol. 2013, 31, 928–933. [Google Scholar] [CrossRef]
- Kaneko, S. In vitro generation of antigen-specific T cells from induced pluripotent stem cells of antigen-specific T cell origin. Methods Mol. Biol. 2016, 1393, 67–73. [Google Scholar]
- Ramos, C.A.; Rouce, R.; Robertson, C.S.; Reyna, A.; Narala, N.; Vyas, G.; Mehta, B.; Zhang, H.; Dakhova, O.; Carrum, G.; et al. In vivo fate and activity of second- versus third-generation CD19-specific CAR-T cells in B cell non-hodgkin’s lymphomas. Mol. Ther. 2018, 26, 2727–2737. [Google Scholar] [CrossRef]
- Petersen, C.T.; Krenciute, G. Next generation CAR T cells for the immunotherapy of high-grade glioma. Front. Oncol. 2019, 9, 69. [Google Scholar] [CrossRef]
- Sahin, A.; Sanchez, C.; Bullain, S.; Waterman, P.; Weissleder, R.; Carter, B.S. Development of third generation anti-egfrviii chimeric T cells and egfrviii-expressing artificial antigen presenting cells for adoptive cell therapy for glioma. PLoS ONE 2018, 13, e0199414. [Google Scholar] [CrossRef]
- June, C.H.; Sadelain, M. Chimeric antigen receptor therapy. N. Engl. J. Med. 2018, 379, 64–73. [Google Scholar] [CrossRef]
- Al-Chami, E.; Tormo, A.; Pasquin, S.; Kanjarawi, R.; Ziouani, S.; Rafei, M. Interleukin-21 administration to aged mice rejuvenates their peripheral T-cell pool by triggering de novo thymopoiesis. Aging Cell 2016, 15, 349–360. [Google Scholar] [CrossRef]
- Fan, Y.; Tajima, A.; Goh, S.K.; Geng, X.; Gualtierotti, G.; Grupillo, M.; Coppola, A.; Bertera, S.; Rudert, W.A.; Banerjee, I.; et al. Bioengineering thymus organoids to restore thymic function and induce donor-specific immune tolerance to allografts. Mol. Ther. 2015, 23, 1262–1277. [Google Scholar] [CrossRef]
- Tajima, A.; Pradhan, I.; Trucco, M.; Fan, Y. Restoration of thymus function with bioengineered thymus organoids. Curr. Stem Cell Rep. 2016, 2, 128–139. [Google Scholar] [CrossRef]
- Akbar, A.N.; Henson, S.M. Are senescence and exhaustion intertwined or unrelated processes that compromise immunity? Nat. Rev. Immunol. 2011, 11, 289–295. [Google Scholar] [CrossRef]
Figure 1.
The priming and inactivation of CD8+ T cells. The interaction between TCRs and the peptide-MHC complex is the first step toward antigen-induced CD8+ T cell activation. This creates a site of extensive contact between T cells and APC, also called immunological synapses, where binding of CD28 on T cells with CD80/CD86 on APCs transduces a pivotal secondary costimulatory signal to complete the priming of naïve CD8+ T cells. In addition, CD4+ T helper (Th) cells when activated by DCs acquire not only the synapse-composed MHC class II and costimulatory molecules (CD54 and CD80), but also the bystander peptide-MHC-I complex from DC and become CD4+ Th-APCs, resulting in direct CD4+ T–CD8+ T cell interactions and subsequently delivery of CD40L signaling to CD40-expressing CD8+ T cells [21]. Furthermore, CD4 Th cells also secrete cytokines, such as IL-2, which promotes the differentiation of naïve CD8+ T cells into effector CTLs and memory CD8+ T cells. CTLs destroy antigen-specific target cells (such as cancer cells or viral infected host cells) via pathways including granule exocytosis, Fas ligand, and TRAIL-mediated apoptosis leading to tumor control or virus clearance and subsequent physiological T-cell inactivation as well as memory CD8+ T-cell formation [33]. Whereas pathological T-cell inactivation or conversion of CTL to CD8+ senescent T cells leads to tumor progression and virus replication.
Figure 1.
The priming and inactivation of CD8+ T cells. The interaction between TCRs and the peptide-MHC complex is the first step toward antigen-induced CD8+ T cell activation. This creates a site of extensive contact between T cells and APC, also called immunological synapses, where binding of CD28 on T cells with CD80/CD86 on APCs transduces a pivotal secondary costimulatory signal to complete the priming of naïve CD8+ T cells. In addition, CD4+ T helper (Th) cells when activated by DCs acquire not only the synapse-composed MHC class II and costimulatory molecules (CD54 and CD80), but also the bystander peptide-MHC-I complex from DC and become CD4+ Th-APCs, resulting in direct CD4+ T–CD8+ T cell interactions and subsequently delivery of CD40L signaling to CD40-expressing CD8+ T cells [21]. Furthermore, CD4 Th cells also secrete cytokines, such as IL-2, which promotes the differentiation of naïve CD8+ T cells into effector CTLs and memory CD8+ T cells. CTLs destroy antigen-specific target cells (such as cancer cells or viral infected host cells) via pathways including granule exocytosis, Fas ligand, and TRAIL-mediated apoptosis leading to tumor control or virus clearance and subsequent physiological T-cell inactivation as well as memory CD8+ T-cell formation [33]. Whereas pathological T-cell inactivation or conversion of CTL to CD8+ senescent T cells leads to tumor progression and virus replication.

Figure 2.
Phenotypical and molecular changes in cellular senescence. A variety of intracellular (DNA damage, oncogenes, etc.) and/or extracellular signals (oxidative stress, chronic inflammation, etc.) can induce cellular senescence. Senescent cells exhibit numerous characteristics including but not limited to cell cycle arrest, increased nuclear p16 and p21 expression, increased lysosomal SA-β-gal activity, shortened telomere, and increased lipofuscin. Senescent cells can also present as a specialized secretory phenotype termed senescence associated secretory phenotype (SASP). Of particular interest, senescent immune cells present with lowered expression of CD28 and CD27 but heightened expression of CD57, KLRG-1, TIGIT, and other NK-cell associated surface receptors.
Figure 2.
Phenotypical and molecular changes in cellular senescence. A variety of intracellular (DNA damage, oncogenes, etc.) and/or extracellular signals (oxidative stress, chronic inflammation, etc.) can induce cellular senescence. Senescent cells exhibit numerous characteristics including but not limited to cell cycle arrest, increased nuclear p16 and p21 expression, increased lysosomal SA-β-gal activity, shortened telomere, and increased lipofuscin. Senescent cells can also present as a specialized secretory phenotype termed senescence associated secretory phenotype (SASP). Of particular interest, senescent immune cells present with lowered expression of CD28 and CD27 but heightened expression of CD57, KLRG-1, TIGIT, and other NK-cell associated surface receptors.

Figure 3.
The heterogeneous functions of CD8+CD28− T cells. CD8+CD28− T cells originate from activated CD8+CD28+ T cells or from interaction with tolerogenic APCs. CD8+CD28− T cells exhibit both cytotoxic and immunoregulatory phenotypes and vary in pathological states such as across different cancer types or inflammatory/autoimmune conditions.
Figure 3.
The heterogeneous functions of CD8+CD28− T cells. CD8+CD28− T cells originate from activated CD8+CD28+ T cells or from interaction with tolerogenic APCs. CD8+CD28− T cells exhibit both cytotoxic and immunoregulatory phenotypes and vary in pathological states such as across different cancer types or inflammatory/autoimmune conditions.

Figure 4.
The many facets of C8+ T cell dysfunction. In comparison to activated effector T cells, T cell anergy, exhaustion, and senescence can be distinguished by their unique expression or lack of expression of surface receptors as well as different levels of intracellular cytokines, such as IL-2.
Figure 4.
The many facets of C8+ T cell dysfunction. In comparison to activated effector T cells, T cell anergy, exhaustion, and senescence can be distinguished by their unique expression or lack of expression of surface receptors as well as different levels of intracellular cytokines, such as IL-2.


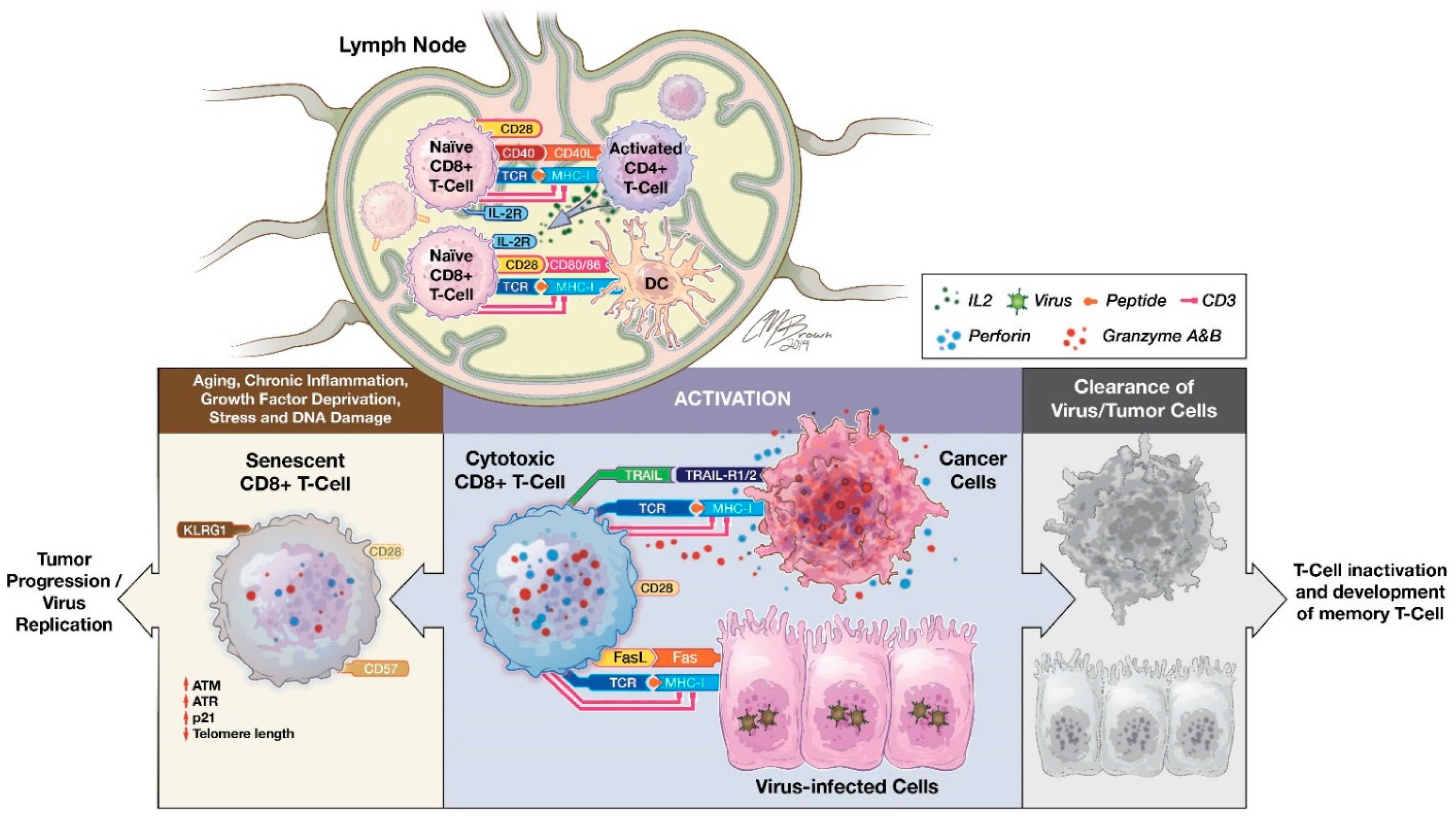{kind=link}
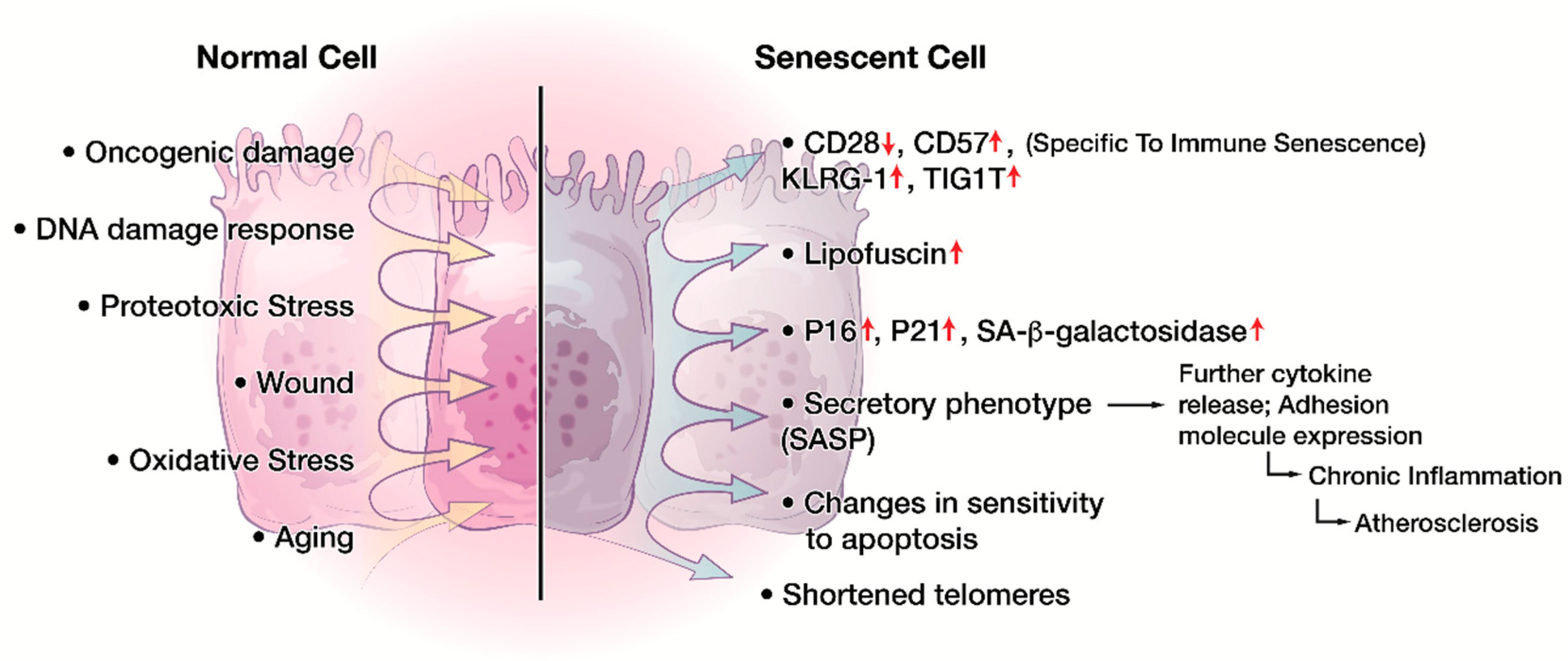{kind=link}
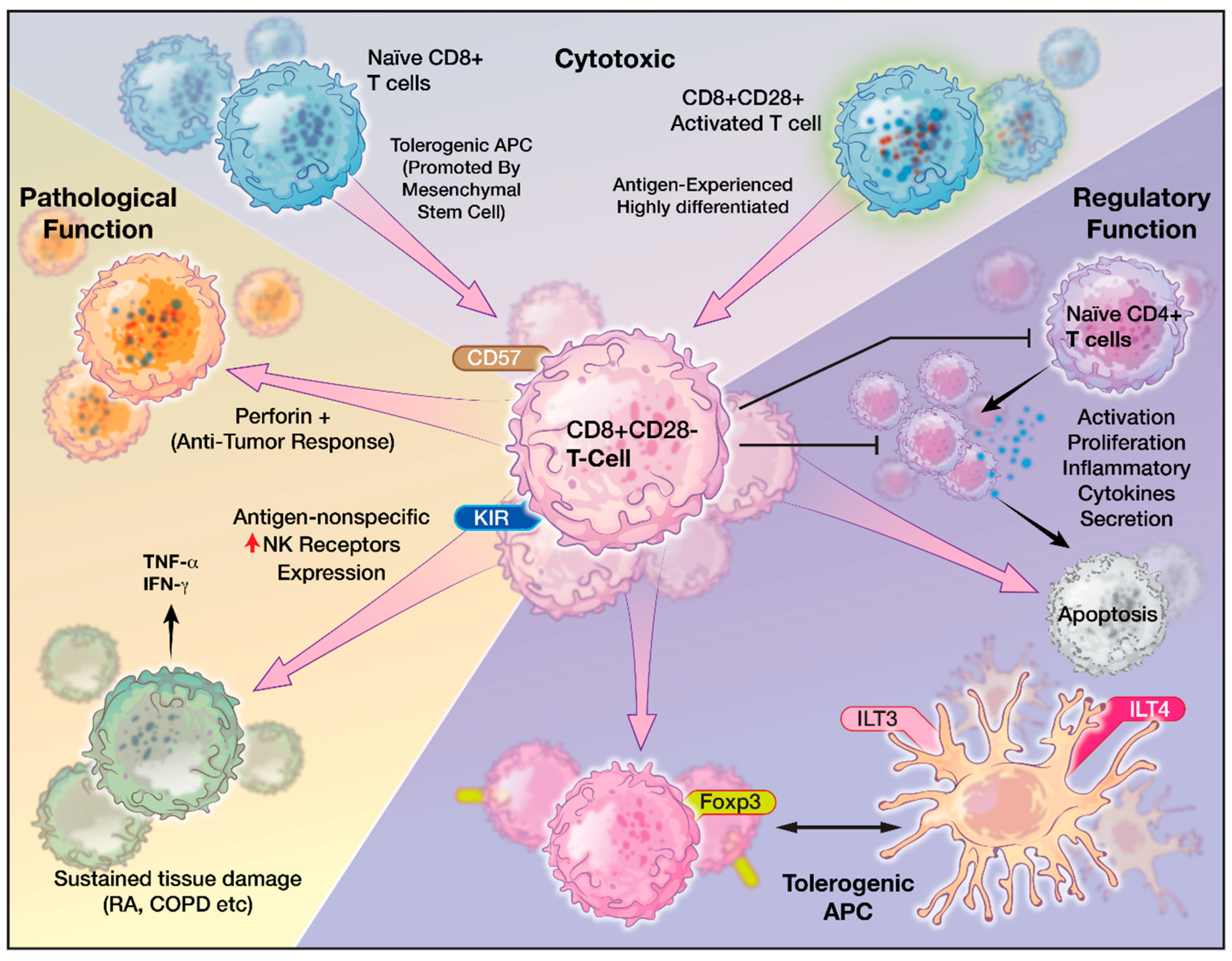{kind=link}
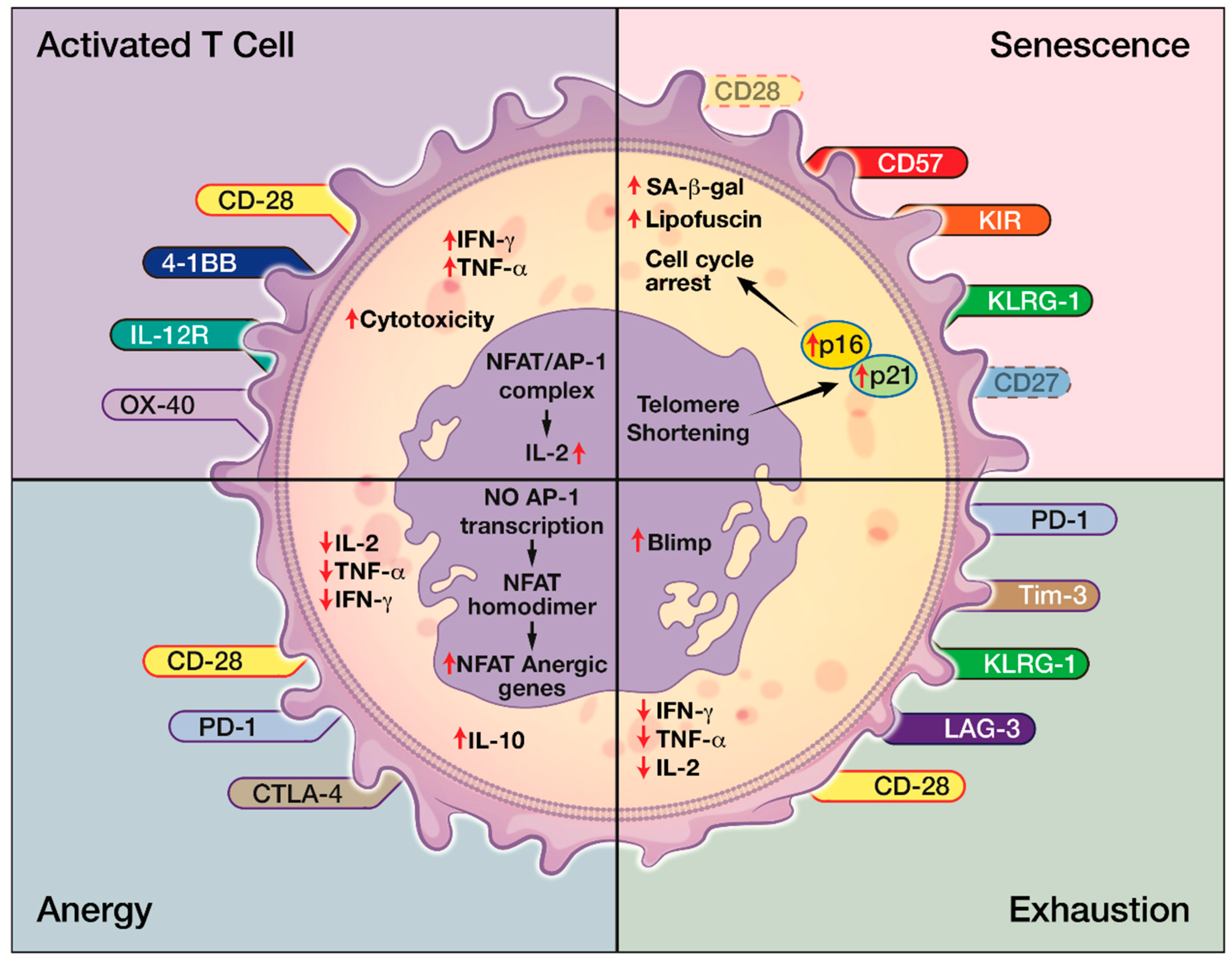{kind=link}
Table 1.
Clinical trials related to the therapeutic use of CD28 manipulation, such as CAR-T cell therapy and monoclonal antibodies.
Table 1.
Clinical trials related to the therapeutic use of CD28 manipulation, such as CAR-T cell therapy and monoclonal antibodies.
| Malignancy | Phase | N | Trial Name | Clinical Trial Identifier | Therapeutics | References |
|---|---|---|---|---|---|---|
| Relapsed or Refractory Acute Lymphoblastic Leukemia | 1 | 5 | Chimeric Antigen Receptor (CAR)-Modified T Cell Therapy in Treating Patients with Acute Lymphoblastic Leukemia | NCT02186860 | Third Gen CAR-T cells containing CD28+CD137 | [71] |
| Glioblastoma | 1 | 17 | CMV-Specific Cytotoxic T Lymphocytes Expressing CAR Targeting HER2 in Patients with GBM (HERT-GBM) | NCT01109095 | Second Gen CMV-selected CAR-T cells against HER2 containing CD28.zeta signaling domain | [72] |
| Rheumatoid Arthritis | 1/2 | 18 | Safety, Tolerability, Pharmacodynamics and Efficacy Study of TAB08 in Patients with Rheumatoid Arthritis | NCT01990157 | TAB08 | |
| Solid Neoplasms | 1 | 38 | Dose Escalation Study of TAB08 in Patients with Advanced Solid Neoplasms (TAB08) | NCT03006029 | TAB08 | [73] |
| Systemic Lupus Erythematosus | 2 | 730 | Safety and Efficacy Study of a Biologic to Treat Systemic Lupus Erythematosus | NCT02265744 | Lulizumab pegol (monoclonal antibody against CD28) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Huff, W.X.; Kwon, J.H.; Henriquez, M.; Fetcko, K.; Dey, M. The Evolving Role of CD8+CD28− Immunosenescent T Cells in Cancer Immunology. Int. J. Mol. Sci. 2019, 20, 2810. https://doi.org/10.3390/ijms20112810
AMA Style
Huff WX, Kwon JH, Henriquez M, Fetcko K, Dey M. The Evolving Role of CD8+CD28− Immunosenescent T Cells in Cancer Immunology. International Journal of Molecular Sciences. 2019; 20(11):2810. https://doi.org/10.3390/ijms20112810
Chicago/Turabian StyleHuff, Wei X., Jae Hyun Kwon, Mario Henriquez, Kaleigh Fetcko, and Mahua Dey. 2019. "The Evolving Role of CD8+CD28− Immunosenescent T Cells in Cancer Immunology" International Journal of Molecular Sciences 20, no. 11: 2810. https://doi.org/10.3390/ijms20112810
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.