T-Cell Accumulation in the Hypertensive Brain: A Role for Sphingosine-1-Phosphate-Mediated Chemotaxis


Abstract
:1. Introduction
2. Results
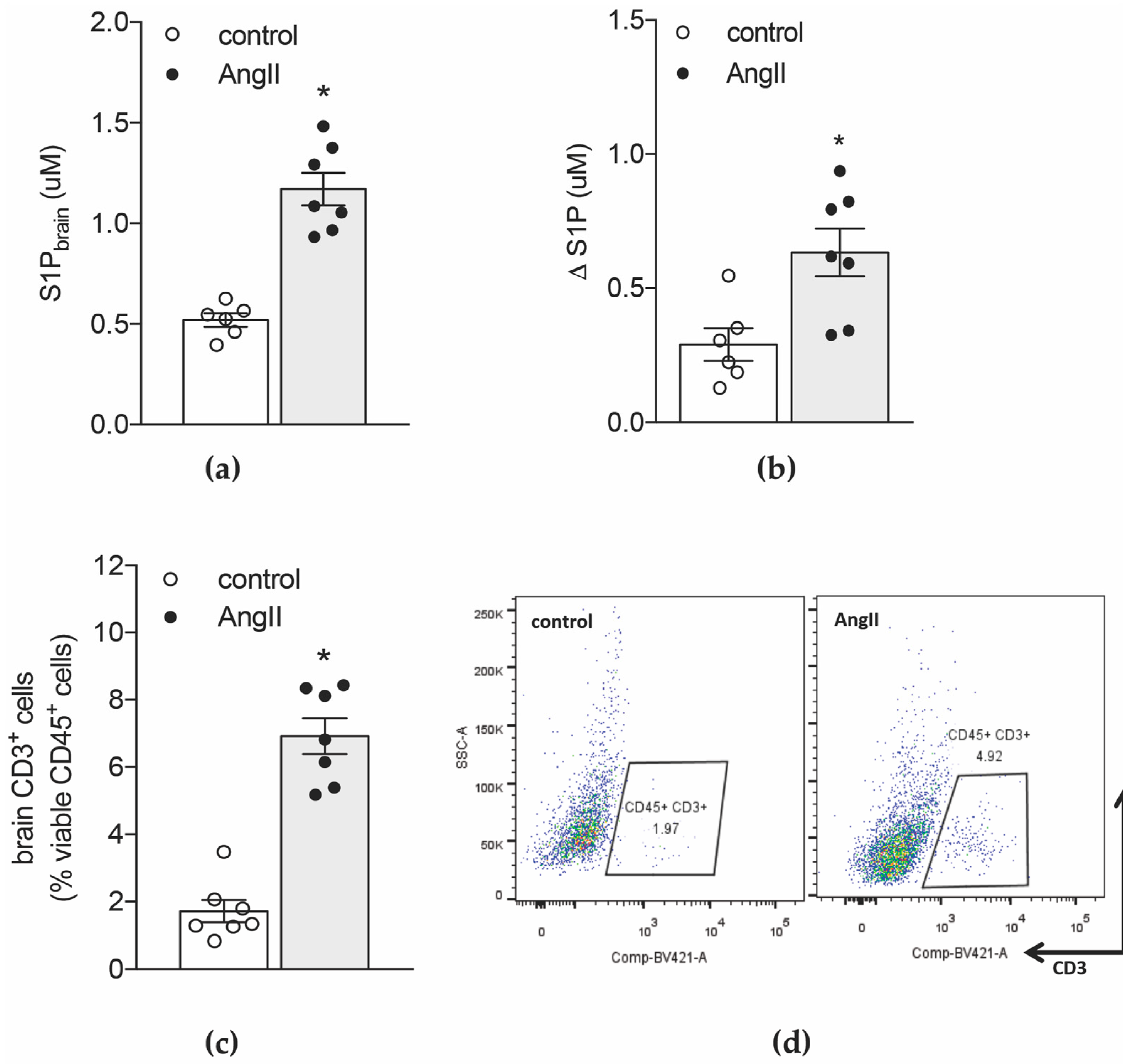
2.1. Hypertension-Associated Cognitive Deficits Link to Elevated S1P Levels in the Brain
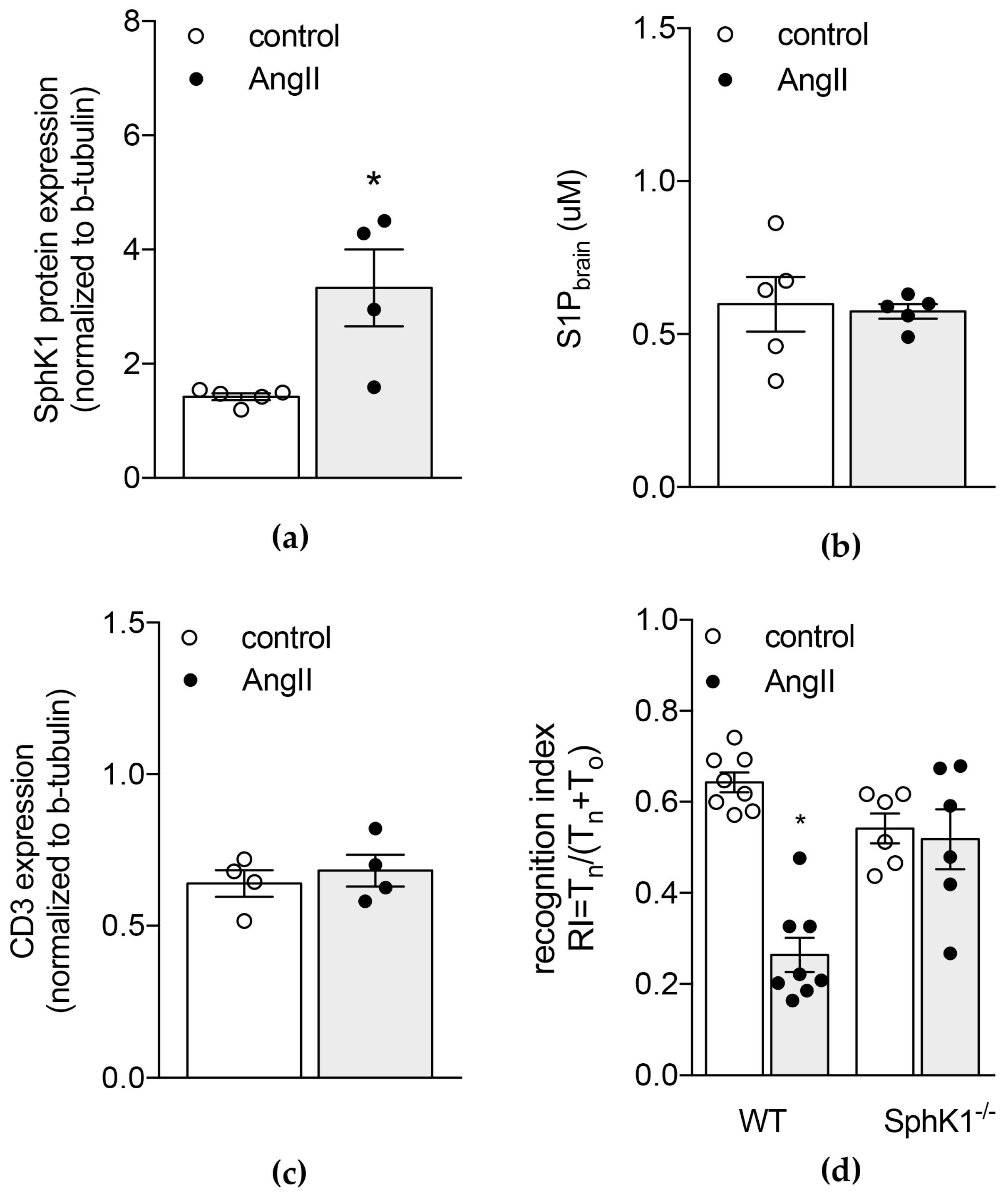
2.2. Genetic Depletion of S1P Generating Enzyme SphK1 Protects from AngII-Induced Increases in Brain S1P Levels and Memory Deficits
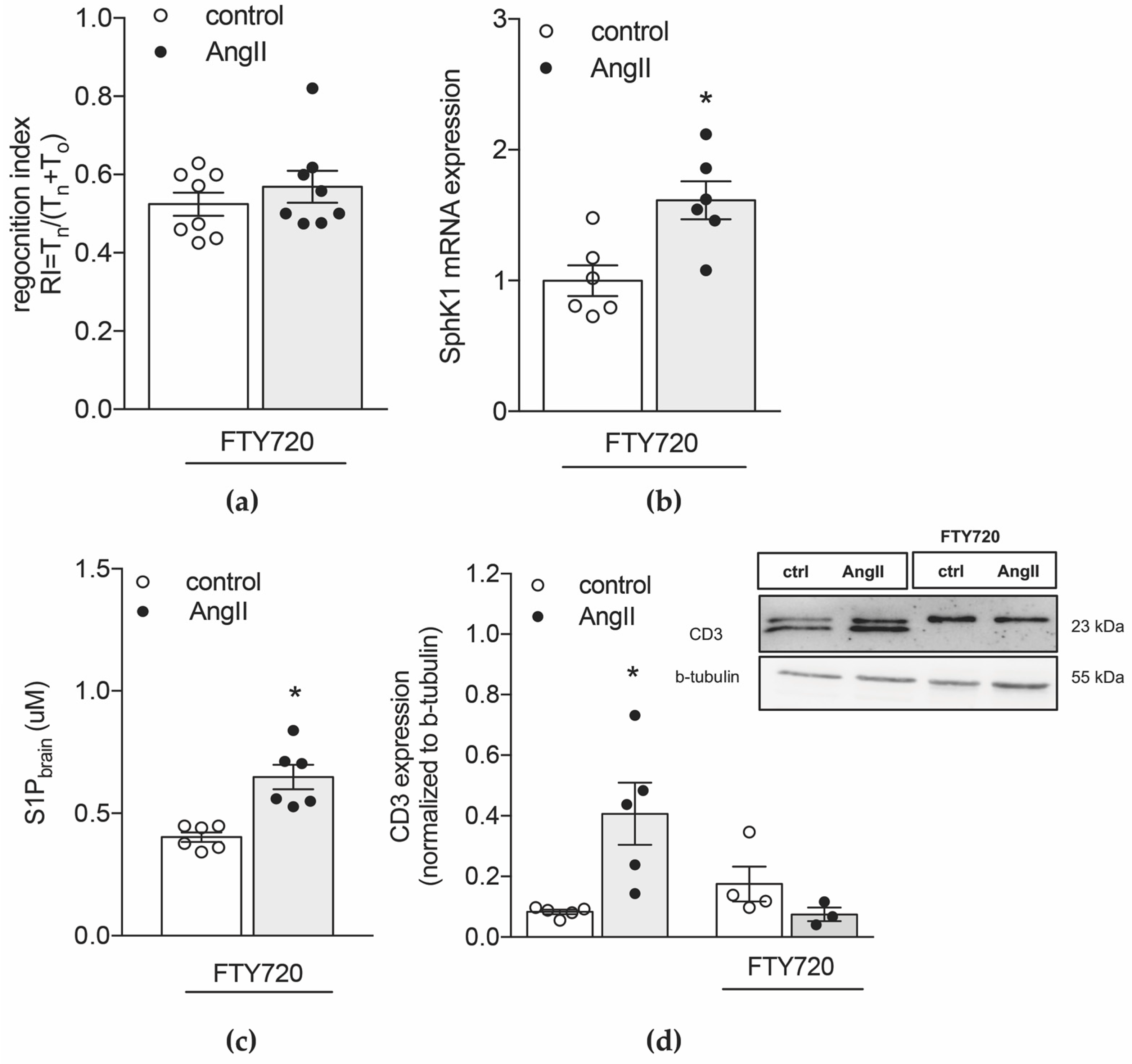
2.3. Inhibiting S1P Chemotaxis Protects from Hypertension-Associated Cognitive Impairment
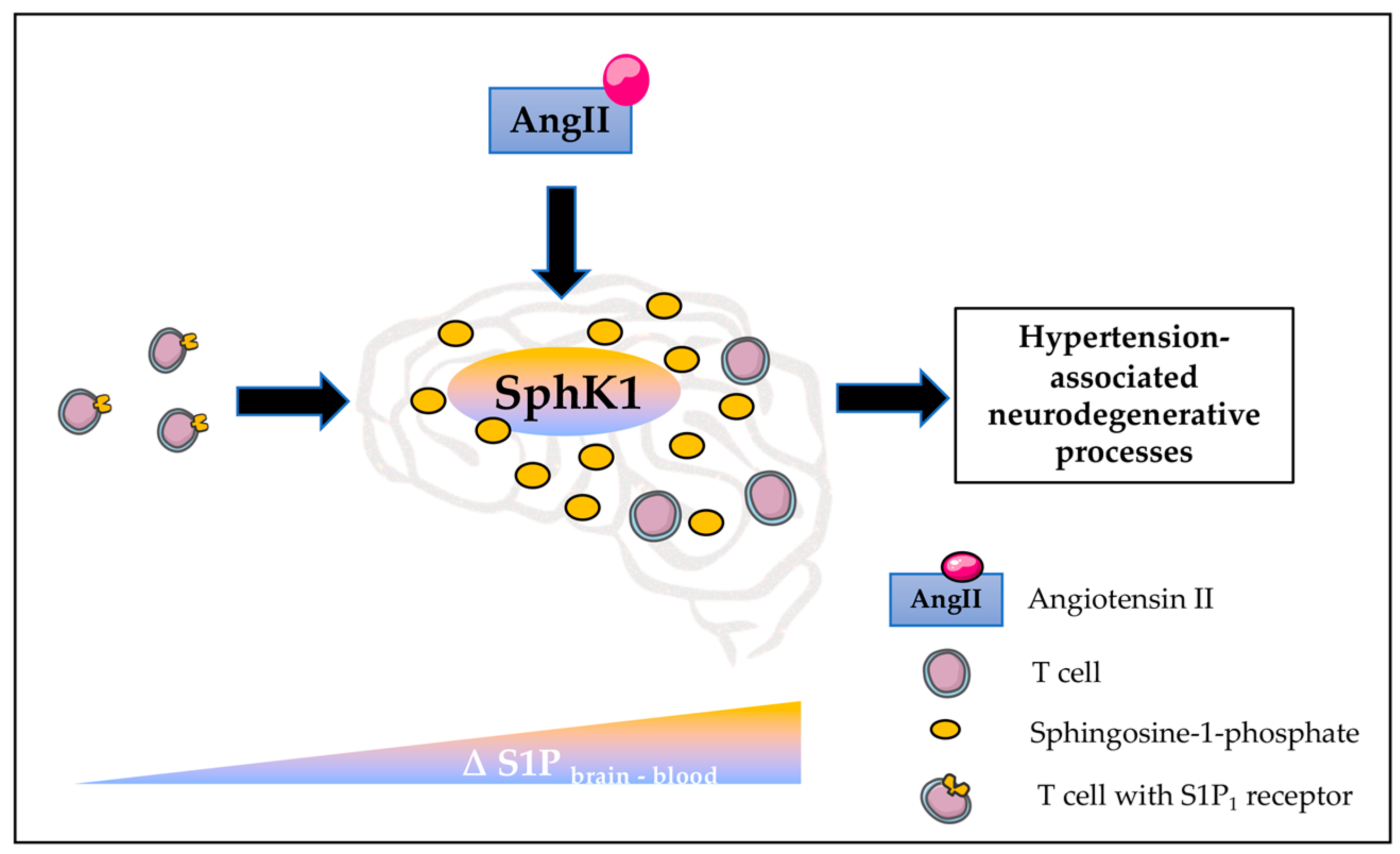
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Animals
4.3. Hypertension Model
4.4. Novel Object Recognition (NOR)
4.5. Mass Spectrometry
4.6. Fluorescence Activated Cell Sorting
4.7. Western Blotting and qPCR
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AngII | Angiotensin II |
| BBB | Blood brain barrier |
| BP | Blood pressure |
| CD3 | Cluster of differentiation 3 |
| CD45 | Cluster of differentiation 45 |
| CVD | Cardiovascular disease |
| FACS | Fluorescence activated cell sorting |
| FTY720 | Fingolimod |
| GFAP | Glial fibrillary acidic protein |
| Iba | Ionized calcium-binding adapter molecule |
| IL | Interleukin |
| NOR | Novel object recognition |
| RI | Recognition index |
| S1P | Sphingosine-1-phosphate |
| SphK1 | Sphingosine kinase 1 |
| SphK2 | Sphingosine kinase 2 |
| S1P1 | Sphingosine receptor type 1 |
| TNFA | Tumour necrosis factor alpha |
| VWF | Van Willebrand factor |
| VCAM-1 | Vascular cell adhesion protein 1 |
| WT | Wild type |
References
- Meissner, A. Hypertension and the Brain: A Risk Factor for More Than Heart Disease. Cereb. Dis. 2016, 42, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Meissner, A.; Minnerup, J.; Soria, G.; Planas, A.M. Structural and functional brain alterations in a murine model of Angiotensin II-induced hypertension. J. Neurochem. 2017, 140, 509–521. [Google Scholar] [CrossRef] [PubMed]
- Igase, M.; Kohara, K.; Miki, T. The Association between Hypertension and Dementia in the Elderly. Int. J. Hypertens. 2012, 2012, 320648. [Google Scholar] [CrossRef] [PubMed]
- Ji, Q.; Cheng, G.; Ma, N.; Huang, Y.; Lin, Y.; Zhou, Q.; Que, B.; Dong, J.; Zhou, Y.; Nie, S. Circulating Th1, Th2, and Th17 Levels in Hypertensive Patients. Dis. Markers 2017, 2017, 7146290. [Google Scholar] [CrossRef] [PubMed]
- Miguel, C.D.; Rudemiller, N.P.; Abais, J.M.; Mattson, D.L. Inflammation and hypertension: New understandings and potential therapeutic targets. Curr. Hypertens. Rep. 2015, 17, 507. [Google Scholar] [CrossRef] [PubMed]
- Don-Doncow, N.; Zhang, Y.; Matuskova, H.; Meissner, A. The emerging alliance of sphingosine-1-phosphate signalling and immune cells: From basic mechanisms to implications in hypertension. Br. J. Pharm. 2018. [Google Scholar] [CrossRef] [PubMed]
- Meissner, A.; Miro, F.; Jimenez-Altayo, F.; Jurado, A.; Vila, E.; Planas, A.M. Sphingosine-1-phosphate signalling-a key player in the pathogenesis of Angiotensin II-induced hypertension. Cardiovasc. Res. 2017, 113, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Itani, H.A.; McMaster, W.G., Jr.; Saleh, M.A.; Nazarewicz, R.R.; Mikolajczyk, T.P.; Kaszuba, A.M.; Konior, A.; Prejbisz, A.; Januszewicz, A.; Norlander, A.E.; et al. Activation of Human T Cells in Hypertension: Studies of Humanized Mice and Hypertensive Humans. Hypertension 2016, 68, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Guoping, P.; Wei, W.; Xiaoyan, L.; Fangping, H.; Zhongqin, C.; Benyan, L. Characteristics of the peripheral T cell immune response of patients at different stages of vascular cognitive impairment. Immunol. Lett. 2015, 168, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Serre-Miranda, C.; Roque, S.; Santos, N.C.; Portugal-Nunes, C.; Costa, P.; Palha, J.A.; Sousa, N.; Correia-Neves, M. Effector memory CD4+ T cells are associated with cognitive performance in a senior population. Neurol. Neuroimmunol. Neuroinflamm. 2015, 2, e54. [Google Scholar] [CrossRef]
- Laurent, C.; Dorothee, G.; Hunot, S.; Martin, E.; Monnet, Y.; Duchamp, M.; Dong, Y.; Legeron, F.P.; Leboucher, A.; Burnouf, S.; et al. Hippocampal T cell infiltration promotes neuroinflammation and cognitive decline in a mouse model of tauopathy. Brain 2017, 140, 184–200. [Google Scholar] [CrossRef] [PubMed]
- Meissner, A.; Visanji, N.P.; Momen, M.A.; Feng, R.; Francis, B.M.; Bolz, S.S.; Hazrati, L.N. Tumor Necrosis Factor-alpha Underlies Loss of Cortical Dendritic Spine Density in a Mouse Model of Congestive Heart Failure. J. Am. Heart Assoc. 2015, 4, e001920. [Google Scholar] [CrossRef] [PubMed]
- Bonow, R.H.; Aïd, S.; Zhang, Y.; Becker, K.G.; Bosetti, F. The brain expression of genes involved in inflammatory response, the ribosome, and learning and memory is altered by centrally injected lipopolysaccharide in mice. Pharm. J. 2009, 9, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Motoki, K.; Kishi, H.; Hori, E.; Tajiri, K.; Nishijo, H.; Muraguchi, A. The direct excitatory effect of IL-1beta on cerebellar Purkinje cell. Biochem. Biophys. Res. Commun. 2009, 379, 665–668. [Google Scholar] [CrossRef] [PubMed]
- Koo, J.W.; Duman, R.S. IL-1beta is an essential mediator of the antineurogenic and anhedonic effects of stress. Proc. Natl. Acad. Sci. USA 2008, 105, 751–756. [Google Scholar] [CrossRef] [PubMed]
- Luan, Y.-Y.; Yao, Y.-M. The Clinical Significance and Potential Role of C-Reactive Protein in Chronic Inflammatory and Neurodegenerative Diseases. Front. Immunol. 2018, 9, 1302. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.; Liu, G.A.; Perez, E.; Rainer, R.D.; Febo, M.; Cruz-Almeida, Y.; Ebner, N.C. Systemic Inflammation Mediates Age-Related Cognitive Deficits. Front. Aging Neurosci. 2018, 10, 236. [Google Scholar] [CrossRef] [PubMed]
- Sommer, A.; Winner, B.; Prots, I. The Trojan horse—Neuroinflammatory impact of T cells in neurodegenerative diseases. Mol. Neurodegener. 2017, 12, 78. [Google Scholar] [CrossRef]
- Browne, T.C.; McQuillan, K.; McManus, R.M.; O’Reilly, J.A.; Mills, K.H.; Lynch, M.A. IFN-gamma Production by amyloid beta-specific Th1 cells promotes microglial activation and increases plaque burden in a mouse model of Alzheimer’s disease. J. Immunol. 2013, 190, 2241–2251. [Google Scholar] [CrossRef] [PubMed]
- Siffrin, V.; Radbruch, H.; Glumm, R.; Niesner, R.; Paterka, M.; Herz, J.; Leuenberger, T.; Lehmann, S.M.; Luenstedt, S.; Rinnenthal, J.L.; et al. In vivo imaging of partially reversible th17 cell-induced neuronal dysfunction in the course of encephalomyelitis. Immunity 2010, 33, 424–436. [Google Scholar] [CrossRef] [PubMed]
- Brochard, V.; Combadière, B.; Prigent, A.; Laouar, Y.; Perrin, A.; Beray-Berthat, V.; Bonduelle, O.; Alvarez-Fischer, D.; Callebert, J.; Launay, J.-M.; et al. Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J. Clin. Investig. 2009, 119, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Meissner, A. S1PR (Sphingosine-1-Phosphate Receptor) Signaling in the Regulation of Vascular Tone and Blood Pressure: Is S1PR1 Doing the Trick? Hypertension 2017, 70, 232–234. [Google Scholar] [CrossRef] [PubMed]
- Jo, E.; Sanna, M.G.; Gonzalez-Cabrera, P.J.; Thangada, S.; Tigyi, G.; Osborne, D.A.; Hla, T.; Parrill, A.L.; Rosen, H. S1P1-selective in vivo-active agonists from high-throughput screening: Off-the-shelf chemical probes of receptor interactions, signaling, and fate. Chem. Biol. 2005, 12, 703–715. [Google Scholar] [CrossRef] [PubMed]
- Mandala, S.; Hajdu, R.; Bergstrom, J.; Quackenbush, E.; Xie, J.; Milligan, J.; Thornton, R.; Shei, G.J.; Card, D.; Keohane, C.; et al. Alteration of lymphocyte trafficking by sphingosine-1-phosphate receptor agonists. Science 2002, 296, 346–349. [Google Scholar] [CrossRef] [PubMed]
- Tarrason, G.; Auli, M.; Mustafa, S.; Dolgachev, V.; Domenech, M.T.; Prats, N.; Dominguez, M.; Lopez, R.; Aguilar, N.; Calbet, M.; et al. The sphingosine-1-phosphate receptor-1 antagonist, W146, causes early and short-lasting peripheral blood lymphopenia in mice. Int. Immunopharmacol. 2011, 11, 1773–1779. [Google Scholar] [CrossRef]
- Park, S.J.; Im, D.S. Sphingosine 1-Phosphate Receptor Modulators and Drug Discovery. Biomol. Ther. 2017, 25, 80–90. [Google Scholar] [CrossRef] [Green Version]
- Meissner, A.; Yang, J.; Kroetsch, J.T.; Sauve, M.; Dax, H.; Momen, A.; Noyan-Ashraf, M.H.; Heximer, S.; Husain, M.; Lidington, D.; et al. Tumor necrosis factor-alpha-mediated downregulation of the cystic fibrosis transmembrane conductance regulator drives pathological sphingosine-1-phosphate signaling in a mouse model of heart failure. Circulation 2012, 125, 2739–2750. [Google Scholar] [CrossRef]
- Yang, J.; Noyan-Ashraf, M.H.; Meissner, A.; Voigtlaender-Bolz, J.; Kroetsch, J.T.; Foltz, W.; Jaffray, D.; Kapoor, A.; Momen, A.; Heximer, S.P.; et al. Proximal cerebral arteries develop myogenic responsiveness in heart failure via tumor necrosis factor-alpha-dependent activation of sphingosine-1-phosphate signaling. Circulation 2012, 126, 196–206. [Google Scholar] [CrossRef]
- Lv, M.; Zhang, D.; Dai, D.; Zhang, W.; Zhang, L. Sphingosine kinase 1/sphingosine-1-phosphate regulates the expression of interleukin-17A in activated microglia in cerebral ischemia/reperfusion. Inflamm. Res. 2016, 65, 551–562. [Google Scholar] [CrossRef]
- Faraco, G.; Brea, D.; Garcia-Bonilla, L.; Wang, G.; Racchumi, G.; Chang, H.; Buendia, I.; Santisteban, M.M.; Segarra, S.G.; Koizumi, K.; et al. Dietary salt promotes neurovascular and cognitive dysfunction through a gut-initiated TH17 response. Nat. Neurosci. 2018, 21, 240–249. [Google Scholar] [CrossRef]
- Oberstein, T.J.; Taha, L.; Spitzer, P.; Hellstern, J.; Herrmann, M.; Kornhuber, J.; Maler, J.M. Imbalance of Circulating Th17 and Regulatory T Cells in Alzheimer’s Disease: A Case Control Study. Front. Immunol. 2018, 9, 1213. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, X.B.; Huang, P.; Huang, M.Y.; Gu, X.J. Change of Peripheral Blood Treg/Thl7 in Cognitive Impairment with Chronic Renal Failure Patients. Cell. Physiol. Biochem. 2018, 45, 281–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Don-Doncow, N.; Zhang, Y.; Rattik, S.; Björkbacka, H.; Meissner, A. Anti-inflammatory Properties of Simvastatin Mediate Improvement of Memory Function in Aged ApoE-/- Mice. Atheroscler. Suppl. 2018, 32, 98. [Google Scholar] [CrossRef]
- Hofmann, M.; Brinkmann, V.; Zerwes, H.G. FTY720 preferentially depletes naive T cells from peripheral and lymphoid organs. Int. Immunopharmacol. 2006, 6, 1902–1910. [Google Scholar] [CrossRef] [PubMed]
- Thangada, S.; Khanna, K.M.; Blaho, V.A.; Oo, M.L.; Im, D.S.; Guo, C.; Lefrancois, L.; Hla, T. Cell-surface residence of sphingosine 1-phosphate receptor 1 on lymphocytes determines lymphocyte egress kinetics. J. Exp. Med. 2010, 207, 1475–1483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frischer, J.M.; Bramow, S.; Dal-Bianco, A.; Lucchinetti, C.F.; Rauschka, H.; Schmidbauer, M.; Laursen, H.; Sorensen, P.S.; Lassmann, H. The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain 2009, 132, 1175–1189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lutz, S.E.; Smith, J.R.; Kim, D.H.; Olson, C.V.L.; Ellefsen, K.; Bates, J.M.; Gandhi, S.P.; Agalliu, D. Caveolin1 Is Required for Th1 Cell Infiltration, but Not Tight Junction Remodeling, at the Blood-Brain Barrier in Autoimmune Neuroinflammation. Cell Rep. 2017, 21, 2104–2117. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.A.; Maltby, S.; Plank, M.W.; Kluge, M.; Nilsson, M.; Foster, P.S.; Walker, F.R. Peripheral immune cells infiltrate into sites of secondary neurodegeneration after ischemic stroke. Brain Behav. Immun. 2018, 67, 299–307. [Google Scholar] [CrossRef]
- Merlini, M.; Kirabali, T.; Kulic, L.; Nitsch, R.M.; Ferretti, M.T. Extravascular CD3+ T Cells in Brains of Alzheimer Disease Patients Correlate with Tau but Not with Amyloid Pathology: An Immunohistochemical Study. Neurodegener. Dis. 2018, 18, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Deng, G.; Carter, J.; Traystman, R.J.; Wagner, D.H.; Herson, P.S. Pro-inflammatory T-lymphocytes rapidly infiltrate into the brain and contribute to neuronal injury following cardiac arrest and cardiopulmonary resuscitation. J. Neuroimmunol. 2014, 274, 132–140. [Google Scholar] [CrossRef] [Green Version]
- Wei, Z.; Spizzo, I.; Diep, H.; Drummond, G.R.; Widdop, R.E.; Vinh, A. Differential phenotypes of tissue-infiltrating T cells during angiotensin II-induced hypertension in mice. PLoS ONE 2014, 9, e114895. [Google Scholar] [CrossRef] [PubMed]
- Saunders, J.A.; Estes, K.A.; Kosloski, L.M.; Allen, H.E.; Dempsey, K.M.; Torres-Russotto, D.R.; Meza, J.L.; Santamaria, P.M.; Bertoni, J.M.; Murman, D.L.; et al. CD4+ regulatory and effector/memory T cell subsets profile motor dysfunction in Parkinson’s disease. J. Neuroimmune Pharm. 2012, 7, 927–938. [Google Scholar] [CrossRef] [PubMed]
- Guzik, T.J.; Hoch, N.E.; Brown, K.A.; McCann, L.A.; Rahman, A.; Dikalov, S.; Goronzy, J.; Weyand, C.; Harrison, D.G. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J. Exp. Med. 2007, 204, 2449–2460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faraco, G.; Sugiyama, Y.; Lane, D.; Garcia-Bonilla, L.; Chang, H.; Santisteban, M.M.; Racchumi, G.; Murphy, M.; Van Rooijen, N.; Anrather, J.; et al. Perivascular macrophages mediate the neurovascular and cognitive dysfunction associated with hypertension. J. Clin. Investig. 2016, 126, 4674–4689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, M.A.; Gibb, D.R.; Picard, F.; Brinkmann, V.; Straume, M.; Ley, K. Transient T cell accumulation in lymph nodes and sustained lymphopenia in mice treated with FTY720. Eur. J. Immunol. 2005, 35, 3570–3580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matloubian, M.; Lo, C.G.; Cinamon, G.; Lesneski, M.J.; Xu, Y.; Brinkmann, V.; Allende, M.L.; Proia, R.L.; Cyster, J.G. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature 2004, 427, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Rivera, J.; Proia, R.L.; Olivera, A. The alliance of sphingosine-1-phosphate and its receptors in immunity. Nat. Rev. Immunol. 2008, 8, 753–763. [Google Scholar] [CrossRef] [Green Version]
- Pham, T.H.; Baluk, P.; Xu, Y.; Grigorova, I.; Bankovich, A.J.; Pappu, R.; Coughlin, S.R.; McDonald, D.M.; Schwab, S.R.; Cyster, J.G. Lymphatic endothelial cell sphingosine kinase activity is required for lymphocyte egress and lymphatic patterning. J. Exp. Med. 2010, 207, 17–27. [Google Scholar] [CrossRef]
- Urtz, N.; Gaertner, F.; von Bruehl, M.L.; Chandraratne, S.; Rahimi, F.; Zhang, L.; Orban, M.; Barocke, V.; Beil, J.; Schubert, I.; et al. Sphingosine 1-Phosphate Produced by Sphingosine Kinase 2 Intrinsically Controls Platelet Aggregation In Vitro and In Vivo. Circ. Res. 2015, 117, 376–387. [Google Scholar] [CrossRef] [Green Version]
- Fukuda, Y.; Kihara, A.; Igarashi, Y. Distribution of sphingosine kinase activity in mouse tissues: Contribution of SPHK1. Biochem. Biophys. Res. Commun. 2003, 309, 155–160. [Google Scholar] [CrossRef]
- Ochi, S.; Oda, M.; Matsuda, H.; Ikari, S.; Sakurai, J. Clostridium perfringens alpha-toxin activates the sphingomyelin metabolism system in sheep erythrocytes. J. Biol. Chem. 2004, 279, 12181–12189. [Google Scholar] [CrossRef] [PubMed]
- Park, S.M.; Angel, C.E.; McIntosh, J.D.; Brooks, A.E.; Middleditch, M.; Chen, C.J.; Ruggiero, K.; Cebon, J.; Rod Dunbar, P. Sphingosine-1-phosphate lyase is expressed by CD68+ cells on the parenchymal side of marginal reticular cells in human lymph nodes. Eur. J. Immunol. 2014, 44, 2425–2436. [Google Scholar] [CrossRef]
- Breart, B.; Ramos-Perez, W.D.; Mendoza, A.; Salous, A.K.; Gobert, M.; Huang, Y.; Adams, R.H.; Lafaille, J.J.; Escalante-Alcalde, D.; Morris, A.J.; et al. Lipid phosphate phosphatase 3 enables efficient thymic egress. J. Exp. Med. 2011, 208, 1267–1278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwab, S.R.; Pereira, J.P.; Matloubian, M.; Xu, Y.; Huang, Y.; Cyster, J.G. Lymphocyte sequestration through S1P lyase inhibition and disruption of S1P gradients. Science 2005, 309, 1735–1739. [Google Scholar] [CrossRef] [PubMed]
- Anelli, V.; Bassi, R.; Tettamanti, G.; Viani, P.; Riboni, L. Extracellular release of newly synthesized sphingosine-1-phosphate by cerebellar granule cells and astrocytes. J. Neurochem. 2005, 92, 1204–1215. [Google Scholar] [CrossRef] [Green Version]
- Kajimoto, T.; Okada, T.; Yu, H.; Goparaju, S.K.; Jahangeer, S.; Nakamura, S. Involvement of sphingosine-1-phosphate in glutamate secretion in hippocampal neurons. Mol. Cell. Biol. 2007, 27, 3429–3440. [Google Scholar] [CrossRef]
- Wilson, P.C.; Fitzgibbon, W.R.; Garrett, S.M.; Jaffa, A.A.; Luttrell, L.M.; Brands, M.W.; El-Shewy, H.M. Inhibition of Sphingosine Kinase 1 Ameliorates Angiotensin II-Induced Hypertension and Inhibits Transmembrane Calcium Entry via Store-Operated Calcium Channel. Mol. Endocrinol. 2015, 29, 896–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, S.; Wei, S.; Wang, X.; Xu, Y.; Xiao, Y.; Liu, H.; Jia, J.; Cheng, J. Sphingosine kinase 1 mediates neuroinflammation following cerebral ischemia. Exp. Neurol. 2015, 272, 160–169. [Google Scholar] [CrossRef]
- Schetters, S.T.T.; Gomez-Nicola, D.; Garcia-Vallejo, J.J.; Van Kooyk, Y. Neuroinflammation: Microglia and T Cells Get Ready to Tango. Front. Immunol. 2018, 8, 1905. [Google Scholar] [CrossRef] [Green Version]
- Moore, A.N.; Kampfl, A.W.; Zhao, X.; Hayes, R.L.; Dash, P.K. Sphingosine-1-phosphate induces apoptosis of cultured hippocampal neurons that requires protein phosphatases and activator protein-1 complexes. Neuroscience 1999, 94, 405–415. [Google Scholar] [CrossRef]
- Chipuk, J.E.; McStay, G.P.; Bharti, A.; Kuwana, T.; Clarke, C.J.; Siskind, L.J.; Obeid, L.M.; Green, D.R. Sphingolipid metabolism cooperates with BAK and BAX to promote the mitochondrial pathway of apoptosis. Cell 2012, 148, 988–1000. [Google Scholar] [CrossRef] [PubMed]
- Riganti, L.; Antonucci, F.; Gabrielli, M.; Prada, I.; Giussani, P.; Viani, P.; Valtorta, F.; Menna, E.; Matteoli, M.; Verderio, C. Sphingosine-1-Phosphate (S1P) Impacts Presynaptic Functions by Regulating Synapsin I Localization in the Presynaptic Compartment. J. Neurosci. 2016, 36, 4624–4634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moon, E.; Han, J.E.; Jeon, S.; Ryu, J.H.; Choi, J.W.; Chun, J. Exogenous S1P Exposure Potentiates Ischemic Stroke Damage That Is Reduced Possibly by Inhibiting S1P Receptor Signaling. Mediat. Inflamm. 2015, 2015, 492659. [Google Scholar] [CrossRef] [PubMed]
- Quarta, S.; Camprubi-Robles, M.; Schweigreiter, R.; Matusica, D.; Haberberger, R.V.; Proia, R.L.; Bandtlow, C.E.; Ferrer-Montiel, A.; Kress, M. Sphingosine-1-Phosphate and the S1P3 Receptor Initiate Neuronal Retraction via RhoA/ROCK Associated with CRMP2 Phosphorylation. Front. Mol. Neurosci. 2017, 10, 317. [Google Scholar] [CrossRef] [PubMed]
- Vojinovic, D.; Adams, H.H.; Jian, X.; Yang, Q.; Smith, A.V.; Bis, J.C.; Teumer, A.; Scholz, M.; Armstrong, N.J.; Hofer, E.; et al. Genome-wide association study of 23,500 individuals identifies 7 loci associated with brain ventricular volume. Nat. Commun. 2018, 9, 3945. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control | AngII | p-Value | SphK1−/− AngII vs. WT AngII (Fold Change) | p-Value | |
|---|---|---|---|---|---|
| Vcam1 | 1.652 ± 0.227 | 1.870 ± 0.189 | 0.841 | 0.48 | 0.057 |
| Tnfa | 0.827 ± 0.267 | 0.431 ± 0.112 | 0.286 | 0.25 | 0.009 |
| Il1b | 1.246 ± 0.153 | 1.346 ± 0.247 | 0.841 | 0.29 | 0.037 |
| Vwf | 4.830 ± 1.672 | 2.859 ± 0.642 | 0.556 | 0.66 | >0.999 |
| Selp | 0.647 ± 0.192 | 0.429 ± 0.187 | 0.413 | 0.19 | 0.003 |
| Control | AngII | p-Value | |
|---|---|---|---|
| WT | 388.3 ± 87.1 | 3069.1 ± 764.9 * | 0.0006 |
| SphK1−/− | 709.2 ± 213.2 | 433.7 ± 37.9 | 0.3939 |
| WT + FTY720 | 372.2 ± 123.8 | 158.7 ± 59.2 | 0.1241 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Don-Doncow, N.; Vanherle, L.; Zhang, Y.; Meissner, A. T-Cell Accumulation in the Hypertensive Brain: A Role for Sphingosine-1-Phosphate-Mediated Chemotaxis. Int. J. Mol. Sci. 2019, 20, 537. https://doi.org/10.3390/ijms20030537
Don-Doncow N, Vanherle L, Zhang Y, Meissner A. T-Cell Accumulation in the Hypertensive Brain: A Role for Sphingosine-1-Phosphate-Mediated Chemotaxis. International Journal of Molecular Sciences. 2019; 20(3):537. https://doi.org/10.3390/ijms20030537
Chicago/Turabian StyleDon-Doncow, Nicholas, Lotte Vanherle, Yun Zhang, and Anja Meissner. 2019. "T-Cell Accumulation in the Hypertensive Brain: A Role for Sphingosine-1-Phosphate-Mediated Chemotaxis" International Journal of Molecular Sciences 20, no. 3: 537. https://doi.org/10.3390/ijms20030537