Augmented Insulin and Leptin Resistance of High Fat Diet-Fed APPswe/PS1dE9 Transgenic Mice Exacerbate Obesity and Glycemic Dysregulation

 , , ,
, , ,  and
and
Abstract
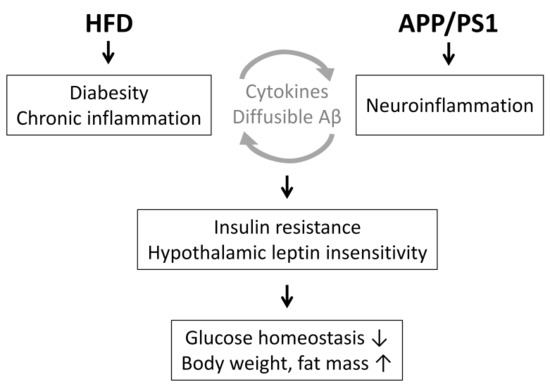
:
1. Introduction
2. Results
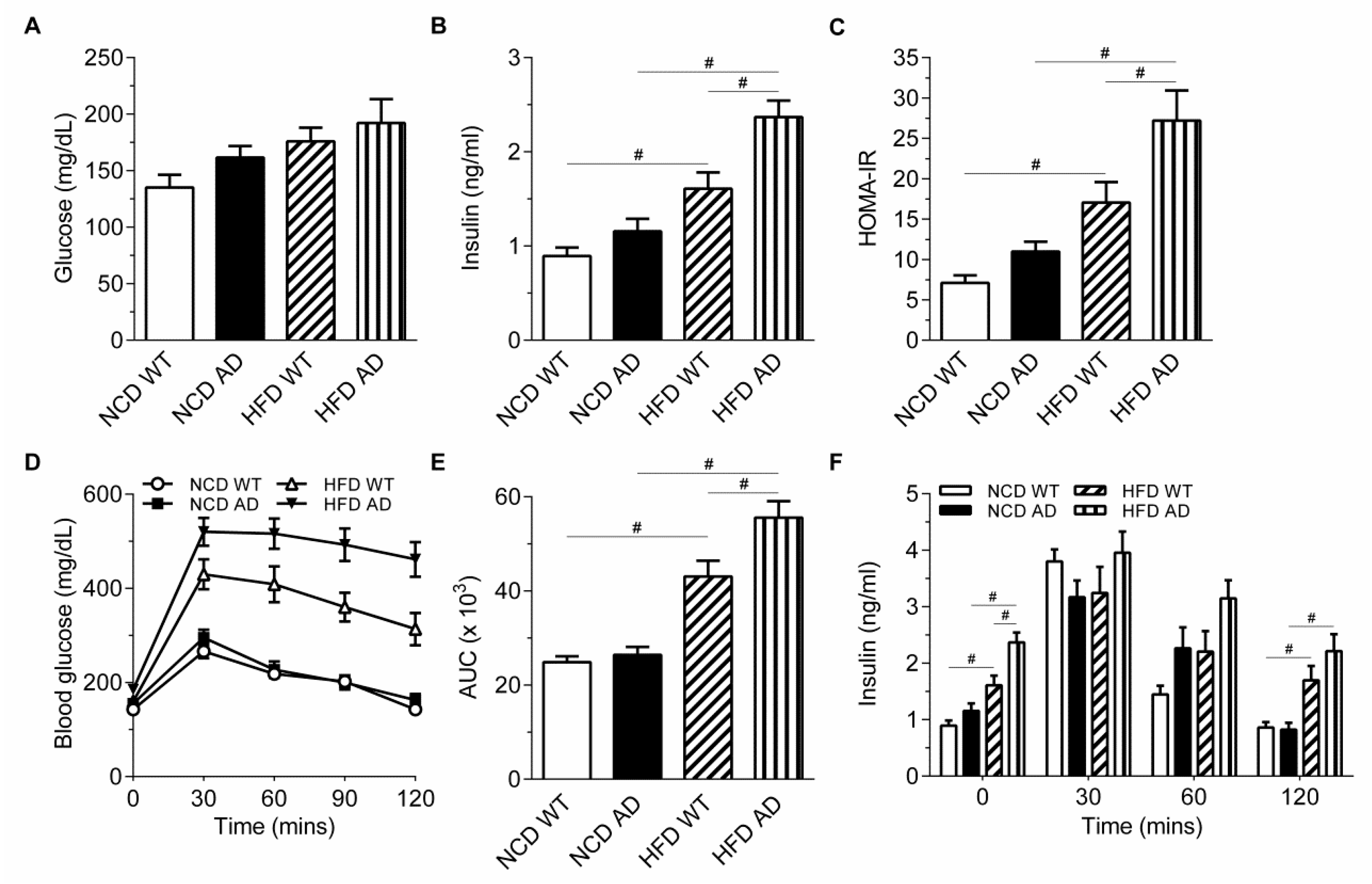
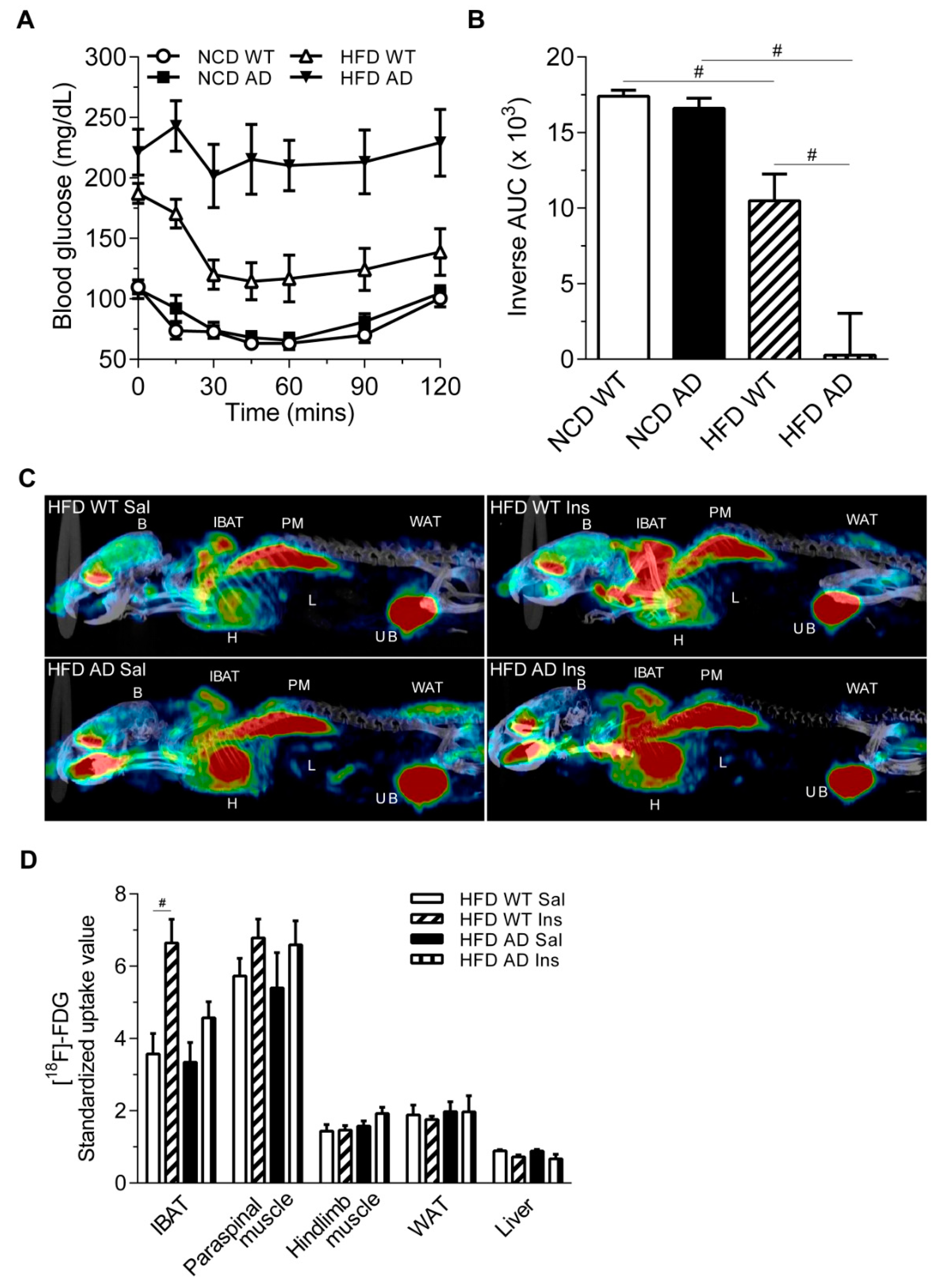
2.1. Alzheimer’s Disease-Related Pathology Interacts with High Fat Diet in Aggravating the Glucose Intolerance and Insulin Resistance of APPswe/PS1dE9 Transgenic Mice
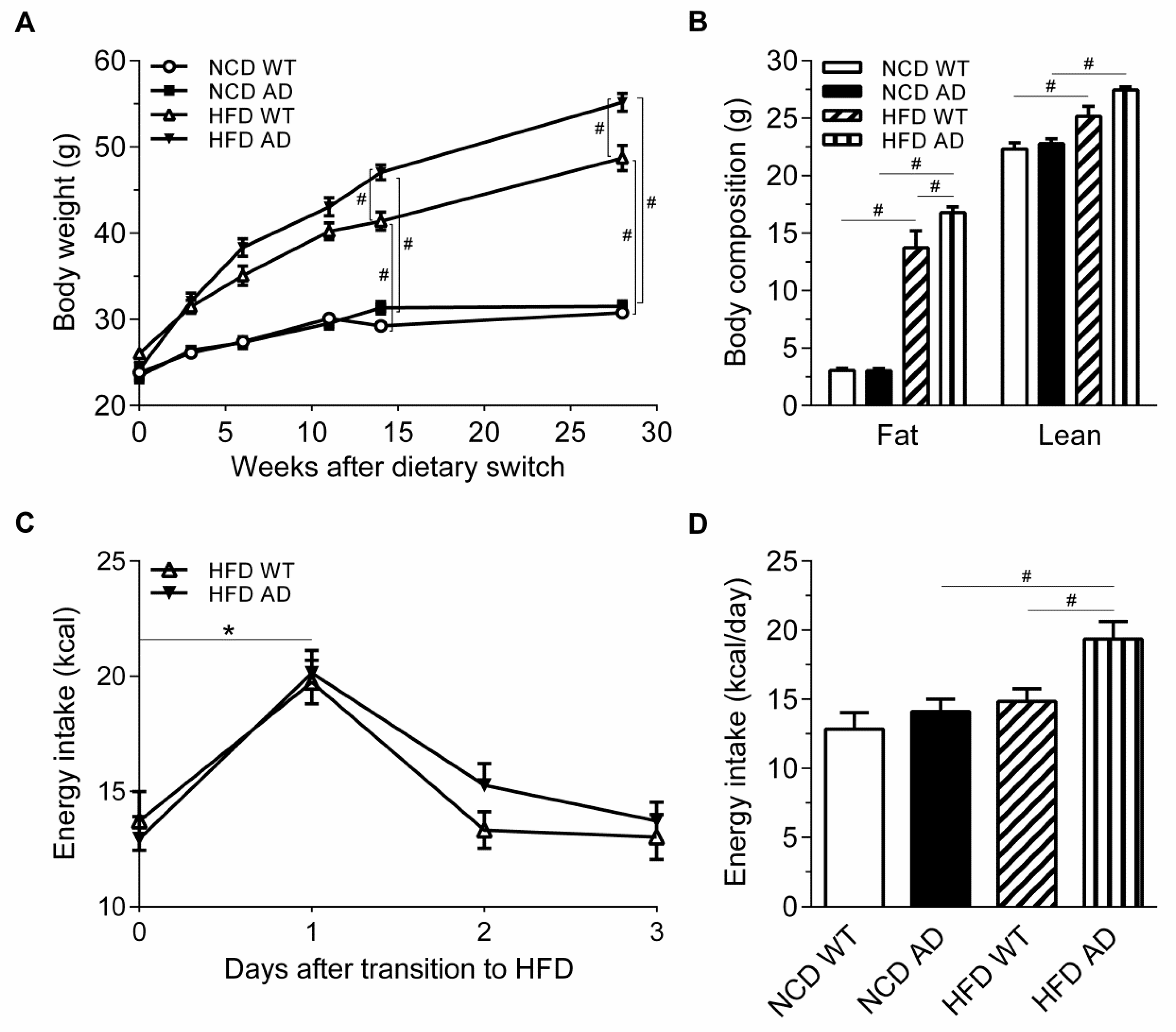
2.2. The Obesity of High Fat Diet APPswe/PS1dE9 Transgenic Mice Is Exacerbated
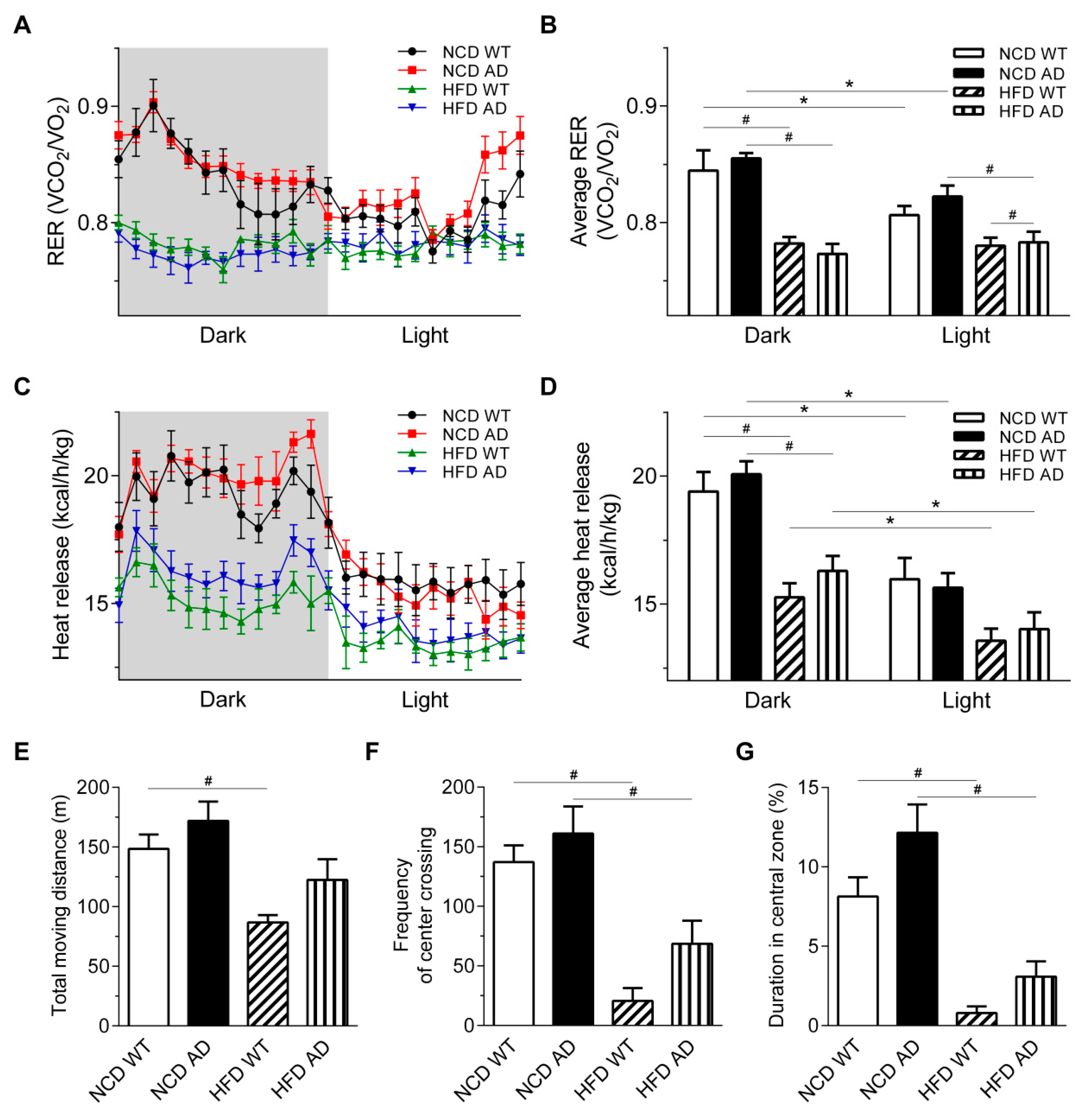
2.3. The Basic Metabolic Rates of High Fat Diet Wild Type and High Fat Diet APPswe/PS1dE9 Transgenic Mice Are Comparable
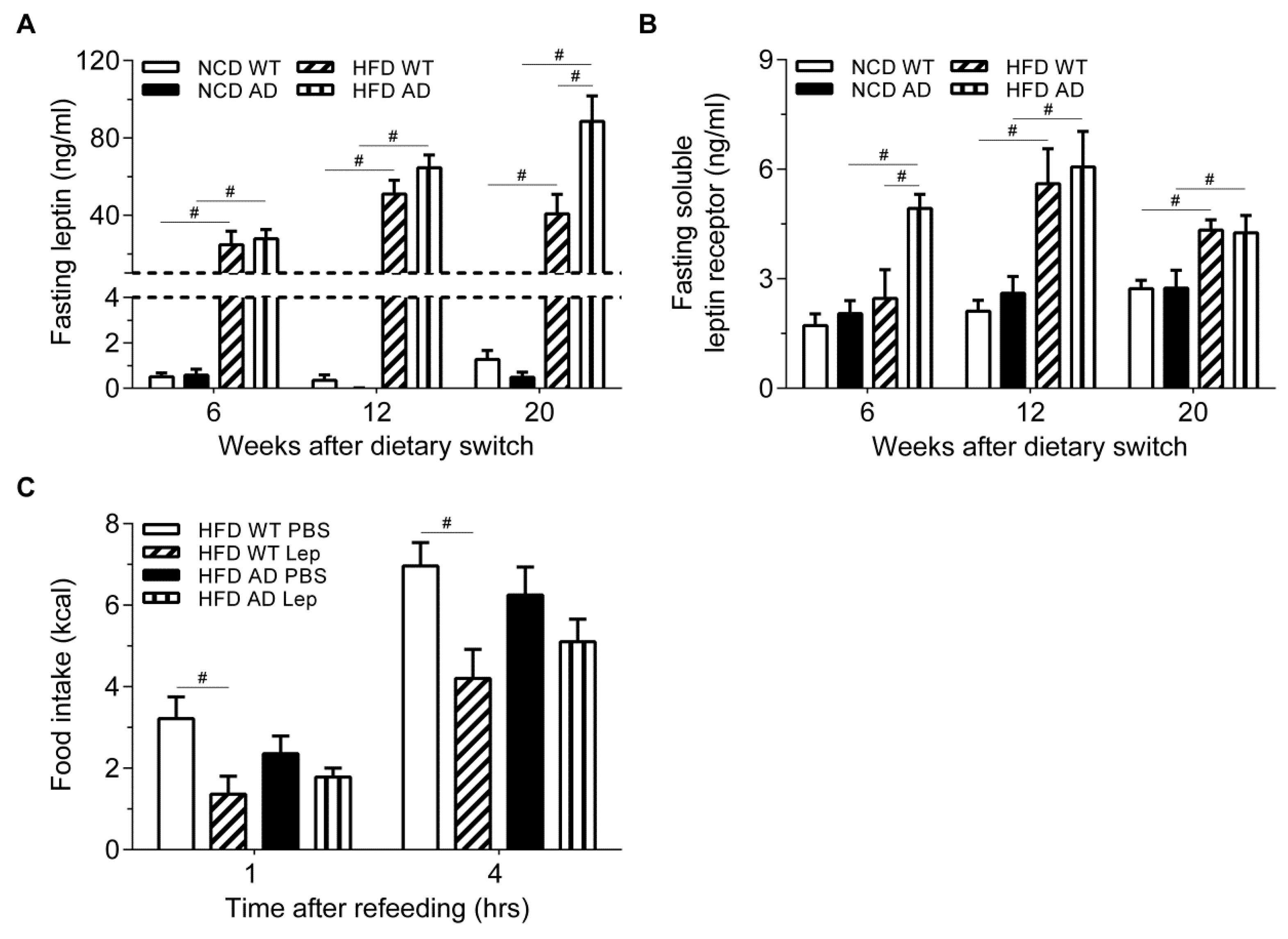
2.4. Leptin-Induced Feeding Suppression in High Fat Diet APPswe/PS1dE9 Transgenic Mice Is Obliterated
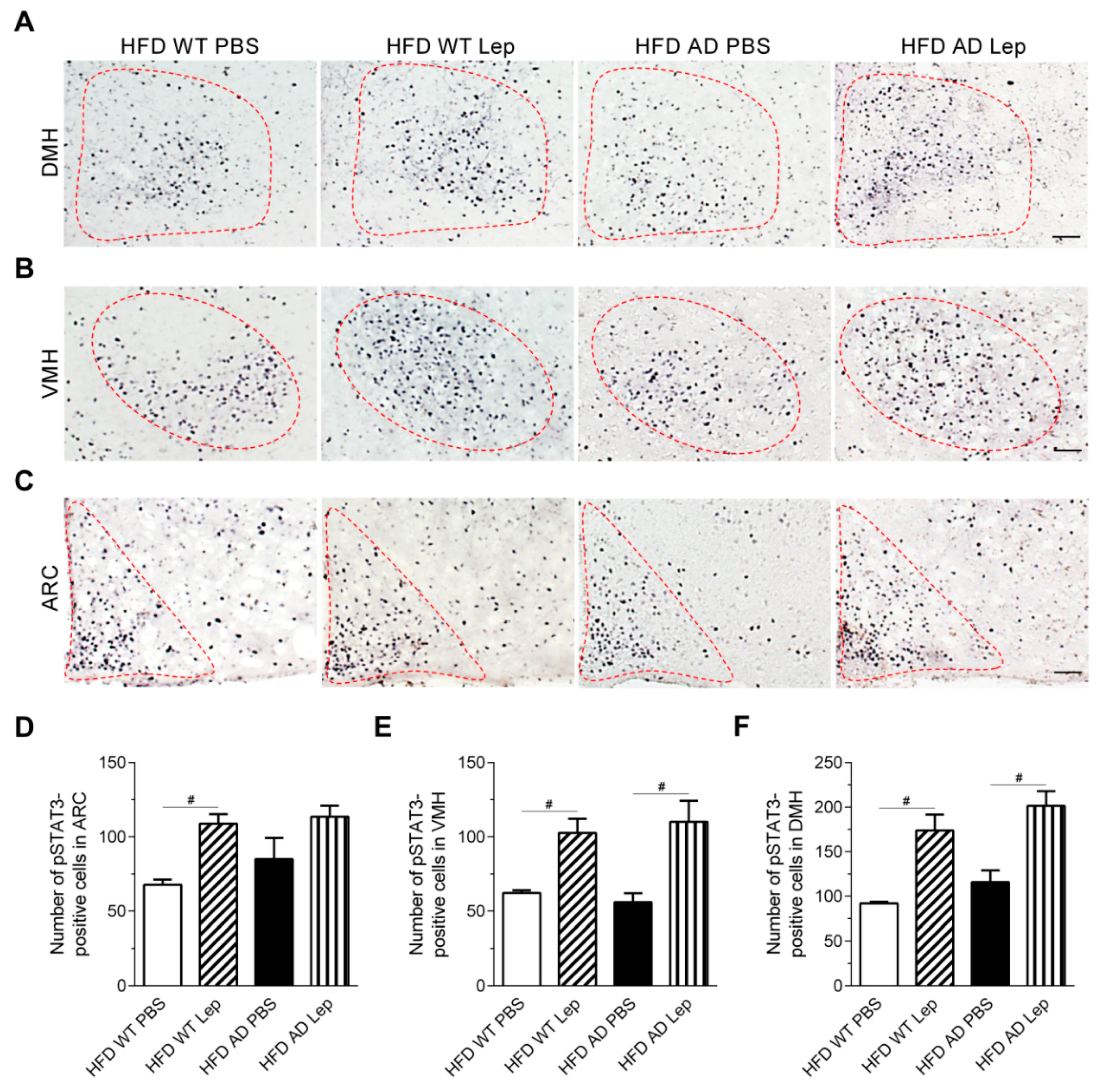
2.5. Leptin Signaling in the Arcuate Nucleus Is Attenuated in High Fat Diet APPswe/PS1dE9 Transgenic Mice
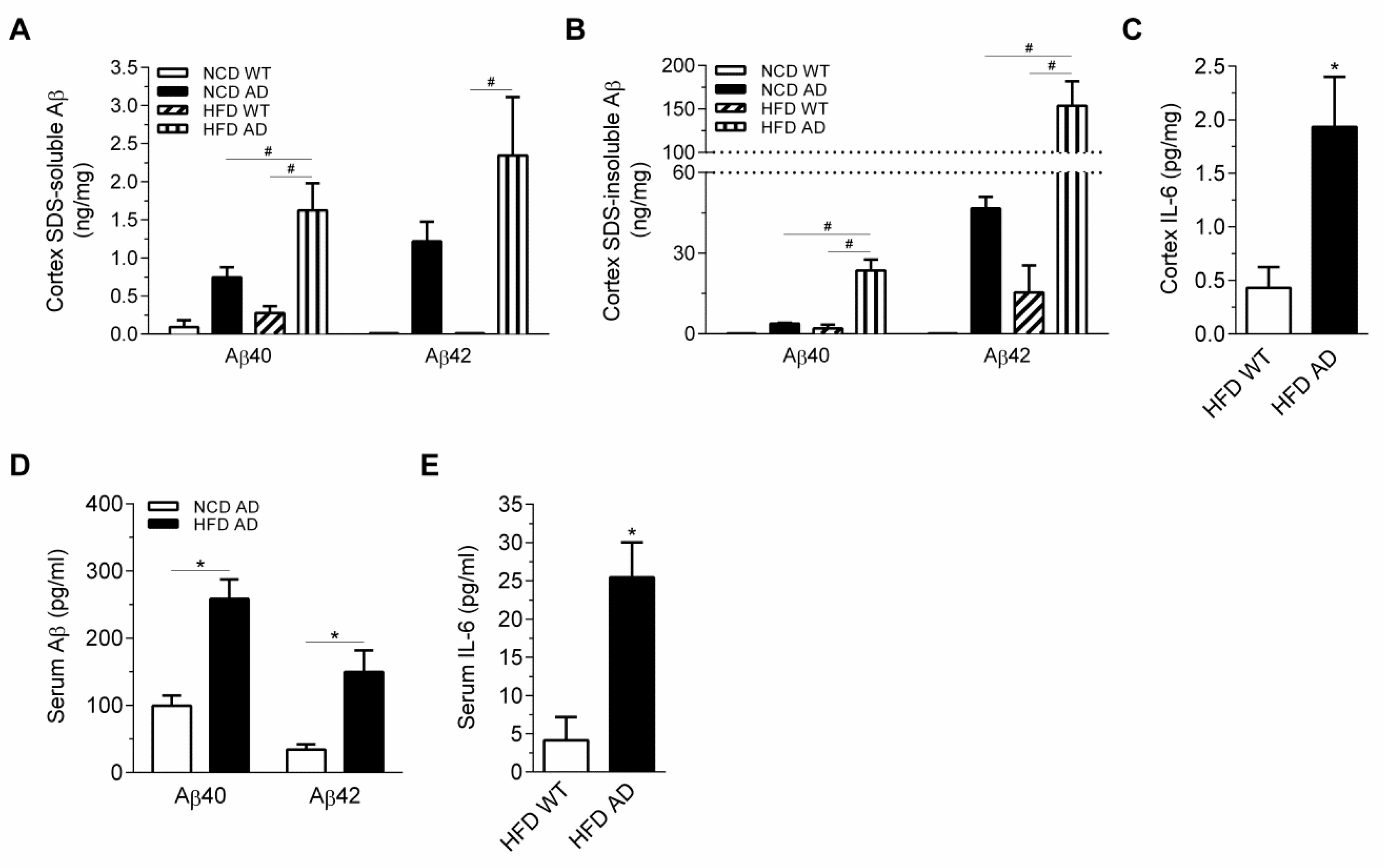
2.6. Elevated Inflammation and Aβ in the Circulation and Cortex, but Not Hypothalamus of High Fat Diet APPswe/PS1dE9 Transgenic Mice
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Glucose Tolerance Test and Insulin Tolerance Test
4.3. In Vivo [18F]-2-fluoro-2-deoxy-d-glucose Positron Emission Tomography Experiment
4.4. Measurement of Biochemical Parameters
4.5. Body Composition and Metabolic Rate Analysis
4.6. Measurement of Feeding Behavior
4.7. Assessment of Locomotor Activity in Open Field
4.8. Immunohistochemistry and Senile Plaque Staining
4.9. Aβ Measurement
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Aβ | β-amyloid |
| AD | Alzheimer’s disease |
| ANOVA | analysis of variance |
| APP | amyloid precursor protein |
| ARC | arcuate nucleus |
| BSB | (trans, trans)-1-bromo-2,5-bis-(3-hydroxycarbonyl-4-hydroxy) styrylbenzene |
| DMH | dorsomedial hypothalamus |
| GFAP | glial fibrillary acidic protein |
| HFD | high-fat diet |
| HOMA-IR | homeostasis model assessment for insulin resistance index |
| HSD | honestly significant difference |
| IBAT | interscapular brown adipose tissue |
| IL-6 | interleukin-6 |
| i.p. | intraperitoneally |
| NCD | normal chow diet |
| RER | respiratory exchange ratio |
| SEM | standard error of mean |
| T2DM | type 2 diabetes mellitus |
| VCO2 | carbon dioxide release |
| VMH | ventromedial hypothalamus |
| VO2 | oxygen consumption |
| WT | wildtype |
References
- Dosunmu, R.; Wu, J.; Basha, M.R.; Zawia, N.H. Environmental and dietary risk factors in Alzheimer’s disease. Expert Rev. Neurother. 2007, 7, 887–900. [Google Scholar] [CrossRef] [PubMed]
- Felice, F.G.D.; Ferreira, S.T. Inflammation, defective insulin signaling, and mitochondrial dysfunction as common molecular denominators connecting type 2 diabetes to Alzheimer disease. Diabetes 2014, 63, 2262–2272. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Rodriguez, J.J.; Ortiz-Barajas, O.; Gamero-Carrasco, C.; de la Rosa, P.R.; Infante-Garcia, C.; Zopeque-Garcia, N.; Lechuga-Sancho, A.M.; Garcia-Alloza, M. Prediabetes-induced vascular alterations exacerbate central pathology in APPswe/PS1dE9 mice. Psychoneuroendocrinology 2014, 48, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Yeh, C.W.; Yeh, S.H.H.; Shie, F.S.; Lai, W.S.; Liu, H.K.; Tzeng, T.T.; Tsay, H.J.; Shiao, Y.J. Impaired cognition and cerebral glucose regulation are associated with astrocyte activation in the parenchyma of metabolically stressed APPswe/PS1dE9 mice. Neurobiol. Aging 2015, 36, 2984–2994. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.C.; Tangney, C.C. Dietary fat composition and dementia risk. Neurobiol. Aging 2014, 35 (Suppl. 2), S59–S64. [Google Scholar] [CrossRef]
- Shie, F.S.; Shiao, Y.J.; Yeh, C.W.; Lin, C.H.; Tzeng, T.T.; Hsu, H.C.; Huang, F.L.; Tsay, H.J.; Liu, H.K. Obesity and hepatic steatosis are associated with elevated serum amyloid beta in metabolically stressed APPswe/PS1dE9 mice. PLoS ONE 2015, 10, e0134531. [Google Scholar] [CrossRef] [PubMed]
- Thériault, P.; ElAli, A.; Rivest, S. High fat diet exacerbates Alzheimer’s disease-related pathology in APPswe/PS1 mice. Oncotarget 2016, 7, 67808–67827. [Google Scholar]
- Park, H.-K.; Ahima, R.S. Physiology of leptin: Energy homeostasis, neuroendocrine function and metabolism. Metab. Clin. Exp. 2015, 64, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Buettner, C.; Pocai, A.; Muse, E.D.; Etgen, A.M.; Myers, M.G.; Rossetti, L. Critical role of STAT3 in leptin’s metabolic actions. Cell Metab. 2006, 4, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Xu, A.W.; Kaelin, C.B.; Takeda, K.; Akira, S.; Schwartz, M.W.; Barsh, G.S. PI3K integrates the action of insulin and leptin on hypothalamic neurons. J. Clin. Investig. 2005, 115, 951–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, D. Neuroinflammation and neurodegeneration in overnutrition-induced diseases. Trends Endocrinol. Metab. 2013, 24, 40–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balland, E.; Dam, J.; Langlet, F.; Caron, E.; Steculorum, S.; Messina, A.; Rasika, S.; Falluel-Morel, A.; Anouar, Y.; Dehouck, B.; et al. Hypothalamic tanycytes are an ERK-gated conduit for leptin into the brain. Cell Metab. 2014, 19, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, M.; Sato, N. Bidirectional interactions between diabetes and Alzheimer’s disease. Neurochem. Int. 2017, 108, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.B.; Tien, A.C.; Boddupalli, G.; Xu, A.W.; Jan, Y.N.; Jan, L.Y. Rapamycin ameliorates age-dependent obesity associated with increased mTOR signaling in hypothalamic POMC neurons. Neuron 2012, 75, 425–436. [Google Scholar] [CrossRef] [PubMed]
- Claret, M.; Smith, M.A.; Knauf, C.; Al-Qassab, H.; Woods, A.; Heslegrave, A.; Piipari, K.; Emmanuel, J.J.; Colom, A.; Valet, P.; et al. Deletion of Lkb1 in pro-opiomelanocortin neurons impairs peripheral glucose homeostasis in mice. Diabetes 2011, 60, 735–745. [Google Scholar] [CrossRef] [PubMed]
- Coupé, B.; Ishii, Y.; Dietrich, M.O.; Komatsu, M.; Horvath, T.L.; Bouret, S.G. Loss of autophagy in pro-opiomelanocortin neurons perturbs axon growth and causes metabolic dysregulation. Cell Metab. 2012, 15, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Loskutova, N.; Honea, R.A.; Brooks, W.M.; Burns, J.M. Reduced limbic and hypothalamic volumes correlate with bone density in early Alzheimer’s disease. J. Alzheimers Dis. 2010, 20, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Ishii, M.; Iadecola, C. Metabolic and non-cognitive manifestations of Alzheimer’s disease: The hypothalamus as both culprit and target of pathology. Cell Metab. 2015, 22, 761–776. [Google Scholar] [CrossRef] [PubMed]
- Adebakin, A.; Bradley, J.; Gumusgoz, S.; Waters, E.J.; Lawrence, C.B. Impaired satiation and increased feeding behaviour in the triple-transgenic Alzheimer’s disease mouse model. PLoS ONE 2012, 7, e45179. [Google Scholar] [CrossRef] [PubMed]
- Kohjima, M.; Sun, Y.; Chan, L. Increased food intake leads to obesity and insulin resistance in the tg2576 Alzheimer’s disease mouse model. Endocrinology 2010, 151, 1532–1540. [Google Scholar] [CrossRef] [PubMed]
- Do, K.; Laing, B.T.; Landry, T.; Bunner, W.; Mersaud, N.; Matsubara, T.; Li, P.; Yuan, Y.; Lu, Q.; Huang, H. The effects of exercise on hypothalamic neurodegeneration of Alzheimer’s disease mouse model. PLoS ONE 2018, 13, e0190205. [Google Scholar] [CrossRef] [PubMed]
- Meakin, P.J.; Jalicy, S.M.; Montagut, G.; Allsop, D.J.P.; Cavellini, D.L.; Irvine, S.W.; McGinley, C.; Liddell, M.K.; McNeilly, A.D.; Parmionova, K.; et al. Bace1-dependent amyloid processing regulates hypothalamic leptin sensitivity in obese mice. Sci. Rep. 2018, 8, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graham, L.C.; Harder, J.M.; Soto, I.; de Vries, W.N.; John, S.W.M.; Howell, G.R. Chronic consumption of a western diet induces robust glial activation in aging mice and in a mouse model of Alzheimer’s disease. Sci. Rep. 2016, 6, 21568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buckman, L.B.; Thompson, M.M.; Lippert, R.N.; Blackwell, T.S.; Yull, F.E.; Ellacott, K.L.J. Evidence for a novel functional role of astrocytes in the acute homeostatic response to high-fat diet intake in mice. Mol. Metab. 2015, 4, 58–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Townsend, K.L.; Tseng, Y.-H. Brown fat fuel utilization and thermogenesis. Trends Endocrinol. Metab. 2014, 25, 168–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, H.; Kastin, A.J.; Hsuchou, H.; Pan, W. Soluble receptor inhibits leptin transport. J. Cell. Physiol. 2007, 214, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Knight, Z.A.; Hannan, K.S.; Greenberg, M.L.; Friedman, J.M. Hyperleptinemia is required for the development of leptin resistance. PLoS ONE 2010, 5, e11376. [Google Scholar] [CrossRef] [PubMed]
- Haim, B.L.; Ceyzeriat, K.; de Sauvage, M.A.C.; Aubry, F.; Auregan, G.; Guillermier, M.; Ruiz, M.; Petit, F.; Houitte, D.; Faivre, E.; et al. The JAK/STAT3 pathway is a common inducer of astrocyte reactivity in Alzheimer’s and Huntington’s diseases. J. Neurosci. 2015, 35, 2817–2829. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.; Fu, A.K.Y.; Ip, F.C.F.; Ng, H.-K.; Hugon, J.; Page, G.; Wang, J.H.; Lai, K.-O.; Wu, Z.; Ip, N.Y. Tyk2/STAT3 signaling mediates b-amyloid-induced neuronal cell death: Implications in Alzheimer’s disease. J. Neurosci. 2010, 30, 6873–6881. [Google Scholar] [CrossRef] [PubMed]
- Marino, J.S.; Xu, Y.; Hill, J.W. Central insulin and leptin-mediated autonomic control of glucose homeostasis. Trends Endocrinol. Metab. 2011, 22, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Clarke, J.R.; Lyra E Silva, N.M.; Figueiredo, C.P.; Frozza, R.L.; Ledo, J.H.; Beckman, D.; Katashima, C.K.; Razolli, D.; Carvalho, B.M.; Frazão, R.; et al. Alzheimer-associated aβ oligomers impact the central nervous system to induce peripheral metabolic deregulation. EMBO Mol. Med. 2015, 7, 190–210. [Google Scholar] [CrossRef] [PubMed]
- Koch, C.E.; Lowe, C.; Pretz, D.; Steger, J.; Williams, L.M.; Tups, A. High-fat diet induces leptin resistance in leptin-deficient mice. J. Neuroendocrinol. 2014, 26, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Hubschle, T.; Thom, E.; Watson, A.; Roth, J.; Klaus, S.; Meyerhof, W. Leptin-induced nuclear translocation of STAT3 immunoreactivity in hypothalamic nuclei involved in body weight regulation. J. Neurosci. 2001, 21, 2413–2424. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, E.; Carmichael, O.; Pasternak, O.; Maier-Hein, K.H.; Decarli, C. Early brain loss in circuits affected by Alzheimer’s disease is predicted by fornix microstructure but may be independent of gray matter. Front. Aging Neurosci. 2014, 6, 106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chondronikola, M.; Volpi, E.; Børsheim, E.; Porter, C.; Annamalai, P.; Enerbäck, S.; Lidell, M.E.; Saraf, M.K.; Labbe, S.M.; Hurren, N.M.; et al. Brown adipose tissue improves whole-body glucose homeostasis and insulin sensitivity in humans. Diabetes 2014, 63, 4089–4099. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Bi, S. Hypothalamic regulation of brown adipose tissue thermogenesis and energy homeostasis. Front. Endocrinol. 2015, 6, 136. [Google Scholar] [CrossRef] [PubMed]
- Roberts-Toler, C.; O’Neill, B.T.; Cypess, A.M. Diet-induced obesity causes insulin resistance in mouse brown adipose tissue. Obesity (Silver Spring) 2015, 23, 1765–1770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrikopoulos, S.; Blair, A.R.; Deluca, N.; Fam, B.C.; Proietto, J. Evaluating the glucose tolerance test in mice. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E1323–E1332. [Google Scholar] [CrossRef] [PubMed]
- Ayala, J.E.; Samuel, V.T.; Morton, G.J.; Obici, S.; Croniger, C.M.; Shulman, G.I.; Wasserman, D.H.; McGuinness, O.P.; NIH Mouse Metabolic Phenotyping Center Consortium. Standard operating procedures for describing and performing metabolic tests of glucose homeostasis in mice. Dis. Model Mech. 2010, 3, 525–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupte, A.A.; Minze, L.J.; Reyes, M.; Ren, Y.; Wang, X.; Brunner, G.; Ghosn, M.; Cordero-Reyes, A.M.; Ding, K.; Pratico, D.; et al. High-fat feeding induced hyperinsulinemia induces cardiac glucose uptake and mitochondria function despite peripheral insulin resistance. Endocrinology 2013, 154, 2650–2662. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.G.; Suyama, S.; Koch, M.; Jin, S.; Argente-Arizon, P.; Argente, J.; Liu, Z.W.; Zimmer, M.R.; Jeong, J.K.; Szigeti-Buck, K.; et al. Leptin signaling in astrocytes regulates hypothalamic neuronal circuits and feeding. Nat. Neurosci. 2014, 17, 908–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loh, K.; Fukushima, A.; Zhang, X.; Galic, S.; Briggs, D.; Enriori, P.J.; Simonds, S.; Wiede, F.; Reichenbach, A.; Hauser, C.; et al. Elevated hypothalamic TCPTP in obesity contributes to cellular leptin resistance. Cell Metab. 2011, 14, 684–699. [Google Scholar] [CrossRef] [PubMed]
- Skovronsky, D.M.; Zhang, B.; Kung, M.P.; Kung, H.F.; Trojanowski, J.Q.; Lee, V.M.Y. In vivo detection of amyloid plaques in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2000, 97, 7609–7614. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolic Index | Interaction Finteraction p Value | Main Effect Fgroup p Value | Simple Main Effect Fgroup p Value | ||||
|---|---|---|---|---|---|---|---|
| Diet | Genotype | NCD | HFD | WT | AD | ||
| Insulin | 2.924 p = 0.100 | 43.499 p < 0.001 | 12.236 p < 0.005 | ||||
| HOMA-IR | 1.726 p = 0.201 | 30.080 p < 0.001 | 8.625 p < 0.01 | ||||
| GTT | 4.287 p < 0.05 | 0.565 p = 0.461 | 6.674 p < 0.05 | 25.876 p < 0.001 | 56.279 p < 0.001 | ||
| ITT | 7.831 p < 0.01 | 1.102 p = 0.309 | 9.742 p < 0.01 | 14.745 p < 0.005 | 33.102 p < 0.001 | ||
| Body weight | 8.403 p < 0.01 | 0.705 p = 0.407 | 13.327 p < 0.005 | 130.958 p < 0.001 | 372.177 p < 0.001 | ||
| Fat mass | 4.624 p < 0.05 | 0.008 p = 0.932 | 3.896 p = 0.084 | 62.659 p < 0.001 | 757.592 p < 0.001 | ||
| Lean mass | 2.677 p = 0.119 | 45.369 p < 0.001 | 6.142 p < 0.05 | ||||
| Daily energy intake | 2.244 p = 0.145 | 11.138 p < 0.005 | 7.124 p < 0.05 | ||||
| Hourly respiratory exchange ratio | |||||||
| Day | 0.585 p = 0.454 | 14.923 p < 0.005 | 1.249 p = 0.278 | ||||
| Night | 0.793 p = 0.385 | 43.524 p < 0.001 | 0.005 p = 0.946 | ||||
| Hourly heat release | |||||||
| Day | 0.350 p = 0.561 | 9.152 p < 0.01 | 0.009 p = 0.927 | ||||
| Night | 0.084 p = 0.775 | 40.638 p < 0.001 | 1.886 p = 0.186 | ||||
| Leptin | 8.584 p < 0.01 | 2.967 p = 0.111 | 8.317 p < 0.05 | 15.082 p < 0.005 | 44.696 p < 0.001 | ||
| Soluble leptin receptor | 0.013 p = 0.909 | 16.322 p < 0.001 | 0.006 p = 0.937 | ||||
| Impact of Insulin and Leptin | Interaction Finteraction p Value | Main Effect Fgroup p Value | |
|---|---|---|---|
| Treatment | Genotype | ||
| Insulin-induced [18F]-FDG uptake | |||
| IBAT | 2.121 p = 0.171 | 11.537 p < 0.01 | 3.291 p = 0.095 |
| Leptin inhibition of energy intake | |||
| 1 h energy intake | 2.374 p = 0.135 | 8.542 p < 0.01 | 0.269 p = 0.609 |
| 4 h energy intake | 1.595 p = 0.218 | 9.388 p < 0.01 | 0.021 p = 0.887 |
| Leptin-induction of pSTAT3 | |||
| ARC | 0.469 p = 0.508 | 14.602 p < 0.005 | 1.433 p = 0.256 |
| VMH | 0.805 p = 0.389 | 39.044 p < 0.001 | 0.009 p = 0.928 |
| DMH | 0.029 p = 0.868 | 48.619 p < 0.001 | 4.599 p = 0.055 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, Y.-H.; Hsu, H.-C.; Kao, P.-C.; Shiao, Y.-J.; Yeh, S.H.-H.; Shie, F.-S.; Hsu, S.-M.; Yeh, C.-W.; Liu, H.-K.; Yang, S.-B.; et al. Augmented Insulin and Leptin Resistance of High Fat Diet-Fed APPswe/PS1dE9 Transgenic Mice Exacerbate Obesity and Glycemic Dysregulation. Int. J. Mol. Sci. 2018, 19, 2333. https://doi.org/10.3390/ijms19082333
Lee Y-H, Hsu H-C, Kao P-C, Shiao Y-J, Yeh SH-H, Shie F-S, Hsu S-M, Yeh C-W, Liu H-K, Yang S-B, et al. Augmented Insulin and Leptin Resistance of High Fat Diet-Fed APPswe/PS1dE9 Transgenic Mice Exacerbate Obesity and Glycemic Dysregulation. International Journal of Molecular Sciences. 2018; 19(8):2333. https://doi.org/10.3390/ijms19082333
Chicago/Turabian StyleLee, Yi-Heng, Hao-Chieh Hsu, Pei-Chen Kao, Young-Ji Shiao, Skye Hsin-Hsien Yeh, Feng-Shiun Shie, Shu-Meng Hsu, Chih-Wen Yeh, Hui-Kang Liu, Shi-Bing Yang, and et al. 2018. "Augmented Insulin and Leptin Resistance of High Fat Diet-Fed APPswe/PS1dE9 Transgenic Mice Exacerbate Obesity and Glycemic Dysregulation" International Journal of Molecular Sciences 19, no. 8: 2333. https://doi.org/10.3390/ijms19082333