Distinct DNA Methylation Profiles in Ovarian Tumors: Opportunities for Novel Biomarkers


 , ,
, ,
Abstract
:
1. Introduction
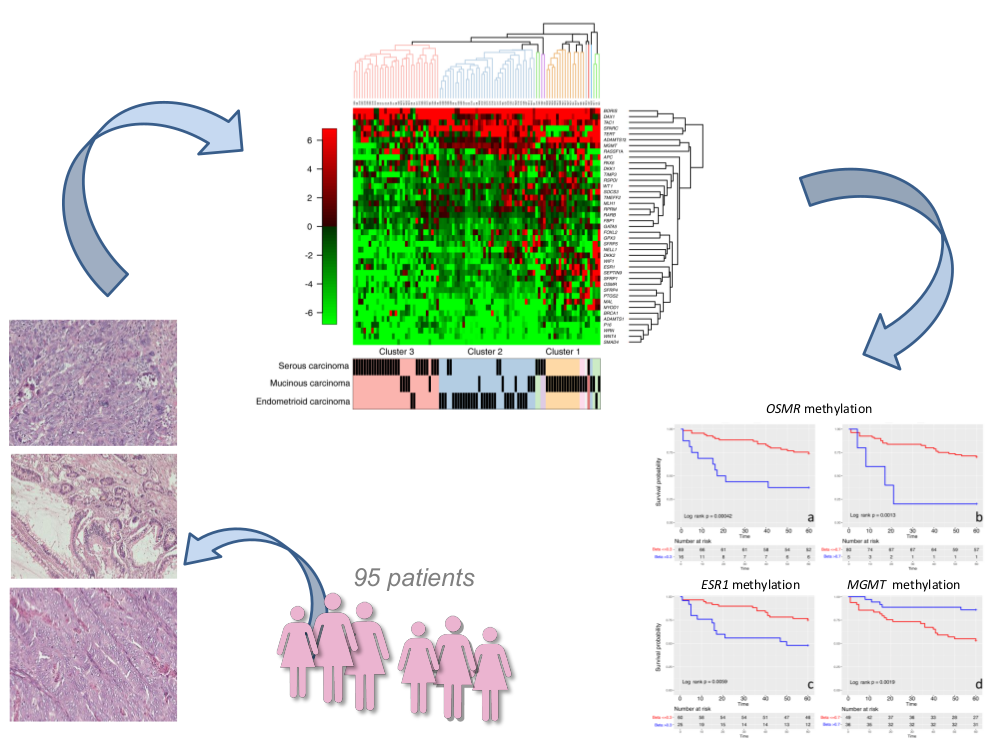
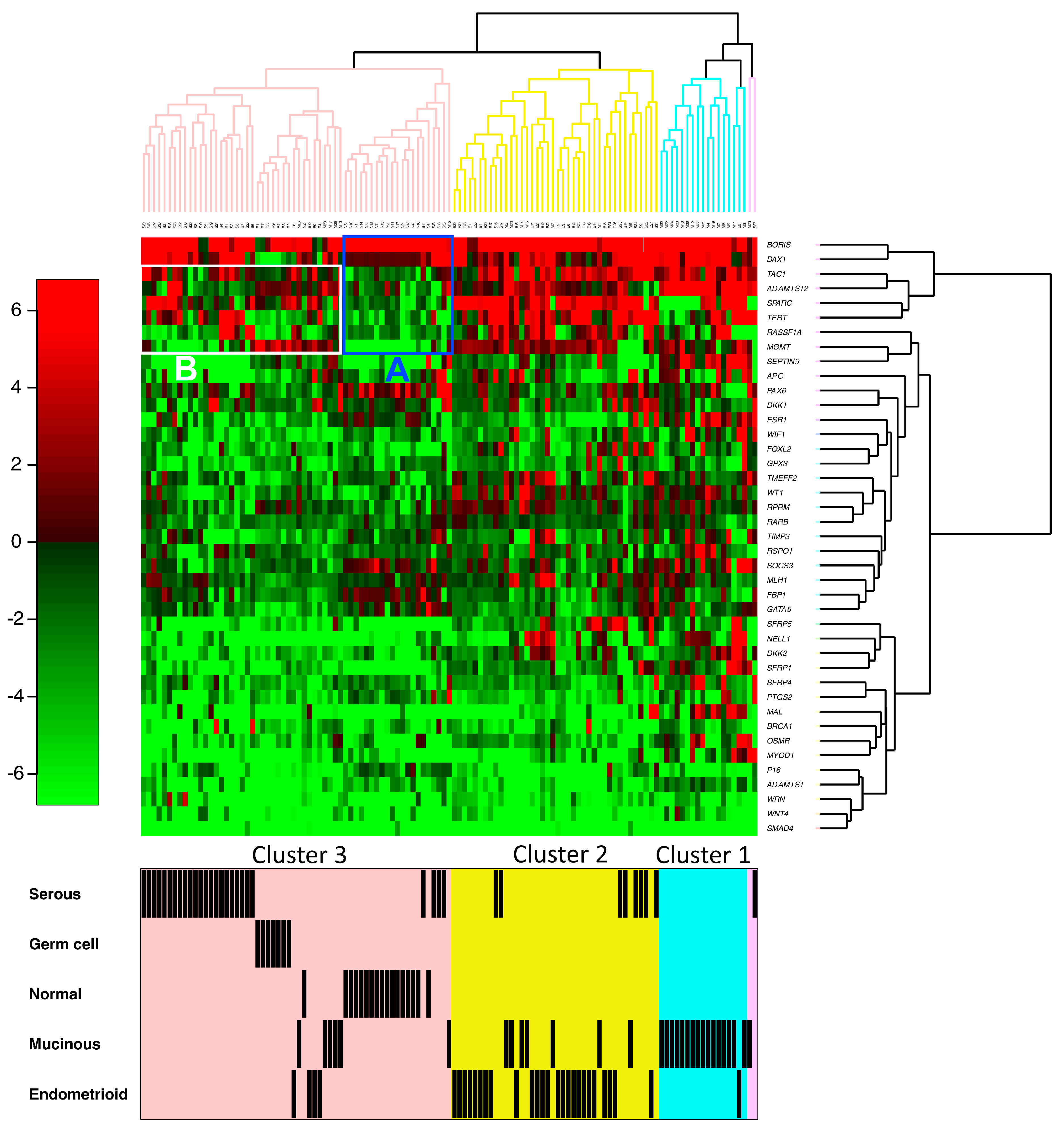
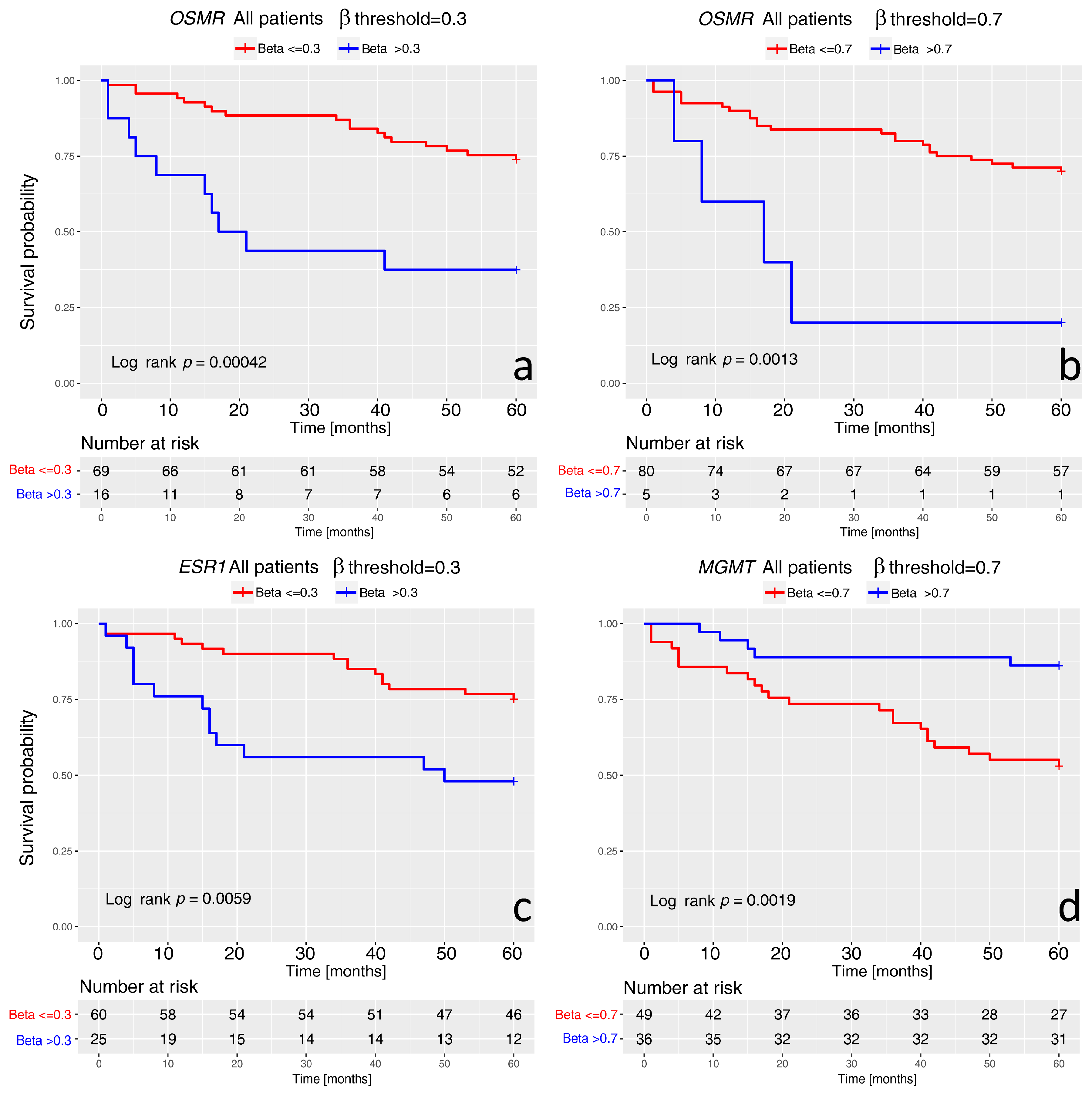
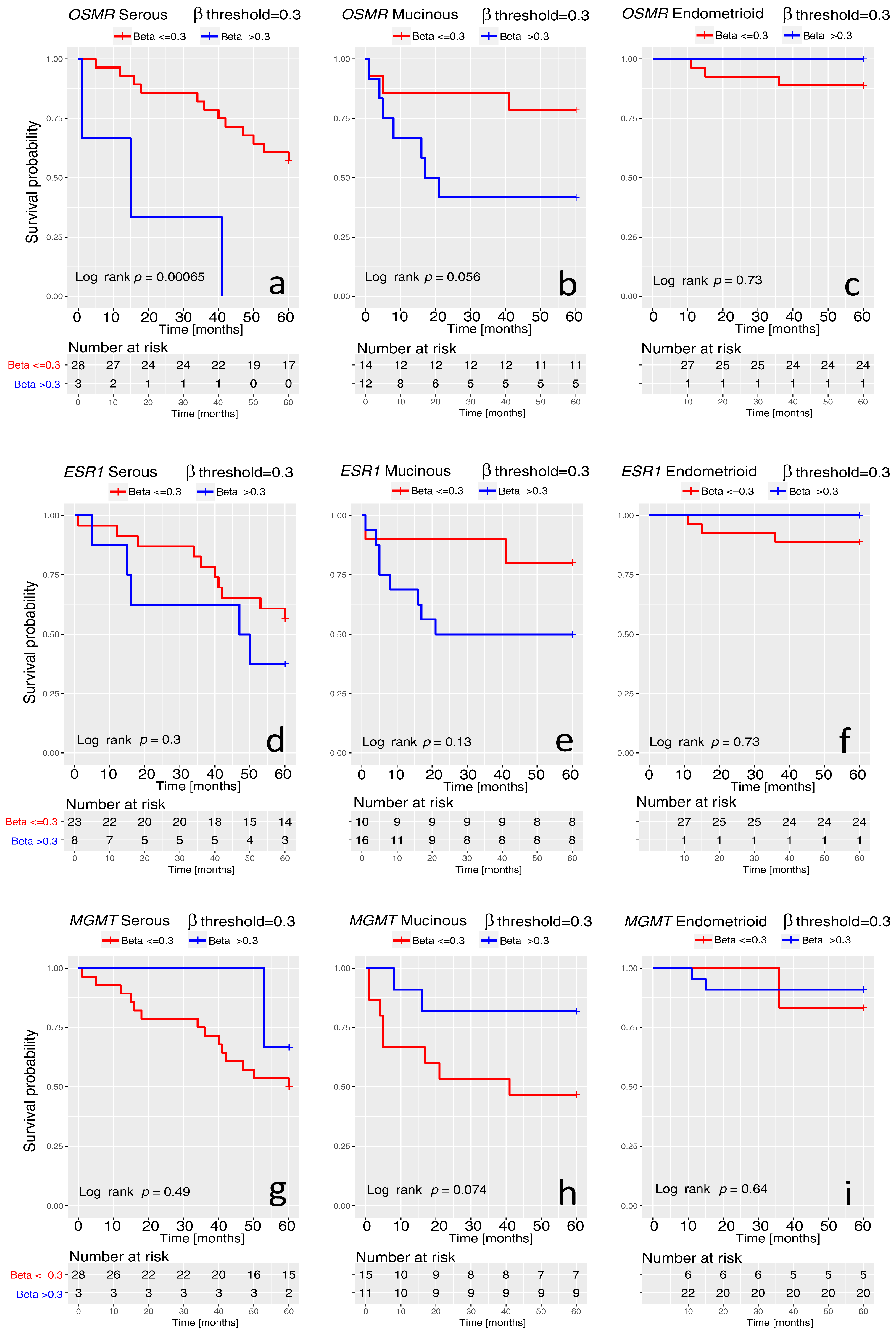
2. Results
3. Discussion
4. Materials and Methods
4.1. Patients and Tissues
4.2. Microdissection and DNA Extraction
4.3. Methylation by Methylation Ligation-Dependent Macroarray (MLM)
4.4. DNA Methylation Unsupervised Hierarchical Clustering and Statistical Analysis
4.5. Genes Investigated
4.6. Survival Analysis
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Dallenbach-Hellweg, G. On the histogenesis and morphology of ovarian carcinomas. J. Cancer Res. Clin. Oncol. 1984, 107, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Auersperg, N. Ovarian surface epithelium as a source of ovarian cancers: Unwarranted speculation or evidence-based hypothesis? Gynecol. Oncol. 2013, 130, 246–251. [Google Scholar] [CrossRef] [PubMed]
- Kurman, R.J.; Shih, I. Molecular pathogenesis and extraovarian origin of epithelial ovarian cancer—Shifting the paradigm. Hum. Pathol. 2011, 42, 918–931. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, R.; Kaye, S.B. Ovarian cancer: Strategies for overcoming resistance to chemotherapy. Nat. Rev. Cancer 2003, 3, 502–516. [Google Scholar] [CrossRef] [PubMed]
- Jemal, A.; Ward, E.; Hao, Y.; Thun, M. Trends in the leading causes of death in the United States. 1970–2002. JAMA 2005, 294, 1255–1259. [Google Scholar] [CrossRef] [PubMed]
- Bas, R.C.; Hennessy, B.; Mills, G.B. The biology of ovarian cancer: New opportunities for translation. Nat. Rev. Cancer 2009, 9, 415–428. [Google Scholar]
- Lorusso, D.; Pietragalla, A.; Mainenti, S.; Di Legge, A.; Amadio, G.; Scambia, G. Emerging drugs for ovarian cancer. Expert Opin. Emerg. Drugs 2010, 15, 635–652. [Google Scholar] [CrossRef] [PubMed]
- Hennessy, B.T.; Coleman, R.L.; Markman, M. Ovarian cancer. Lancet 2009, 374, 1371–1382. [Google Scholar] [CrossRef]
- Kwon, M.J.; Shin, Y.K. Epigenetic regulation of cancer-associated genes in ovarian cancer. Int. J. Mol. Sci. 2011, 12, 983–1008. [Google Scholar] [CrossRef] [PubMed]
- Senturk, E.; Cohen, S.; Dottino, P.R.; Martignetti, J.A. A critical re-appraisal of BRCA1 methylation studies in ovarian cancer. Gynecol. Oncol. 2010, 119, 376–383. [Google Scholar] [CrossRef] [PubMed]
- Montavon, C.; Gloss, B.S.; Warton, K.; Barton, C.A.; Statham, A.L.; Scurry, J.P.; Tabor, B.; Nguyen, T.V.; Qu, W.; Samimi, G.; et al. Prognostic and diagnostic significance of DNA methylation patterns in high grade serous ovarian cancer. Gynecol. Oncol. 2012, 124, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Shih, I.; Chen, L.; Wang, C.C.; Gu, J.; Davidson, B.; Cope, L.; Kurman, R.J.; Xuan, J.; Wang, T.L. Distinct DNA methylation profiles in ovarian serous neoplasms and their implications in ovarian carcinogenesis. Am. J. Obstet. Gynecol. 2010, 203, 584.e1–584.e22. [Google Scholar] [CrossRef] [PubMed]
- Yoon, M.S.; Suh, D.S.; Choi, K.U.; Sol, M.Y.; Shin, D.H.; Park, W.Y.; Lee, J.H.; Jeong, S.M.; Kim, W.G.; Shin, N.R. High-throughput DNA hypermethylation profiling in different ovarian epithelial cancer subtypes using universal bead array. Oncol. Rep. 2010, 24, 917–925. [Google Scholar] [PubMed]
- Guilleret, I.; Losi, L.; Chelbi, S.T.; Fonda, S.; Bougel, S.; Saponaro, S.; Gozzi, G.; Alberti, L.; Braunschweig, R.; Benhattar, J. DNA methylation profiling of esophageal adenocarcinoma using Methylation Ligation-dependent Macroarray (MLM). Biochem. Biophys. Res. Commun. 2016, 479, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Gloss, B.S.; Samini, G. Epigenetic biomarkers in epithelial ovarian cancer. Cancer Lett. 2014, 342, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Huhtinen, K.; Kaipio, K.; Mikkonen, P.; Aittomäki, V.; Lindell, R.; Hynninen, J.; Auranen, A.; Grénman, S.; Lehtonen, R.; et al. Identification of Prognostic Groups in High-Grade Serous Ovarian Cancer Treated with Platinum-Taxane Chemotherapy. Cancer Res. 2015, 75, 2987–2998. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.Y.; Liao, Y.P.; Wang, H.C.; Chen, Y.C.; Huang, R.L.; .Wang, Y.C.; Yuan, C.C.; Lai, H.C. An epigenetic signature of adhesion molecules predicts poor prognosis of ovarian cancer patient. Oncotarget 2017, 8, 53432–53439. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.H.; Chen, C.M.; Strathdee, G.; Harnsomburana, J.; Shyu, C.R.; Rahmatpanah, F.; Shi, H.; Ng, S.W.; Yan, P.S.; Nephew, K.P.; et al. Methylation microarray analysis of late-stage ovarian carcinomas distinguishes progression-free survival in patients and identifies candidate epigenetic markers. Clin. Cancer Res. 2002, 8, 2246–2252. [Google Scholar] [PubMed]
- Prakash, B.; Makarla, P.B.; Saboorian, M.H.; Ashfaq, R.; Toyooka, K.O.; Toyooka, S.; Minna, J.D.; Gazdar, A.F.; Schorge, J.O. Promoter hypermethylation profile of ovarian epithelial neoplasms. Clin. Cancer Res. 2005, 11, 5365–5369. [Google Scholar]
- Houshdaran, S.; Hawley, S.; Palmer, C.; Campan, M.; Olsen, M.N.; Ventura, A.P.; Knudsen, B.S.; Drescher, C.W.; Urban, N.D.; Brown, P.O.; et al. DNA methylation profiles of ovarian epithelial carcinoma tumors and cell lines. PLoS ONE 2010, 5, e9359. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Fridley, B.L.; Song, H.; Lawrenson, K.; Cunningham, J.M.; Ramus, S.J.; Cicek, M.S.; Tyrer, J.; Stram, D.; Larson, M.C.; et al. Epigenetic analysis leads to identification of HNF1B as a subtype-specific susceptibility gene for ovarian cancer. Nat. Commun. 2013, 4, 1628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolbe, D.L.; DeLoia, J.A.; Porter-Gill, P.; Strange, M.; Petrykowska, H.M.; Guirguis, A.; Krivak, T.C.; Brody, L.C.; Elnitski, L. Differential analysis of ovarian and endometrial cancers identifies a methylator phenotype. PLoS ONE 2012, 7, e32941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keita, M.; Wang, Z.Q.; Pelletier, J.F.; Bachvarova, M.; Plante, M.; Gregoire, J.; Renaud, M.C.; Mes-Masson, A.M.; Paquet, É.R.; Bachvarov, D. Global methylation profiling in serous ovarian cancer is indicative for distinct aberrant DNA methylation signatures associated with tumor aggressiveness and disease progression. Gynecol. Oncol. 2013, 128, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Niskakoski, A.; Kaur, S.; Staff, S.; Renkonen-Sinisalo, L.; Lassus, H.; Järvinen, H.J.; Mecklin, J.P.; Bützow, R.; Peltomäki, P. Epigenetic analysis of sporadic and Lynch-associated ovarian cancers reveals histology-specific patterns of DNA methylation. Epigenetics 2014, 9, 1577–1587. [Google Scholar] [CrossRef] [PubMed]
- Earp, M.A.; Cunningham, J.M. DNA methylation changes in epithelial ovarian cancer histotypes. Genomics 2015, 106, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Qiao, B.; Zhang, Z.; Li, Y. Association of MGMT promoter methylation with tumorigenesis features in patients with ovarian cancer: A systematic meta-analysis. Mol. Genet. Genom. Med. 2018, 6, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Gozzi, G.; Chelbi, S.T.; Manni, P.; Alberti, L.; Fonda, S.; Saponaro, S.; Fabbiani, L.; Rivasi, F.; Benhattar, J.; Losi, L. Promoter methylation and downregulated expression of the TBX15 gene in ovarian carcinoma. Oncol. Lett. 2016, 12, 2811–2816. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Xue, X.; Li, W.; Wang, Q.; Han, L.; Brunson, T.; Xu, W.; Chambers-Harris, I.; Wang, Q.; Li, X.; et al. Heterogeneous DNA methylation status in same-cell subpopulations of ovarian cancer tissues. Tumour Biol. 2017, 39, 1010428317701650. [Google Scholar] [CrossRef] [PubMed]
- Alberti, L.; Renaud, S.; Losi, L.; Leyvraz, S.; Benhattar, J. High expression of hTERT and stemness genes in BORIS/CTCFL positive cells isolated from embryonic cancer cells. PLoS ONE 2014, 9, e109921. [Google Scholar] [CrossRef] [PubMed]
- Alberti, L.; Losi, L.; Leyvraz, S.; Benhattar, J. Different Effects of BORIS/CTCFL on Stemness Gene Expression, Sphere Formation and Cell Survival in Epithelial Cancer Stem Cells. PLoS ONE 2015, 10, e0132977. [Google Scholar] [CrossRef] [PubMed]
- Guilleret, I.; Yan, P.; Grange, F.; Braunschweig, R.; Bosman, F.T.; Benhattar, J. Hypermethylation of the human telomerase catalytic subunit (hTERT) gene correlates with telomerase activity. Int. J. Cancer 2012, 101, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Renaud, S.; Loukinov, D.; Bosman, F.T.; Lobanenkov, V.; Benhattar, J. CTCF binds the proximal exonic region of hTERT and inhibits its transcription. Nucleic Acids Res. 2005, 33, 6850–6860. [Google Scholar] [CrossRef] [PubMed]
- Renaud, S.; Loukinov, D.; Alberti, L.; Vostrov, A.; Kwon, Y.W.; Bosman, F.T.; Lobanenkov, V.; Benhattar, J. BORIS/CTCFL-mediated transcriptional regulation of the hTERT telomerase gene in testicular and ovarian tumor cells. Nucleic Acids Res. 2011, 39, 862–873. [Google Scholar] [CrossRef] [PubMed]
- Julsing, J.R.; Peters, G.J. Methylation of DNA repair genes and the efficacy of DNA targeted anticancer treatment. Oncol. Discov. 2014, 2, 3. [Google Scholar] [CrossRef]
- Gao, D.; Herman, J.G.; Guo, M. The clinical value of aberrant epigenetic changes of DNA damage repair genes in human cancer. Oncotarget 2016, 7, 37331–37346. [Google Scholar] [CrossRef] [PubMed]
- Lheureux, S.; Lai, Z.; Dougherty, B.A.; Runswick, S.; Hodgson, D.R.; Timms, K.M.; Lanchbury, J.S.; Kaye, S.; Gourley, C.; Bowtell, D.; et al. Long-Term Responders on Olaparib Maintenance in High-Grade Serous Ovarian Cancer: Clinical and Molecular Characterization. Clin. Cancer Res. 2017, 23, 4086–4094. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.D.; Clamp, A.R.; Evans, D.G.R.; Edmondson, R.J.; Jayson, G.C. PARP inhibitors in platinum-sensitive high-grade serous ovarian cancer. Cancer Chemother. Pharmacol. 2018, 81, 647–658. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Cragun, D.; Thompson, Z.; Coppola, D.; Nicosia, S.V.; Akbari, M.; Zhang, S.; McLaughlin, J.; Narod, S.; Schildkraut, J.; et al. Association between IHC and MSI testing to identify mismatch repair-deficient patients with ovarian cancer. Genet. Test. Mol. Biomark. 2014, 18, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Shilpa, V.; Bhagat, R.; Premalata, C.S.; Pallavi, V.R.; Krishnamoorthy, L. Microsatellite instability, promoter methylation and protein expression of the DNA mismatch repair genes in epithelial ovarian cancer. Genomics 2014, 104, 257–263. [Google Scholar]
- Watanabe, Y.; Ueda, H.; Etoh, T.; Koike, E.; Fujinami, N.; Mitsuhashi, A.; Hoshiai, H. A change in promoter methylation of hMLH1 is a cause of acquired resistance to platinum-based chemotherapy in epithelial ovarian cancer. Anticancer Res. 2007, 27, 1449–1452. [Google Scholar] [PubMed]
- Viale, G.; Trapani, D.; Curigliano, G. Mismatch Repair Deficiency as a Predictive Biomarker for Immunotherapy Efficacy. BioMed Res. Int. 2017, 2017, 4719194. [Google Scholar] [CrossRef] [PubMed]
- Christmann, M.; Verbeek, B.; Roos, W.P.; Kaina, B. O-6-Methylguanine-DNA methyltransferase (MGMT) in normal tissues and tumors: Enzyme activity, promoter methylation and immunohistochemistry. Biochim. Biophys. Acta 2011, 1816, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Roh, H.J.; Suh, D.S.; Choi, K.U.; Yoo, H.J.; Joo, W.D.; Yoon, M.S. Inactivation of O⁶-methyguanine-DNA methyltransferase by promoter hypermethylation: Association of epithelial ovarian carcinogenesis in specific histological types. J. Obstet. Gynaecol. Res. 2011, 37, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Chmelařová, M.; Křepinská, E.; Spaček, J.; Laco, J.; Nekvindová, J.; Palička, V. Methylation analysis of tumour suppressor genes in ovarian cancer using MS-MLPA. Folia Biol. (Praha) 2012, 58, 246–250. [Google Scholar] [PubMed]
- Zhao, Y.H.; Wang, Z.F.; Cao, C.J.; Weng, H.; Xu, C.S.; Li, K.; Li, J.L.; Lan, J.; Zeng, X.T.; Li, Z.Q. The Clinical Significance of O-6-Methylguanine-DNA Methyltransferase Promoter Methylation Status in Adult Patients with Glioblastoma: A Meta-analysis. Front. Neurol. 2018, 9, 127. [Google Scholar] [CrossRef] [PubMed]
- Teodoridis, J.M.; Hall, J.; Marsh, S.; Kannall, H.D.; Smyth, C.; Curto, J.; Siddiqui, N.; Gabra, H.; McLeod, H.L.; Strathdee, G.; et al. CpG island methylation of DNA damage response genes in advanced ovarian cancer. Cancer Res. 2005, 65, 8961–8967. [Google Scholar] [CrossRef] [PubMed]
- Polakis, P. Wnt signaling in cancer. Cold Spring Harb. Perspect. Biol. 2012, 4, a008052. [Google Scholar] [CrossRef] [PubMed]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt signaling in cancer. Oncogene 2017, 36, 1461–1473. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; He, B.; You, L.; Jablons, D.M. Roles of secreted frizzled-related proteins in cancer. Acta Pharmacol. Sin. 2007, 28, 1499–1504. [Google Scholar] [CrossRef] [PubMed]
- Caburet, S.; Georges, A.; L’Hote, D.; Todeschini, A.L.; Benayoun, B.A.; Veitia, R.A. The transcription factor FOXL2: At the crossroads of ovarian physiology and pathology. Mol. Cell. Endocrinol. 2012, 356, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Abir, R.; Ao, A.; Jin, S.; Barnett, M.; Van den Hurk, R.; Freimann, S.; Fisch, B. Immunocytochemical detection and reverse transcription polymerase chain reaction expression of oncostatin M (OSM) and its receptor (OSMRβ) in human fetal and adult ovaries. Fertil. Steril. 2005, 83, 1188–1196. [Google Scholar] [CrossRef] [PubMed]
- Kirn, V.; Shi, R.; Heublein, S.; Knabl, J.; Guenthner-Biller, M.; Andergassen, U.; Fridrich, C.; Malter, W.; Harder, J.; Friese, K.; et al. Estrogen receptor promoter methylation predicts survival in low-grade ovarian carcinoma patients. J. Cancer Res. Clin. Oncol. 2014, 140, 1681–1687. [Google Scholar] [CrossRef] [PubMed]
- Prat, J. FIGO Committee on Gynecologic Oncology. Staging classification for cancer of the ovary, fallopian tube, and peritoneum. Int. J. Gynaecol. Obstet. 2014, 124, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Silverberg, S.G. Histopathologic grading of ovarian carcinoma: A review and proposal. Int. J. Gynecol. Pathol. 2000, 19, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Malpica, A.; Deavers, M.T.; Lu, K.; Bodurka, D.C.; Atkinson, E.N.; Gershenson, D.M.; Silva, E.G. Grading ovarian serous carcinoma using a two-tier system. Am. J. Surg. Pathol. 2004, 28, 496–504. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2015; Available online: http://www.R-project.org/ (accessed on 21 May 2018).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Neoplasia | Number | Mean Age | Grade | Stage |
|---|---|---|---|---|
| Serous carcinoma | 35 | 58 | Low grade: 5 High grade: 30 | I = 11 |
| II = 4 | ||||
| Min = 37 | III = 19 | |||
| Max = 87 | IV = 1 | |||
| Mucinous carcinoma | 30 | 59 | I = 15 | I = 24 |
| Min = 30 | II = 8 | II = 3 | ||
| Max = 85 | III = 7 | III = 3 | ||
| Endometrioid carcinoma | 30 | 60 | I = 14 | I = 22 |
| Min = 36 | II = 9 | II = 5 | ||
| Max = 88 | III = 7 | III =3 | ||
| Germ cell tumor | 7 | 25 | - | I = 6 |
| Min = 12 | ||||
| II = 1 | ||||
| Max = 33 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Losi, L.; Fonda, S.; Saponaro, S.; Chelbi, S.T.; Lancellotti, C.; Gozzi, G.; Alberti, L.; Fabbiani, L.; Botticelli, L.; Benhattar, J. Distinct DNA Methylation Profiles in Ovarian Tumors: Opportunities for Novel Biomarkers. Int. J. Mol. Sci. 2018, 19, 1559. https://doi.org/10.3390/ijms19061559
Losi L, Fonda S, Saponaro S, Chelbi ST, Lancellotti C, Gozzi G, Alberti L, Fabbiani L, Botticelli L, Benhattar J. Distinct DNA Methylation Profiles in Ovarian Tumors: Opportunities for Novel Biomarkers. International Journal of Molecular Sciences. 2018; 19(6):1559. https://doi.org/10.3390/ijms19061559
Chicago/Turabian StyleLosi, Lorena, Sergio Fonda, Sara Saponaro, Sonia T. Chelbi, Cesare Lancellotti, Gaia Gozzi, Loredana Alberti, Luca Fabbiani, Laura Botticelli, and Jean Benhattar. 2018. "Distinct DNA Methylation Profiles in Ovarian Tumors: Opportunities for Novel Biomarkers" International Journal of Molecular Sciences 19, no. 6: 1559. https://doi.org/10.3390/ijms19061559