Biochemical Analysis of Two Single Mutants that Give Rise to a Polymorphic G6PD A-Double Mutant

 , , , , , ,
, , , , , ,
Abstract
:
1. Introduction
2. Results and Discussion
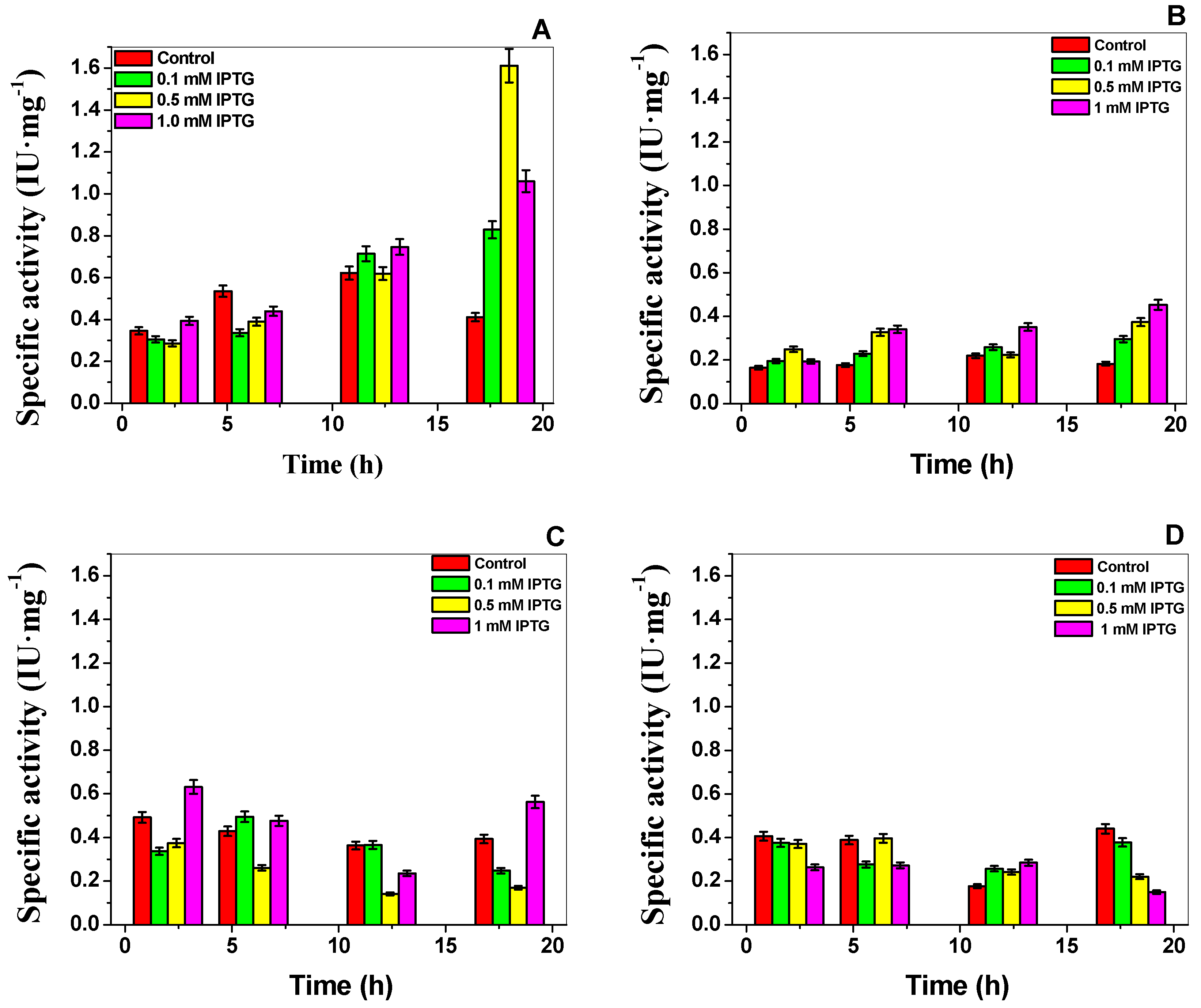
2.1. Construction, Expression, and Purification of Recombinant G6PD Variants
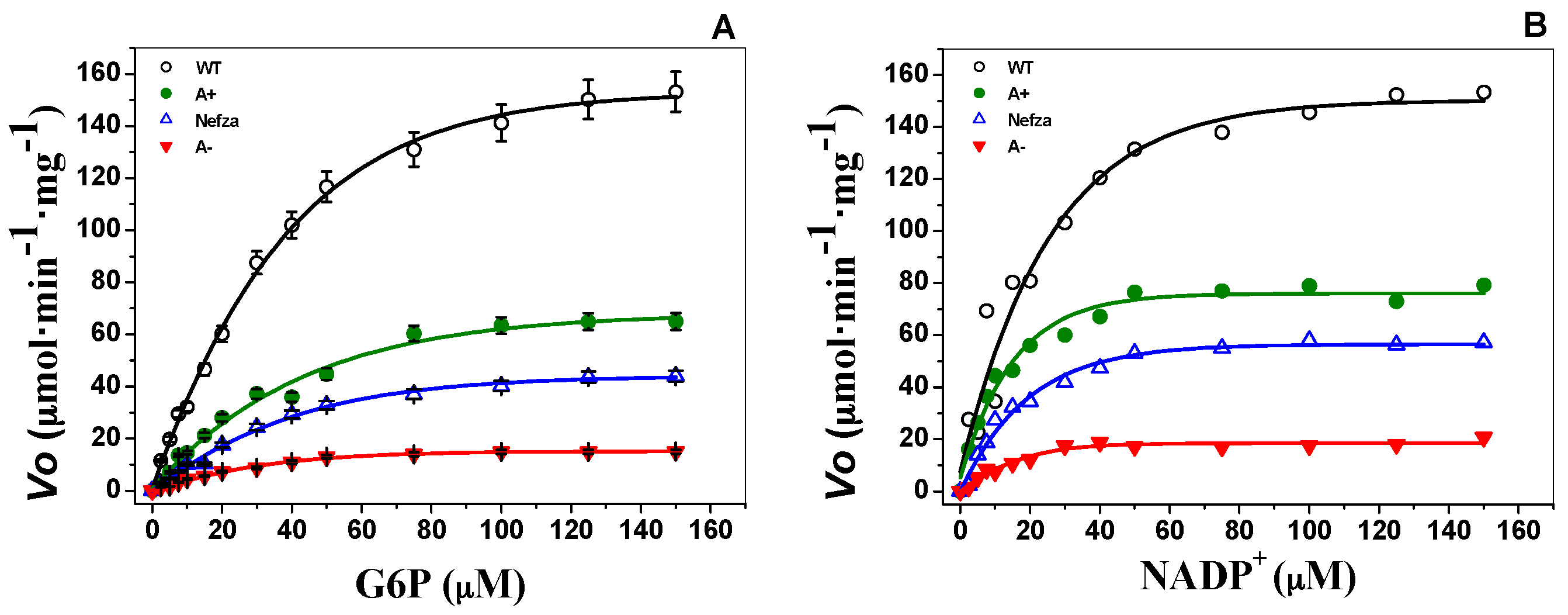
2.2. Measurement of Steady-State Kinetic Parameters
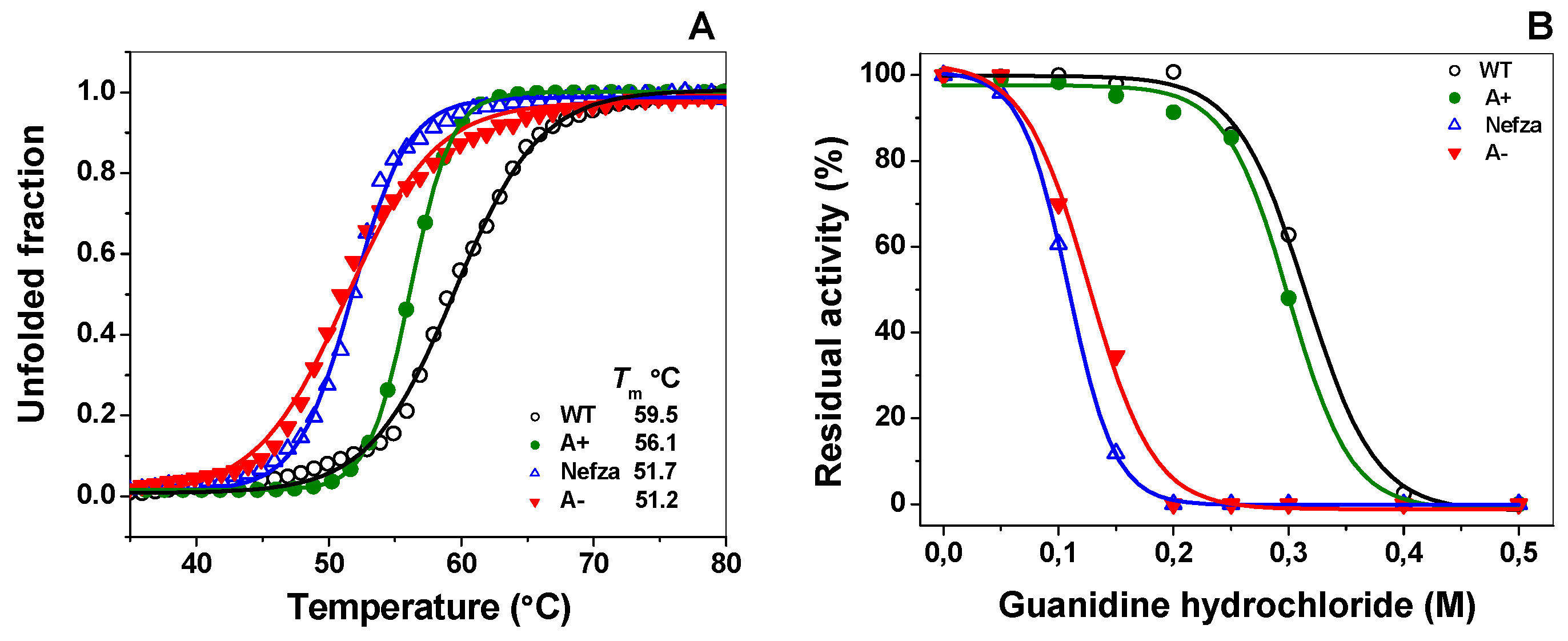
2.3. Evaluation of Protein Stability
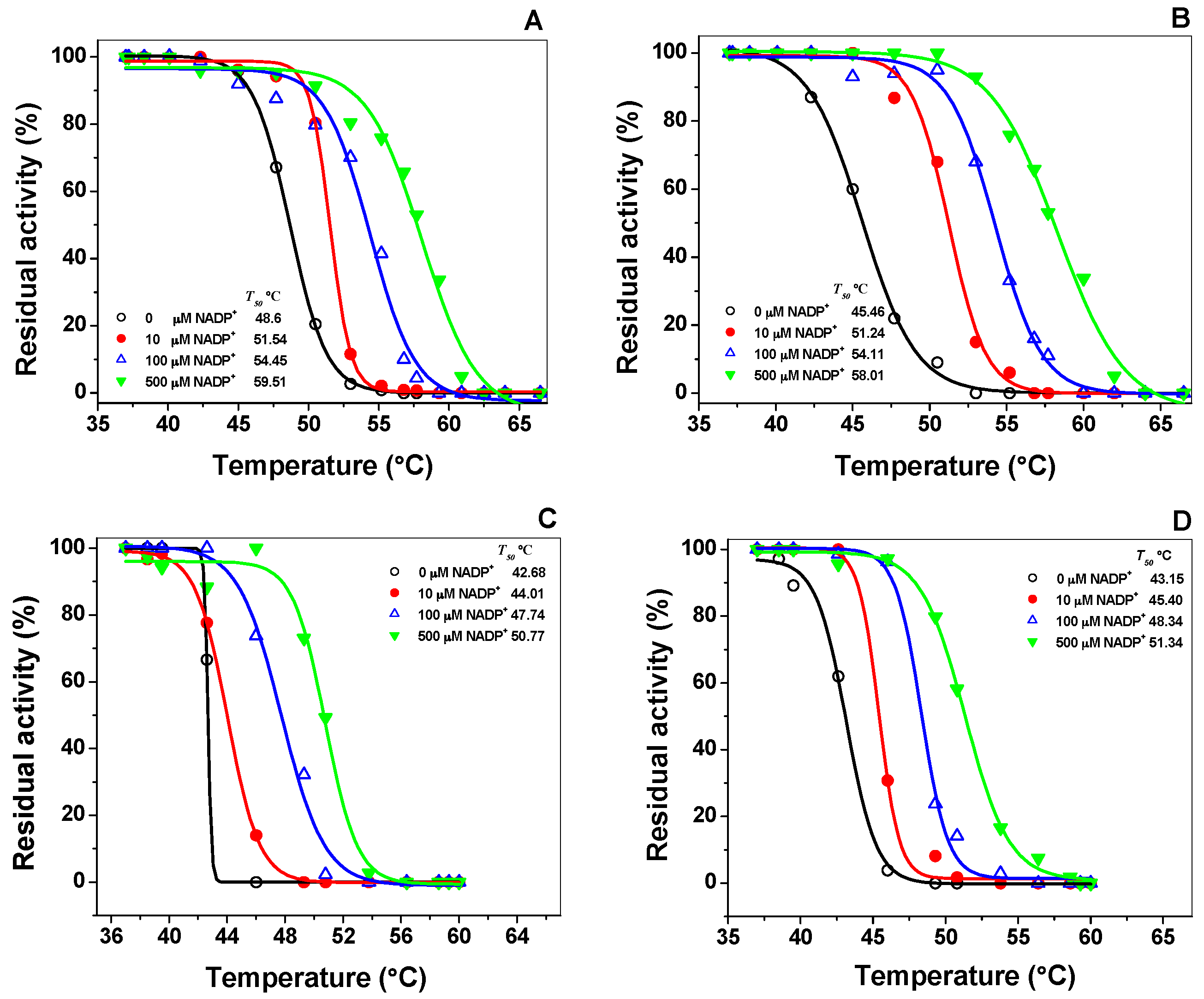
2.3.1. Thermostability Analysis
2.3.2. Thermal Stability of Recombinant Human G6PD Enzymes
2.3.3. Stability of G6PD Variants in the Presence of Guanidine Hydrochloride (Gdn-HCl)
2.4. Spectroscopic Characterization
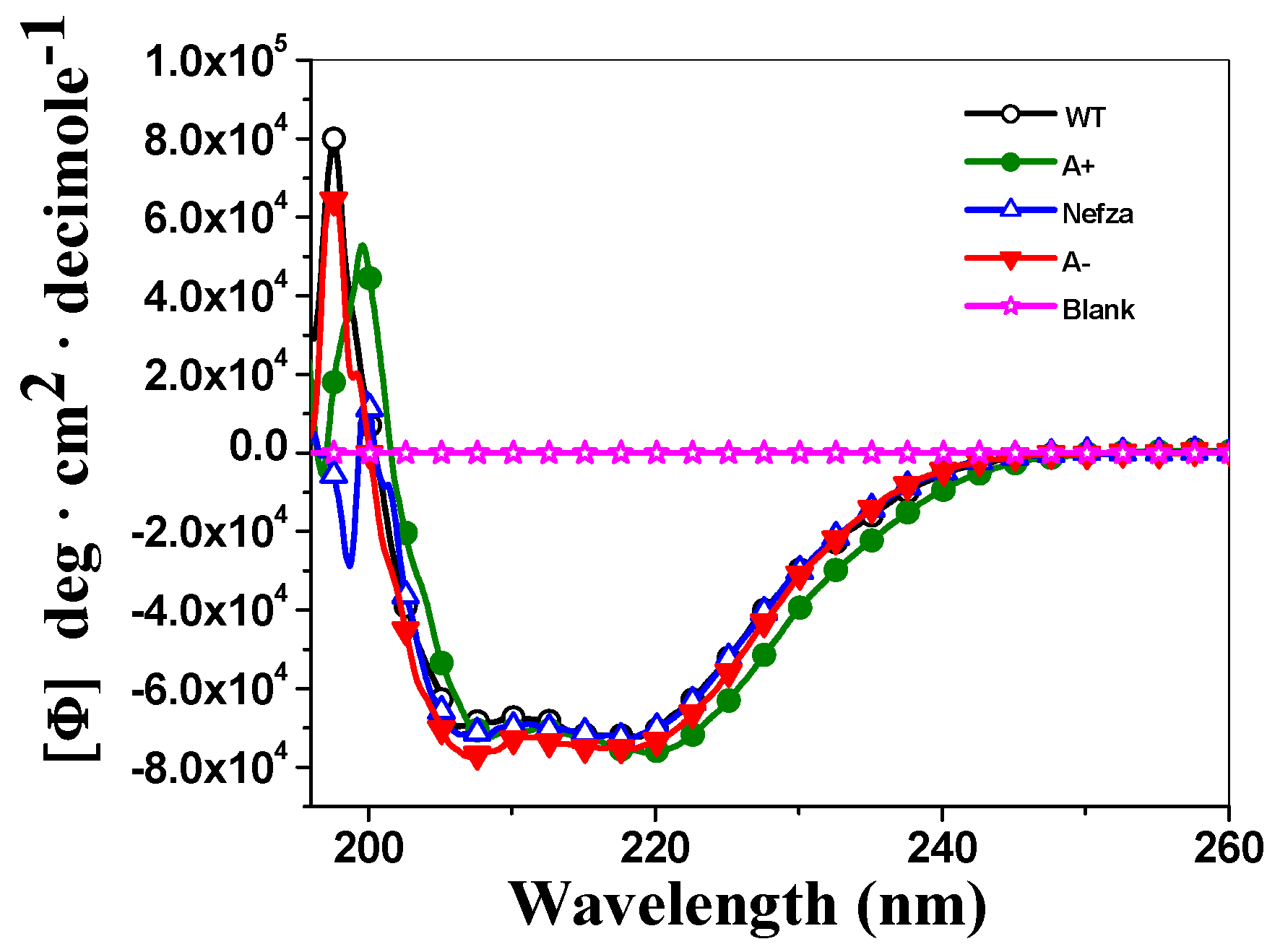
2.4.1. Circular Dichroism (CD) Analysis
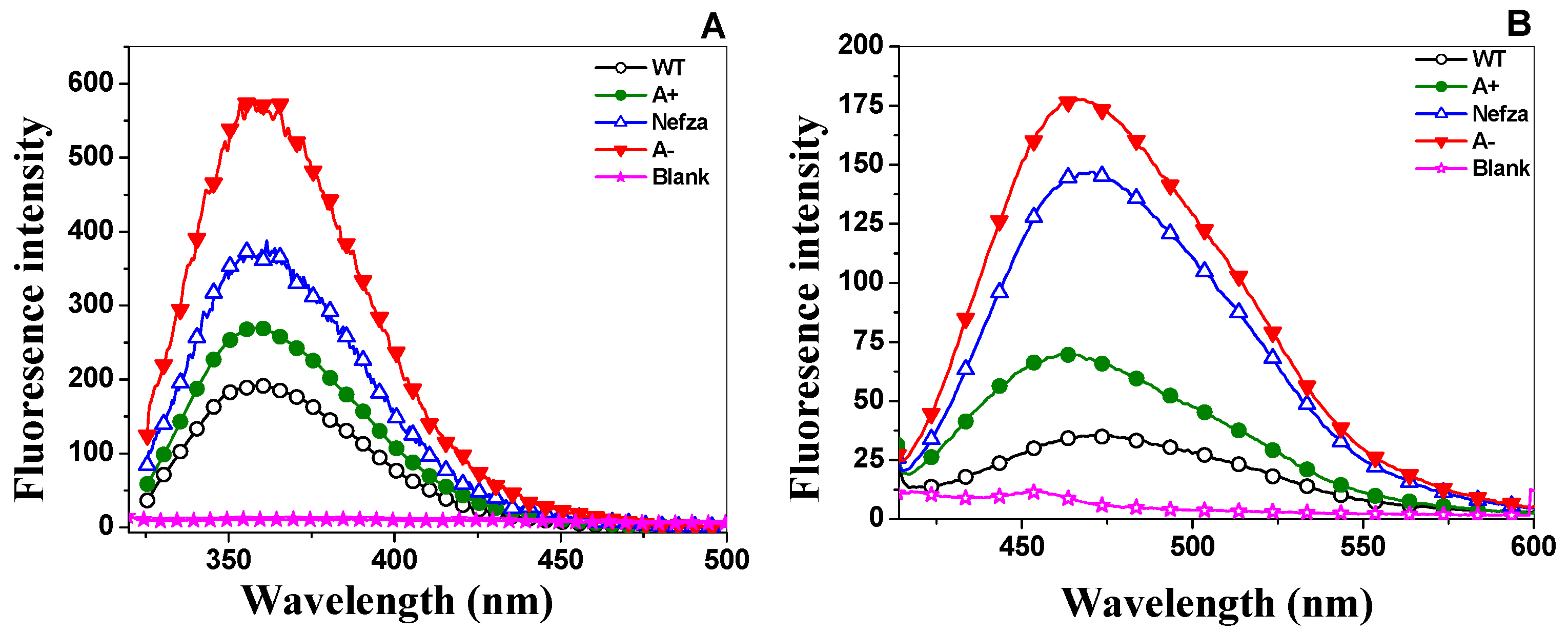
2.4.2. Intrinsic Fluorescence and 8-Anilinonaphthalene-1-Sulfonate (ANS) Binding Analysis
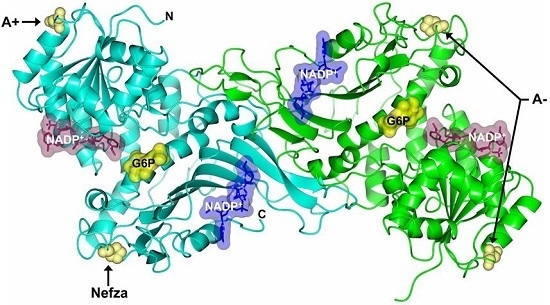
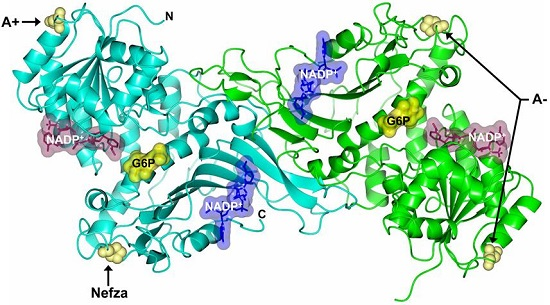
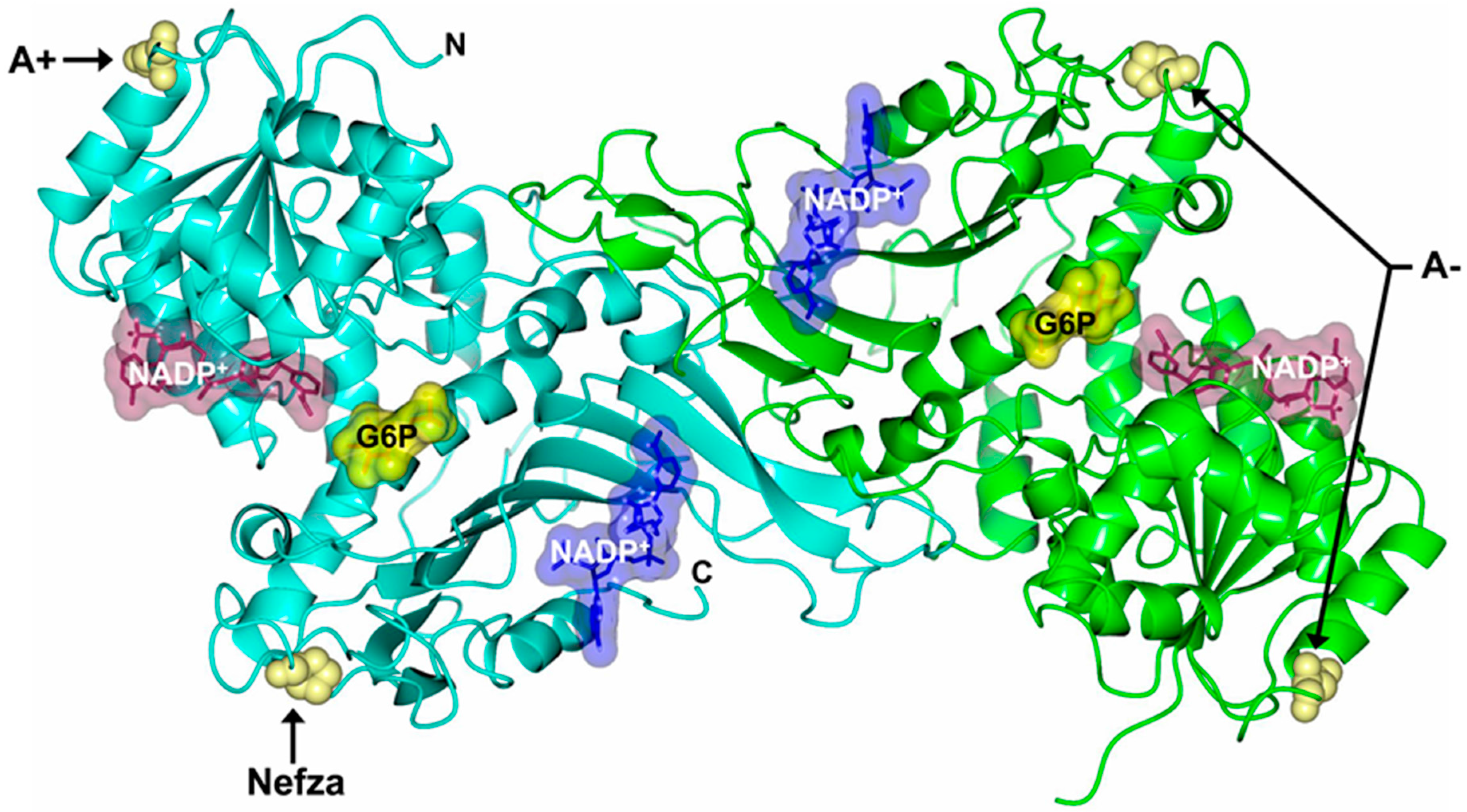
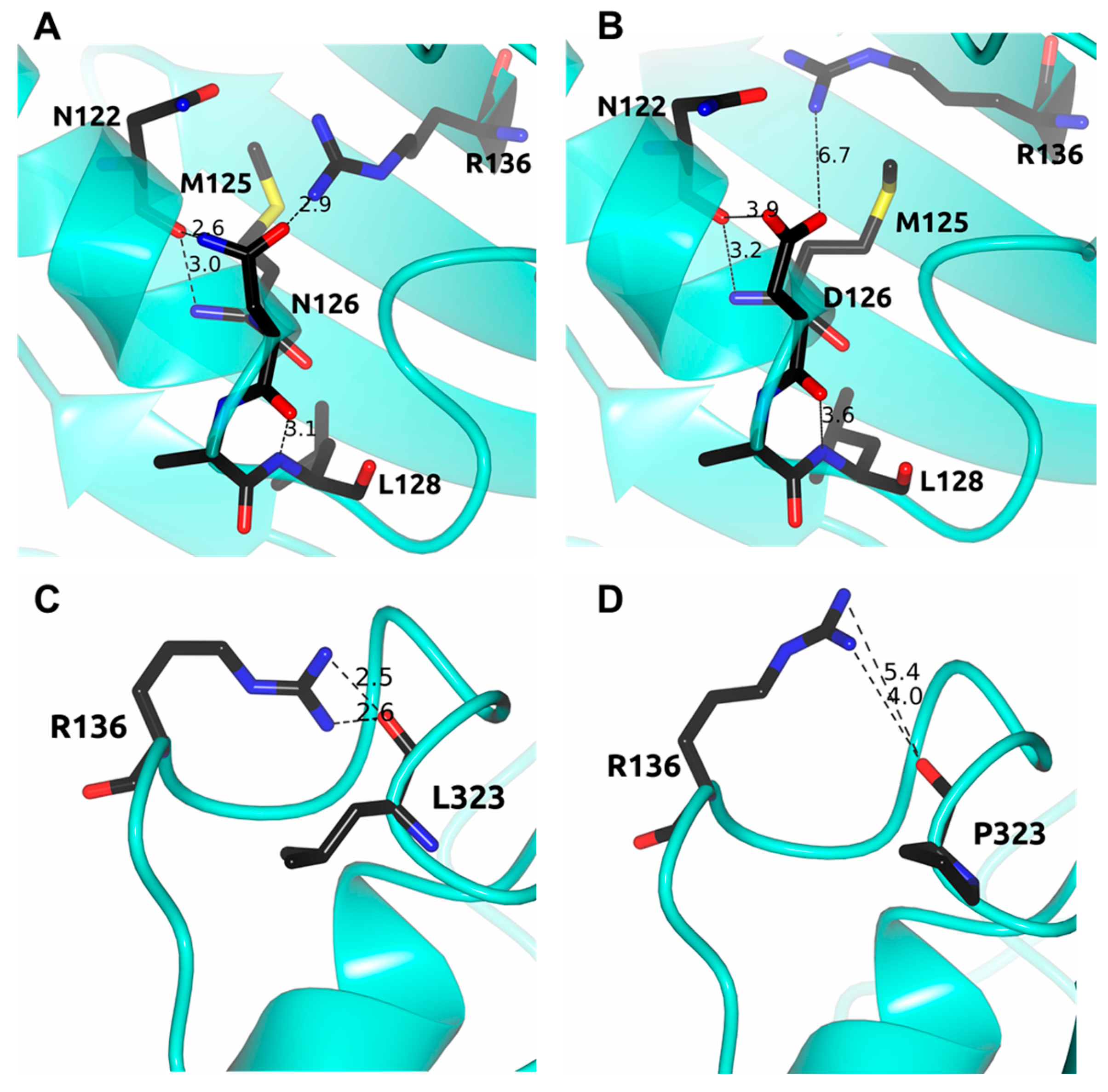
2.5. In Silico Mutagenesis and Computer Modeling
3. Materials and Methods
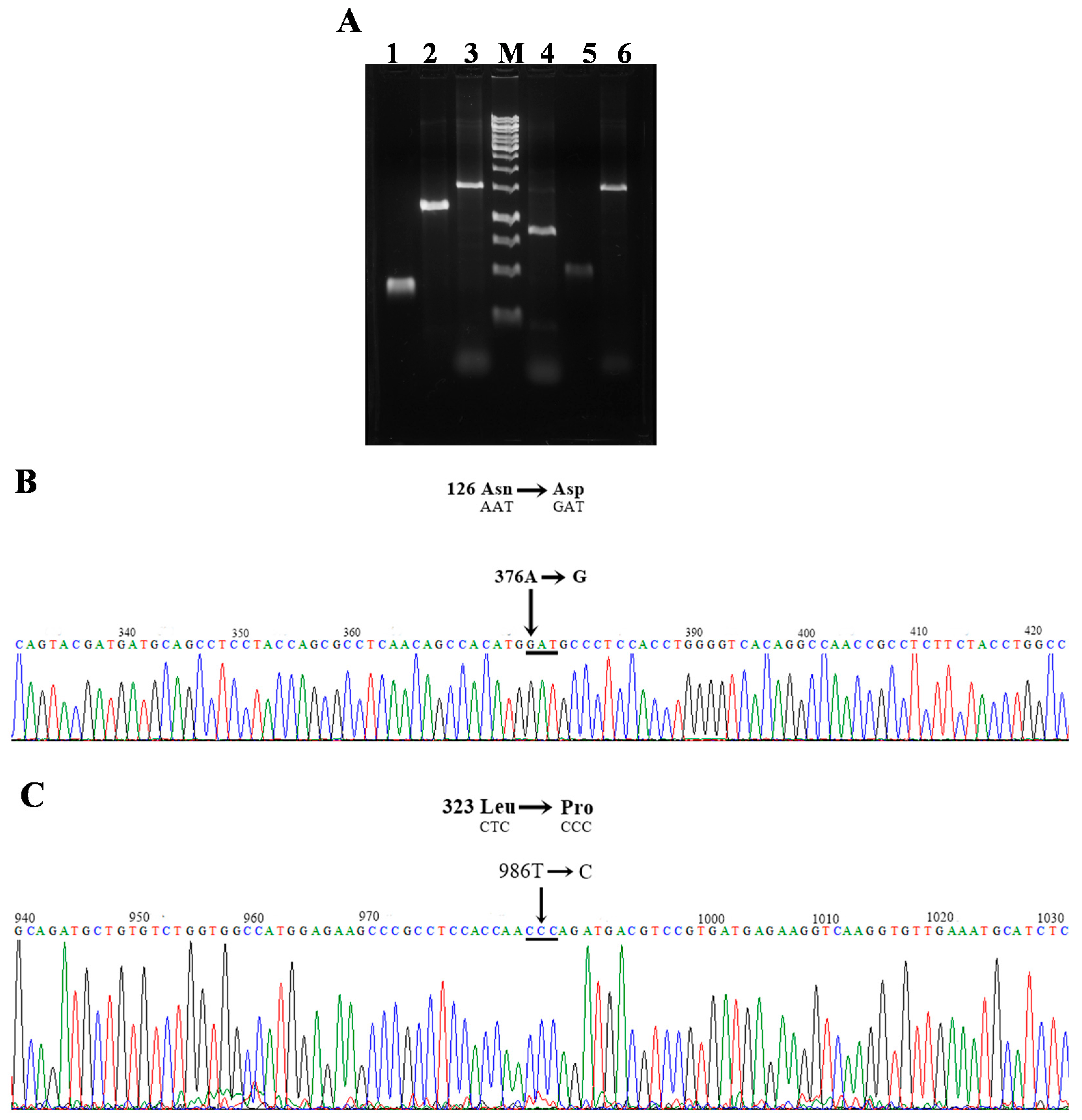
3.1. Construction of Recombinant G6PD by Site-Directed Mutagenesis
3.2. Expression and Purification of Recombinant Human G6PD Enzymes
3.3. Measurement of Steady-State Kinetic Parameters
3.4. Evaluation of Protein Stability
3.4.1. Thermal Inactivation Analysis
3.4.2. Thermal Stability of Recombinant Human G6PD Enzymes
3.4.3. Stability of G6PD Variants in the Presence of Guanidine Hydrochloride (Gdn-HCl)
3.5. Spectroscopic Characterization
3.5.1. Circular Dichroism (CD) Analysis
3.5.2. Intrinsic Fluorescence and 8-Anilinonaphthalene-1-Sulfonate (ANS) Binding Analysis
3.6. In Silico Mutagenesis and Modeling
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Luzzato, L.; Metha, A.; Vulliamy, T. Glucose-6-phosphate dehydrogenase deficiency. In The Metabolic and Molecular Basis of Inherited Disease, 8th ed.; Scriver, C.R., Beaudet, A.L., Sly, W.S., Valle, D., Eds.; McGraw-Hill: Columbus, OH, USA, 2001; pp. 4517–4553. [Google Scholar]
- Gaetani, G.F.; Galiano, S.; Canepa, L.; Ferraris, A.M.; Kirkman, H.N. Catalase and glutathione peroxidase are equally active in detoxification of hydrogen peroxide in human erythrocytes. Blood 1989, 73, 334–339. [Google Scholar] [PubMed]
- Pai, G.S.; Sprenkle, J.A.; Do, T.T.; Mareni, C.E.; Migeon, B.R. Localization of loci for hypoxanthine phosphoribosyltransferase and glucose-6-phosphate dehydrogenase and biochemical evidence of nonrandom X chromosome expression from studies of a human X-autosome translocation. Proc. Natl. Acad. Sci. USA 1980, 77, 2810–2813. [Google Scholar] [CrossRef] [PubMed]
- Cappellini, D.; Fiorelli, G. Glucose-6-phosphate dehydrogenase deficiency. Lancet 2008, 606, 64–74. [Google Scholar] [CrossRef]
- Mason, P.J.; Bautista, J.M.; Gilsanz, F. G6PD deficiency: The genotype-phenotype association. Blood Rev. 2007, 5, 267–283. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Manzo, S.; Marcial-Quino, J.; Vanoye-Carlo, A.; Serrano-Posada, H.; Ortega-Cuellar, D.; González-Valdez, A.; Castillo-Rodríguez, R.A.; Hernández-Ochoa, B.; Sierra-Palacios, E.; Rodríguez-Bustamante, E.; et al. Glucose-6-Phosphate Dehydrogenase: Update and Analysis of New Mutations around the World. Int. J. Mol. Sci. 2016, 17, 2069. [Google Scholar] [CrossRef] [PubMed]
- Minucci, A.; Moradkhani, K.; Hwang, M.; Zuppi, C.; Giardina, B.; Capoluongo, P. Glucose-6-phosphate dehydrogenase (G6PD) mutations database: Review of the “old” and update of the new mutations. Blood Cells Mol. Dis. 2012, 48, 154–165. [Google Scholar] [CrossRef] [PubMed]
- Vaca, G.; Arambula, E.; Esparza, A. Molecular heterogeneity of glucose-6-phosphate dehydrogenase deficiency in Mexico: Overall results of a 7-year project. Blood Cells Mol. Dis. 2002, 28, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Takizawa, T.; Yoneyama, Y.; Miwa, S.; Yoshida, A. A single nucleotide base transition is the basis of the common human glucose-6-phosphate dehydrogenase variant (A+). Genomics 1987, 1, 228–231. [Google Scholar] [CrossRef]
- Beutler, E.; Kuhl, W.; Vives-Corrons, J.; Prchal, J. Molecular heterogeneity of glucose-6-phosphate dehydrogenase A−. Blood 1989, 74, 2550–2555. [Google Scholar] [PubMed]
- Luzzatto, L. Human erythrocyte glucose-6-phosphate dehydrogenase deficiency. In Metabolic and Molecular Bases of Inherited Disease, 6th ed.; Scriver, C.R., Baudet, A.L., Sly, W.S., Valle, D., Eds.; McGraw-Hill: New York, NY, USA, 1989; pp. 2237–2265. [Google Scholar]
- Adil, M.; Malahat, B.; Serhat, E.; Rabia, C.; Sezen, C.; Serap, Y.; Serkan, Y.; Emrah, S.; Nehir, O. Glucose-6-Phosphate Dehydrogenase Deficiency and Malaria: A Method to Detect Primaquine-Induced Hemolysis in vitro. In Dehydrogenases; INTECH: Charlotte, NC, USA, 2012; pp. 65–90. [Google Scholar]
- Zhao, X.; Li, Z.; Zhang, X. G6PD-MutDB: A mutation and phenotype database of glucose-6-phosphate (G6PD) deficiency. J. Bioinform Comput. Biol. 2010, 8, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Luzzatto, L.; Nannelli, C.; Notaro, R. Glucose-6-Phosphate Dehydrogenase Deficiency. Hematol. Oncol. Clin. N. Am. 2016, 30, 373–393. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Manzo, S.; Terrón-Hernández, J.; De la Mora-De la Mora, I.; González-Valdez, A.; Marcial-Quino, J.; García-Torres, I.; Vanoye-Carlo, A.; López-Velázquez, G.; Hernández-Alcantara, G.; Oria-Hernández, J.; et al. The stability of G6PD is affected by mutations with different clinical phenotypes. Int. J. Mol. Sci. 2014, 15, 21179–21201. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.T.; Lam, V.M.; Engel, P.C. Marked decrease in specific activity contributes to disease phenotype in two human glucose-6-phosphate dehydrogenase mutants, G6PDUnion and G6PDAndalus. Hum. Mutat. 2005, 26, 284–293. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Manzo, S.; Marcial-Quino, J.; Ortega-Cuellar, D.; Serrano-Posada, H.; González-Valdez, H.; Vanoye-Carlo, A.; Hernández-Ochoa, B.; Sierra-Palacios, E.; Castillo-Villanueva, A.; Reyes-Vivas, H. Functional and Biochemical Analysis of Glucose-6-Phosphate Dehydrogenase (G6PD) Variants: Elucidating the Molecular Basis of G6PD Deficiency. Catalysts 2017, 7, 135. [Google Scholar] [CrossRef]
- Boonyuen, U.; Chamchoy, K.; Swangsri, T.; Junkree, T.; Day, M.; White, N.; Imwong, M. A trade-off between catalytic activity and protein stability determines the clinical manifestations of glucose-6-phosphate dehydrogenase (G6PD) deficiency. Int. J. Biol. Macromol. 2017, 104, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Au, S.W.N.; Gover, S.; Lam, V.M.S.; Adams, M. Human glucose-6-phosphate dehydrogenase: The crystal structure reveals a structural NADP+ molecule and provides insights into enzyme deficiency. Structure 2000, 8, 293–303. [Google Scholar] [CrossRef]
- Kotaka, M.; Gover, S.; Vandeputte-Rutten, L.; Au, S.W.N.; Lam, V.M.S.; Adams, M.J. Structural studies of glucose-6-phosphate and NADP+ binding to human glucose-6-phosphate dehydrogenase. Acta Crystallogr. 2005, D61, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.T.; Chan, T.F.; Lam, V.; Engel, P. What is the role of the second “structural” NADP-binding site in human glucose-6-phosphate dehydrogenase? Protein Sci. 2008, 17, 1403–1411. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Choi, M.Y.; Au, S.W.; Au, D.M.; Lam, V.M.S.; Engel, P.C. Purification and detailed study of two clinically different human glucose 6-phosphate dehydrogenase variants, G6PD (Plymouth) and G6PD (Mahidol): Evidence for defective protein folding as the basis of disease. Mol. Genet. Metab. 2008, 93, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Manzo, S.; Marcial-Quino, J.; Vanoye-Carlo, A.; Serrano-Posada, H.; González-Valdez, A.; Martínez-Rosas, V.; Hernández-Ochoa, B.; Sierra-Palacios, E.; Castillo-Rodríguez, R.A.; Cuevas-Cruz, M.; et al. Functional and biochemical characterization of three recombinant human Glucose-6-Phosphate Dehydrogenase mutants: Zacatecas, Vanua-Lava and Viangchan. Int. J. Mol. Sci. 2016, 17, 787. [Google Scholar] [CrossRef] [PubMed]
- Boonyuen, U.; Chamchoy, K.; Swangsri, T.; Saralamba, T.; Day, N.P.J.; Imwong, M. Detailed functional analysis of two clinical glucose-6-phosphate dehydrogenase (G6PD) variants, G6PDViangchan and G6PDViangchan + Mahidol: Decreased stability and catalytic efficiency contribute to the clinical phenotype. Mol. Genet. Metab. 2016, 2, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Manzo, S.; Marcial-Quino, J.; Vanoye-Carlo, A.; Enríquez-Flores, S.; De la Mora-De la Mora, I.; González-Valdez, A.; García-Torres, I.; Martínez-Rosas, V.; Sierra-Palacios, E.; Lazcano-Pérez, F.; et al. Mutations of glucose-6-phosphate dehydrogenase durham, Santa-Maria and A+ variants are associated with loss functional and structural stability of the protein. Int. J. Mol. Sci. 2015, 16, 28657–28668. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.T.; Lam, V.M.S.; Engel, P.C. Functional properties of two mutants of human glucose 6-phosphate dehydrogenase, R393G and R393H, corresponding to the clinical variants G6PD Wisconsin and Nashville. Biochim. Biophys. Acta 2006, 1762, 767–774. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.; Chandra, S.; Suthar, M.K.; Doharey, P.K.; Siddiqi, M.I.; Saxena, J.K. NADP+ binding effects tryptophan accessibility, folding and stability of recombinant B. malayi G6PD. Int. J. Biol. Macromol. 2016, 85, 645–654. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Manzo, S.; Terrón-Hernández, J.; de la Mora-de la Mora, I.; García Torres, I.; López-Velázquez, G.; Reyes-Vivas, H.; Oria-Hernández, J. Cloning, expression, purification and characterization of His-tagged human glucose-6-phosphate dehydrogenase: A simplified method for protein yield. Protein J. 2013, 32, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Datsenko, K.A.; Wanner, B.L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 2000, 97, 6640–6645. [Google Scholar] [CrossRef] [PubMed]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [PubMed]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. 2010, 66, 486–501. [Google Scholar]
- Krieger, E.; Joo, K.; Lee, J.; Lee, J.; Raman, S.; Thompson, J.; Tyka, M.; Baker, D.; Karplus, K. Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: Four approaches that performed well in CASP8. Proteins Struct. Funct. Bioinform. 2009, 77, 114–122. [Google Scholar] [CrossRef] [PubMed]
- McNicholas, S.; Potterton, E.; Wilson, K.S.; Noble, M.E.M. Presenting your structures: The CCP4mg molecular-graphics software. Acta Crystallogr. 2011, 67, 386–394. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| G6PD | Total Protein (mg) | Specific Activity (IU·mg−1) | Total Activity (IU) | Yield (%) |
|---|---|---|---|---|
| WT | 4.8 | 224 | 1075 | 61 |
| A+ | 3.6 | 114 | 410 | 43 |
| Nefza | 3.52 | 62 | 218 | 28 |
| A− | 3.53 | 22 | 77 | 24 |
| Kinetic Constants | WT-G6PD | Mutants | ||
|---|---|---|---|---|
| A+ | Nefza | A− | ||
| Km·G6P (μM) | 38.4 ± 4.1 | 56.4 ± 5.5 | 50.4 ± 6.2 | 33.8 ± 3.5 |
| Km·NADP+ (μM) | 6.1 ± 1.2 | 12.9 ± 1.4 | 16.4 ± 1.6 | 14.3 ± 2.1 |
| kcat (s−1) | 230 ± 7.6 | 114 ± 3.2 | 126 ± 2.8 | 35.8 ± 3 |
| kcat/Km·G6P (μM−1·s−1) | 5.9 ± 0.6 | 2.0 ± 0.1 | 2.4 ± 0.2 | 1.1 ± 0.1 |
| kcat/Km·NADP+ (μM−1·s−1) | 37.3 ± 3.1 | 8.7 ± 0.7 | 7.5 ± 0.6 | 2.5 ± 0.2 |
| Strain E. coli | Relevant Characteristic(s) or Sequence | Source and/or Reference |
| BW25113 | F−, DE(araD-araB)567, lacZ4787(del)::rrnB-3, LAM−, rph-1, DE(rhaD-rhaB)568, hsdR514 | [29] |
| BL21(DE3)Δzwf::kanr | F− ompT gal dcm lon hsdSB(rB− mB−) λ(DE3 [lacI lacUV5-T7 gene 1 ind1 sam7 nin5]) Δzwf-777::kan. | [15] |
| Plasmids | ||
| pET-HisTEVP-G6PD | pETg6pd carrying the human G6PD gene, AmpR | [28] |
| PjetG6PD A+ | pJET 1.2 plasmid carrying the human G6PD gene with a Asn126Asp mutation in the G6PD protein, AmpR | This study |
| PjetG6PD Nefza | pJET 1.2 plasmid carrying the human G6PD gene with a L323P mutation in the G6PD protein, AmpR | This study |
| pJETG6PD A− | pJET 1.2 plasmid carrying the human G6PD gene with a double mutation of Asn126Asp + Leu323Pro in the G6PD protein, AmpR | This study |
| pETG6PD A+ | pET-3a carrying the human G6PD gene with a Asn126Asp mutation in the G6PD protein, AmpR | This study |
| pETG6PD Nefza | pET-3a carrying the human G6PD gene with an Leu323Pro mutation in the G6PD protein, AmpR | This study |
| pETG6PD A− | pET-3a carrying the human G6PD gene with a double mutation of Asn126Asp + Leu323Pro in the G6PD protein, AmpR | This study |
| Mutagenesis | Primer Sequence | |
| A+ forward | 5′-AGCCACATGGATGCCCTCCAC-3′ | This study |
| A+ reverse | 5′-GTGGAGGGCATCCATGTGGCT-3′ | This study |
| Nefza forward | 5′-TCCACCAACCCAGATGACGT-3′ | This study |
| Nefza reverse | 5′-ACGTCATCTGGGTTGGTGGA-3′ | This study |
| Oligonucleotides for sequencing | Primer Sequence | |
| Flanking NdeI forward | 5′-CGACAGCCATATGGCAGAG-3′ | This study |
| Flanking Bpu reverse | 5′-TGCGCTGAGCTCAGAGCTT-3′ | This study |
| Internal G6PD forward | 5′-GGCCAACTGCCTCTTCTAC-3′ | This study |
| Internal G6PD reverse | 5′-GAGAAGGTCAAGATGTTGAAATG-3′ | This study |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramírez-Nava, E.J.; Ortega-Cuellar, D.; Serrano-Posada, H.; González-Valdez, A.; Vanoye-Carlo, A.; Hernández-Ochoa, B.; Sierra-Palacios, E.; Hernández-Pineda, J.; Rodríguez-Bustamante, E.; Arreguin-Espinosa, R.; et al. Biochemical Analysis of Two Single Mutants that Give Rise to a Polymorphic G6PD A-Double Mutant. Int. J. Mol. Sci. 2017, 18, 2244. https://doi.org/10.3390/ijms18112244
Ramírez-Nava EJ, Ortega-Cuellar D, Serrano-Posada H, González-Valdez A, Vanoye-Carlo A, Hernández-Ochoa B, Sierra-Palacios E, Hernández-Pineda J, Rodríguez-Bustamante E, Arreguin-Espinosa R, et al. Biochemical Analysis of Two Single Mutants that Give Rise to a Polymorphic G6PD A-Double Mutant. International Journal of Molecular Sciences. 2017; 18(11):2244. https://doi.org/10.3390/ijms18112244
Chicago/Turabian StyleRamírez-Nava, Edson Jiovany, Daniel Ortega-Cuellar, Hugo Serrano-Posada, Abigail González-Valdez, America Vanoye-Carlo, Beatriz Hernández-Ochoa, Edgar Sierra-Palacios, Jessica Hernández-Pineda, Eduardo Rodríguez-Bustamante, Roberto Arreguin-Espinosa, and et al. 2017. "Biochemical Analysis of Two Single Mutants that Give Rise to a Polymorphic G6PD A-Double Mutant" International Journal of Molecular Sciences 18, no. 11: 2244. https://doi.org/10.3390/ijms18112244