Sodium Hydrosulfide Prevents Myocardial Dysfunction through Modulation of Extracellular Matrix Accumulation and Vascular Density

Abstract
:1. Introduction
2. Results and Discussion
2.1. NaHS Inhibited Fibrosis in the Border Zone of Infarcted Myocardium


2.2. NaHS Mitigated Type I and III Collagen as well as Matrix Metalloproteinases-9 (MMP-9) Expression
2.3. NaHS Modulated CSE and Heme Oxygenase-1 Expression in the Border Zone of Infarcted Myocardium


2.4. NaHS Promoted the Growth of New Vessels in the Border Zone of Infarcted Myocardium

2.5. NaHS Prevented Cardiac Dysfunction


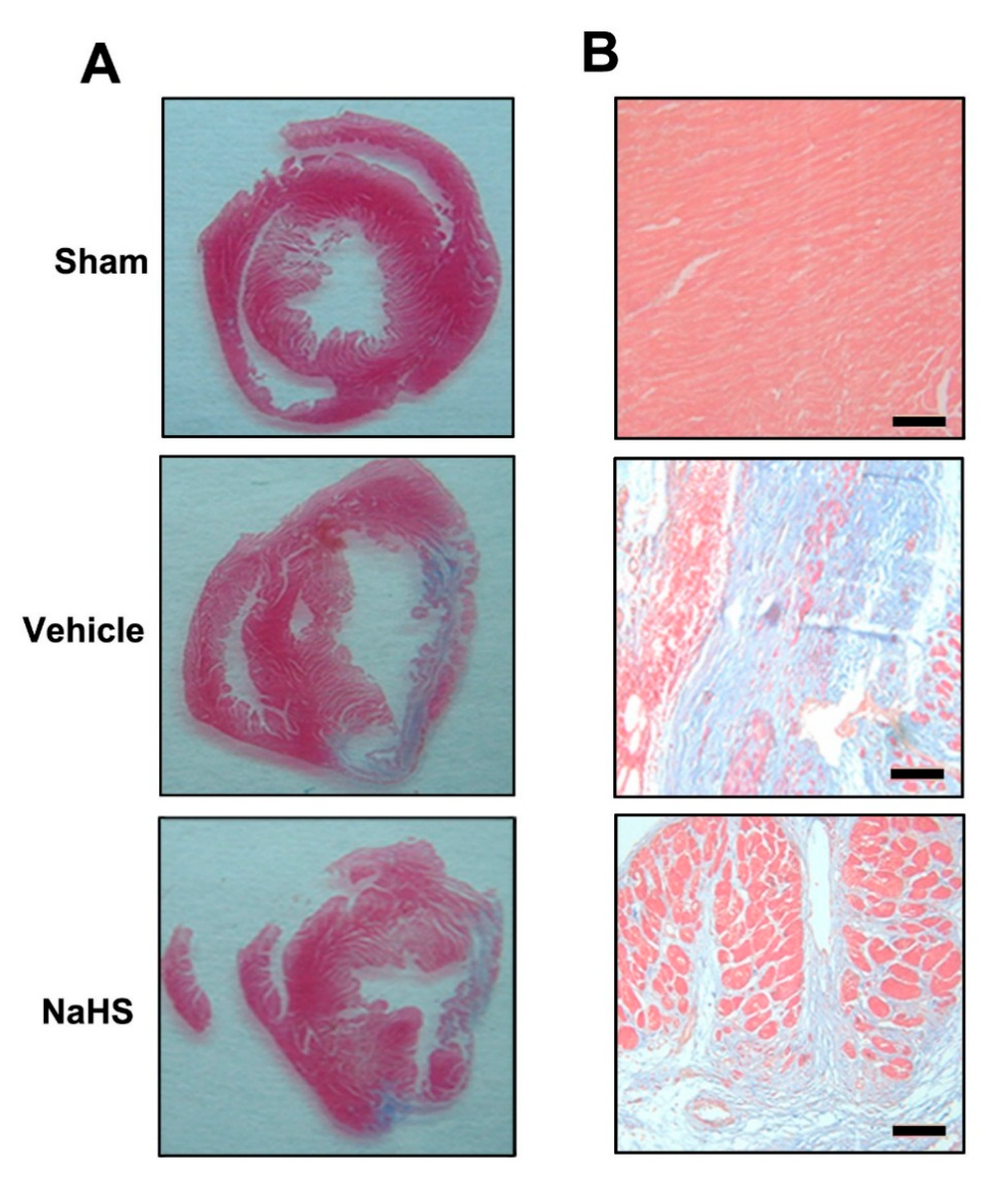{kind=link}
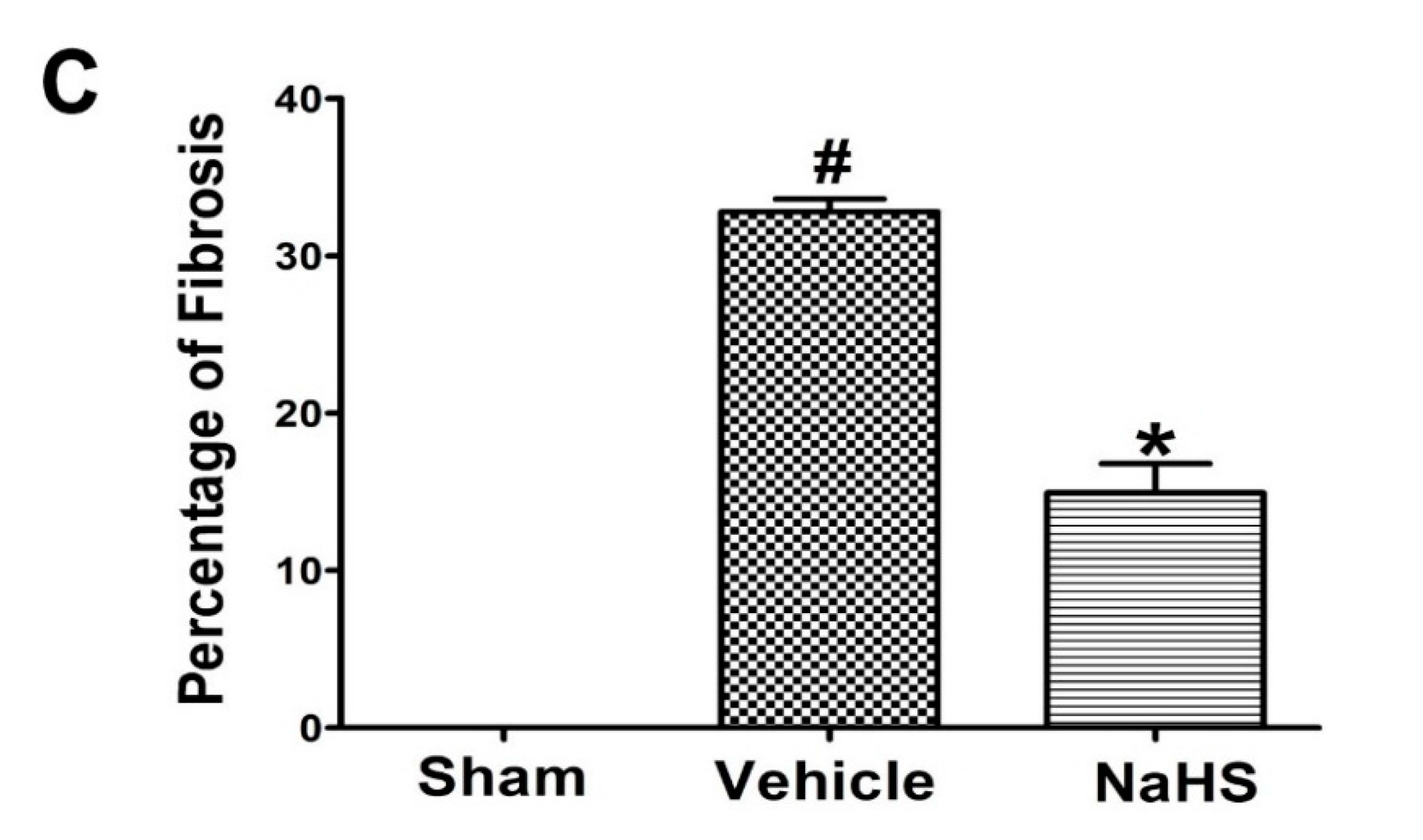{kind=link}
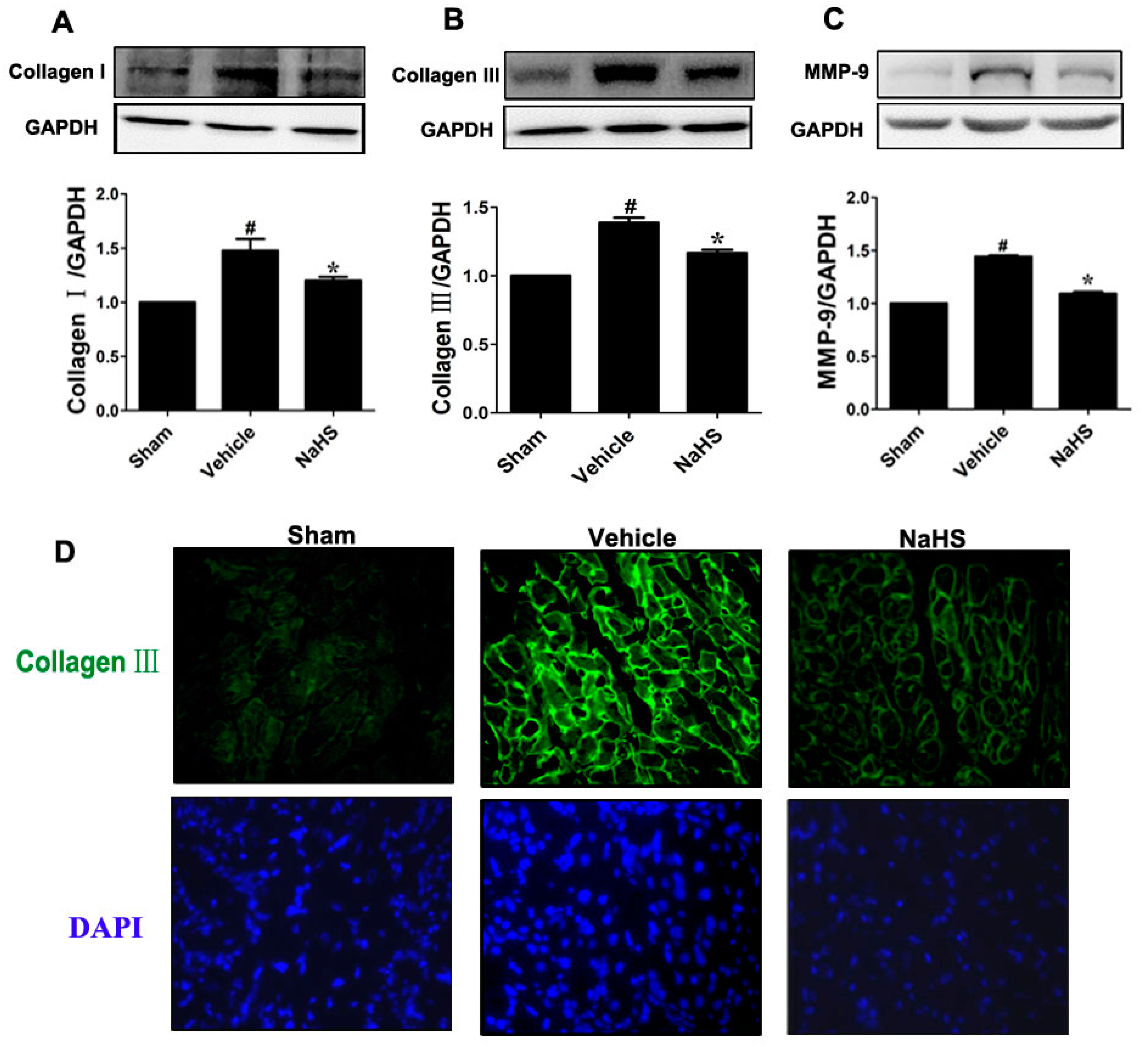{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Sham (n = 7) | Vehicle (n = 8) | NaHS (n = 8) |
|---|---|---|---|
| LVIDd (cm) | 0.57 ± 0.02 | 0.73 ± 0.03 # | 0.71 ± 0.03 |
| LVIDs (cm) | 0.3 ± 0.02 | 0.51 ± 0.06 # | 0.41 ± 0.05 * |
| R-R (s) | 0.15 ± 0.01 | 0.15 ± 0.01 | 0.16 ± 0.01 |
| LVEDV (mL) | 0.42 ± 0.04 | 0.88 ± 0.05 # | 0.81 ± 0.14 |
| LVESV (mL) | 0.07 ± 0.01 | 0.35 ± 0.06 # | 0.19 ± 0.05 * |
| EF (%) | 84.0 ± 0.30 | 63.0 ± 0.20 # | 78.0 ± 0.30 * |
| FS (%) | 47.37 ± 3.44 | 28.55 ± 1.17 # | 39.45 ± 3.11 * |
| HR (bpm) | 393 ± 16 | 415 ± 16 | 396 ± 13 |
2.6. Discussion
3. Methods
3.1. Animal Care
3.2. Chemicals and Antibodies
3.3. Generation of Chronic MI and Drug Treatment
3.4. Echocardiography
3.5. Sample Preparation for Histological and Morphometric Analysis
3.6. Immunohistochemistry and Immunofluorescence Analysis
3.7. Western Blot Analysis
3.8. Statistical Analysis
4. Conclusions
Acknowledgments
Author Contributions
Abbreviation
| CSE | cystathionine γ-lyase |
| MI | myocardial infarction |
| MMPs | matrix metalloproteinases |
| HO-1 | heme oxygenase-1 |
| VEGF | vascular endothelial growth factor |
| NaHS | Sodium hydrosulfide |
| H2S | Hydrogen sulfide |
| LVIDd | left ventricular internal dimension diastole |
| LVIDs | left ventricular internal dimension systole |
| LVEDV | left ventricular end-diastolic volume |
| LVESV | left ventricular end-systolic volume |
| EF | ejection fraction |
| FS | fractional shortening |
Conflicts of Interest
References
- Inglis, S.C.; Bebchuk, J.; Al-Suhaim, S.A.; Case, J.; Pfeffer, M.A.; Solomon, S.D.; Hou, Y.R.; Pitt, B.; Dargie, H.J.; Ford, I.; et al. Peripheral artery disease and outcomes after myocardial infarction: An individual-patient meta-analysis of 28,771 patients in CAPRICORN, EPEHESUS, OPTIMAAL and VALIANT. Int. J. Cardiol. 2013, 168, 1094–1101. [Google Scholar]
- Golomb, B.A.; Dang, T.T.; Criqui, M.H. Peripheral arterial disease: Morbidity and mortality implications. Circulation 2006, 114, 688–699. [Google Scholar] [CrossRef] [PubMed]
- Halade, G.V.; Jin, Y.F.; Lindsey, M.L. Matrix metalloproteinase (MMP)-9: A proximal biomarker for cardiac remodeling and a distal biomarker for inflammation. Pharmacol. Ther. 2013, 139, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Saparov, A.; Chen, C.W.; Beckman, S.A.; Wang, Y.; Huard, J. The role of antioxidation and immunomodulation in postnatal multipotent stem cell-mediated cardiac repair. Int. J. Mol. Sci. 2013, 14, 16258–16279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konstam, M.A.; Kramer, D.G.; Patel, A.R.; Maron, M.S.; Udelson, J.E. Left ventricular remodeling in heart failure: Current concepts in clinical significance and assessment. JACC Cardiovasc. Imaging 2011, 4, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.L.; Liu, X.H.; Shen, Y.Q.; Wang, N.Z.; Xu, J.; Wu, D.; Xiong, Q.H.; Deng, H.Y.; Huang, G.Y.; Zhu, Y.Z. Inhibition of NADPH oxidase 4-related signaling by sodium hydrosulfide attenuates myocardial fibrotic response. Int. J. Cardiol. 2013, 168, 3770–3778. [Google Scholar] [CrossRef] [PubMed]
- Spinale, F.G.; Villarreal, F. Targeting matrix metalloproteinases in heart disease: Lessons from endogenous inhibitors. Biochem. Pharmacol. 2014, 90, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.L.; Liu, X.H.; Gong, Q.H.; Yang, H.B.; Zhu, Y.Z. Role of cystathionine γ-lyase/hydrogen sulfide pathway in cardiovascular disease: A novel therapeutic strategy? Antioxid. Redox Signal. 2012, 17, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.Z.; Wang, Z.J.; Ho, P.; Loke, Y.Y.; Zhu, Y.C.; Huang, S.H.; Tan, C.S.; Whiteman, M.; Lu, J.; Moore, P.K. Hydrogen sulfide and its possible roles in myocardial ischemia in experimental rats. J. Appl. Physiol. 2007, 102, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhao, X.; Jin, H.; Wei, H.; Li, W.; Bu, D.; Tang, X.; Ren, Y.; Tang, C.; Du, J. Role of hydrogen sulfide in the development of atherosclerotic lesions in apolipoprotein E knockout mice. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Zhang, J.; Lu, Y.; Wang, R. The vasorelaxant effect of H2S as a novel endogenous gaseous KATP channel opener. EMBO J. 2001, 20, 6008–6016. [Google Scholar] [CrossRef] [PubMed]
- Szabo, C. Hydrogen sulphide and its therapeutic potential. Nat. Rev. Drug Discov. 2007, 6, 917–935. [Google Scholar] [CrossRef] [PubMed]
- Hibuya, N.; Koike, S.; Tanaka, M.; Ishigami-Yuasa, M.; Kimura, Y.; Ogasawara, Y.; Fukui, K.; Nagahara, N.; Kimura, H. A novel pathway for the production of hydrogen sulfide from d-cysteine in mammalian cells. Nat. Commun. 2013, 4, 1366–. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H. Signaling molecules: Hydrogen sulfide and polysulfide. Antioxid. Redox Signal. 2014. [Google Scholar] [CrossRef]
- Polhemus, D.J.; Kondo, K.; Bhushan, S.; Bir, S.C.; Kevil, C.G.; Murohara, T.; Lefer, D.J.; Calvert, J.W. Hydrogen sulfide attenuates cardiac dysfunction after heart failure via induction of angiogenesis. Circ. Heart Fail. 2013, 6, 1077–1086. [Google Scholar] [CrossRef] [PubMed]
- Snijder, P.M.; Frenay, A.S.; de Boer, R.A.; Pasch, A.; Hillebrands, J.; Leuvenink, H.G.; van Goor, H. Exogenous administration of thiosulfate, a donor of hydrogen sulfide, attenuates Angiotensin II-induced hypertensive heart disease in rats. Br. J. Pharmacol. 2014. [Google Scholar] [CrossRef]
- Polhemus, D.J.; Lefer, D.J. Emergence of hydrogen sulfide as an endogenous gaseous signaling molecule in cardiovascular disease. Circ. Res. 2014, 114, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Guo, W.; Wang, Z.; Zhang, Y.; Zhong, L.; Zhu, Y. Protective effects of hydrogen sulfide in hypoxic human umbilical vein endothelial cells: A possible mitochondria-dependent pathway. Int. J. Mol. Sci. 2013, 14, 13093–13108. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.L.; Liu, X.H.; Gong, Q.H.; Wu, D.; Zhu, Y.Z. Hydrogen sulfide attenuated tumor necrosis factor-α-induced inflammatory signaling and dysfunction in vascular endothelial cells. PLoS One 2011, 6, e19766. [Google Scholar] [CrossRef] [PubMed]
- Mustafa, A.K.; Sikka, G.; Gazi, S.K.; Steppan, J.; Jung, S.M.; Bhunia, A.K.; Barodka, V.M.; Gazi, F.K.; Barrow, R.K.; Wang, R.; et al. Hydrogen sulfide as endothelium-derived hyperpolarizing factor sulfhydrates potassium channels. Circ. Res. 2011, 109, 1259–1268. [Google Scholar]
- Wang, X.; Wang, Q.; Guo, W.; Zhu, Y.Z. Hydrogen sulfide attenuates cardiac dysfunction in a rat model of heart failure: A mechanism through cardiac mitochondrial protection. Biosci. Rep. 2011, 31, 87–98. [Google Scholar] [CrossRef]
- Liu, X.H.; Pan, L.L.; Deng, H.Y.; Xiong, Q.H.; Wu, D.; Huang, G.; Gong, Q.H.; Zhu, Y.Z. Leonurine (SCM-198) attenuates myocardial fibrotic response via inhibition of NADPH oxidase 4. Free Radic. Biol. Med. 2013, 54, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Dobaczewski, M.; Gonzalez-Quesada, C.; Frangogiannis, N.G. The extracellular matrix as a modulator of the inflammatory and reparative response following myocardial infarction. J. Mol. Cell. Cardiol. 2010, 48, 504–511. [Google Scholar] [CrossRef] [PubMed]
- Kondo, K.; Bhushan, S.; King, A.L.; Prabhu, S.D.; Hamid, T.; Koenig, S.; Murohara, T.; Predmore, B.L.; Gojon, G., Sr.; Gojon, G., Jr.; et al. H2S protects against pressure overload-induced heart failure via upregulation of endothelial nitric oxide synthase. Circulation 2013, 127, 1116–1127. [Google Scholar]
- Wang, Q.; Liu, H.R.; Mu, Q.; Rose, P.; Zhu, Y.Z. S-propargyl-cysteine protects both adult rat hearts and neonatal cardiomyocytes from ischemia/hypoxia injury: The contribution of the hydrogen sulfide-mediated pathway. J. Cardiovasc. Pharmacol. 2009, 54, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Shen, Z.; Miao, L.; Xin, X.; Lin, S.; Zhu, Y.; Guo, W.; Zhu, Y.Z. miRNA-30 family inhibition protects against cardiac ischemic injury by regulating cystathionine-γ-lyase expression. Antioxid. Redox Signal. 2014. [Google Scholar] [CrossRef]
- Calvert, J.W.; Jha, S.; Gundewar, S.; Elrod, J.W.; Ramachandran, A.; Pattillo, C.B.; Kevil, C.G.; Lefer, D.J. Hydrogen sulfide mediates cardioprotection through Nrf2 signaling. Circ. Res. 2009, 105, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Kovacic, D.; Glavnik, N.; Marinsek, M.; Zagozen, P.; Rovan, K.; Goslar, T.; Mars, T.; Podbregar, M. Total plasma sulfide in congestive heart failure. J. Card. Fail. 2012, 18, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Burchfield, J.S.; Xie, M.; Hill, J.A. Pathological ventricular remodeling Mechanisms: Part 1 of 2. Circulation 2013, 128, 388–400. [Google Scholar] [CrossRef] [PubMed]
- Heusch, G.; Libby, P.; Gersh, B.; Yellon, D.; Bohm, M.; Lopaschuk, G.; Opie, L. Cardiovascular remodelling in coronary artery disease and heart failure. Lancet 2014, 383, 1933–1943. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, T.A.; Iyer, R.P.; Ghasemi, O.; Lopez, E.F.; Levin, D.B.; Zhang, J.; Zamilpa, R.; Chou, Y.M.; Jin, Y.F.; Lindsey, M.L. Aliskiren and valsartan mediate left ventricular remodeling post-myocardial infarction in mice through MMP-9 effects. J. Mol. Cell. Cardiol. 2014, 72, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Shinde, A.V.; Frangogiannis, N.G. Fibroblasts in myocardial infarction: A role in inflammation and repair. J. Mol. Cell. Cardiol. 2014, 70, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.J.; Wang, M.J.; Moore, P.K.; Jin, H.M.; Yao, T.; Zhu, Y.C. The novel proangiogenic effect of hydrogen sulfide is dependent on Akt phosphorylation. Cardiovasc. Res. 2007, 76, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Kan, J.; Guo, W.; Huang, C.; Bao, G.; Zhu, Y.; Zhu, Y.Z. S-propargyl-cysteine, a novel water-soluble modulator of endogenous hydrogen sulfide, promotes angiogenesis through activation of signal transducer and activator of transcription 3. Antioxid. Redox Signal. 2014, 20, 2303–2316. [Google Scholar] [CrossRef] [PubMed]
- Bir, S.C.; Kolluru, G.K.; McCarthy, P.; Shen, X.; Pardue, S.; Pattillo, C.B.; Kevil, C.G. Hydrogen sulfide stimulates ischemic vascular remodeling through nitric oxide synthase and nitrite reduction activity regulating hypoxia-inducible factor-1alpha and vascular endothelial growth factor-dependent angiogenesis. J. Am. Heart Assoc. 2012, 1, e004093. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Wu, T.; Laham, R.J.; Johnson, R.B.; Douglas, P.; Li, J.; Sellke, F.W.; Bunting, S.; Simons, M.; Post, M.J. Efficacy of intracoronary or intravenous VEGF165 in a pig model of chronic myocardial ischemia. J. Am. Coll. Cardiol. 2001, 37, 616–623. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.M.; Li, J.; Liu, L.; Fan, L.; Li, X.Y.; Wang, Y.T.; Abraham, N.G.; Cao, J. Effects of heme oxygenase-1 upregulation on blood pressure and cardiac function in an animal model of hypertensive myocardial infarction. Int. J. Mol. Sci. 2013, 14, 2684–2706. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Hamid, T.; Keith, R.J.; Zhou, G.; Partridge, C.R.; Xiang, X.; Kingery, J.R.; Lewis, R.K.; Li, Q.; Rokosh, D.G.; et al. Cardioprotective and antiapoptotic effects of heme oxygenase-1 in the failing heart. Circulation 2010, 121, 1912–1925. [Google Scholar]
- Penumathsa, S.V.; Koneru, S.; Zhan, L.; John, S.; Menon, V.P.; Prasad, K.; Maulik, N. Secoisolariciresinol diglucoside induces neovascularization-mediated cardioprotection against ischemia-reperfusion injury in hypercholesterolemic myocardium. J. Mol. Cell. Cardiol. 2008, 44, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.M.; Pae, H.O.; Park, J.E.; Lee, Y.C.; Woo, J.M.; Kim, N.H.; Choi, Y.K.; Lee, B. S.; Kim, S R.; Chung, H.T. Heme oxygenase in the regulation of vascular biology: From molecular mechanisms to therapeutic opportunities. Antioxid. Redox Signal. 2011, 14, 137–167. [Google Scholar]
- Kim, J.H.; Lee, K.S.; Lee, D.K.; Kim, J.; Kwak, S.N.; Ha, K.S.; Choe, J.; Won, M.H.; Cho, B.R.; Jeoung, D.; et al. Hypoxia-responsive microRNA-101 promotes angiogenesis via heme oxygenase-1/vascular endothelial growth factor axis by targeting Cullin 3. Antioxid. Redox Signal. 2014, 21, 2469–2482. [Google Scholar]
- Kweider, N.; Fragoulis, A.; Rosen, C.; Pecks, U.; Rath, W.; Pufe, T.; Wruck, C.J. Interplay between vascular endothelial growth factor (VEGF) and nuclear factor erythroid 2-related factor-2 (Nrf2): Implications for preeclampsia. J. Biol. Chem. 2011, 286, 42863–42872. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pan, L.-L.; Wang, X.-L.; Wang, X.-L.; Zhu, Y.-Z. Sodium Hydrosulfide Prevents Myocardial Dysfunction through Modulation of Extracellular Matrix Accumulation and Vascular Density. Int. J. Mol. Sci. 2014, 15, 23212-23226. https://doi.org/10.3390/ijms151223212
Pan L-L, Wang X-L, Wang X-L, Zhu Y-Z. Sodium Hydrosulfide Prevents Myocardial Dysfunction through Modulation of Extracellular Matrix Accumulation and Vascular Density. International Journal of Molecular Sciences. 2014; 15(12):23212-23226. https://doi.org/10.3390/ijms151223212
Chicago/Turabian StylePan, Li-Long, Xian-Li Wang, Xi-Ling Wang, and Yi-Zhun Zhu. 2014. "Sodium Hydrosulfide Prevents Myocardial Dysfunction through Modulation of Extracellular Matrix Accumulation and Vascular Density" International Journal of Molecular Sciences 15, no. 12: 23212-23226. https://doi.org/10.3390/ijms151223212