
Variation among the Complete Chloroplast Genomes of the Sumac Species Rhus chinensis: Reannotation and Comparative Analysis

1
School of Geosciences, Qinghai Normal University, Xining 810008, China
2
School of Life Science, Shanxi University, 92 Wucheng Rd., Taiyuan 030006, China
3
Department of Botany, National Museum of Natural History, Smithsonian Institution, Washington, DC 20013, USA
4
Academy of Plateau Science and Sustainability, Qinghai Normal University, Xining 810016, China
5
Key Laboratory of Biodiversity Formation Mechanism and Comprehensive Utilization of Qinghai-Tibetan Plateau in Qinghai Province, School of Life Sciences, Qinghai Normal University, Xining 810008, China
*
Authors to whom correspondence should be addressed.
Genes 2022, 13(11), 1936; https://doi.org/10.3390/genes13111936
Submission received: 10 September 2022
/
Revised: 19 October 2022
/
Accepted: 21 October 2022
/
Published: 25 October 2022
(This article belongs to the Special Issue Advances in Evolution of Plant Organelle Genome)
Abstract
:The sumac Rhus chinensis Mill. is an economically and ecologically important shrub or tree species in the family of Anacardiaceae with a wide distribution in East to Southeast Asia. We assembled the complete chloroplast genome of 159,187 bp in length and the GC content of 37.8%. The genome encoded 132 genes, including 86 protein-coding genes, 37 tRNA genes, 8 rRNA genes, and 1 pseudogene, and 77 SSRs were identified as well as the interval regions, totaling 46,425 bp in length. The mauve alignment revealed one gene rearrangement among the Rhus species. All the SSRs were divided into five types, most of which consisted of mono- and tri- repeat motifs. Our genome exhibited the longest size and more annotated genes compared to the three other genomes of R. chinensis reported in GenBank. We also discovered some relatively highly variable regions in the complete chloroplast genomes of the Rhus species. The ML phylogenetic analysis of the available chloroplast sequences of the Anacardiaceae well supported the monophyly of each tribe and each genus; the tribe Rhoideae was close to the tribe Anacardiaceae with a high support of 100%, and they then grouped with the tribe Spondiadeae. R. chinensis was sister to R. potaninii, and they then grouped with the species R. typhina.
1. Introduction
The sumac genus Rhus (Anacardiaceae) contains c. 35 species mainly distributed in temperate and sub-tropical regions [1] and is widely recognized in the north temperate zone for its adaptability to various climate and soil conditions [2,3]. R. chinensis is a common deciduous sumac tree that is widely distributed in Asia, including China, India, Vietnam, Korea, and Japan [4]. This species is an important medicinal plant containing various pharmacologically active constituents, which have been used for medicinal purposes such as anticancer, antiviral, antimicrobial, and anti-inflammatory as well as being used as a revegetation plant for ecological restoration due to its cold tolerance and easy multiplication [5,6,7,8]. In particular, as the primary host plant of several species of Chinese sumac aphids, R. chinensis hosted the aphids which induced galls, which are a source of traditional Chinese medicine [9]. The galls are economically very important because they are rich in tannins, which were extracted and synthesized to be utilized earlier in the rubber industry and for improving leather quality, while at present they are used in various fields, e.g., medicine, food, dye, chemical, and military industries [9,10]. However, in recent decades, their saplings have been repeatedly destroyed by anthropogenic activities, which severely restricted their natural regeneration. The number of wild R. chinensis plants is decreasing, and their natural distribution range is gradually shrinking [11].
Most of the previous studies on R. chinensis mainly focused on its pharmacology, phytochemical, biological activities, biological features, and biogeographic diversification [12,13,14,15,16]. In the case of the molecular analysis, the chloroplast fragments (ndhF, rbcL, trnL-F, and trnC-trnD) and nuclear ITS sequences were applied as markers to study the genetic diversity and phylogenetic relationships of the Rhus species [17,18,19,20]. The analysis of the nuclear ITS region and the chloroplast fragments indicated that Rhus was monophyletic, but the relationships among these genera of the Rhus complex were not well resolved [18,19,20]. Thus, obtaining more information on the genome of the Rhus species is essential to further study their delimitation, genetic diversity, phylogenetic relationships, and evolutionary history [21].
The chloroplasts are well known to be key organelles in plants with crucial functions in photosynthesis and biosynthesis [22]. Moreover, compared with nuclear and mitochondrial genomes, the chloroplast genomes in angiosperms are highly conserved in structure, gene content, and organization [23,24]. In particular, for its exhibiting variations at the intra-species level, the genome has been widely used for the study of systematics and evolution among different populations [25,26]. In recent years, more and more complete chloroplast genomes in plants have been reported [27,28,29], which provide more valuable information and screen more new markers with potentially high resolution for more comprehensive research in the future to better resolve the phylogenetic relationships and intraspecific diversity of the Rhus species. At present, there were three complete chloroplast genomes of R. chinensis reported [21,30,31]. However, we found that these three genomes showed large differences in both sequence length, from 149,011 bp to 158,809 bp, and number of genes, from 126 to 130, which is very unusual for different accessions of the same species. Hence, it is important to obtain the high-quality genome sequences, especially in the case of organellar genomes, for the analysis of genetic diversity and phylogenetics [29].
In the present study, we sequenced and assembled a complete chloroplast genome of R. chinensis and analyzed its nucleotide composition, gene organization, and structure, especially in comparison with the three reported chloroplast genomes of R. chinensis and other Rhus species downloaded from GenBank (https://www.ncbi.nlm.nih.gov/GenBank, accessed on 29 September 2021). We aimed to accurately assemble and validate the complete chloroplast genome of the species R. chinensis in order to further examine the genetic variation among these chloroplast genomes of R. chinensis accessions and other Rhus species. Furthermore, the phylogeny of the Anacardiaceae species was constructed for the purpose of clarifying the relationships between different species, genera, and tribes.
2. Materials and Methods
2.1. Sample, DNA Extraction, and Sequencing
We collected fresh leaves of R. chinensis at Wufeng county (30°11′10.514″ N, 111°5′42.342″ E; alt. 824 m), Hubei, China, on 6 October 2014. The leaf samples were dried in a timely manner using silica gel, and the total genomic DNA of R. chinensis was extracted using the Plant Genomic DNA Kit (TIANGEN) following its instructions. The quality and quantity of the extracted DNA were examined using Nanodrop 2000. The qualified total DNA was used for library construction and sequencing by using the shotgun genome skimming method on an Illumina HiSeq 4000 platform [32]. The paired-end (PE) reads of 2 × 150 bp (insert size of 400 bp) were generated and the qualified DNA sequences were obtained after filtering out low-quality and adapter-contaminated reads. A total of 8.4 G clean reads was used for assembling the genome. The specimen was stored at the Herbarium of School of Life Science, Shanxi University, China (voucher no: Ren_P1966).
2.2. Chloroplast Genome Assembly and Annotation
The clean reads were decompressed in Terminal, and then the complete chloroplast genome was assembled using the de novo method by the program GetOrganelle with kmers 21, 45, 65, 85, and 105 [33]. Additionally, we assembled the genome by mapping it with the chloroplast genome of R. chinensis (KX447140) and Pistacia weinmanniifolia (NC_037471) as references. The annotations of chloroplast genome were conducted by the PGA software [34] and the Geneious Prime software (version 11.0.3; https://www.geneious.com, accessed on 29 September 2021) using the reported genome sequences of Rhus species as references (GenBank accession Nos: KX447140, MF351625, and NC_037471). In addition, we searched the homologous sequences of the unannotated or ambiguous region by BLAST in GenBank to attempt annotate these regions. Start/stop codons and intron/exon borders of protein coding genes with mistakes were detected by translating the sequences. In addition, the transfer RNA (tRNA) genes were verified by the online tRNAscan-SE v2.0 software [35]. The physical map of the complete chloroplast genome of R. chinensis was generated utilizing the OGDRAW program (https://chlorobox.mpimp-golm.mpg.de/OGDraw.html, accessed on 1 December 2021) [36] to demonstrate its structural characteristics.
2.3. Sequence Analysis
We performed the statistics on the general information of the complete chloroplast genome of R. chinensis, including the total genome length, the gene number and size, the base content, and the lengths of exons and introns in genes using the Geneious Prime software (version 11.0.3; Created by the Biomatters development team Copyright; New Zealand).
The relative synonymous codon usage (RSCU) ratio was obtained by the programs MEGA7.0 and CodonW. MISA was used to detect the microsatellites, also called simple sequence repeats (SSRs), i.e., mono-, di-, tri-, tetra-, penta-, and hexa-nucleotide repeats, with thresholds of ten, six, and three repeat units for mononucleotide SSRs, di- and tri-nucleotide SSRs, and tetra-, penta-, and hexa-nucleotide SSRs, respectively [37].
2.4. Genome Comparison
The complete chloroplast genome of R. chinensis was compared with those of its related species Rhus to detect the genomic variation. Multiple genome alignment among complete chloroplast genomes of Rhus species was conducted through the Mauve program to detect evolutionary events such as rearrangement and inversion.
The borders of large single-copy (LSC), small single-copy (SSC), and inverted repeat (IR) regions were visually displayed and compared among Rhus species using BLAST, Find Repeats regions, and IRscope (http://irscope.shinapps.io/irapp/, accessed on 10 December 2021).
We used two datasets, the sequences of four complete chloroplast genomes of R. chinensis, and the other sequences of six complete chloroplast genomes of Rhus species excluding R. chinensis (accession Nos: KX447140 and MF351625) to detect the hotspots of intraspecific and species divergence, and analyze sequence divergence using the MAFFT online service (https://mafft.cbrc.jp/alignment/server/, accessed on 2 January 2022). We also analyzed the nucleotide diversity (Pi) using the DnaSP software with the step size of 200 bp and the window length of 800 bp [38].
2.5. Phylogenetic Reconstruction
All the protein-coding genes from the complete chloroplast genomes were used to construct the phylogenetic tree of the family Anacardiaceae. The detailed species information used in this study is shown in Table 1. Each protein-coding gene was aligned using MAFFT version 7 [39,40] and then concatenated as a dataset by Geneious v11.0.3 for phylogenetic analysis.
The phylogenetic relationship of 21 Anacardiaceae species including six genera was constructed with two Burseraceae species, namely Commiphora gileadensis and Boswellia sacra, as outgroups. We performed the construction of the maximum likelihood tree using the RAxML program under the GTRGAMMA model with 1000 bootstrap replicates [41].
3. Results and Discussion
3.1. General Features of the Complete Chloroplast Genome of R. chinensis
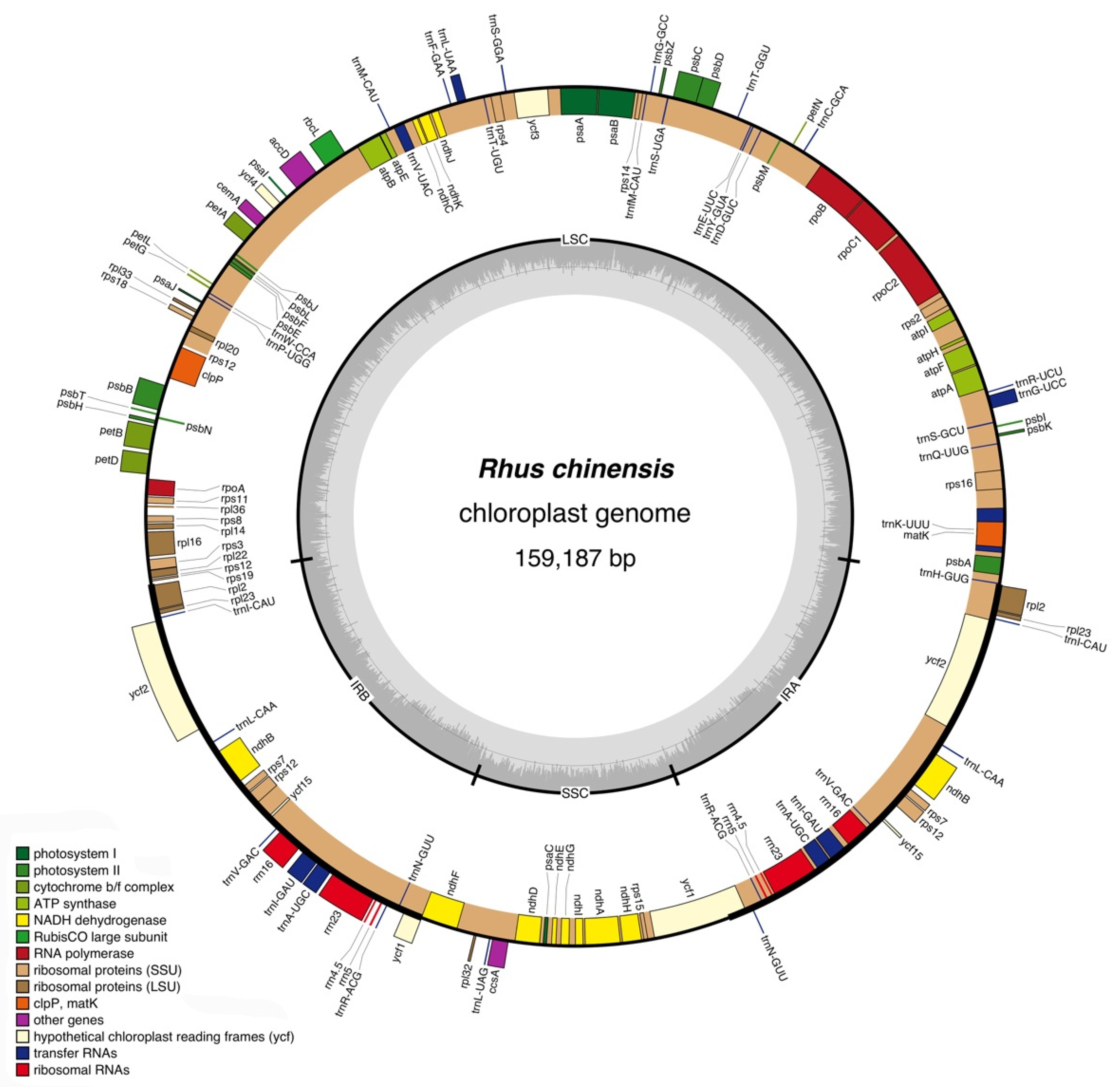
The assembled complete chloroplast genome of R. chinensis totaled 159,187 bp in length, comprising an LSC region of 87,961 bp, an SSC region of 18,522 bp, and a pair of IR (IRA/IRB) regions of 26,506 bp. The genome contained 132 genes with 86 protein-coding genes, 37 tRNAs, 8 rRNAs, and 1 pseudogene, among which 60 protein-coding genes were in the LSC region, and 11 in the SSC region. All the protein-coding genes started with the common initiation codon ATG. There were 22 tRNAs and 1 tRNA in the LSC and SSC region, respectively. The IR region duplicated eight protein-coding genes and seven tRNAs. All rRNA genes were located in the IR region. The location and organization of the complete chloroplast genome of R. chinensis are shown in Table 2, and the diagram is shown in Figure 1. The genome sequence was submitted to GenBank with the accession No. OP326720.
The GC content of the complete chloroplast genome totaled 37.8%, which was much lower than the AT content (about 62%). The GC content (42.9%) in the IR regions was the highest among those of the overall genome (37.8%), the LSC (35.9%), and the SSC regions (32.6%), which may result from the presence of GC-rich rRNAs and tRNAs in these regions. The statistics of the base composition showed that the third codon position had the highest AT content (69.5%), whereas the first codon position had the lowest AT content (53.6%), with 62.2% at the second codon position. Our results are consistent with the previous reports on the plant chloroplast genomes [31,42].
There were 21 one-intron-containing genes, including 13 protein-coding genes and 8 tRNA genes, whereas 2 protein-coding genes, i.e., ycf3 and clpP, possessed two introns (Table 3). Among the 21 genes, the shortest intron was found in the trnL-UAA gene with 450 bp in length, whereas the longest was found in the trnK-UUU gene with 2599 bp and contained the protein-coding gene matK, which are the general characteristics in other land plants [43]. The gene rps12 was trans-spliced with the duplicated 3′ end in the IRs and the 5′ end located in the LSC region, as previously reported in other plants [44].
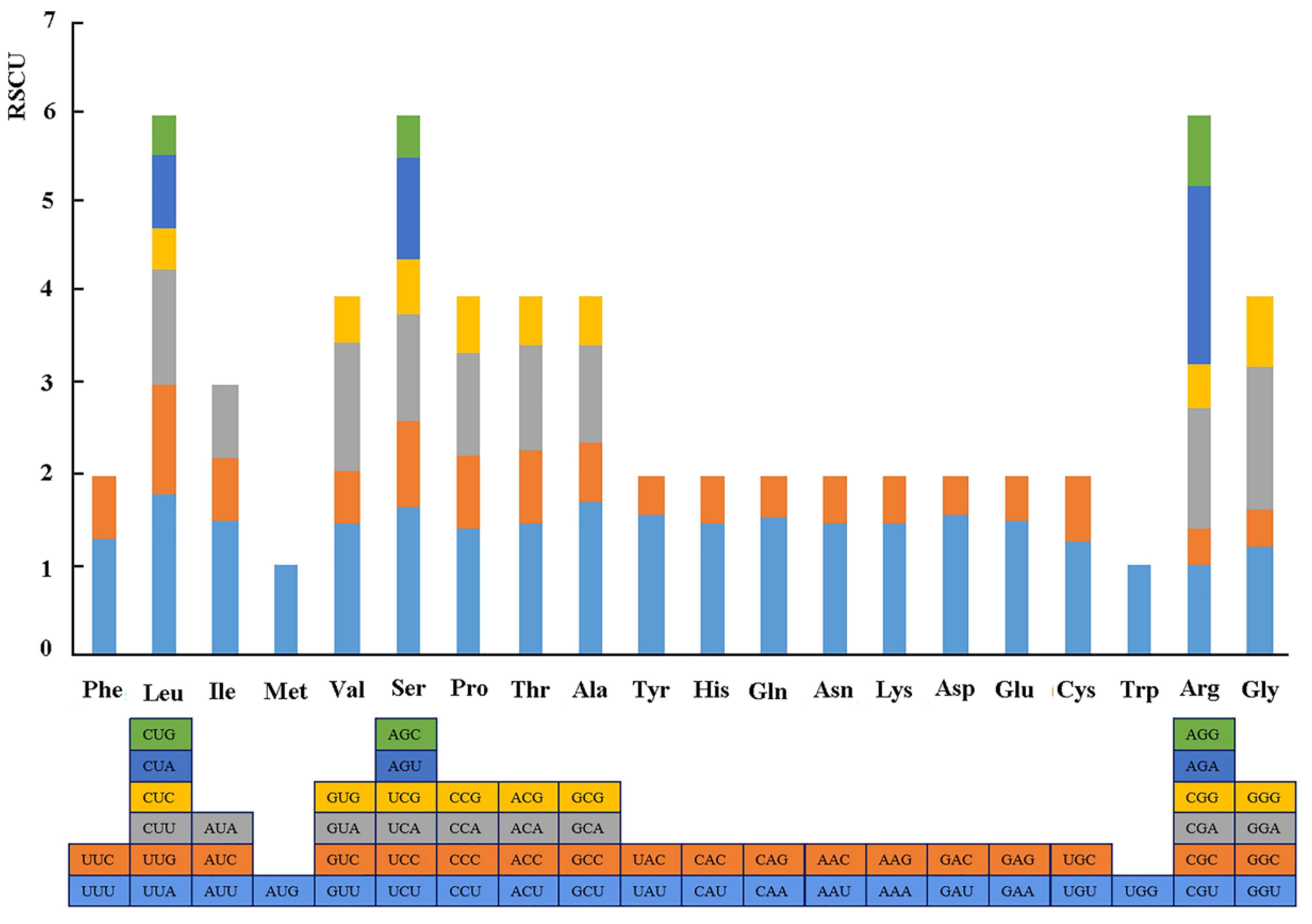
The results of the relative synonymous codon usage (RSCU), one of the commonly used parameters to measure codon usage bias, reveal that the 86 protein-coding genes of R. chinensis plastome included 26,654 codons (Figure 2). Among these codons, Leu was the most abundant amino acid, at 2805 (10.5%), while Cys was the least abundant, at 312 (1.2%). The 31 codons with RSCU values >1 mostly ended with A/T (U), while the 31 other codons having RSCU values <1 mostly ended with G/C. The remaining two, Met and Tyr, showed no biased usage (RSCU = 1).
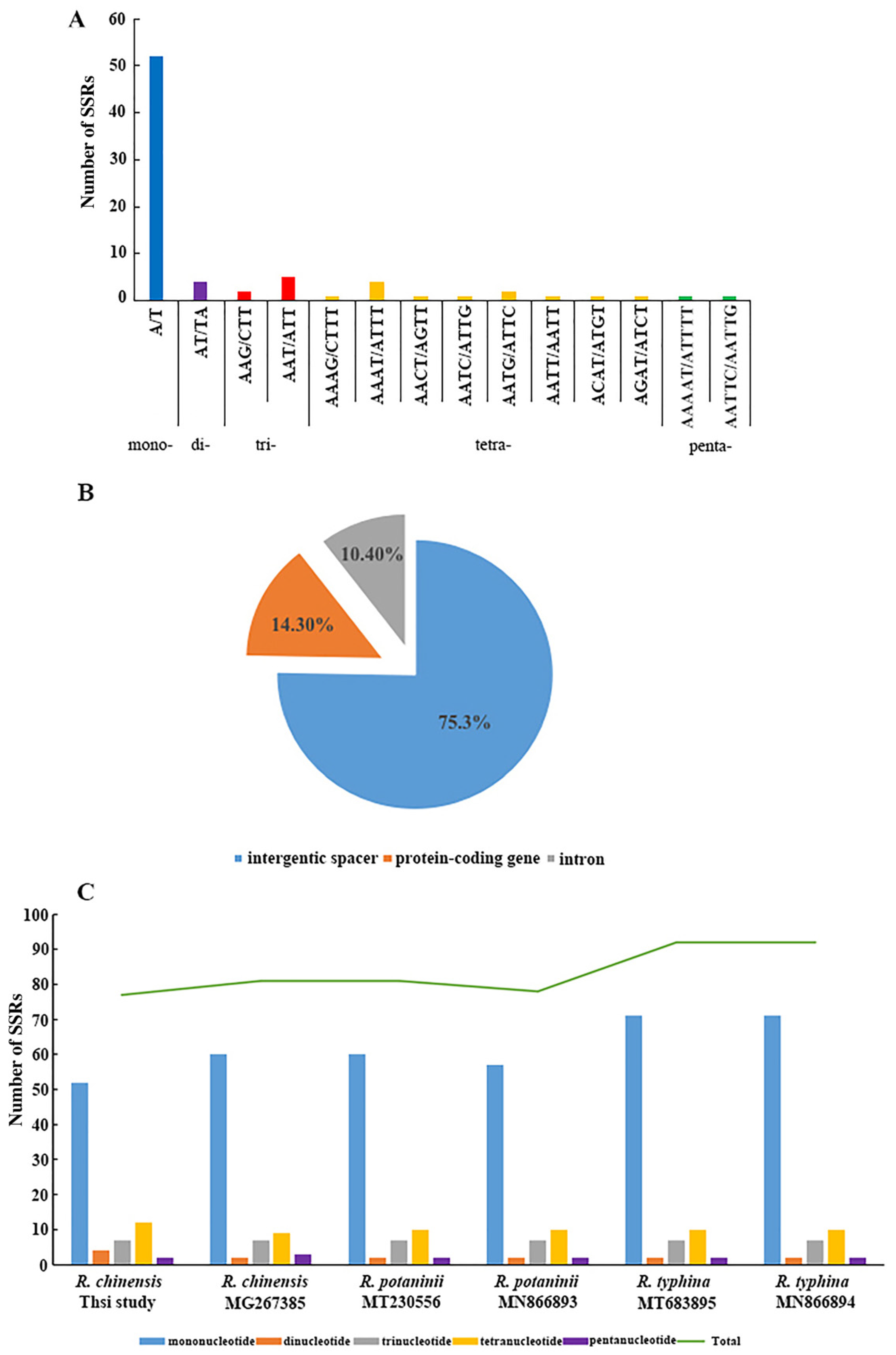
We performed the SSR analysis using the MISA program, and the results are shown in Figure 3. A total of 77 perfect microsatellites were identified in the chloroplast genome of our R. chinensis sample, which included 52 mononucleotides, 4 dinucleotides, 7 trinucleotides, 12 tetranucleotides, and 2 pentanucleotides, whereas the hexanucleotide repeats were not detected (Figure 3A). Among these SSRs, all mononucleotides were composed of A/T and all dinucleotides were composed of AT/TA, whereas the rest of the SSRs also had a high A/T content. Our findings are consistent with the previous reports, which showed that the SSRs in the chloroplast genome were composed of polyA or polyT repeats, while they rarely contained tandem G or C repeats. In the total SSRs loci, the repeats located in the LSC region were much higher (68.83%) than those in the SSC (18.18%) and IR (12.99%) regions (see Figure 3B). Most of these SSRs (75.4%) were located in intergenic regions (IGS), and the rest (24.6%) were located in protein-coding regions and tRNA genes (eight SSRs in introns).
3.2. Comparison among R. chinensis Accessions
We downloaded the three complete chloroplast genomes of R. chinensis from GenBank, which were from Gangwon (accession No. KX447140) in Korea, Shandong (accession No. MF351625) and Anhui (accession No. MG267385) in China, and examined their variation in the different accessions of the R. chinensis species. By comparison, we found great differences among these genomes, not only in the nucleotide variation but also in the length and gene numbers. Therefore, we re-annotated all the complete chloroplast genomes downloaded from GenBank to avoid the mistakes from the annotation. As a result, the sample with GenBank (accession No. MG267385) obtained more annotated genes. The diversity and variation of four complete chloroplast genomes of R. chinensis are shown in Table 4. We can see that the genomes ranged from 149,011 bp to 159,187 bp in length with the longest difference being of 10,176 bp between our current sample and the accession with No. KX447140. Our current sample exhibited the longest genome size and the most annotated genes compared to the other three accessions, and the sample with accession No. KX447140 had the shortest genome size and the least genes. All the chloroplast genomes displayed the quadripartite structure with a pair of IRs (16,602–26,550 bp) separated by the LSC (87,045–97,246 bp) and SSC (18,522–18,674 bp) regions, while they exhibited obvious differences in genome size and in the lengths of the LSC and IR regions. All four individuals varied in the number of genes, with the total gene number ranging from 126 to 132, the total protein-coding genes ranging from 82 to 86, and the total tRNA genes ranging from 36 to 37. All four complete chloroplast genomes contained eight rRNA genes. The overall GC content in each complete chloroplast genome was approximately 37.9%.
We examined the gene rearrangement of four complete chloroplast genomes of R. chinensis by alignment using the Mauve software implemented in Geneious (Figure 4A). All the four genomes had the same gene arrangement except for the missing regions, and the differences in genome length and in number of genes were mainly from the IR region. The gene organization and order in the IR region are shown in Figure 4B. Only one rpl2 intron-containing gene was observed in the IR region of the accessions KX447140 and MF351625, while the four genes rpl2, rpl23, trnI-CAU, and ycf2 had two copies in the two R. chinensis samples, our current study, and the accession No. MG267385. Furthermore, two more genes, namely rpl22 and rps19, were only annotated in our sample. The lengths of exons and introns in intron-carrying genes also exhibited some slight changes among four complete chloroplast genomes, and the length variations in the introns were greater than those in the exons (Table 3). In general, the exon lengths were conserved in the four R. chinensis individuals except for the ndhA and atpF genes.
We compared the nucleotide diversity in the total, LSC, SSC, and IR regions of the four complete chloroplast genomes of R. chinensis (Table 4). In total, 1436 variable sites (0.89%), including 93 parsimony-informative sites (0.06%), were examined in the chloroplast genomes. The LSC and SSC regions contributed 29 and 20 informative sites, respectively, while the IR regions only contributed 11 informative sites. Among these regions, the IR regions exhibited the least nucleotide diversity (0.00156) and the SSC region exhibited the highest divergence (0.00783).
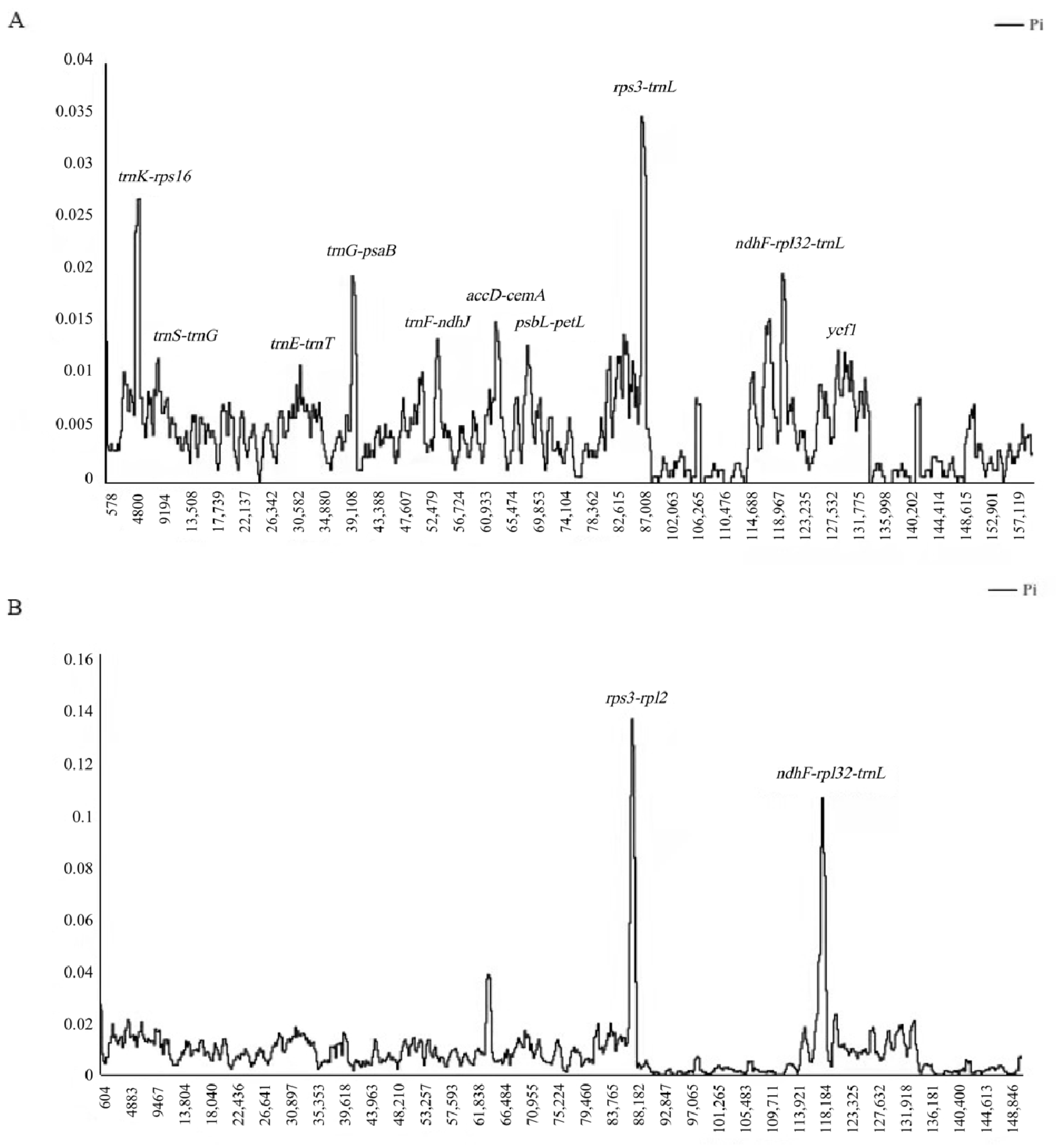
Genome-wide comparative analyses among the four individuals of R. chinensis showed that the non-coding region exhibited a higher nucleotide divergence than the coding region and the SC region with higher divergence than the IR regions, respectively. The peak with the highest degree (Pi: 0.035) of difference was located between the rps3 and trnL genes. By sequence alignment, there were missing fragments between the two rpl22 and ycf2 genes, including four genes: rps19, rpl2, rpl23, and trnI-CAU (Figure 5A). Furthermore, the nucleotide diversity (pi) value within 800 bp was calculated to estimate the sequence divergence level. The pi values varied from 0 to 0.035 for the four R. chinensis chloroplast genomes. With the exception of the rps3-trnL region, we identified 10 hotspot regions for genome divergence, 7 of which (trnK-rps16, trnS-trnG, trnE-trnT, trnG-psaB, trnF-ndhJ, accD-cemA, and psbL-petL) were located in the LSC region, and 3 (ndhF-rpl32-trnL, ccsA-ndhD, and ycf1) in the SSC region, while only 1 ycf1 was located in the coding region. Interestingly, the highly variable regions of R. chinensis were different from those previously reported for designing phylogenetic trees and the species identification of Rhus, such as trnL-trnF, trnC-trnD, and rbcL [17,18,19,20]. Thus, by analyzing the variable regions of the chloroplast genomes, we were able to identify some molecular markers, and these hotspot regions could be utilized as potential markers to reconstruct the phylogeny and plant identification.
3.3. Comparison among the Rhus Species
We downloaded the four complete chloroplast genomes of Rhus potaninii and R. typhina from GenBank, which were from Beijing (accession No. MN866893), Shaanxi (accession No. MT230556), Shandong (accession No. MT083895), and an unknown location (accession No. MN866894). Combined with R. chinensis, we conducted a comparative analysis to test their features and variation excluding two R. chinensis sequences (accession No. MF351625 and KX447140) due to their large missing regions (the sequences need to be confirmed). All the six Rhus chloroplast genomes ranged from 160,254 bp to 158,809 bp in length and contained a pair of inverted repeat regions (IRs: 26,475–26,550 bp), which were separated by a small single-copy region (SSC: 18,522–19,453 bp) and a large single-copy region (LSC: 87,045–87,789 bp). The number of the total genes in the chloroplast genomes ranged from 130 to 133, the number of total protein-coding genes ranged from 85 to 86, and all the genomes contained 37 tRNA genes and 8 rRNA genes, among which the 2 protein coding genes, namely rpl22 and rps19, were present in our R. chinensis, R. potaninii (accession Nos: MN866893 and MT230556), and R. typhina (accession No. MN866894 and MT083895) samples, but not in R. chinensis with accession No. MG267385.
In order to determine the level of sequence divergence, we compared nucleotide diversity in the whole genome, the LSC, SSC, and IR regions of six complete chloroplast genomes of the Rhus species. As shown in Table 4, in total, we examined 2931 variable sites (1.8%), including 1621 parsimony-informative sites (0.99%). The LSC and SSC regions contributed 1075 and 435 informative sites, respectively, while the IR regions only contributed 61 informative sites. The nucleotide diversity (Pi) among six Rhus species’ chloroplast genomes is 0.00841. The analysis of nucleotide diversity identified two significant peaks: rps3-rpl2 (Pi = 0.13533) and ndhF-rpl32-trnL (Pi = 0.10533), which are shown in Figure 5B. The first peak, rps3-rpl2, reflected the sequence variations that occur in R. chinensis (accession No. MG267385). The second one, ndhF-rpl32-trnL, indicated the sequence variations occurring in R. typhina (accession No. MN866894 and MT083895). These regions can be used as a source of potential barcodes for the identification of the Rhus species as well as resources for inferring the phylogenetic relationships of the genus.
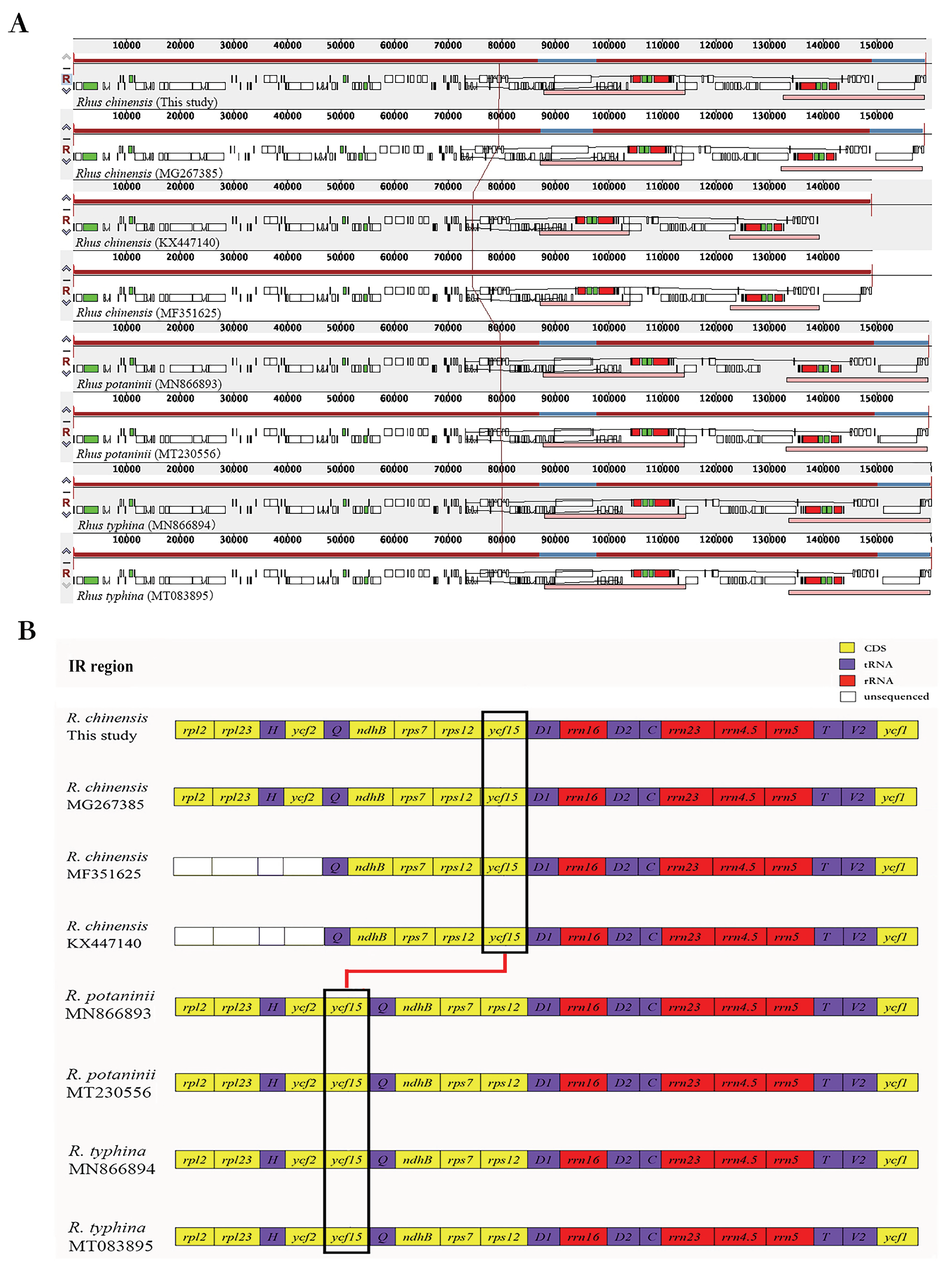
The mauve alignment revealed that all six species were relatively conserved. All three species, R. chinensis, R. potaninii, and R. typhina, revealed a syntenic structure, and no large-area and multi-segment gene rearrangement was detected in the cpDNA sequences. In particular, we found that the position of the ycf15 gene was different in R. chinensis than in other Rhus species. The ycf15 gene in the IR region was located between the rps12 gene and the trnV-GAC gene in R. chinensis; however, it was positioned between the ycf2 gene and the trnL-CAA gene in the species R. potaninii and R. typhina, which showed the gene rearrangement.
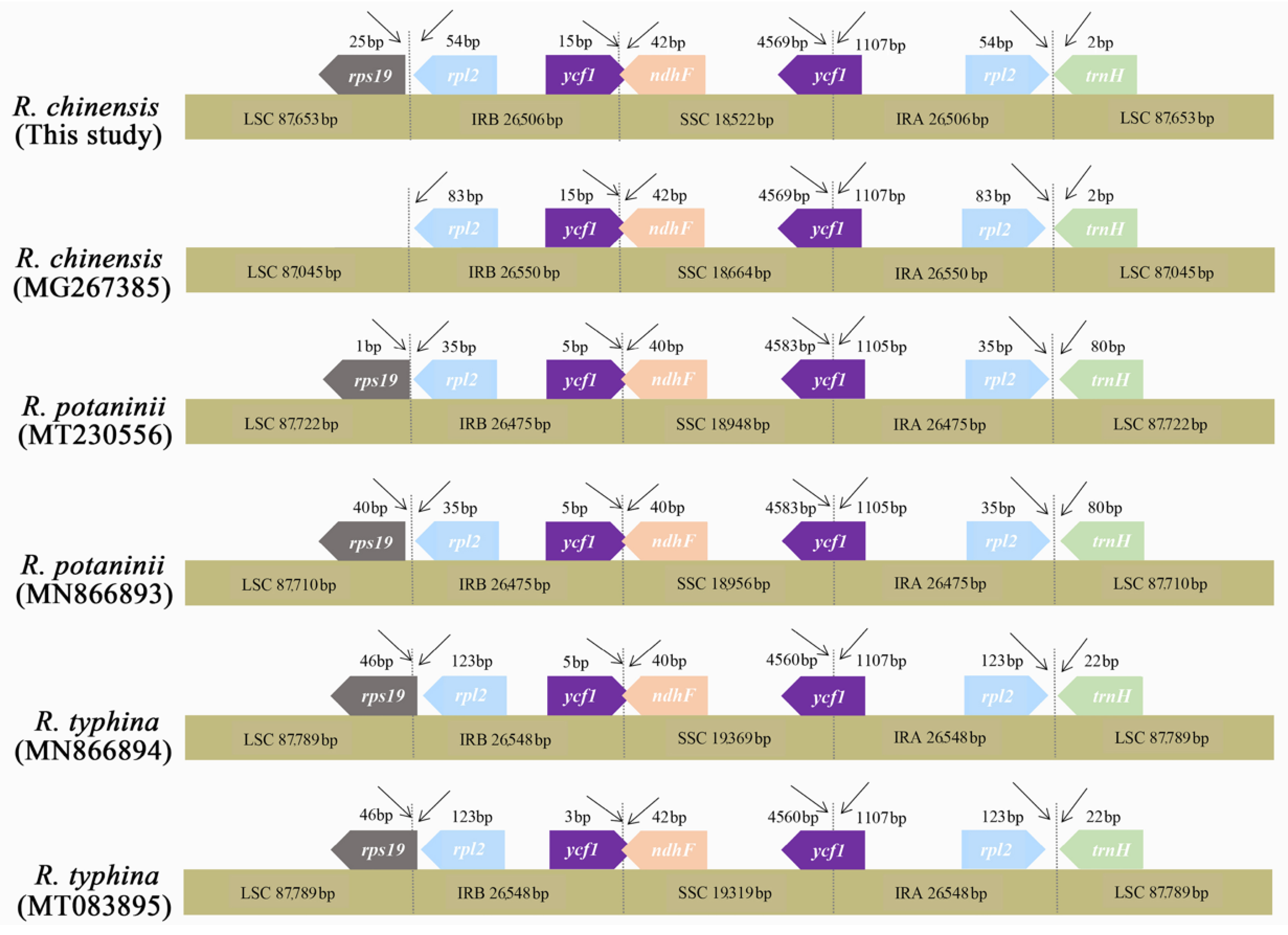
We compared the exact IR boundary positions and the adjacent genes of six Rhus species by IRscope, and the detailed comparison for the four genomic boundaries (LSC/IRA, LSC/IRB, SSC/IRA, and SSC/IRB) are shown in Figure 6. The lengths of the LSC, IR, and SSC regions were similar among the six Rhus genomes, and the IR organization was also highly conserved with minor variances for expansions and contractions. The functional ycf1 gene crossed the IRA/SSC boundary to create the ycf1 pseudogene fragment at the IRB region in all the genomes, which means that the ycf1 pseudogene overlapped with the ndhF gene in the SSC and IRa junctions with a stretch of 30 to 42 bp. The trnH and rpl2 genes were entirely located within the LSC and IR regions, respectively. The rps19 gene of R. chinensis and R. potaninii was entirely located in the LSC region, but it was positioned at the boundary between LSC and IRB in the two R. typhina accessions.
There have been many studies and reports on the expansion and contraction of the IR region, and the main view on the mechanism of the slighter IR region expansion is that it may be caused by gene transfer to different regions, while the larger IR region expansion may be realized by a double-strand break repair (DSBR) mechanism [45,46].
Although the chloroplast genome has a nearly collinear gene order in most land plants, the changes in the genome such as gene loss [47], expansion, and contraction at the borders of the IR regions cause size variations [42]. Our results likewise indicate that the downstream sequence of IRb/SSC was conserved and that the ndhF gene was adjacent to the IRb/SSC boundary, consistent with the general pattern described in angiosperms [48].
Barrett et al. (2020) found the loss of rps19 and rpl22 genes from the chloroplast genome of R. chinensis (accession No. MG267385) [49], while the two genes were annotated in our current study. With the acquisition of the new complete chloroplast genome of R. chinensis, more complete genomic information indicated that the rps19 gene was present in the R. chinensis sample and that there were some minor differences in its location compared to other Rhus species.
As shown in Figure 3C, the SSRs analysis for six Rhus species showed that the number of SSRs in R. typhina was the highest (92), while in R. chinensis it was the lowest (57). These SSRs are similar and divided into five types of microsatellites, i.e., mononucleotide, dinucleotide, trinucleotide, tetranucleotide, and pentanucleotide, most of which consisted of mono- and tri-repeat motifs. However, hexanucleotide repeats were not detected in the six Rhus species. The current results are consistent with the recent studies, which exhibited that the SSRs detected in the chloroplast genome of angiosperms were usually composed of A or T repeats and rarely comprised tandem G and C repeats [50]. Therefore, SSRs extended a greater contribution to the ‘AT’ diversity of the Rhus chloroplast genomes, as previously reported in different species [51]. These analyses also revealed that approximately 75% of the SSRs were determined in non-coding regions. Microsatellites being distributed throughout the genome, the SSRs in the chloroplast genome can be highly variable at the intra-specific level and may be used as genetic markers in population genetics and evolutionary studies, and in particular, polymorphic SSRs can be used to study genetic diversity, population structure, and biogeography within and between groups [52,53].
3.4. Phylogenetic Analysis
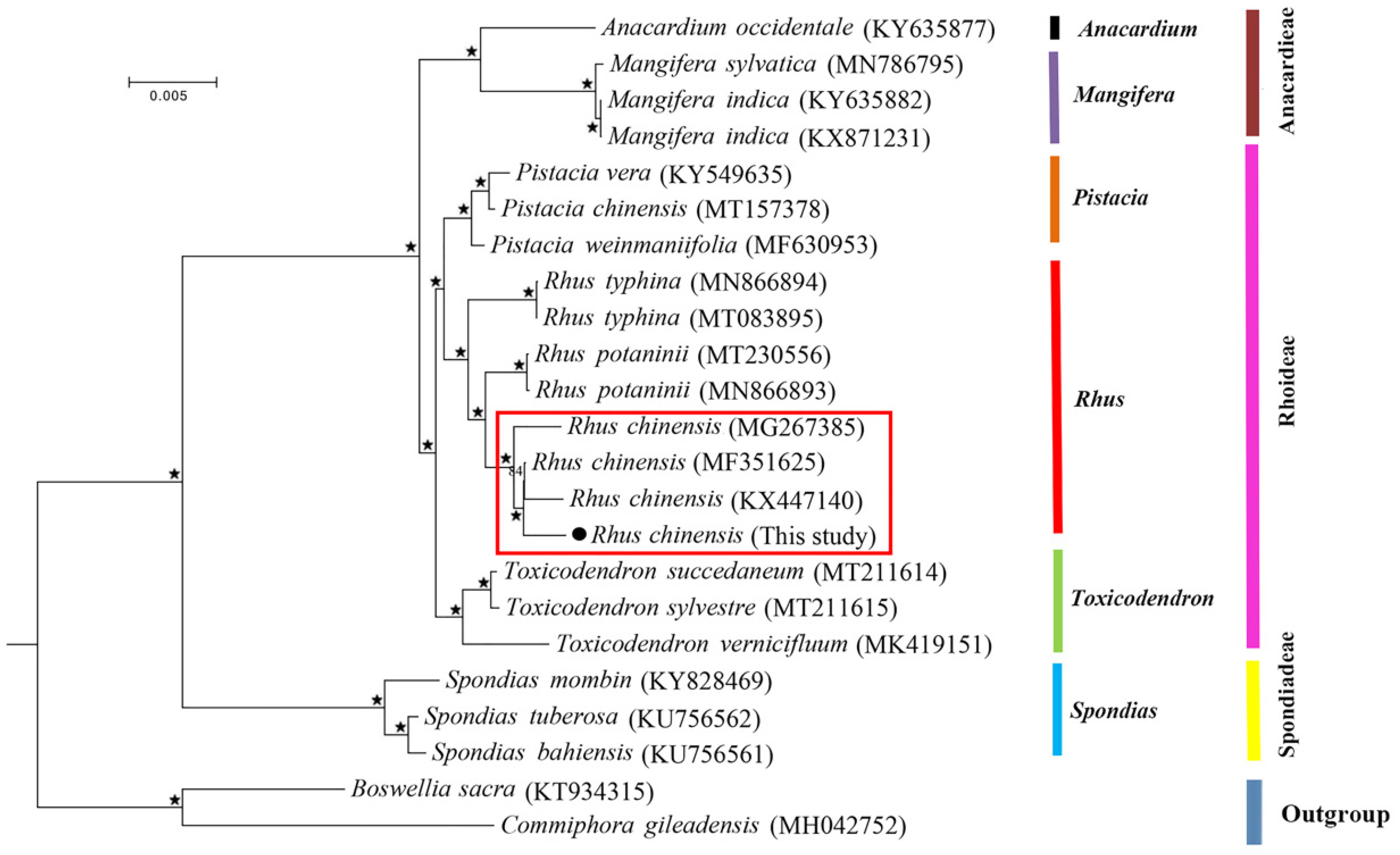
The complete chloroplast genomes provided valuable information in plant phylogenies due to their highly conserved structure and higher evolutionary rate when compared to the mitochondrial genome [54]. In recent decades, numerous analyses on the comparison of plastid protein-coding genes [55] and complete genome sequences [56] have been conducted to answer the phylogenetic disposition at deep nodes and the evolutionary relationships among angiosperms were further revealed. In this study, in order to present the relationships among the Anacardiaceae species, we conducted an ML phylogenetic tree based on all the 86 protein-coding genes from the complete chloroplast genomes with B. sacra and C. gileadensis in the family Burseraceae as outgroups. The multiple alignment of protein-coding sequences possessed 70,234 bp nucleotide sites, of which 4452 were variable and 2796 were parsimony informative. The ML tree is shown in Figure 7. The ML phylogenetic analysis supported the monophyly of each tribe and each genus well, and the samples were largely classified into three groups. The tribe Rhoideae was close to the tribe Anacardieae with a high support of 100%, and they then grouped with the tribe Spondiadeae. In the tribe Rhoideae clade, the genus Rhus was sister to Pistacia with a high support of 100%, and they then grouped with the Toxicodendron species, consistent with the findings from a previous analysis [57]. In the genus Rhus, our sample was closely clustered with the three other R. chinensis individuals, forming a sister to Rhus potaninii, which then grouped with R. typhina.
Chloroplast genome sequence has been widely used to reconstruct the phylogenetic relationships among plant lineages [58,59]. The current phylogenetic analysis showed the relationships between the species in the Anacardiaceae family. As an economic and ecological species, the phylogenetic relationship of R. chinensis is also a base for further edible and medicinal research. The more complete chloroplast genomes of the Rhus species would provide valuable genetic information for the further conservation and evolutionary research.
4. Conclusions
In this study, we assembled the complete chloroplast genome sequence of Rhus chinensis and identified its basic structure and gene content. After comparing this newly sequenced chloroplast genome with closely related species, we found some differences in genome size, GC content, gene number, and order between species. We also observed the contraction and expansion of the IR boundaries. Our findings demonstrate that most SSRs were A/T rich and located in the intergenic regions (IGS). The analysis of nucleotide divergence of the chloroplast genome sequences exhibited higher levels in the non-coding region than in the coding region. Ten highly variable regions were identified, which will potentially provide plastid markers for further taxonomic, phylogenetic, and population genetic studies in the Rhus genus. Furthermore, our analysis also determined gene rearrangement, that is, the position of the ycf15 gene was different in R. chinensis and other Rhus species. The phylogenetic trees constructed using all protein-coding genes supported the monophyly of each tribe, and Rhus was sister to Pistacia, which then grouped with Toxicodendron. The results obtained in this study are expected to provide valuable genetic resources to perform species identification, molecular breeding, and intraspecific diversity of the Rhus species.
Author Contributions
Formal analysis, methodology, data curation, writing—original draft, software, Y.X.; writing—review and editing, J.W.; writing—review and editing, X.S.; conceptualization, funding acquisition, investigation, project administration, resources, supervision, writing—review and editing, Z.R. All authors have read and agreed to the published version of the manuscript.
Funding
This study was partially supported by the National Natural Science Foundation of China (31870366, 31170359), Shanxi International Science and Technology Cooperation Project (201803D421051), Research Project Supported by Shanxi Scholarship Council of China (2020-018), and the National High Technology Research and Development ‘863′ Program (2014AA021802).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data which supports the findings of this study are openly available in GenBank of NCBI at https://www.ncbi.nlm.nih.gov (accessed on 30 August 2022), GenBank accession number: No. OP326720.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Rayne, S.; Mazza, G. Biological activities of extracts from sumac (Rhus spp.): A review. Plant Foods Hum. Nutr. 2007, 62, 165–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barkley, F.A. A monographic study of Rhus and its immediate allies in North and Central America, including the West Indies. Ann. Mo. Bot. Gard. 1937, 24, 265–498. [Google Scholar] [CrossRef]
- Andres-Hernandez, A.R.; Terrazas, T.; Salazar, G.; Ochoterena, H. Phylogenetic analysis based on structural and combined analyses of Rhus s.s. (Anacardiaceae). Bot. J. Linn. Soc. 2014, 176, 452–468. [Google Scholar] [CrossRef] [Green Version]
- Barfod, M. Anacardiaceae. In Flora of China; Science Press: Beijing, China, 2008; p. 11. [Google Scholar]
- Chen, J.C.; Ho, T.Y.; Chang, Y.S.; Wu, S.L.; Li, C.C.; Hsiang, C.Y. Identification of Escherichia coli enterotoxin inhibitors from traditional medicinal herbs by in silico, in vitro, and in vivo analyses. J. Ethnopharmacol. 2009, 121, 372. [Google Scholar] [CrossRef] [PubMed]
- Djakpo, O.; Yao, W. Rhus chinensis and Galla chinensis—Folklore to modern evidence: Review. Phytother. Res. 2010, 24, 1739–1747. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.Y.; Oh, C.H. Growth and spatial distribution characteristics of Rhus javanica populations sowed on cut-slopes. J. Korean Soc. Environ. Restor. Technol. 2015, 18, 39–49. [Google Scholar] [CrossRef] [Green Version]
- Nam, S.J.; Yeo, W.J.; Choi, J.H.; Kim, N.C. Development of revegetation method using forest topsoils for ecological restoration of the slopes (I). J. Korean Soc. Environ. Restor. Technol. 2004, 7, 110–119. [Google Scholar]
- Zhang, G.X.; Qiao, G.X.; Zhong, T.S.; Zhang, W.Y. Fauna Sinica Insecta. Homoptera: Mindaridae and Pemphigidae; Science Press: Beijing, China, 1999; p. 14. [Google Scholar]
- Yang, Z.X.; Chen, X.M.; Havill, N.P.; Feng, Y.; Chen, H. Phylogeny of Rhus gall aphids (Hemiptera: Pemphigidae) based on combined molecular analysis of nuclear EF1 and mitochondrial COII genes. Entomol. Sci. 2010, 13, 351–357. [Google Scholar] [CrossRef]
- Su, T.C. Protection and utilization of wild Anacardiaceae plant resources in the mountainous areas of Eastern Liaoning. J. Liaoning For. Sci. Technol. 2016, 3, 59–60. [Google Scholar]
- Yang, X.Y.; Wang, J.; Li, C.; Ren, Z.M.; Ma, W.L. Cloning, expression and characterization of chalcone isomerase from medicinal plant Chinese sumac (Rhus chinensis). Chin. J. Chin. Mat. Med. 2019, 44, 3253–3260. [Google Scholar] [CrossRef]
- Ye, M.; Wen, X.; He, D.Q.; Wu, X.; Lai, W.F.; Zhang, X.Q.; Lin, Y.; Xu, W.; Li, X.W. Dammarane-type triterpenoids from the roots of Rhus chinensis and their preventive effects on zebrafish heart failure and thrombosis. J. Nat. Prod. 2020, 83, 362–373. [Google Scholar] [CrossRef]
- Ma, W.L.; Wu, M.; Wu, Y.; Ren, Z.M.; Zhong, Y. Cloning and characterisation of a phenylalanine ammonia-lyase gene from Rhus chinensis. Plant Cell. Rep. 2013, 32, 1179–1190. [Google Scholar] [CrossRef]
- Liang, Y.K.; Zhang, Y.; Wen, J.; Su, X.; Ren, Z.M. Evolutionary history of Rhus chinensis (Anacardiaceae) from the temperate and subtropical zones of China based on cpDNA and nuclear DNA sequences and ecological niche model. Front. Genet. 2019, 10, 171. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Yang, Z.X.; Chen, X.M.; Liu, P.; Tang, Y.F. Effects of Chinese gallnut on photosynthetic characteristics and total nitrogen content of Rhus chinensis. Acta Ecol. Sin. 2013, 33, 6876–6884. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.K.; Kim, M.J.; Heo, K. Phylogeny of Korean Rhus spp. based on ITS and rbcL sequences. J. Korean Med. Sci. 2004, 12, 60–66. [Google Scholar]
- Miller, A.J.; Young, D.A.; Wen, J. Phylogeny and biogeography of Rhus (Anacardiaceae) based on ITS sequences data. Int. J. Plant Sci. 2001, 162, 1401–1407. [Google Scholar] [CrossRef]
- Yi, T.S.; Miller, A.J.; Wen, J. Phylogenetic and bio-geographic diversification of Rhus (Anacardiaceae) in the Northern Hemisphere. Mol. Phylogenet. Evol. 2004, 33, 861–879. [Google Scholar] [CrossRef]
- Yi, T.S.; Miller, A.J.; Wen, J. Phylogeny of Rhus (Anacardiaceae) based on sequences of nuclear Nia-i3 intron and chloroplast trnC-trnD. Syst. Bot. 2007, 32, 379–391. [Google Scholar] [CrossRef]
- Kim, I.; Park, J.Y.; Lee, Y.S.; Joh, H.J.; Kang, S.J.; Murukarthick, J.M.; Lee, H.O.; Hur, Y.J.; Kim, Y.; Kim, K.H.; et al. The complete chloroplast genome sequence and intra-species diversity of Rhus chinensis. Plant. Breed. Biotechnol. 2017, 5, 243–251. [Google Scholar] [CrossRef] [Green Version]
- Neuhaus, H.; Emes, M. No photosynthetic metabolism in plastids. Annu. Rev. Plant Biol. 2000, 51, 111–140. [Google Scholar] [CrossRef]
- Henry, R.J. Importance of plant diversity. In Plant Diversity and Evolution: Genotypic and Phenotypic Variation in Higher Plants; CABI Publishing: Cambridge, MA, USA, 2005; pp. 1–5. [Google Scholar]
- Raubeson, L.A.; Jansen, R.K. Chloroplast genomes of plants. In Plant Diversity and Evolution: Genotypic and Phenotypic Variation in Higher Plants; CABI Publishing: Cambridge, MA, USA, 2005; pp. 45–68. [Google Scholar]
- Cheng, Y.J.; De Vicente, M.C.; Meng, H.J.; Guo, W.W.; Tao, N.G.; Deng, X.X. A set of primers for analyzing chloroplast DNA diversity in Citrus and related genera. Tree Physiol. 2005, 25, 661–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, W.P.; Liu, J.; Yu, J.; Wang, L.; Zhou, S.L. Highly variable chloroplast markers for evaluating plant phylogeny at low taxonomic levels and for DNA barcoding. PLoS ONE 2012, 7, e35071. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.N.; Zhu, Z.C.; Lu, Z.K.; Ding, Z.; Zhang, C.; Luan, F.S. The complete chloroplast genome sequence of the Sechium edule (Jacq.) Swartz. (Cucurbitaceae). Mitochondrial DNA Part B 2021, 6, 97–98. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.X.; Movahedi, A.; Yang, W.G.; Xu, D.Z.; Jiang, C.B.; Xie, J.G.; Zhang, Y. The complete chloroplast genome and characteristics analysis of Musa basjoo Siebold. Mol. Biol. Rep. 2021, 48, 7113–7125. [Google Scholar] [CrossRef] [PubMed]
- Skuza, L.; Gastineau, R.; Sielska, A. The complete chloroplast genome of Secale sylvestre (Poaceae: Triticeae). J. Appl. Genet. 2022, 63, 115–117. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Kim, I.; Kim, J.K.; Park, J.Y.; Joh, H.J.; Park, H.S.; Lee, H.O.; Lee, S.C.; Hur, Y.J.; Yang, T.J. The complete chloroplast genome sequence of Rhus chinensis Mill (Anacardiaceae). Mitochondrial DNA Part B 2016, 1, 696–697. [Google Scholar] [CrossRef] [Green Version]
- Zuo, R.H.; Jiang, P.; Sun, C.B.; Chen, C.W.; Lou, X.J. Analysis of the chloroplast genome characteristics of Rhus chinensis by de novo sequencing. J. Biotechnol. 2020, 36, 772–781. [Google Scholar] [CrossRef]
- Zimmer, E.A.; Wen, J. Using nuclear gene data for plant phylogenetics: Progress and prospects II next-gen approaches. J. Syst. Evol. 2015, 53, 371–379. [Google Scholar] [CrossRef]
- Jin, J.J.; Yu, W.B.; Yang, J.B.; Song, Y.; Depamphilis, C.W.; Yi, T.S.; Li, D.Z. GetOrganelle: A fast and versatile toolkit for accurate denovo assembly of organelle genomes. Genome Biol. 2020, 21, 241. [Google Scholar] [CrossRef]
- Qu, X.J.; Moore, M.J.; Li, D.Z.; Yi, T.S. PGA: A software package for rapid, accurate, and flexible batch annotation of plastomes. Plant Methods. 2019, 15, 50. [Google Scholar] [CrossRef] [Green Version]
- Schattner, P.; Brooks, A.N.; Lowe, T.M. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 2005, 33, 686–689. [Google Scholar] [CrossRef]
- Lohse, O.; Drechsel, O.; Kahlau, S.; Bock, R. Organellar genome DRAW-a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 2013, 41, 575–581. [Google Scholar] [CrossRef]
- MISA-Microsatellite Identification Tool. 2010. Available online: http://pgrc.ipk-gatersleben.de/misa/ (accessed on 27 September 2010).
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. Mafft online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2017, 20, 1160–1166. [Google Scholar] [CrossRef] [Green Version]
- Shigehiro, K.; Zmasek, C.M.; Osamu, N.; Kazutaka, K. Aleaves facilitates on-demand exploration of metazoan gene family trees on mafft sequence alignment server with enhanced interactivity. Nucleic Acids Res. 2013, 41, 22–28. [Google Scholar] [CrossRef] [Green Version]
- Stamatakis, A. Raxml version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [Green Version]
- Saina, J.K.; Li, Z.Z.; Gichira, A.W.; Liao, Y.Y. The complete chloroplast genome sequence of tree of heaven (Ailanthus altissima Mill) (Sapindales: Simaroubaceae), an important pantropical tree. Int. J. Mol. Sci. 2018, 19, 929. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.Y.; Li, C.; Miao, H.M.; Xiong, S.J. Insights from the complete chloroplast genome into the evolution of Sesamum indicum L. PLoS ONE 2013, 8, e80508. [Google Scholar] [CrossRef]
- Zhou, T.; Chen, C.; Wei, Y.; Chang, Y.; Bai, G.; Li, Z.; Kanwal, N.; Zhao, G. Comparative transcriptome and chloroplast genome analyses of two related Dipteronia species. Front. Plant Sci. 2016, 7, 1512. [Google Scholar] [CrossRef] [Green Version]
- Goulding, S.E.; Wolfe, K.H.; Olmstead, R.G.; Morden, C.W. Ebb and flow of the chloroplast inverted repeat. Mol. Gen. Genet. 1996, 252, 195–206. [Google Scholar] [CrossRef]
- Wang, R.J.; Cheng, C.L.; Chang, C.C.; Chaw, S.M. Dynamics and evolution of the inverted repeat-large single copy junctions in the chloroplast genomes of monocots. BMC Evol. Biol. 2008, 8, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, P.C.; Zhang, Y.Z.; Geng, H.M.; Chen, S.L. The complete chloroplast genome sequence of Gentiana lawrencei var. farreri (Gentianaceae) and comparative analysis with its congeneric species. PeerJ 2016, 4, e2540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.J.; Lee, H.L. Complete chloroplast genome sequences from Korean ginseng (Panax schinseng Nees) and comparative analysis of sequence evolution among 17 vascular plants. DNA Res. 2004, 11, 247–261. [Google Scholar] [CrossRef] [PubMed]
- Barrett, C.F. Plastid genomes of the North American Rhus integrifolia-ovata complex and phylogenomic implications of inverted repeat structural evolution in Rhus L. PeerJ 2020, 8, e9315. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.P.; Xu, C.; Cheng, T.; Lin, K.; Zhou, S.L. Sequencing angiosperm plastid genomes made easy: A complete set of universal primers and a case study on the phylogeny of Saxifragales. Genome Biol. Evol. 2013, 5, 989–997. [Google Scholar] [CrossRef] [Green Version]
- Dong, W.P.; Xu, C.; Li, D.L.; Jin, X.B.; Lu, Q.; Suo, Z.L. Comparative analysis of the complete chloroplast genome sequences in psammophytic Haloxylon species (Amaranthaceae). PeerJ 2016, 4, e2699. [Google Scholar] [CrossRef] [Green Version]
- Kaur, S.; Panesar, P.S.; Bera, M.B.; Kaur, V. Simple sequence repeat markers in genetic divergence and marker-assisted selection of rice cultivars: A review. Crit. Rev. Food Sci. Nutr. 2015, 55, 41–49. [Google Scholar] [CrossRef]
- Yang, Y.; Zhou, T.; Duan, D.; Yang, J.; Feng, L.; Zhao, G. Comparative analysis of the complete chloroplast genomes of five Quercus species. Front. Plant Sci. 2016, 7, 959. [Google Scholar] [CrossRef] [Green Version]
- Chaney, L.; Mangelson, R.; Ramaraj, T.; Jellen, E.N.; Maughan, P.J. The complete chloroplast genome sequences for four Amaranthus species (Amaranthaceae). Appl. Plant Sci. 2016, 4, 1600063. [Google Scholar] [CrossRef] [Green Version]
- Moore, M.J.; Soltis, P.S.; Bell, C.D.; Burleigh, J.G.; Soltis, D.E. Phylogenetic analysis of 83 plastid genes further resolves the early diversification of eudicots. Proc. Natl. Acad. Sci. USA 2010, 107, 4623–4628. [Google Scholar] [CrossRef] [Green Version]
- Dobes, C.; Paule, J. A comprehensive chloroplast DNA-based phylogeny of the genus Potentilla (Rosaceae): Implications for its geographic origin, phylogeography and generic circumscription. Mol. Phylogenet. Evol. 2010, 56, 156–175. [Google Scholar] [CrossRef]
- Pan, Y.J.; Feng, J.L.; Nie, L.P.; Cui, Y.X.; Yang, C.H.; Lin, Y.; Yao, H. The complete chloroplast genome sequence of Rhus potaninii. Mitochondrial DNA Part B 2020, 3, 2425–2426. [Google Scholar] [CrossRef]
- Sun, L.; Fang, L.; Zhang, Z.; Chang, X.; Penny, D.; Zhong, B. Chloroplast phylogenomic inference of green algae relationships. Sci. Rep. 2016, 6, 20528. [Google Scholar] [CrossRef]
- Du, Y.P.; Bi, Y.; Yang, F.P.; Zhang, M.F.; Chen, X.Q.; Xue, J.; Zhang, X.H. Complete chloroplast genome sequences of Lilium: Insights into evolutionary dynamics and phylogenetic analyses. Sci. Rep. 2017, 7, 5751. [Google Scholar] [CrossRef]
Figure 1.
Gene map of the complete chloroplast genome of R. chinensis. The genes inside and outside of the circle are transcribed in clockwise and counterclockwise directions, respectively. The light and darker gray in the inner circle correspond to AT and GC content, respectively.
Figure 1.
Gene map of the complete chloroplast genome of R. chinensis. The genes inside and outside of the circle are transcribed in clockwise and counterclockwise directions, respectively. The light and darker gray in the inner circle correspond to AT and GC content, respectively.

Figure 2.
Relative synonymous codon usage (RSCU) of the complete chloroplast genome of R. chinensis in this study.
Figure 2.
Relative synonymous codon usage (RSCU) of the complete chloroplast genome of R. chinensis in this study.

Figure 3.
SSRs in the chloroplast genome of Rhus species. (A) Frequency of identified SSR motifs in R. chinensis. Mono-: Mononucleotide, Di-: Dinucleotide, Tri-: Trinucleotide, Tetra-: Tetranucleotide, Penta-: Pentanucleotide, Hexa-: Hexanucleotide. (B) Location distribution of all the SSR motifs in R. chinensis. (C) Number of different SSR types detected in complete chloroplast genomes of Rhus.
Figure 3.
SSRs in the chloroplast genome of Rhus species. (A) Frequency of identified SSR motifs in R. chinensis. Mono-: Mononucleotide, Di-: Dinucleotide, Tri-: Trinucleotide, Tetra-: Tetranucleotide, Penta-: Pentanucleotide, Hexa-: Hexanucleotide. (B) Location distribution of all the SSR motifs in R. chinensis. (C) Number of different SSR types detected in complete chloroplast genomes of Rhus.

Figure 4.
Gene arrangement of eight chloroplast genomes of Rhus species. (A) Complete genomes. (B) IR regions. The gene ycf15 in the black frame changed position (rearranged) in the chloroplast genome of Rhus species.
Figure 4.
Gene arrangement of eight chloroplast genomes of Rhus species. (A) Complete genomes. (B) IR regions. The gene ycf15 in the black frame changed position (rearranged) in the chloroplast genome of Rhus species.

Figure 5.
Analysis of nucleotide diversity of the Rhus chloroplast genomes. (A) Four individuals (window length: 800 bp; step size: 200 bp). X-axis: position of the midpoint of a window; Y-axis: nucleotide diversity of each window. (B) Six chloroplast genomes of Rhus except R. chinensis (accession Nos: KX447140 and MF351625) (window length: 800 bp; step size: 200 bp). X-axis: position of the midpoint of a window; Y-axis: nucleotide diversity of each window.
Figure 5.
Analysis of nucleotide diversity of the Rhus chloroplast genomes. (A) Four individuals (window length: 800 bp; step size: 200 bp). X-axis: position of the midpoint of a window; Y-axis: nucleotide diversity of each window. (B) Six chloroplast genomes of Rhus except R. chinensis (accession Nos: KX447140 and MF351625) (window length: 800 bp; step size: 200 bp). X-axis: position of the midpoint of a window; Y-axis: nucleotide diversity of each window.

Figure 6.
Comparison of the SC/IR junction regions for complete chloroplast genomes of Rhus species. The number above the gene features indicates the distance between the ends of the genes and the border sites. Features are not to scale.
Figure 6.
Comparison of the SC/IR junction regions for complete chloroplast genomes of Rhus species. The number above the gene features indicates the distance between the ends of the genes and the border sites. Features are not to scale.

Figure 7.
Maximum likelihood (ML) phylogeny of Rhus and other species of Anacardiaceae based on all 86 protein-coding genes with B. sacra and C. gileadensis as outgroups (stars represent nodes with 100% bootstrap values; the clade in the red frame shows the four accessions from the species R. chinensis).
Figure 7.
Maximum likelihood (ML) phylogeny of Rhus and other species of Anacardiaceae based on all 86 protein-coding genes with B. sacra and C. gileadensis as outgroups (stars represent nodes with 100% bootstrap values; the clade in the red frame shows the four accessions from the species R. chinensis).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Information of complete chloroplast genomes of Anacardiaceae species used in this study.
| Species | Family | Accession No. | Size (bp) |
|---|---|---|---|
| R. chinensis | Anacardiaceae | This Study | 159,187 |
| R. chinensis | Anacardiaceae | KX447140 | 149,011 |
| R. chinensis | Anacardiaceae | MF351625 | 149,094 |
| R. chinensis | Anacardiaceae | MG267385 | 158,809 |
| R. potaninii | Anacardiaceae | MT230556 | 159,620 |
| R. potaninii | Anacardiaceae | MN866893 | 159,616 |
| R. typhina | Anacardiaceae | MT083895 | 160,204 |
| R. typhina | Anacardiaceae | MN866894 | 160,254 |
| Pistacia chinensis | Anacardiaceae | MT157378 | 160,596 |
| Pistacia vera | Anacardiaceae | KY549635 | 160,674 |
| P. weinmanniifolia | Anacardiaceae | MF630953 | 160,767 |
| Toxicodendron succedaneum | Anacardiaceae | MT211614 | 150,710 |
| Toxicodendron sylvestre | Anacardiaceae | MT211615 | 159,600 |
| Toxicodendron vernicifluum | Anacardiaceae | MK419151 | 159,571 |
| Mangifera indica | Anacardiaceae | KX871231 | 157,780 |
| Mangifera indica | Anacardiaceae | KY635882 | 157,780 |
| Mangifera sylvatica | Anacardiaceae | MN786795 | 158,106 |
| Anacardium occidentale | Anacardiaceae | KY635877 | 172,199 |
| Spondias tuberosa | Anacardiaceae | KU756562 | 162,039 |
| Spondias bahiensis | Anacardiaceae | KU756561 | 162,218 |
| Spondias mombin | Anacardiaceae | KY828469 | 162,302 |
| B. sacra | Burseraceae | KT934315 | 160,543 |
| C. gileadensis | Burseraceae | MH042752 | 160,268 |
Table 2.
Characteristics of the complete chloroplast genomes of Rhus species.
| Species | R. chinensis | R. potaninii | R. typhina | |||||
|---|---|---|---|---|---|---|---|---|
| Accession No. | OP326720 | KX447140 | MF351625 | MG267385 | MN866893 | MT230556 | MN866894 | MT083895 |
| Location | Hubei | Gangwon | Shandong | Anhui | Beijing | Shaanxi | Unknown | Shandong |
| Size (bp) | 159,187 | 149,011 | 149,094 | 158,809 | 159,616 | 159,620 | 160,254 | 160,204 |
| LSC (bp) | 87,653 | 96,882 | 97,246 | 87,045 | 87,710 | 87,722 | 87,789 | 87,789 |
| SSC (bp) | 18,522 | 18,674 | 18,644 | 18,664 | 18,956 | 18,948 | 19,453 | 19,319 |
| IR (bp) | 26,506 | 16,741 | 16,602 | 26,550 | 26,475 | 26,475 | 26,506 | 26,548 |
| No. of total genes | 132 | 126 | 126 | 130 | 131 | 133 | 130 | 130 |
| Protein-coding genes | 86 | 82 | 82 | 85 | 86 | 86 | 85 | 85 |
| tRNAs | 37 | 36 | 36 | 37 | 37 | 37 | 37 | 37 |
| rRNAs | 8 | 8 | 8 | 8 | 8 | 8 | 8 | 8 |
| Overall GC content (%) | 37.8 | 37.8 | 37.9 | 37.9 | 37.9 | 37.9 | 37.8 | 37.8 |
| GC content of LSC (%) | 35.9 | 36.2 | 36.2 | 36.0 | 36.0 | 36.0 | 35.8 | 35.8 |
| GC content of SSC (%) | 32.6 | 32.7 | 32.7 | 32.6 | 32.6 | 32.6 | 32.5 | 32.5 |
| GC content of IR (%) | 42.9 | 45.4 | 45.5 | 42.6 | 43.0 | 43.0 | 43.0 | 43.0 |
Table 3.
Lengths of exons and introns of coding genes in the complete chloroplast genomes of four R. chinensis accessions.
Table 3.
Lengths of exons and introns of coding genes in the complete chloroplast genomes of four R. chinensis accessions.
| Gene | Exon Ⅰ (bp) | Intron Ⅰ (bp) | Exon Ⅱ (bp) | Intron Ⅱ (bp) | Exon Ⅲ (bp) | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 1 | 2 | 3 | 4 | 1 | 2 | 3 | 4 | 1 | 2 | 3 | 4 | 1 | 2 | 3 | 4 | |
| rps16 | 226 | 226 | 227 | 226 | 894 | 899 | 896 | 882 | 38 | 38 | 40 | 38 | ||||||||
| atpF | 411 | 748 | 748 | 760 | 747 | 156 | 156 | 144 | 156 | |||||||||||
| rpoC1 | 1626 | 760 | 760 | 760 | 755 | 435 | ||||||||||||||
| ycf3 | 155 | 813 | 797 | 797 | 798 | 226 | 732 | 735 | 733 | 734 | 126 | |||||||||
| clpP | 228 | 631 | 653 | 638 | 637 | 291 | 291 | 292 | 291 | 773 | 776 | 768 | 775 | 69 | 69 | 71 | 69 | |||
| petB | 6 | 785 | 786 | 785 | 772 | 642 | ||||||||||||||
| petD | 8 | 741 | 724 | 741 | 750 | 475 | ||||||||||||||
| rpl16 | 402 | 1068 | 1071 | 1068 | 1133 | 9 | ||||||||||||||
| rpl2 | 393 | 662 | 662 | 662 | 661 | 435 | ||||||||||||||
| ndhB | 777 | 681 | 756 | |||||||||||||||||
| rps12 | 232 | 536 | 26 | |||||||||||||||||
| ndhA | 541 | 1120 | 1120 | 1129 | 1124 | 467 | 467 | 551 | 467 | |||||||||||
| rps12 | 26 | 536 | 232 | |||||||||||||||||
| ndhB | 756 | 681 | 777 | |||||||||||||||||
| rpl2 | 434 | — | — | 434 | 665 | — | — | 664 | 391 | — | — | 391 | ||||||||
| trnK-UUU | 35 | 2599 | 2593 | 2596 | 2596 | 37 | ||||||||||||||
| trnG-UCC | 23 | 714 | 713 | 713 | 713 | 47 | ||||||||||||||
| trnL-UAA | 37 | 450 | 50 | |||||||||||||||||
| trnV-UAC | 37 | 585 | 39 | |||||||||||||||||
| trnA-UGC | 35 | 841 | 38 | |||||||||||||||||
| trnI-GAU | 35 | 950 | 949 | 950 | 940 | 42 | ||||||||||||||
| trnI-GAU | 42 | 950 | 949 | 950 | 940 | 35 | ||||||||||||||
| trnA-UGC | 38 | 841 | 35 | |||||||||||||||||
Notes: 1, This study; 2, KX447140; 3, MF351625; 4, MG267385.
Table 4.
Variable site of complete chloroplast genomes of four R. chinensis accessions and six Rhus species.
Table 4.
Variable site of complete chloroplast genomes of four R. chinensis accessions and six Rhus species.
| Species | Region | Total Sites | Variable Sites | Informative Sites | Nucleotide Diversity |
|---|---|---|---|---|---|
| R. chinensis | Large single-copy region | 98,636 | 969 | 29 | 0.00584 |
| Small single-copy region | 18,751 | 281 | 20 | 0.00783 | |
| Inverted repeat region | 27,478 | 48 | 11 | 0.00156 | |
| Complete cp genome | 160,884 | 1436 | 93 | 0.00502 | |
| Six Rhus species | Large single-copy region | 89,280 | 1743 | 1075 | 0.00958 |
| Small single-copy region | 19,572 | 616 | 435 | 0.01614 | |
| Inverted repeat region | 27,432 | 109 | 61 | 0.00184 | |
| Complete cp genome | 162,196 | 2931 | 1621 | 0.00841 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Xu, Y.; Wen, J.; Su, X.; Ren, Z. Variation among the Complete Chloroplast Genomes of the Sumac Species Rhus chinensis: Reannotation and Comparative Analysis. Genes 2022, 13, 1936. https://doi.org/10.3390/genes13111936
AMA Style
Xu Y, Wen J, Su X, Ren Z. Variation among the Complete Chloroplast Genomes of the Sumac Species Rhus chinensis: Reannotation and Comparative Analysis. Genes. 2022; 13(11):1936. https://doi.org/10.3390/genes13111936
Chicago/Turabian StyleXu, Yujie, Jun Wen, Xu Su, and Zhumei Ren. 2022. "Variation among the Complete Chloroplast Genomes of the Sumac Species Rhus chinensis: Reannotation and Comparative Analysis" Genes 13, no. 11: 1936. https://doi.org/10.3390/genes13111936
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.