
Genetic Characterization, Current Model Systems and Prognostic Stratification in PAX Fusion-Negative vs. PAX Fusion-Positive Rhabdomyosarcoma



Abstract
:1. Introduction
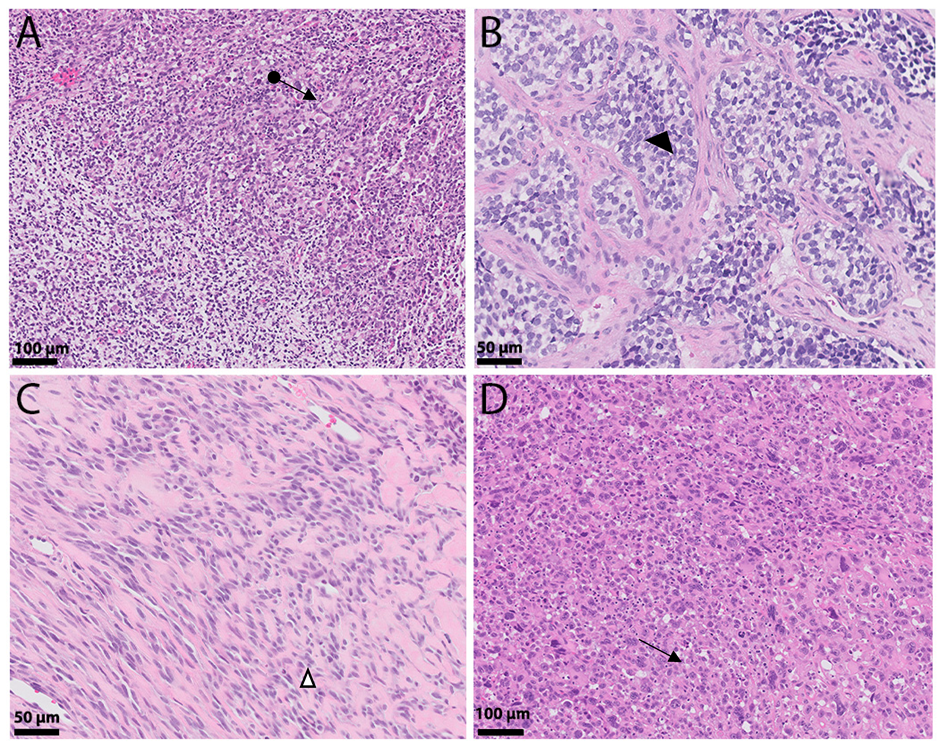
2. Histologic Sub-Classification
2.1. ERMS
2.2. ARMS
2.3. SCRMS
2.4. PRMS
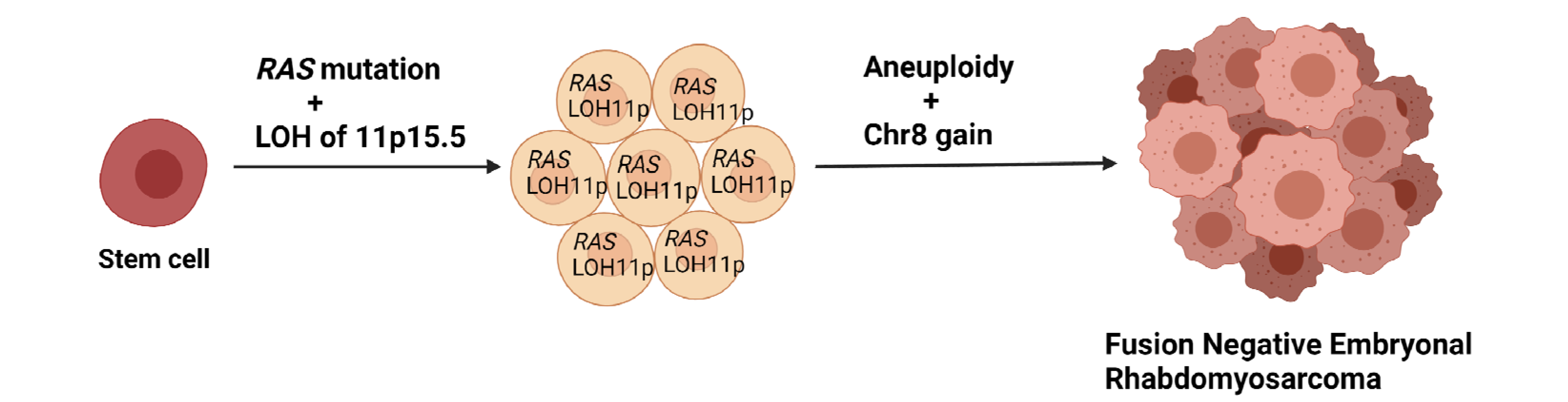
3. Genetics of ERMS
4. Epigenetics
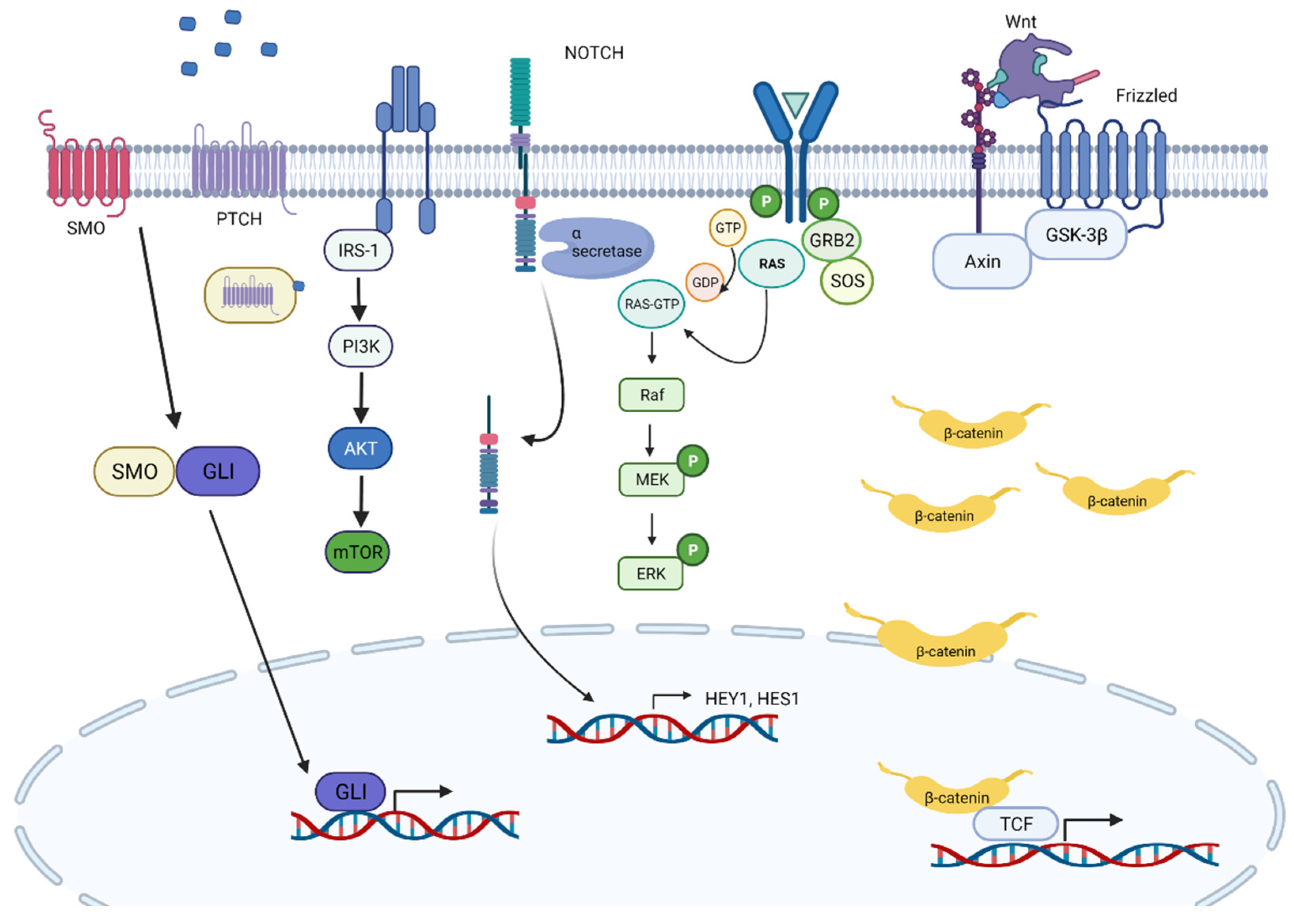
5. Signaling Pathways in RMS
6. Rhabdomyosarcoma Cell Lines
7. Mouse Models
8. Risk Stratification
9. Conclusions
Funding
Conflicts of Interest
References
- Perez, E.A.; Kassira, N.; Cheung, M.C.; Koniaris, L.G.; Neville, H.L.; Sola, J. Rhabdomyosarcoma in children: A SEER population based study. J. Surg. Res. 2011, 170, e243–e251. [Google Scholar] [CrossRef]
- Raney, R.B.; Maurer, H.M.; Anderson, J.R.; Andrassy, R.J.; Donaldson, S.S.; Qualman, S.J.; Wharam, M.D.; Wiener, E.S.; Crist, W.M. The Intergroup Rhabdomyosarcoma Study Group (IRSG): Major lessons from the IRS-I through IRS-IV studies as background for the current IRS-V treatment protocols. Sarcoma 2001, 5, 9–15. [Google Scholar] [CrossRef]
- Chen, E.; Ricciotti, R.; Futran, N.; Oda, D. Head and neck rhabdomyosarcoma: Clinical and pathologic characterization of seven cases. Head Neck Pathol. 2016, 11, 321–326. [Google Scholar] [CrossRef] [Green Version]
- Tapscott, S.J.; Thayer, M.J.; Weintraub, H. Deficiency in rhabdomyosarcomas of a factor required for MyoD activity and myogenesis. Science 1993, 259, 1450–1453. [Google Scholar] [CrossRef]
- WHO. Classification of Tumours of Soft Tissue and Bone; WHO: Geneva, Switzerland, 2021. [Google Scholar]
- Zhu, L.; Sun, Y.; Wang, X.; Wang, L.; Zhang, S.; Meng, Q.; Wang, X. Survival stratification in childhood rhabdomyosarcoma of the extremities: A derivation and validation study. Sci. Rep. 2020, 10, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Parham, D.M.; Ellison, D.A. Rhabdomyosarcomas in adults and children: An update. Arch. Pathol. Lab. Med. 2006, 130, 1454–1465. [Google Scholar] [CrossRef]
- Teot, L.A.; Schneider, M.; Thorner, A.R.; Tian, J.; Chi, Y.-Y.; Ducar, M.; Lin, L.; Wlodarski, M.; Grier, H.E.; Fletcher, C.D.M.; et al. Clinical and mutational spectrum of highly differentiated, paired box 3:forkhead box protein o1 fusion-negative rhabdomyosarcoma: A report from the Children’s Oncology Group. Cancer 2018, 124, 1973–1981. [Google Scholar] [CrossRef]
- Shenoy, A.; Alvarez, E.; Chi, Y.-Y.; Li, M.; Shern, J.F.; Khan, J.; Hiniker, S.M.; Granberg, C.F.; Hawkins, D.S.; Parham, D.M.; et al. The prognostic significance of anaplasia in childhood rhabdomyosarcoma: A report from the Children’s Oncology Group. Eur. J. Cancer 2020, 143, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Rudzinski, E.R. Histology and fusion status in rhabdomyosarcoma. Am. Soc. Clin. Oncol. Educ. Book 2013, 33, 425–428. [Google Scholar] [CrossRef] [PubMed]
- Barr, F.G.; Galili, N.; Holick, J.; Biegel, J.A.; Rovera, G.; Emanuel, B.S. Rearrangement of the PAX3 paired box gene in the paediatric solid tumour alveolar rhabdomyosarcoma. Nat. Genet. 1993, 3, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.J.; D’Cruz, C.M.; Lovell, M.A.; Biegel, J.A.; Barr, F.G. Fusion of PAX7 to FKHR by the variant t(1;13)(p36;q14) translocation in alveolar rhabdomyosarcoma. Cancer Res. 1994, 54, 2869–2872. [Google Scholar]
- Galili, N.; Davis, R.J.; Fredericks, W.J.; Mukhopadhyay, S.; Rauscher, F.J.; Emanuel, B.S.; Rovera, G.; Barr, F.G. Fusion of a fork head domain gene to PAX3 in the solid tumour alveolar rhabdomyosarcoma. Nat. Genet. 1993, 5, 230–235. [Google Scholar] [CrossRef]
- Sorensen, P.H.; Lynch, J.C.; Qualman, S.J.; Tirabosco, R.; Lim, J.F.; Maurer, H.M.; Bridge, J.A.; Crist, W.M.; Triche, T.J.; Barr, F.G. PAX3-FKHR and PAX7-FKHR gene fusions are prognostic indicators in alveolar rhabdomyosarcoma: A report from the Children’s Oncology Group. J. Clin. Oncol. 2002, 20, 2672–2679. [Google Scholar] [CrossRef]
- Rudzinski, E.R.; Anderson, J.R.; Chi, Y.-Y.; Gastier-Foster, J.M.; Astbury, C.; Barr, F.G.; Skapek, S.X.; Hawkins, D.S.; Weigel, B.J.; Pappo, A.; et al. Histology, fusion status, and outcome in metastatic rhabdomyosarcoma: A report from the Children’s Oncology Group. Pediatr. Blood Cancer 2017, 64, e26645. [Google Scholar] [CrossRef] [PubMed]
- Duan, F.; Smith, L.M.; Gustafson, D.M.; Zhang, C.; Dunlevy, M.J.; Gastier-Foster, J.M.; Barr, F.G. Genomic and clinical analysis of fusion gene amplification in rhabdomyosarcoma: A report from the Children’s Oncology Group. Genes Chromosom. Cancer 2012, 51, 662–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Guzman, M.A.; Pezanowski, D.; Patel, D.; Hauptman, J.; Keisling, M.; Hou, S.J.; Papenhausen, P.R.; Pascasio, J.M.; Punnett, H.H.; et al. FOXO1–FGFR1 fusion and amplification in a solid variant of alveolar rhabdomyosarcoma. Mod. Pathol. 2011, 24, 1327–1335. [Google Scholar] [CrossRef] [Green Version]
- Szuhai, K.; de Jong, D.; Leung, W.Y.; Fletcher, C.D.M.; Hogendoorn, P. Transactivating mutation of the MYOD1 gene is a frequent event in adult spindle cell rhabdomyosarcoma. J. Pathol. 2014, 232, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Agaram, N.P.; Laquaglia, M.P.; Alaggio, R.; Zhang, L.; Fujisawa, Y.; Ladanyi, M.; Wexler, L.H.; Antonescu, C.R. MYOD1-mutant spindle cell and sclerosing rhabdomyosarcoma: An aggressive subtype irrespective of age. A reappraisal for molecular classification and risk stratification. Mod. Pathol. 2018, 32, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Leuschner, I.; Newton, W.A.; Schmidt, D.; Sachs, N.; Asmar, L.; Hamoudi, A.; Harms, D.; Maurer, H.M. Spindle cell variants of embryonal rhabdomyosarcoma in the paratesticular region. Am. J. Surg. Pathol. 1993, 17, 221–230. [Google Scholar] [CrossRef]
- Nascimento, A.F.; Fletcher, C.D.M. Spindle cell rhabdomyosarcoma in adults. Am. J. Surg. Pathol. 2005, 29, 1106–1113. [Google Scholar] [CrossRef]
- Gavino, A.C.P.; Spears, M.D.; Peng, Y. Sclerosing spindle cell rhabdomyosarcoma in an adult: Report of a new case and review of the literature. Int. J. Surg. Pathol. 2008, 18, 394–397. [Google Scholar] [CrossRef]
- Kuhnen, C.; Herter, P.; Leuschner, I.; Mentzel, T.; Druecke, D.; Jaworska, M.; Johnen, G. Sclerosing pseudovascular rhabdomyosarcoma—Immunohistochemical, ultrastructural, and genetic findings indicating a distinct subtype of rhabdomyosarcoma. Virchows Archiv. 2006, 449, 572–578. [Google Scholar] [CrossRef] [PubMed]
- Stock, N.; Chibon, F.; Binh, M.B.N.; Terrier, P.; Michels, J.J.; Valo, I.; Robin, Y.M.; Guillou, L.; Ranchère-Vince, D.; Decouvelaere, A.-V.; et al. Adult-type rhabdomyosarcoma: Analysis of 57 cases with clinicopathologic description, identification of 3 morphologic patterns and prognosis. Am. J. Surg. Pathol. 2009, 33, 1850–1859. [Google Scholar] [CrossRef] [PubMed]
- Furlong, M.A.; Mentzel, T.; Fanburg-Smith, J.C. Pleomorphic rhabdomyosarcoma in adults: A clinicopathologic study of 38 cases with emphasis on morphologic variants and recent skeletal muscle-specific markers. Mod. Pathol. 2001, 14, 595–603. [Google Scholar] [CrossRef]
- Gaffney, E.F.; Dervan, P.A.; Fletcher, C.D. Pleomorphic rhabdomyosarcoma in adulthood. Analysis of 11 cases with definition of diagnostic criteria. Am. J. Surg. Pathol. 1993, 17, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Hollowood, K.; Fletcher, C.D. Rhabdomyosarcoma in adults. Semin. Diagn. Pathol. 1994, 11, 47–57. [Google Scholar]
- Arnold, M.A.; Anderson, J.R.; Gastier-Foster, J.M.; Barr, F.G.; Skapek, S.X.; Hawkins, D.S.; Raney, R.B.J.; Parham, D.M.; Teot, L.A.; Rudzinski, E.R.; et al. Histology, fusion status, and outcome in alveolar rhabdomyosarcoma with low-risk clinical features: A report from the Children’s Oncology Group. Pediatr. Blood Cancer 2016, 63, 634–639. [Google Scholar] [CrossRef] [Green Version]
- Skapek, S.X.; Ferrari, A.; Gupta, A.A.; Lupo, P.J.; Butler, E.; Shipley, J.; Barr, F.G.; Hawkins, D.S. Rhabdomyosarcoma. Nat. Rev. Dis. Prim. 2019, 5, 1–19. [Google Scholar] [CrossRef]
- Williamson, D.; Missiaglia, E.; Pritchard-Jones, K.; Oberlin, O.; Shipley, J.; Delattre, O.; De Reyniès, A.; Pierron, G.; Thuille, B.; Palenzuela, G.; et al. Fusion gene-negative alveolar rhabdomyosarcoma is clinically and molecularly indistinguishable from embryonal rhabdomyosarcoma. J. Clin. Oncol. 2010, 28, 2151–2158. [Google Scholar] [CrossRef] [Green Version]
- Shern, J.F.; Chen, L.; Badgett, T.; Getz, G.; Chmielecki, J.; Mora, J.; Anderson, J.R.; Skapek, S.X.; Barr, F.G.; Meyerson, M.; et al. Comprehensive genomic analysis of rhabdomyosarcoma reveals a landscape of alterations affecting a common genetic axis in fusion-positive and fusion-negative tumors. Cancer Discov. 2014, 4, 216–231. [Google Scholar] [CrossRef] [Green Version]
- Skapek, S.X.; Anderson, J.; Barr, F.G.; Bridge, J.A.; Gastier-Foster, J.M.; Parham, D.M.; Rudzinski, E.R.; Triche, T.; Hawkins, D.S. PAX-FOXO1 fusion status drives unfavorable outcome for children with rhabdomyosarcoma: A children’s oncology group report. Pediatr. Blood Cancer 2013, 60, 1411–1417. [Google Scholar] [CrossRef] [Green Version]
- Missiaglia, E.; Williamson, D.; Chisholm, J.; Wirapati, P.; Pierron, G.; Petel, F.; Concordet, J.-P.; Thway, K.; Oberlin, O.; Pritchard-Jones, K.; et al. PAX3/FOXO1 fusion gene status is the key prognostic molecular marker in rhabdomyosarcoma and significantly improves current risk stratification. J. Clin. Oncol. 2012, 30, 1670–1677. [Google Scholar] [CrossRef]
- Bridge, J.A.; Liu, J.; Qualman, S.J.; Suijkerbuijk, R.; Wenger, G.; Zhang, J.; Wan, X.; Baker, K.S.; Sorensen, P.; Barr, F.G. Genomic gains and losses are similar in genetic and histologic subsets of rhabdomyosarcoma, whereas amplification predominates in embryonal with anaplasia and alveolar subtypes. Genes Chromosom. Cancer 2001, 33, 310–321. [Google Scholar] [CrossRef] [PubMed]
- Davicioni, E.; Anderson, M.J.; Finckenstein, F.G.; Lynch, J.C.; Qualman, S.J.; Shimada, H.; Schofield, D.E.; Buckley, J.D.; Meyer, W.H.; Sorensen, P.H.; et al. Molecular classification of rhabdomyosarcoma—Genotypic and phenotypic determinants of diagnosis: A report from the Children’s Oncology Group. Am. J. Pathol. 2009, 174, 550–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gripp, K.W.; Lin, A.E. Costello syndrome: A Ras/mitogen activated protein kinase pathway syndrome (rasopathy) resulting from HRAS germline mutations. Genet. Med. 2012, 14, 285–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishimura, R.; Takita, J.; Sato-Otsubo, A.; Kato, M.; Koh, K.; Hanada, R.; Tanaka, Y.; Kato, K.; Maeda, D.; Fukayama, M.; et al. Characterization of genetic lesions in rhabdomyosarcoma using a high-density single nucleotide polymorphism array. Cancer Sci. 2013, 104, 856–864. [Google Scholar] [CrossRef]
- Pandita, A.; Zielenska, M.; Thomer, P.; Bayani, J.; Godbout, R.; Greenberg, M.; Squire, J.A. Application of comparative genomic hybridization, spectral karyotyping, and microarray analysis in the identification of subtype-specific patterns of genomic changes in rhabdomyosarcoma. Neoplasia 1999, 1, 262–275. [Google Scholar] [CrossRef] [Green Version]
- Bridge, J.A.; Liu, J.; Weibolt, V.; Baker, K.S.; Perry, D.; Kruger, R.; Qualman, S.; Barr, F.; Sorensen, P.; Triche, T.; et al. Novel genomic imbalances in embryonal rhabdomyosarcoma revealed by comparative genomic hybridization and fluorescence in situ hybridization: An Intergroup Rhabdomyosarcoma Study. Genes Chromosom. Cancer 2000, 27, 337–344. [Google Scholar] [CrossRef]
- Gordon, T.; McManus, A.; Anderson, J.; Min, T.; Swansbury, J.; Pritchard-Jones, K. Cytogenetic abnormalities in 42 rhabdomyosarcomata: A United Kingdom Cancer Cytogenetics Group Study. Med. Pediatr. Oncol. 2001, 36, 259–267. [Google Scholar] [CrossRef]
- Paulson, V.; Chandler, G.; Rakheja, D.; Galindo, R.L.; Wilson, K.; Amatruda, J.F.; Cameron, S. High-resolution array CGH identifies common mechanisms that drive embryonal rhabdomyosarcoma pathogenesis. Genes Chromosom. Cancer 2011, 50, 397–408. [Google Scholar] [CrossRef]
- Boerma, E.G.; Siebert, R.; Kluin, P.M.; Baudis, M. Translocations involving 8q24 in Burkitt lymphoma and other malignant lymphomas: A historical review of cytogenetics in the light of today’s knowledge. Leukemia 2008, 23, 225–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bridge, J.A.; Meloni, A.M.; Neff, J.R.; DeBoer, J.; Pickering, D.; Dalence, C.; Jeffrey, B.; Sandberg, A.A. Deletion 5q in desmoid tumor and fluorescence in situ hybridization for chromosome 8 and/or 20 copy number. Cancer Genet. Cytogenet. 1996, 92, 150–151. [Google Scholar] [CrossRef]
- Sandberg, A.A. Updates on the cytogenetics and molecular genetics of bone and soft tissue tumors: Liposarcoma. Cancer Genet. Cytogenet. 2004, 155, 1–24. [Google Scholar] [CrossRef]
- Fletcher, J.A.; Kozakewich, H.P.; Schoenberg, M.L.; Morton, C.C. Cytogenetic findings in pediatric adipose tumors: Consistent rearrangement of chromosome 8 in lipoblastoma. Genes Chromosom. Cancer 1993, 6, 24–29. [Google Scholar] [CrossRef]
- Scrable, H.; Witte, D.; Shimada, H.; Seemayer, T.; Wang-Wuu, S.; Soukup, S.; Koufos, A.; Houghton, P.; Lampkin, B.; Cavenee, W. Molecular differential pathology of rhabdomyosarcoma. Genes Chromosom. Cancer 1989, 1, 23–35. [Google Scholar] [CrossRef]
- Shern, J.F.; Yohe, M.E.; Khan, J. Pediatric rhabdomyosarcoma. Crit. Rev. Oncog. 2015, 20, 227–243. [Google Scholar] [CrossRef] [Green Version]
- Ping, A.J.; E Reeve, A.; Law, D.J.; Young, M.R.; Boehnke, M.; Feinberg, A. Genetic linkage of Beckwith-Wiedemann syndrome to 11p15. Am. J. Hum. Genet. 1989, 44, 720–723. [Google Scholar]
- Rainier, S.; Dobry, C.J.; Feinberg, A. Loss of imprinting in hepatoblastoma. Cancer Res. 1995, 55, 1836–1838. [Google Scholar]
- Chen, L.; Shern, J.F.; Wei, J.S.; Yohe, M.E.; Song, Y.K.; Hurd, L.; Liao, H.; Catchpoole, D.; Skapek, S.X.; Barr, F.G.; et al. Clonality and evolutionary history of rhabdomyosarcoma. PLoS Genet. 2015, 11, e1005075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yohe, M.E.; Gryder, B.E.; Shern, J.F.; Song, Y.K.; Chou, H.-C.; Sindiri, S.; Mendoza, A.; Patidar, R.; Zhang, X.; Guha, R.; et al. MEK inhibition induces MYOG and remodels super-enhancers in RAS-driven rhabdomyosarcoma. Sci. Transl. Med. 2018, 10, eaan4470. [Google Scholar] [CrossRef] [Green Version]
- Pylayeva-Gupta, Y.; Grabocka, E.; Bar-Sagi, D. RAS oncogenes: Weaving a tumorigenic web. Nat. Rev. Cancer 2011, 11, 761–774. [Google Scholar] [CrossRef] [Green Version]
- Shern, J.F.; Selfe, J.; Izquierdo, E.; Patidar, R.; Chou, H.-C.; Song, Y.K.; Yohe, M.E.; Sindiri, S.; Wei, J.; Wen, X.; et al. Genomic classification and clinical outcome in rhabdomyosarcoma: A report from an international consortium. J. Clin. Oncol. 2021, 39, 2859–2871. [Google Scholar] [CrossRef]
- Chen, X.; Stewart, E.; Shelat, A.; Qu, C.; Bahrami, A.; Hatley, M.; Wu, G.; Bradley, C.; McEvoy, J.; Pappo, A.; et al. Targeting oxidative stress in embryonal rhabdomyosarcoma. Cancer Cell 2013, 24, 710–724. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Guo, W.; Shen, J.K.; Mankin, H.J.; Hornicek, F.J.; Duan, Z. Rhabdomyosarcoma: Advances in molecular and cellular biology. Sarcoma 2015, 2015, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Marchesi, I.; Fiorentino, F.P.; Rizzolio, F.; Giordano, A.; Bagella, L. The ablation of EZH2 uncovers its crucial role in rhabdomyosarcoma formation. Cell Cycle 2012, 11, 3828–3836. [Google Scholar] [CrossRef] [Green Version]
- Sparmann, A.; Van Lohuizen, M. Polycomb silencers control cell fate, development and cancer. Nat. Rev. Cancer 2006, 6, 846–856. [Google Scholar] [CrossRef]
- Sun, W.; Chatterjee, B.; Shern, J.F.; Patidar, R.; Song, Y.; Wang, Y.; Walker, R.L.; Pawel, B.R.; Linardic, C.M.; Houghton, P.; et al. Relationship of DNA methylation to mutational changes and transcriptional organization in fusion-positive and fusion-negative rhabdomyosarcoma. Int. J. Cancer 2018, 144, 2707–2717. [Google Scholar] [CrossRef] [PubMed]
- Stewart, E.; McEvoy, J.; Wang, H.; Chen, X.; Honnell, V.; Ocarz, M.; Gordon, B.; Dapper, J.; Blankenship, K.; Yang, Y.; et al. Identification of therapeutic targets in rhabdomyosarcoma through integrated genomic, epigenomic, and proteomic analyses. Cancer Cell 2018, 34, 411–426.e19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Benavente, C.; McEvoy, J.; Flores-Otero, J.; Ding, L.; Chen, X.; Ulyanov, A.; Wu, G.; Wilson, M.; Wang, J.; et al. A novel retinoblastoma therapy from genomic and epigenetic analyses. Nature 2012, 481, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Pugh, T.; Weeraratne, S.D.; Archer, T.C.; Krummel, D.A.P.; Auclair, D.; Bochicchio, J.; Carneiro, M.O.; Carter, S.L.; Cibulskis, K.; Erlich, R.L.; et al. Medulloblastoma exome sequencing uncovers subtype-specific somatic mutations. Nature 2012, 488, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Drummond, C.; Hanna, J.; Garcia, M.R.; Devine, D.J.; Heyrana, A.J.; Finkelstein, D.; Rehg, J.E.; Hatley, M.E. Hedgehog pathway drives fusion-negative rhabdomyosarcoma initiated from non-myogenic endothelial progenitors. Cancer Cell 2018, 33, 108–124.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hahn, H.; Wojnowski, L.; Zimmer, A.M.; Hall, J.; Miller, G.; Zimmer, A. Rhabdomyosarcomas and radiation hypersensitivity in a mouse model of Gorlin syndrome. Nat. Med. 1998, 4, 619–622. [Google Scholar] [CrossRef] [PubMed]
- Hatley, M.; Tang, W.; Garcia, M.R.; Finkelstein, D.; Millay, D.P.; Liu, N.; Graff, J.; Galindo, R.L.; Olson, E.N. A mouse model of rhabdomyosarcoma originating from the adipocyte lineage. Cancer Cell 2012, 22, 536–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.-W.; Moskowitz, M.; Sims, J. Sonic hedgehog inversely regulates the expression of angiopoietin-1 and angiopoietin-2 in fibroblasts. Int. J. Mol. Med. 2007, 19. [Google Scholar] [CrossRef]
- Kashi, V.P.; Hatley, M.E.; Galindo, R.L. Probing for a deeper understanding of rhabdomyosarcoma: Insights from complementary model systems. Nat. Rev. Cancer 2015, 15, 426–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voronova, A.; Coyne, E.; Al Madhoun, A.; Fair, J.V.; Bosiljcic, N.; St-Louis, C.; Li, G.; Thurig, S.; Wallace, V.A.; Wiper-Bergeron, N.; et al. Hedgehog Signaling Regulates MyoD Expression and Activity. J. Biol. Chem. 2013, 288, 4389–4404. [Google Scholar] [CrossRef] [Green Version]
- Zibat, A.; Missiaglia, E.; Rosenberger, A.; Pritchard-Jones, K.; Shipley, J.; Hahn, H.; Fulda, S. Activation of the hedgehog pathway confers a poor prognosis in embryonal and fusion gene-negative alveolar rhabdomyosarcoma. Oncogene 2010, 29, 6323–6330. [Google Scholar] [CrossRef] [Green Version]
- Satheesha, S.; Manzella, G.; Bovay, A.; Casanova, E.A.; Bode, P.K.; Belle, R.; Feuchtgruber, S.; Jaaks, P.; Dogan, N.; Koscielniak, E.; et al. Targeting hedgehog signaling reduces self-renewal in embryonal rhabdomyosarcoma. Oncogene 2015, 35, 2020–2030. [Google Scholar] [CrossRef] [Green Version]
- Bauer, J.; Cuvelier, N.; Ragab, N.; Simon-Keller, K.; Nitzki, F.; Geyer, N.; Botermann, D.S.; Elmer, D.P.; Rosenberger, A.; Rando, T.A.; et al. Context-dependent modulation of aggressiveness of pediatric tumors by individual oncogenic RAS isoforms. Oncogene 2021, 40, 4955–4966. [Google Scholar] [CrossRef]
- Schuster-Gossler, K.; Cordes, R.; Gossler, A. Premature myogenic differentiation and depletion of progenitor cells cause severe muscle hypotrophy in Delta1 mutants. Proc. Natl. Acad. Sci. USA 2006, 104, 537–542. [Google Scholar] [CrossRef] [Green Version]
- Vasyutina, E.; Lenhard, D.C.; Wende, H.; Erdmann, B.; Epstein, J.A.; Birchmeier, C. RBP-J (Rbpsuh) is essential to maintain muscle progenitor cells and to generate satellite cells. Proc. Natl. Acad. Sci. USA 2007, 104, 4443–4448. [Google Scholar] [CrossRef] [Green Version]
- Belyea, B.C.; Naini, S.; Bentley, R.C.; Linardic, C.M. Inhibition of the Notch-Hey1 axis blocks embryonal rhabdomyosarcoma tumorigenesis. Clin. Cancer Res. 2011, 17, 7324–7336. [Google Scholar] [CrossRef] [Green Version]
- Brunelli, S.; Relaix, F.; Baesso, S.; Buckingham, M.; Cossu, G. Beta catenin-independent activation of MyoD in presomitic mesoderm requires PKC and depends on Pax3 transcriptional activity. Dev. Biol. 2007, 304, 604–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borello, U.; Berarducci, B.; Murphy, P.; Bajard, L.; Buffa, V.; Piccolo, S.; Buckingham, M.; Cossu, G. The Wnt/β-catenin pathway regulates Gli-mediatedMyf5expression during somitogenesis. Development 2006, 133, 3723–3732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tenente, I.M.; Hayes, M.N.; Ignatius, M.S.; McCarthy, K.; Yohe, M.; Sindiri, S.; Gryder, B.; Oliveira, M.L.; Ramakrishnan, A.; Tang, Q.; et al. Myogenic regulatory transcription factors regulate growth in rhabdomyosarcoma. eLife 2017, 6, 35. [Google Scholar] [CrossRef] [Green Version]
- Hutcheson, D.A.; Zhao, J.; Merrell, A.; Haldar, M.; Kardon, G. Embryonic and fetal limb myogenic cells are derived from developmentally distinct progenitors and have different requirements for -catenin. Genes Dev. 2009, 23, 997–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, B.; Rao, K.S.; Rao, C.M. Ubiquitin–proteasome-mediated degradation and synthesis of MyoD is modulated by αB-crystallin, a small heat shock protein, during muscle differentiation. Biochim. Biophys. Acta (BBA)-Bioenergy 2010, 1803, 288–299. [Google Scholar] [CrossRef] [Green Version]
- Tremblay, A.M.; Missiaglia, E.; Galli, G.; Hettmer, S.; Urcia, R.; Carrara, M.; Judson, R.N.; Thway, K.; Nadal, G.; Selfe, J.; et al. The hippo transducer YAP1 transforms activated satellite cells and is a potent effector of embryonal rhabdomyosarcoma formation. Cancer Cell 2014, 26, 273–287. [Google Scholar] [CrossRef] [Green Version]
- Slemmons, K.K.; Yeung, C.; Baumgart, J.T.; Juarez, J.O.M.; McCalla, A.; Helman, L.J. Targeting hippo-dependent and hippo-independent YAP1 signaling for the treatment of childhood rhabdomyosarcoma. Cancer Res. 2020, 80, 3046–3056. [Google Scholar] [CrossRef] [PubMed]
- Ebi, H.; Corcoran, R.B.; Singh, A.; Chen, Z.; Song, Y.; Lifshits, E.; Ryan, D.P.; Meyerhardt, J.A.; Benes, C.; Settleman, J.; et al. Receptor tyrosine kinases exert dominant control over PI3K signaling in human KRAS mutant colorectal cancers. J. Clin. Investig. 2011, 121, 4311–4321. [Google Scholar] [CrossRef]
- Flanigan, J.C.; Jilaveanu, L.B.; Chiang, V.L.; Kluger, H.M. Advances in therapy for melanoma brain metastases. Clin. Dermatol. 2013, 31, 264–281. [Google Scholar] [CrossRef]
- Weisberg, E.; Nonami, A.; Chen, Z.; Nelson, E.; Chen, Y.; Liu, F.; Cho, H.; Zhang, J.; Sattler, M.; Mitsiades, C.; et al. Upregulation of IGF1R by mutant RAS in leukemia and potentiation of RAS signaling inhibitors by small-molecule inhibition of IGF1R. Clin. Cancer Res. 2014, 20, 5483–5495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- E Martin, D.; Hall, M.N. The expanding TOR signaling network. Curr. Opin. Cell Biol. 2005, 17, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Renshaw, J.; Taylor, K.R.; Bishop, R.; Valenti, M.; Brandon, A.D.H.; Gowan, S.; Eccles, S.A.; Ruddle, R.R.; Johnson, L.D.; Raynaud, F.; et al. Dual blockade of the PI3K/AKT/mTOR (AZD8055) and RAS/MEK/ERK (AZD6244) pathways synergistically inhibits rhabdomyosarcoma cell growth in vitro and in vivo. Clin. Cancer Res. 2013, 19, 5940–5951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guenther, M.K.; Graab, U.; Fulda, S. Synthetic lethal interaction between PI3K/Akt/mTOR and Ras/MEK/ERK pathway inhibition in rhabdomyosarcoma. Cancer Lett. 2013, 337, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Mascarenhas, L.; Chi, Y.-Y.; Hingorani, P.; Anderson, J.R.; Lyden, E.R.; Rodeberg, D.A.; Indelicato, D.J.; Kao, S.; Dasgupta, R.; Spunt, S.L.; et al. Randomized phase II trial of bevacizumab or temsirolimus in combination with chemotherapy for first relapse rhabdomyosarcoma: A report from the children’s oncology group. J. Clin. Oncol. 2019, 37, 2866–2874. [Google Scholar] [CrossRef]
- Hinson, A.R.; Jones, R.; Crose, L.E.; Belyea, B.C.; Barr, F.G.; Linardic, C.M. Human rhabdomyosarcoma cell lines for rhabdomyosarcoma research: Utility and pitfalls. Front Oncol. 2013, 3, 183. [Google Scholar] [CrossRef] [Green Version]
- Linardic, C.M.; Downie, D.L.; Qualman, S.; Bentley, R.C.; Counter, C.M. Genetic modeling of human rhabdomyosarcoma. Cancer Res. 2005, 65, 4490–4495. [Google Scholar] [CrossRef] [Green Version]
- Jacks, T.; Remington, L.; Williams, B.; Schmitt, E.M.; Halachmi, S.; Bronson, R.T.; Weinberg, R.A. Tumor spectrum analysis in p53-mutant mice. Curr. Biol. 1994, 4, 1–7. [Google Scholar] [CrossRef]
- Taulli, R.; Scuoppo, C.; Bersani, F.; Accornero, P.; Forni, P.E.; Miretti, S.; Grinza, A.; Allegra, P.; Schmitt-Ney, M.; Crepaldi, T.; et al. Validation of met as a therapeutic target in alveolar and embryonal rhabdomyosarcoma. Cancer Res. 2006, 66, 4742–4749. [Google Scholar] [CrossRef] [Green Version]
- Sharp, R.; Recio, J.A.; Jhappan, C.; Otsuka, T.; Liu, S.; Yu, Y.; Liu, W.; Anver, M.; Navid, F.; Helman, L.J.; et al. Synergism between INK4a/ARF inactivation and aberrant HGF/SF signaling in rhabdomyosarcomagenesis. Nat. Med. 2002, 8, 1276–1280. [Google Scholar] [CrossRef]
- Kappler, R.; Bauer, R.; Calzada-Wack, J.; Rosemann, M.; Hemmerlein, B.; Hahn, H. Profiling the molecular difference between Patched- and p53-dependent rhabdomyosarcoma. Oncogene 2004, 23, 8785–8795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.; Kawagoe, R.; Sasai, K.; Li, Y.; Russell, H.R.; Curran, T.; McKinnon, P.J. Loss of suppressor-of-fused function promotes tumorigenesis. Oncogene 2007, 26, 6442–6447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanola, A.; Rossi, S.; Faggi, F.; Monti, E.; Fanzani, A. Rhabdomyosarcomas: An overview on the experimental animal models. J. Cell. Mol. Med. 2012, 16, 1377–1391. [Google Scholar] [CrossRef] [PubMed]
- Langenau, D.M.; Keefe, M.D.; Storer, N.Y.; Guyon, J.R.; Kutok, J.L.; Le, X.; Goessling, W.; Neuberg, D.S.; Kunkel, L.M.; Zon, L.I. Effects of RAS on the genesis of embryonal rhabdomyosarcoma. Genes Dev. 2007, 21, 1382–1395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKinnon, T.; Venier, R.; Yohe, M.; Sindiri, S.; Gryder, B.E.; Shern, J.F.; Kabaroff, L.; Dickson, B.; Schleicher, K.; Chouinard-Pelletier, G.; et al. Functional screening of FGFR4-driven tumorigenesis identifies PI3K/mTOR inhibition as a therapeutic strategy in rhabdomyosarcoma. Oncogene 2018, 37, 2630–2644. [Google Scholar] [CrossRef]
- Fleischmann, A.; Jochum, W.; Eferl, R.; Witowsky, J.; Wagner, E.F. Rhabdomyosarcoma development in mice lacking Trp53 and Fos: Tumor suppression by the Fos protooncogene. Cancer Cell. 2003, 4, 477–482. [Google Scholar] [CrossRef] [Green Version]
- Nanni, P.; Nicoletti, G.; De Giovanni, C.; Croci, S.; Astolfi, A.; Landuzzi, L.; DI Carlo, E.; Iezzi, M.; Musiani, P.; Lollini, P.-L. Development of rhabdomyosarcoma in HER-2/neu transgenic p53 mutant mice. Cancer Res. 2003, 63, 2728–2732. [Google Scholar] [PubMed]
- Tsumura, H.; Yoshida, T.; Saito, H.; Imanaka-Yoshida, K.; Suzuki, N. Cooperation of oncogenic K-ras and p53 deficiency in pleomorphic rhabdomyosarcoma development in adult mice. Oncogene 2006, 25, 7673–7679. [Google Scholar] [CrossRef]
- Doyle, B.; Morton, J.; Delaney, D.W.; Ridgway, R.A.; Wilkins, J.A.; Sansom, O.J. p53 mutation and loss have different effects on tumourigenesis in a novel mouse model of pleomorphic rhabdomyosarcoma. J. Pathol. 2010, 222, 129–137. [Google Scholar] [CrossRef]
- Nitzki, F.; Tolosa, E.; Cuvelier, N.; Frommhold, A.; Salinas-Riester, G.; Johnsen, S.A.; Fernandez-Zapico, M.E.; Hahn, H. Overexpression of mutant Ptch in rhabdomyosarcomas is associated with promoter hypomethylation and increased Gli1 and H3K4me3 occupancy. Oncotarget 2015, 6, 9113–9124. [Google Scholar] [CrossRef] [Green Version]
- Feber, A.; Guilhamon, P.; Lechner, M.; Fenton, T.; Wilson, G.A.; Thirlwell, C.; Morris, T.J.; Flanagan, A.M.; E Teschendorff, A.; Kelly, J.D.; et al. Using high-density DNA methylation arrays to profile copy number alterations. Genome Biol. 2014, 15, R30. [Google Scholar] [CrossRef] [Green Version]
- Jin, K.; Teng, L.; Shen, Y.; He, K.; Xu, Z.; Li, G. Patient-derived human tumour tissue xenografts in immunodeficient mice: A systematic review. Clin. Transl. Oncol. 2010, 12, 473–480. [Google Scholar] [CrossRef]
- Murayama, T.; Gotoh, N. Patient-derived xenograft models of breast cancer and their application. Cells 2019, 8, 621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pompili, L.; Porru, M.; Caruso, C.; Biroccio, A.; Leonetti, C. Patient-derived xenografts: A relevant preclinical model for drug development. J. Exp. Clin. Cancer Res. 2016, 35, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dehner, C.; Moon, C.I.; Zhang, X.; Zhou, Z.; Miller, C.; Xu, H.; Wan, X.; Yang, K.; Mashl, J.; Gosline, S.J.; et al. Chromosome 8 gain is associated with high-grade transformation in MPNST. JCI Insight 2021, 6. [Google Scholar] [CrossRef] [PubMed]
- Carrió, M.; Mazuelas, H.; Richaud-Patin, Y.; Gel, B.; Terribas, E.; Rosas, I.; Jimenez-Delgado, S.; Biayna, J.; Vendredy, L.; Blanco, I.; et al. Reprogramming captures the genetic and tumorigenic properties of neurofibromatosis type 1 plexiform neurofibromas. Stem Cell Rep. 2019, 12, 639–641. [Google Scholar] [CrossRef] [PubMed]
- Papapetrou, E.P. Patient-derived induced pluripotent stem cells in cancer research and precision oncology. Nat. Med. 2016, 22, 1392–1401. [Google Scholar] [CrossRef] [PubMed]
- Chal, J.; Al Tanoury, Z.; Hestin, M.; Gobert, B.; Aivio, S.; Hick, A.; Cherrier, T.; Nesmith, A.P.; Parker, K.K.; Pourquie, O. Generation of human muscle fibers and satellite-like cells from human pluripotent stem cells in vitro. Nat. Protoc. 2016, 11, 1833–1850. [Google Scholar] [CrossRef] [Green Version]
- Maffioletti, S.M.; Gerli, M.F.M.; Ragazzi, M.; Dastidar, S.; Benedetti, S.; Loperfido, M.; Vandendriessche, T.; Chuah, M.K.; Tedesco, F.S. Efficient derivation and inducible differentiation of expandable skeletal myogenic cells from human ES and patient-specific iPS cells. Nat. Protoc. 2015, 10, 941–958. [Google Scholar] [CrossRef]
- Ortuño-Costela, M.D.C.; Cerrada, V.; García-López, M.; Arenas, J.; Martínez, J.; Lucia, A.; Garesse, R.; Gallardo, M.E. Derivation of an aged mouse induced pluripotent stem cell line, IISHDOi005-A. Stem Cell Res. 2019, 36, 101418. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.J.; Holder, D.D.; Pawel, B.R.; Zhang, C.; Barr, F.G. High expression of the PAX3-FKHR oncoprotein is required to promote tumorigenesis of human myoblasts. Am. J. Pathol. 2009, 175, 2600–2608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yohe, M.E.; Heske, C.M.; Stewart, E.; Adamson, P.C.; Ahmed, N.; Antonescu, C.R.; Chen, E.; Collins, N.; Ehrlich, A.; Galindo, R.L.; et al. Insights into pediatric rhabdomyosarcoma research: Challenges and goals. Pediatr. Blood Cancer 2019, 66, e27869. [Google Scholar] [CrossRef] [PubMed]
- Meza, J.L.; Anderson, J.; Pappo, A.S.; Meyer, W.H. Analysis of prognostic factors in patients with nonmetastatic rhabdomyosarcoma treated on intergroup rhabdomyosarcoma studies III and IV: The Children’s Oncology Group. J. Clin. Oncol. 2006, 24, 3844–3851. [Google Scholar] [CrossRef]
- Seitz, G.; Dantonello, T.M.; Inform, C.I.D.; Blumenstock, M.G.; Godzinski, J.; Klingebiel, T.; Schuck, A.; Leuschner, I.; Koscielniak, E.; Fuchs, J.; et al. Treatment efficiency, outcome and surgical treatment problems in patients suffering from localized embryonal bladder/prostate rhabdomyosarcoma: A report from the cooperative soft tissue sarcoma trial CWS-96. Pediatr. Blood Cancer 2010, 56, 718–724. [Google Scholar] [CrossRef]
- Maurer, H.M.; Gehan, E.A.; Beltangady, M.; Crist, W.; Dickman, P.S.; Donaldson, S.S.; Fryer, C.; Hammond, D.; Hays, D.M.; Herrmann, J.; et al. The Intergroup Rhabdomyosarcoma Study-II. Cancer 1993, 71, 1904–1922. [Google Scholar] [CrossRef]
- Crist, W.; Gehan, E.A.; Ragab, A.H.; Dickman, P.S.; Donaldson, S.S.; Fryer, C.; Hammond, D.; Hays, D.M.; Herrmann, J.; Heyn, R. The third intergroup rhabdomyosarcoma study. J. Clin. Oncol. 1995, 13, 610–630. [Google Scholar] [CrossRef]
- Crist, W.M.; Anderson, J.R.; Meza, J.L.; Fryer, C.; Raney, R.B.; Ruymann, F.B.; Breneman, J.; Qualman, S.J.; Wiener, E.; Wharam, M.; et al. Intergroup rhabdomyosarcoma study-IV: Results for patients with nonmetastatic disease. J. Clin. Oncol. 2001, 19, 3091–3102. [Google Scholar] [CrossRef]
- Oberlin, O.; Rey, A.; Lyden, E.; Bisogno, G.; Stevens, M.; Meyer, W.H.; Carli, M.; Anderson, J.R. Prognostic factors in metastatic rhabdomyosarcomas: Results of a pooled analysis from United States and European Cooperative Groups. J. Clin. Oncol. 2008, 26, 2384–2389. [Google Scholar] [CrossRef] [Green Version]
- Arndt, C.A.S. Risk stratification of rhabdomyosarcoma: A moving target. Am. Soc. Clin. Oncol. Educ. Book 2013, 415–419. [Google Scholar] [CrossRef]
- Hibbitts, E.; Chi, Y.; Hawkins, D.S.; Barr, F.G.; Bradley, J.A.; Dasgupta, R.; Meyer, W.H.; Rodeberg, D.A.; Rudzinski, E.R.; Spunt, S.L.; et al. Refinement of risk stratification for childhood rhabdomyosarcoma using FOXO1 fusion status in addition to established clinical outcome predictors: A report from the Children’s Oncology Group. Cancer Med. 2019, 8, 6437–6448. [Google Scholar] [CrossRef] [Green Version]
- Walterhouse, D.O.; Pappo, A.S.; Meza, J.L.; Breneman, J.C.; Hayes-Jordan, A.A.; Parham, D.M.; Cripe, T.P.; Anderson, J.R.; Meyer, W.H.; Hawkins, D.S. Shorter-duration therapy using vincristine, dactinomycin, and lower-dose cyclophosphamide with or without radiotherapy for patients with newly diagnosed low-risk rhabdomyosarcoma: A report from the soft tissue sarcoma Committee of the Children’s Oncology Group. J. Clin. Oncol. 2014, 32, 3547–3552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yechieli, R.L.; Mandeville, H.C.; Hiniker, S.M.; Bernier-Chastagner, V.; McGovern, S.; Scarzello, G.; Wolden, S.; Cameron, A.; Breneman, J.; Fajardo, R.D.; et al. Rhabdomyosarcoma. Pediatr. Blood Cancer 2021, 68, e28254. [Google Scholar] [CrossRef] [PubMed]
- Faria, P.; Beckwith, J.B.; Mishra, K.; Zuppan, C.; Weeks, D.A.; Breslow, N.; Green, D.M. Focal versus diffuse anaplasia in wilms tumor—New definitions with prognostic significance. Am. J. Surg. Pathol. 1996, 20, 909–920. [Google Scholar] [CrossRef] [PubMed]
- Qualman, S.J.; Lynch, J.C.; Bridge, J.A.; Parham, D.M.; Teot, L.A.; Meyer, W.H.; Pappo, A.S. Prevalence and clinical impact of anaplasia in childhood rhabdomyosarcoma. Cancer 2008, 113, 3242–3247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hettmer, S.; Archer, N.M.; Somers, G.R.; Novokmet, A.; Wagers, A.J.; Diller, L.; Rodriguez-Galindo, C.; Teot, L.A.; Malkin, D. Anaplastic rhabdomyosarcoma inTP53germline mutation carriers. Cancer 2013, 120, 1068–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Genes/Targeted Pathways | Genetic Modification in Mouse | FN-RMS | References |
|---|---|---|---|
| RAS | Expression of KRAS G12D expression (±background of heterozygous or homozygous p53) | UPS with myogenic features | [96] |
| FGFR4 | Murine myoblasts expressing FGFR4V550E | RMS | [97] |
| P53 | Knock-out of p53 in C57BL/6 | RMS | [90] |
| Trp53/FOS double knock-out in 129Sv X C57BL/6 | ERMS | [98] | |
| Constitutively expressed HER2/neu in Balb/c with p53+/− via MMTV promotor | ERMS | [99] | |
| KRAS G12V conditional expression in adult Balb/c, expressed from Rag2 promotor | ERMS | [100] | |
| Transgenic KRASG12V expression with concurrent knock-out of p53/gain of p53R172H mutant in C57B16J/S129 mice | PRMS | [101] | |
| HGF | HGF/SF Overexpression of HGF in albino FVB/N mice | PRMS with lung metastases | [91,92] |
| Sonic hedgehog | Inactivation of Ptch mutations | RMS | [102] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dehner, C.A.; Armstrong, A.E.; Yohe, M.; Shern, J.F.; Hirbe, A.C. Genetic Characterization, Current Model Systems and Prognostic Stratification in PAX Fusion-Negative vs. PAX Fusion-Positive Rhabdomyosarcoma. Genes 2021, 12, 1500. https://doi.org/10.3390/genes12101500
Dehner CA, Armstrong AE, Yohe M, Shern JF, Hirbe AC. Genetic Characterization, Current Model Systems and Prognostic Stratification in PAX Fusion-Negative vs. PAX Fusion-Positive Rhabdomyosarcoma. Genes. 2021; 12(10):1500. https://doi.org/10.3390/genes12101500
Chicago/Turabian StyleDehner, Carina A., Amy E. Armstrong, Marielle Yohe, Jack F. Shern, and Angela C. Hirbe. 2021. "Genetic Characterization, Current Model Systems and Prognostic Stratification in PAX Fusion-Negative vs. PAX Fusion-Positive Rhabdomyosarcoma" Genes 12, no. 10: 1500. https://doi.org/10.3390/genes12101500