Homology-Free Detection of Transposable Elements Unveils Their Dynamics in Three Ecologically Distinct Rhodnius Species

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sequencing Data
2.2. Annotation and Quantification of the Repeat Content
2.3. Comparisons of Shared TE Families
3. Results
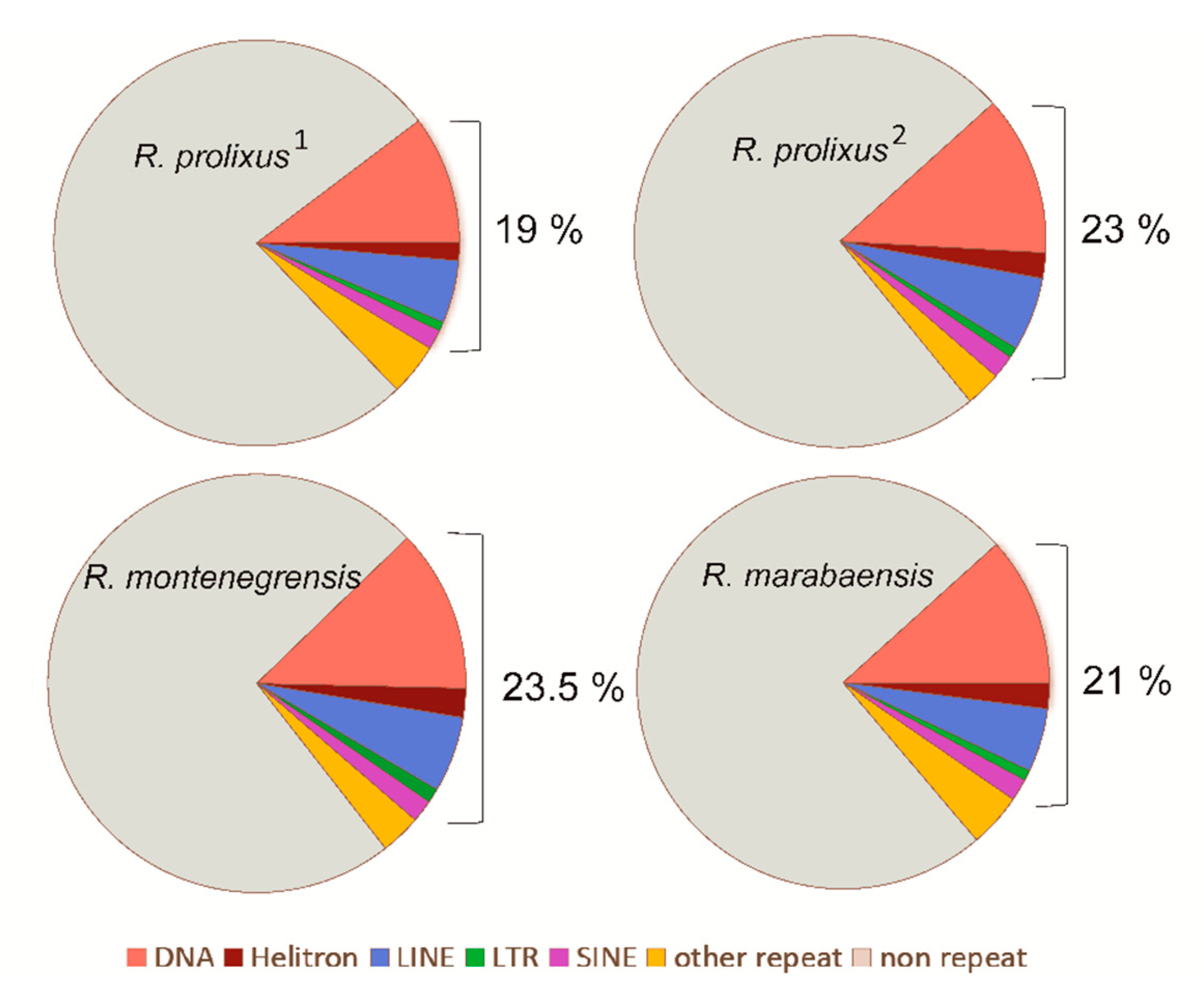
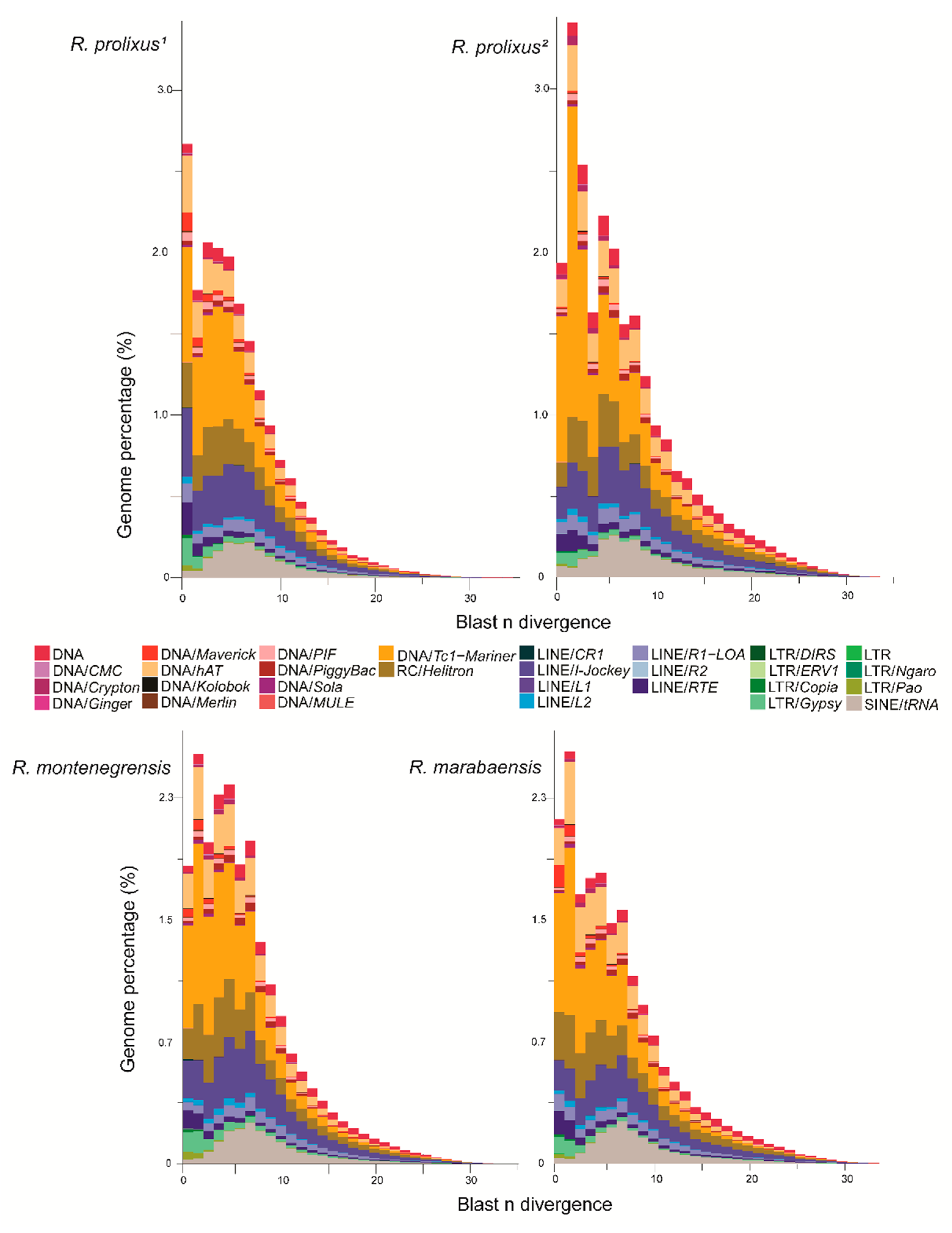
3.1. Proportions and Divergence of TE Superfamilies in Rhodnius Species
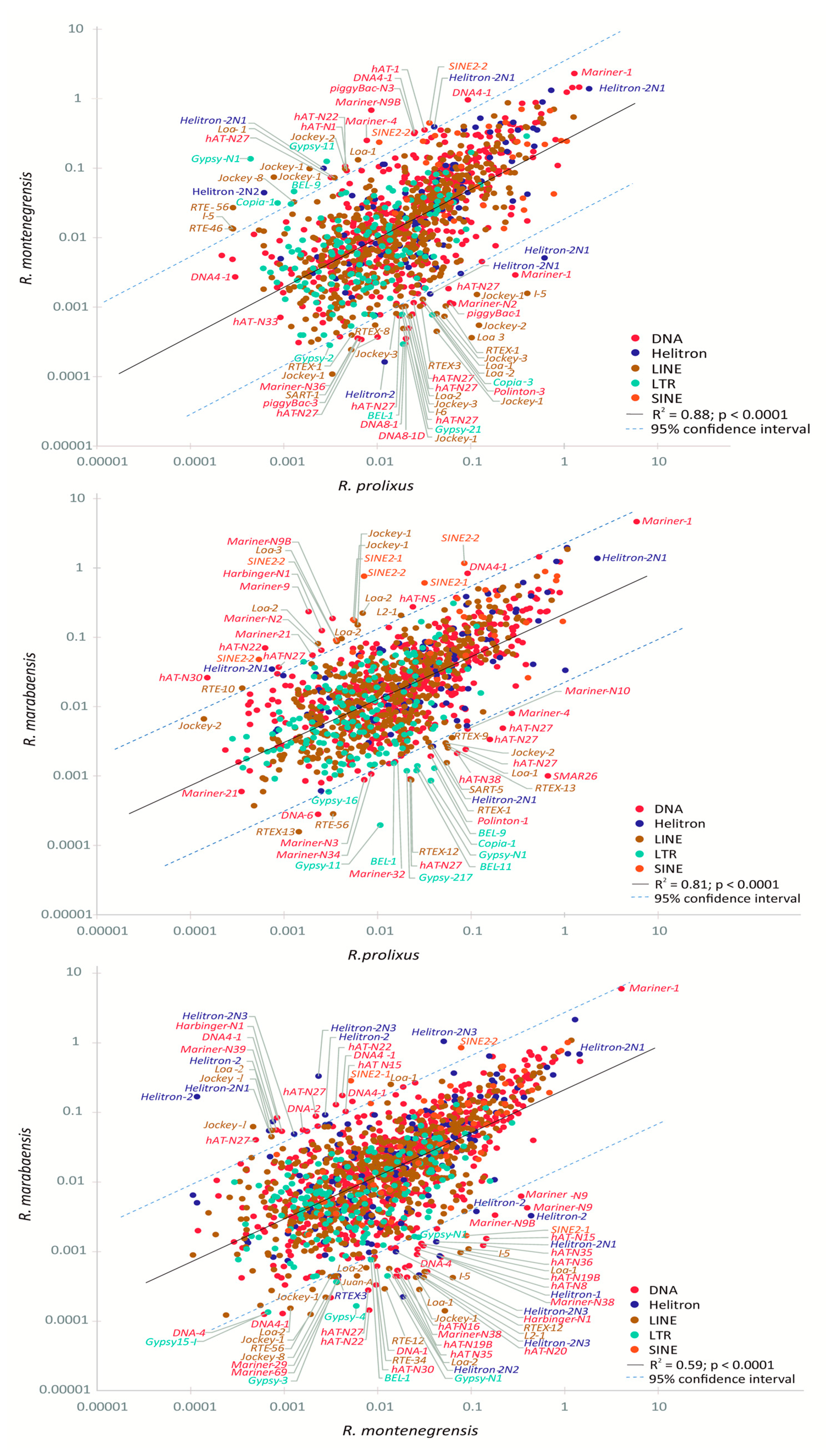
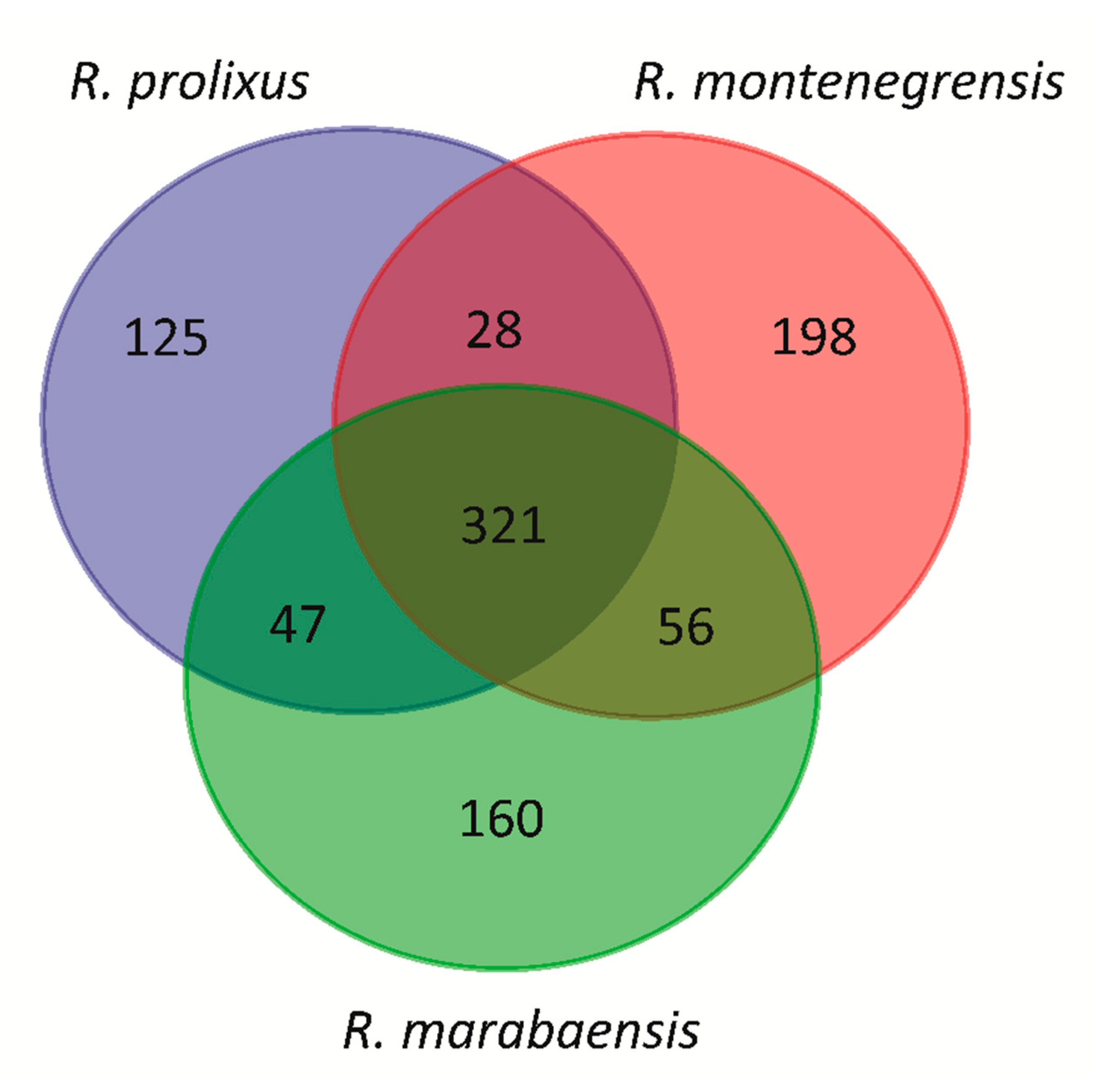
3.2. Relative Proportions of TE Families Shared between the Rhodnius Species
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Maumus, F.; Quesneville, H. Deep investigation of Arabidopsis thaliana junk DNA reveals a continuum between repetitive elements and genomic dark matter. PLoS ONE 2014, 9, e94101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raffaele, S.; Kamoun, S. Genome evolution in filamentous plant pathogens: Why bigger can be better. Nat. Rev. Microbiol. 2012, 10, 417–430. [Google Scholar] [CrossRef] [PubMed]
- Hua-Van, A.; Le Rouzic, A.; Maisonhaute, C.; Capy, P. Abundance, distribution and dynamics of retrotransposable elements and transposons: Similarities and differences. Cytogenet. Genome Res. 2005, 110, 426–440. [Google Scholar] [CrossRef] [PubMed]
- Paterson, A.H.; Bowers, J.E.; Bruggmann, R.; Dubchak, I.; Grimwood, J.; Gundlach, H.; Haberer, G.; Hellsten, U.; Mitros, T.; Poliakov, A.; et al. The Sorghum bicolor genome and the diversification of grasses. Nature 2009, 457, 551–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasyukova, E.G.; Nuzhdin, S.V.; Morozova, T.V.; Mackay, T.F. Accumulation of transposable elements in the genome of Drosophila melanogaster is associated with a decrease in fitness. J. Hered. 2004, 95, 284–290. [Google Scholar] [CrossRef] [Green Version]
- Payer, L.M.; Burns, K.H. Transposable elements in human genetic disease. Nat. Rev. Genet. 2019, 20, 760–772. [Google Scholar] [CrossRef]
- Carareto, C.M.; Hernandez, E.H.; Vieira, C. Genomic regions harboring insecticide resistance-associated Cyp genes are enriched by transposable element fragments carrying putative transcription factor binding sites in two sibling Drosophila species. Gene 2014, 537, 93–99. [Google Scholar] [CrossRef] [Green Version]
- Lopes, F.R.; Jjingo, D.; da Silva, C.R.; Andrade, A.C.; Marraccini, P.; Teixeira, J.B.; Carazzolle, M.F.; Pereira, G.A.; Pereira, L.F.; Vanzela, A.L.; et al. Transcriptional activity, chromosomal distribution and expression effects of transposable elements in Coffea genomes. PLoS ONE 2013, 8, e78931. [Google Scholar] [CrossRef] [Green Version]
- van de Lagemaat, L.N.; Landry, J.R.; Mager, D.L.; Medstrand, P. Transposable elements in mammals promote regulatory variation and diversification of genes with specialized functions. Trends Genet. 2003, 19, 530–536. [Google Scholar] [CrossRef]
- Flabet, M.; Vieira, C. Evolvability, epigenetics and transposable elements. Biomol. Concepts 2011, 2, 333–341. [Google Scholar]
- Wicker, T.; Sabot, F.; Hua-Van, A.; Bennetzen, J.L.; Capy, P.; Chalhoub, B.; Flavell, A.; Leroy, P.; Morgante, M.; Panaud, O.; et al. A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 2007, 8, 973–982. [Google Scholar] [CrossRef] [PubMed]
- Bohne, A.; Brunet, F.; Galiana-Arnoux, D.; Schultheis, C.; Volff, J.N. Transposable elements as drivers of genomic and biological diversity in vertebrates. Chromosome Res. 2008, 16, 203–215. [Google Scholar] [CrossRef] [PubMed]
- Marino-Ramirez, L.; Lewis, K.C.; Landsman, D.; Jordan, I.K. Transposable elements donate lineage-specific regulatory sequences to host genomes. Cytogenet. Genome Res. 2005, 110, 333–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horns, F.; Petit, E.; Hood, M.E. Massive expansion of Gypsy-like retrotransposons in Microbotryum Fungi. Genome Biol. Evol. 2017, 9, 363–371. [Google Scholar] [CrossRef] [Green Version]
- de Boer, J.G.; Yazawa, R.; Davidson, W.S.; Koop, B.F. Bursts and horizontal evolution of DNA transposons in the speciation of pseudotetraploid salmonids. BMC Genom. 2007, 8, 422. [Google Scholar] [CrossRef] [Green Version]
- Pace, J.K.; Feschotte, C. The evolutionary history of human DNA transposons: Evidence for intense activity in the primate lineage. Genome Res. 2007, 17, 422–432. [Google Scholar] [CrossRef] [Green Version]
- Pritham, E.J.; Feschotte, C. Massive amplification of rolling-circle transposons in the lineage of the bat Myotis lucifugus. Proc. Natl. Acad. Sci. USA 2007, 104, 1895–1900. [Google Scholar] [CrossRef] [Green Version]
- Ray, D.A.; Feschotte, C.; Pagan, H.J.; Smith, J.D.; Pritham, E.J.; Arensburger, P.; Atkinson, P.W.; Craig, N.L. Multiple waves of recent DNA transposon activity in the bat, Myotis lucifugus. Genome Res. 2008, 18, 717–728. [Google Scholar] [CrossRef] [Green Version]
- Volff, J.N.; Korting, C.; Meyer, A.; Schartl, M. Evolution and discontinuous distribution of Rex3 retrotransposons in fish. Mol. Biol. Evol. 2001, 18, 427–431. [Google Scholar] [CrossRef] [Green Version]
- Volff, J.N.; Korting, C.; Schartl, M. Multiple lineages of the non-LTR retrotransposon Rex1 with varying success in invading fish genomes. Mol. Biol. Evol. 2000, 17, 1673–1684. [Google Scholar] [CrossRef] [Green Version]
- Belyayev, A. Bursts of transposable elements as an evolutionary driving force. J. Evol. Biol. 2014, 27, 2573–2584. [Google Scholar] [CrossRef] [PubMed]
- da Rosa, J.A.; Rocha, C.S.; Gardim, S.; Pinto, M.C.; Mendonça, V.J.; Ferreira Filho, J.C.R.; de Carvalho, E.O.C.; Camargo, L.M.A.; de Oliveira, J.; Nascimento, J.D.; et al. Description of Rhodnius montenegrensis sp. nov. (Hemiptera: Reduviidae: Triatominae) from the state of Rondonia, Brazil. 2012, 3478, 62–76. [Google Scholar]
- Souza, E.S.; Von Atzingen, N.C.B.; Furtado, M.B.; Oliveira, J.; Nascimento, J.D.; Vendrami, D.P.; Sueli Gardim, S.; da Rosa, J.A. Description of Rhodnius marabaensis sp. n. (Hemiptera, Reduviidade, Triatominae) from Pará State, Brazil. ZooKeys 2016, 621, 45–62. [Google Scholar]
- Harry, M.; Galindez, I.; Cariou, M.L. Isozyme variability and differentiation between Rhodnius prolixus, R. robustus and R. pictipes, vectors of Chagas disease in Venezuela. Med. Vet. Entomol. 1992, 6, 37–43. [Google Scholar] [CrossRef]
- Monteiro, F.A.; Barrett, T.V.; Fitzpatrick, S.; Cordon-Rosales, C.; Feliciangeli, D.; Beard, C.B. Molecular phylogeography of the Amazonian Chagas disease vectors Rhodnius prolixus and R. robustus. Mol. Ecol. 2003, 12, 997–1006. [Google Scholar] [CrossRef]
- Monteiro, F.A.; Escalante, A.A.; Beard, C.B. Molecular tools and triatomine systematics: A public health perspective. Trends Parasitol. 2001, 17, 344–347. [Google Scholar] [CrossRef]
- Monteiro, F.A.; Weirauch, C.; Felix, M.; Lazoski, C.; Abad-Franch, F. Evolution, Systematics, and Biogeography of the Triatominae, Vectors of Chagas Disease. Adv. Parasitol. 2018, 99, 265–344. [Google Scholar]
- Carod-Artal, F.J. American trypanosomiasis. Handb. Clin. Neurol. 2013, 114, 103–123. [Google Scholar]
- Pavan, M.G.; Correa-Antonio, J.; Peixoto, A.A.; Monteiro, F.A.; Rivas, G.B. Rhodnius prolixus and R. robustus (Hemiptera: Reduviidae) nymphs show different locomotor patterns on an automated recording system. Parasites Vector 2016, 9, 239. [Google Scholar] [CrossRef] [Green Version]
- Pavan, M.G.; Mesquita, R.D.; Lawrence, G.G.; Lazoski, C.; Dotson, E.M.; Abubucker, S.; Mitreva, M.; Randall-Maher, J.; Monteiro, F.A. A nuclear single-nucleotide polymorphism (SNP) potentially useful for the separation of Rhodnius prolixus from members of the Rhodnius robustus cryptic species complex (Hemiptera: Reduviidae). Infect. Genet. Evol. 2013, 14, 426–433. [Google Scholar] [CrossRef] [Green Version]
- Mesquita, R.D.; Vionette-Amaral, R.J.; Lowenberger, C.; Rivera-Pomar, R.; Monteiro, F.A.; Minx, P.; Spieth, J.; Carvalho, A.B.; Panzera, F.; Lawson, D.; et al. Genome of Rhodnius prolixus, an insect vector of Chagas disease, reveals unique adaptations to hematophagy and parasite infection. Proc. Natl. Acad. Sci. USA 2015, 112, 14936–14941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Medina, R.D.; Granzotto, A.; Ribeiro, J.M.; Carareto, C.M. Transposition burst of mariner-like elements in the sequenced genome of Rhodnius prolixus. Insect Biochem. Mol. Biol. 2016, 69, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Dujardin, J.P.; Munoz, M.; Chavez, T.; Ponce, C.; Moreno, J.; Schofield, C.J. The origin of Rhodnius prolixus in Central America. Med. Vet. Entomol. 1998, 12, 113–115. [Google Scholar] [CrossRef] [PubMed]
- Lopez, G.; Moreno, J. Genetic variability and differentiation between populations of Rhodnius prolixus and R. pallescens, vectors of Chagas’ disease in Colombia. Memorias do Instituto Oswaldo Cruz 1995, 90, 353–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goubert, C.; Modolo, L.; Vieira, C.; ValienteMoro, C.; Mavingui, P.; Boulesteix, M. De novo assembly and annotation of the Asian tiger mosquito (Aedes albopictus) repeatome with dnaPipeTE from raw genomic reads and comparative analysis with the yellow fever mosquito (Aedes aegypti). Genome Biol. Evol. 2015, 7, 1192–1205. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Li, L.; Myers, J.R.; Marth, G.T. ART: A next-generation sequencing read simulator. Bioinformatics 2012, 28, 593–594. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [Green Version]
- Platt, R.N., II; Blanco-Berdugo, L.; Ray, D.A. Accurate transposable element annotation is vital when analyzing new genome assemblies. Genome Biol. Evol. 2016, 8, 403–410. [Google Scholar] [CrossRef] [Green Version]
- Green, P. Whole-genome disassembly. Proc. Natl. Acad. Sci. USA 2002, 99, 4143–4144. [Google Scholar] [CrossRef] [Green Version]
- Filée, J.; Rouault, J.D.; Harry, M.; Hua-Van, A. Mariner transposons are sailing in the genome of the blood-sucking bug Rhodnius prolixus. BMC Genom. 2015, 16, 1061. [Google Scholar] [CrossRef] [Green Version]
- Feschotte, C.; Swamy, L.; Wessler, S.R. Genome-wide analysis of mariner-like transposable elements in rice reveals complex relationships with stowaway miniature inverted repeat transposable elements (MITEs). Genetics 2003, 163, 747–758. [Google Scholar] [PubMed]
- Zhang, H.H.; Xu, H.E.; Shen, Y.H.; Han, M.J.; Zhang, Z. The origin and evolution of six miniature inverted-repeat transposable elements in Bombyx mori and Rhodnius prolixus. Genome Biol. Evol. 2013, 5, 2020–2031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osanai-Futahashi, M.; Suetsugu, Y.; Mita, K.; Fujiwara, H. Genome-wide screening and characterization of transposable elements and their distribution analysis in the silkworm, Bombyx mori. Insect Biochem. Mol. Biol. 2008, 38, 1046–1057. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.G.; Eisen, M.B.; Smith, D.R.; Bergman, C.M.; Oliver, B.; Markow, T.A.; Kaufman, T.C.; Kellis, M.; Gelbart, W. Evolution of genes and genomes on the Drosophila phylogeny. Nature 2007, 450, 203–218. [Google Scholar]
- Wang, S.; Lorenzen, M.D.; Beeman, R.W.; Brown, S.J. Analysis of repetitive DNA distribution patterns in the Tribolium castaneum genome. Genome Biol. 2008, 9, R61. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Medina, R.D.; Ribeiro, J.M.; Carareto, C.M.; Velasque, L.; Struchiner, C.J. Losing identity: Structural diversity of transposable elements belonging to different classes in the genome of Anopheles gambiae. BMC Genom. 2012, 13, 272. [Google Scholar] [CrossRef] [Green Version]
- Elliott, T.A.; Gregory, T.R. Do larger genomes contain more diverse transposable elements? BMC Evol. Biol. 2015, 15, 69. [Google Scholar] [CrossRef] [Green Version]
- Wallau, G.L.; Capy, P.; Loreto, E.; Hua-Van, A. Genomic landscape and evolutionary dynamics of mariner transposable elements within the Drosophila genus. BMC Genom. 2014, 15, 727. [Google Scholar] [CrossRef] [Green Version]
- Lerat, E.; Burlet, N.; Biemont, C.; Vieira, C. Comparative analysis of transposable elements in the melanogaster subgroup sequenced genomes. Gene 2011, 473, 100–109. [Google Scholar] [CrossRef]
- Staton, S.E.; Bakken, B.H.; Blackman, B.K.; Chapman, M.A.; Kane, N.C.; Tang, S.; Ungerer, M.C.; Knapp, S.J.; Rieseberg, L.H.; Burke, J.M. The sunflower (Helianthus annuus L.) genome reflects a recent history of biased accumulation of transposable elements. Plant J. 2012, 72, 142–153. [Google Scholar] [CrossRef]
- Warren, I.A.; Naville, M.; Chalopin, D.; Levin, P.; Berger, C.S.; Galiana, D.; Volff, J.N. Evolutionary impact of transposable elements on genomic diversity and lineage-specific innovation in vertebrates. Chromosome Res. 2015, 23, 505–531. [Google Scholar] [CrossRef] [PubMed]
- Pinsker, W.; Haring, E.; Hagemann, S.; Miller, W.J. The evolutionary life history of P transposons: From horizontal invaders to domesticated neogenes. Chromosoma 2001, 110, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Dotto, B.R.; Carvalho, E.L.; Silva, A.F.; Duarte Silva, L.F.; Pinto, P.M.; Ortiz, M.F.; Wallau, G.L. HTT-DB: Horizontally transferred transposable elements database. Bioinformatics 2015, 31, 2915–2917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallau, G.L.; Capy, P.; Loreto, E.; Le Rouzic, A.; Hua-Van, A. VHICA, a New Method to Discriminate between Vertical and Horizontal Transposon Transfer: Application to the Mariner Family within Drosophila. Mol. Biol. Evol. 2016, 33, 1094–1109. [Google Scholar] [CrossRef] [Green Version]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castro, M.R.J.; Goubert, C.; Monteiro, F.A.; Vieira, C.; Carareto, C.M.A. Homology-Free Detection of Transposable Elements Unveils Their Dynamics in Three Ecologically Distinct Rhodnius Species. Genes 2020, 11, 170. https://doi.org/10.3390/genes11020170
Castro MRJ, Goubert C, Monteiro FA, Vieira C, Carareto CMA. Homology-Free Detection of Transposable Elements Unveils Their Dynamics in Three Ecologically Distinct Rhodnius Species. Genes. 2020; 11(2):170. https://doi.org/10.3390/genes11020170
Chicago/Turabian StyleCastro, Marcelo R. J., Clément Goubert, Fernando A. Monteiro, Cristina Vieira, and Claudia M. A. Carareto. 2020. "Homology-Free Detection of Transposable Elements Unveils Their Dynamics in Three Ecologically Distinct Rhodnius Species" Genes 11, no. 2: 170. https://doi.org/10.3390/genes11020170