Altered Expression Ratio of Actin-Binding Gelsolin Isoforms Is a Novel Hallmark of Mitochondrial OXPHOS Dysfunction

 , ,
, ,  and
and 
Abstract
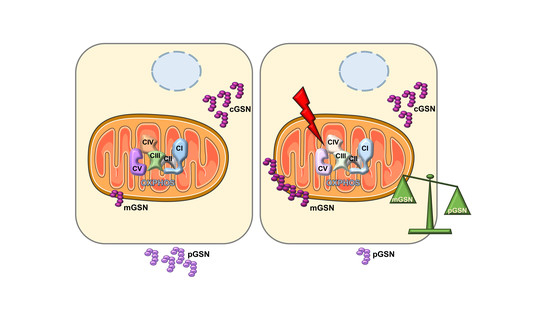
:
1. Introduction
2. Materials and Methods
2.1. Cell Cultures and Treatments
2.2. Blood Samples
2.3. Peripheral Blood Mononuclear Cells and Plasma Separation
2.4. Mitochondrial Respiration
2.5. Intracellular ROS Levels
2.6. Isolation of Mitochondrial Fractions from Human Cells
2.7. Purification of Secreted Proteins from Cultured Cells
2.8. SDS-PAGE Electrophoresis
2.9. Blue Native Electrophoresis and In-Gel Activity Assays
2.10. Antibody Detection
2.11. GSN ELISA Analyses
2.12. Data Analysis
3. Results
3.1. Cytoplasmic GSN Is Upregulated and Targeted to Mitochondria as a General Response to OXPHOS Deficiency
3.2. Plasma GSN Levels Are Significantly Reduced in the Secretomes from Human Cybrids with OXPHOS Deficiency
3.3. Plasma GSN Levels Are Significantly Reduced in Primary Fibroblast Cultures with OXPHOS Deficiency
3.4. Increased Mitochondrial-to-Plasma GSN Ratio in Human Cellular Models of OXPHOS Deficiency
3.5. The Mitochondrial-to-Plasma GSN Ratio Can Be Modulated by Genetic Manipulation of OXPHOS-Deficient Cell Lines
3.6. Oxidative Stress Does Not Trigger the Upregulation and Mitochondrial Location of Cytoplasmic Gelsolin in Human Cybrids
3.7. Mitochondrial-to-Plasma GSN Ratio Increases in Blood Samples from Patients with Mitochondrial OXPHOS Disease
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Reid, R.A.; Moyle, J.; Mitchell, P. Synthesis of adenosine triphosphate by a protonmotive force in rat liver mitochondria. Nature 1966, 212, 257–258. [Google Scholar] [CrossRef] [PubMed]
- McCormick, E.M.; Zolkipli-Cunningham, Z.; Falk, M.J. Mitochondrial disease genetics update. Curr. Opin. Pediatrics 2018, 30, 714–724. [Google Scholar] [CrossRef] [PubMed]
- Koliaki, C.; Roden, M. Alterations of Mitochondrial Function and Insulin Sensitivity in Human Obesity and Diabetes Mellitus. Annu. Rev. Nutr. 2016, 36, 337–367. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Franquesa, A.; Patti, M.-E. Insulin Resistance and Mitochondrial Dysfunction. Adv. Exp. Med. and Biol. 2017, 982, 465–520. [Google Scholar]
- Mulder, H. Transcribing β-cell mitochondria in health and disease. Mol. Metab. 2017, 6, 1040–1051. [Google Scholar] [CrossRef]
- Kawamata, H.; Manfredi, G. Proteinopathies and OXPHOS dysfunction in neurodegenerative diseases. J. Cell Biol. 2017, 216, 3917–3929. [Google Scholar] [CrossRef]
- Area-Gomez, E.; Guardia-Laguarta, C.; Schon, E.A.; Przedborski, S. Mitochondria, OxPhos, and neurodegeneration: Cells are not just running out of gas. J. Clin. Investig. 2019, 129, 34–45. [Google Scholar] [CrossRef] [Green Version]
- Sica, V.; Bravo-San Pedro, J.M.; Stoll, G.; Kroemer, G. Oxidative phosphorylation as a potential therapeutic target for cancer therapy. Int. J. Cancer 2019, 146, 10–17. [Google Scholar] [CrossRef]
- Roth, K.G.; Mambetsariev, I.; Kulkarni, P.; Salgia, R. The Mitochondrion as an Emerging Therapeutic Target in Cancer. Trends Mol. Med. 2020, 26, 119–134. [Google Scholar] [CrossRef]
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial diseases. Nat. Rev. Dis. Primers 2016, 2, 1–22. [Google Scholar] [CrossRef]
- Morán, M.; Marín-Buera, L.; Carmen Gil-Borlado, M.; Rivera, H.; Blázquez, A.; Seneca, S.; Vázquez-López, M.; Arenas, J.; Martín, M.A.; Ugalde, C. Cellular pathophysiological consequences of BCS1L mutations in mitochondrial complex III enzyme deficiency. Hum. Mutat. 2010, 31, 930–941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marín-Buera, L.; García-Bartolomé, A.; Morán, M.; López-Bernardo, E.; Cadenas, S.; Hidalgo, B.; Sánchez, R.; Seneca, S.; Arenas, J.; Martín, M.A.; et al. Differential proteomic profiling unveils new molecular mechanisms associated with mitochondrial complex III deficiency. J. Proteom. 2015, 113, 38–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagener, N.; Ackermann, M.; Funes, S.; Neupert, W. A Pathway of Protein Translocation in Mitochondria Mediated by the AAA-ATPase Bcs1. Mol. Cell 2011, 44, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Vizarra, E.; Bugiani, M.; Goffrini, P.; Carrara, F.; Farina, L.; Procopio, E.; Donati, A.; Uziel, G.; Ferrero, I.; Zeviani, M. Impaired complex III assembly associated with BCS1L gene mutations in isolated mitochondrial encephalopathy. Hum. Mol. Genet. 2007, 16, 1241–1252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, H.L.; Stossel, T.P. Control of cytoplasmic actin gel-sol transformation by gelsolin, a calcium-dependent regulatory protein. Nature 1979, 281, 583–586. [Google Scholar] [CrossRef]
- Li, G.H.; Arora, P.D.; Chen, Y.; McCulloch, C.A.; Liu, P. Multifunctional roles of gelsolin in health and diseases. Med. Res. Rev. 2012, 32, 999–1025. [Google Scholar] [CrossRef]
- Feldt, J.; Schicht, M.; Garreis, F.; Welss, J.; Schneider, U.W.; Paulsen, F. Structure, regulation and related diseases of the actin-binding protein gelsolin. Expert Rev. Mol. Med. 2018, 20. [Google Scholar] [CrossRef]
- Yin, H.L.; Kwiatkowski, D.J.; Mole, J.E.; Cole, F.S. Structure and biosynthesis of cytoplasmic and secreted variants of gelsolin. J. Biol. Chem. 1984, 259, 5271–5276. [Google Scholar]
- Janmey, P.A.; Stossel, T.P. Gelsolin-polyphosphoinositide interaction. Full expression of gelsolin-inhibiting function by polyphosphoinositides in vesicular form and inactivation by dilution, aggregation, or masking of the inositol head group. J. Biol. Chem. 1989, 264, 4825–4831. [Google Scholar]
- Kwiatkowski, D.J.; Stossel, T.P.; Orkin, S.H.; Mole, J.E.; Colten, H.R.; Yin, H.L. Plasma and cytoplasmic gelsolins are encoded by a single gene and contain a duplicated actin-binding domain. Nature 1986, 323, 455–458. [Google Scholar] [CrossRef]
- Allen, P.G. Functional consequences of disulfide bond formation in gelsolin. FEBS Lett. 1997, 401, 89–94. [Google Scholar] [CrossRef] [Green Version]
- Lind, S.E.; Smith, D.B.; Janmey, P.A.; Stossel, T.P. Role of plasma gelsolin and the vitamin D-binding protein in clearing actin from the circulation. J. Clin. Investig. 1986, 78, 736–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, W.M.; Galbraith, R.M.; Galbraith, R.M. The extracellular actin-scavenger system and actin toxicity. N. Engl. J. Med. 1992, 326, 1335–1341. [Google Scholar] [PubMed]
- Piktel, E.; Levental, I.; Durnaś, B.; Janmey, P.; Bucki, R. Plasma Gelsolin: Indicator of Inflammation and Its Potential as a Diagnostic Tool and Therapeutic Target. Int. J. Mol. Sci. 2018, 19, 2516. [Google Scholar] [CrossRef] [Green Version]
- Maury, C.P. Gelsolin-related amyloidosis. Identification of the amyloid protein in Finnish hereditary amyloidosis as a fragment of variant gelsolin. J. Clin. Investig. 1991, 87, 1195–1199. [Google Scholar] [CrossRef]
- Solomon, J.P.; Page, L.J.; Balch, W.E.; Kelly, J.W. Gelsolin amyloidosis: Genetics, biochemistry, pathology and possible strategies for therapeutic intervention. Crit. Rev. Biochem. Mol. Biol. 2012, 47, 282–296. [Google Scholar] [CrossRef] [Green Version]
- Khatri, N.; Sagar, A.; Peddada, N.; Choudhary, V.; Chopra, B.S.; Garg, V.; Garg, R.; Ashish. Plasma Gelsolin Levels Decrease in Diabetic State and Increase upon Treatment with F-Actin Depolymerizing Versions of Gelsolin. J. Diabetes Res. 2014, 2014, 152075. [Google Scholar] [CrossRef]
- Peddada, N.; Sagar, A.; Garg, R. Plasma gelsolin: A general prognostic marker of health. Med Hypotheses 2012, 78, 203–210. [Google Scholar] [CrossRef]
- Ahn, J.S.; Jang, I.-S.; Kim, D.-I.; Cho, K.A.; Park, Y.H.; Kim, K.; Kwak, C.S.; Chul Park, S. Aging-associated increase of gelsolin for apoptosis resistance. Biochem. Biophys. Res. Commun. 2003, 312, 1335–1341. [Google Scholar] [CrossRef]
- Ji, L.; Chauhan, A.; Muthaiyah, B.; Wegiel, J.; Chauhan, V. Gelsolin Levels are Increased in the Brain as a Function of Age During Normal Development in Children That are Further Increased in Down Syndrome. Alzheimer Dis. Assoc. Disord. 2009, 23, 319–322. [Google Scholar] [CrossRef] [Green Version]
- Li, G.H.; Shi, Y.; Chen, Y.; Sun, M.; Sader, S.; Maekawa, Y.; Arab, S.; Dawood, F.; Chen, M.; De Couto, G.; et al. Gelsolin regulates cardiac remodeling after myocardial infarction through DNase I-mediated apoptosis. Circ. Res. 2009, 104, 896–904. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.B.; Zhabyeyev, P.; Chen, X.; Wang, F.; Paul, M.; Fan, D.; McLean, B.A.; Basu, R.; Zhang, P.; Shah, S.; et al. PI3Kα-regulated gelsolin activity is a critical determinant of cardiac cytoskeletal remodeling and heart disease. Nat. Commun. 2018, 9, 5390. [Google Scholar] [CrossRef] [PubMed]
- Mahalka, A.K.; Maury, C.P.J.; Kinnunen, P.K.J. 1-Palmitoyl-2-(9′-oxononanoyl)- sn -glycero-3-phosphocholine, an Oxidized Phospholipid, Accelerates Finnish Type Familial Gelsolin Amyloidosis in Vitro. Biochemistry 2011, 50, 4877–4889. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.; Chauhan, A.; Wegiel, J.; Essa, M.M.; Chauhan, V. Gelsolin is Proteolytically Cleaved in the Brains of Individuals with Alzheimer’s Disease. J. Alzheimer’s Dis. 2009, 18, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.; Chauhan, A.; Chauhan, V. Calcium induces expression of cytoplasmic gelsolin in SH-SY5Y and HEK-293 cells. Neurochem. Res. 2010, 35, 1075–1082. [Google Scholar] [CrossRef]
- Ji, L.; Chauhan, A.; Chauhan, V. Upregulation of cytoplasmic gelsolin, an amyloid-beta-binding protein, under oxidative stress conditions: Involvement of protein kinase C. J. Alzheimer’s Dis. JAD 2010, 19, 829–838. [Google Scholar] [CrossRef]
- Greijer, A.E.; van der Groep, P.; Kemming, D.; Shvarts, A.; Semenza, G.L.; Meijer, G.A.; van de Wiel, M.A.; Belien, J.A.M.; van Diest, P.J.; van der Wall, E. Up-regulation of gene expression by hypoxia is mediated predominantly by hypoxia-inducible factor 1 (HIF-1). J. Pathol. 2005, 206, 291–304. [Google Scholar] [CrossRef]
- Li, Q.; Ye, Z.; Wen, J.; Ma, L.; He, Y.; Lian, G.; Wang, Z.; Wei, L.; Wu, D.; Jiang, B. Gelsolin, but not its cleavage, is required for TNF-induced ROS generation and apoptosis in MCF-7 cells. Biochem. Biophys. Res. Commun. 2009, 385, 284–289. [Google Scholar] [CrossRef]
- Tochhawng, L.; Deng, S.; Pugalenthi, G.; Kumar, A.P.; Lim, K.H.; Tan, T.Z.; Yang, H.; Hooi, S.C.; Goh, Y.C.; Maciver, S.K.; et al. Gelsolin-Cu/ZnSOD interaction alters intracellular reactive oxygen species levels to promote cancer cell invasion. Oncotarget 2016, 7, 52832–52848. [Google Scholar] [CrossRef]
- García-Bartolomé, A.; Peñas, A.; Marín-Buera, L.; Lobo-Jarne, T.; Pérez-Pérez, R.; Morán, M.; Arenas, J.; Martín, M.A.; Ugalde, C. Respiratory chain enzyme deficiency induces mitochondrial location of actin-binding gelsolin to modulate the oligomerization of VDAC complexes and cell survival. Hum. Mol. Genet. 2017, 26, 2493–2506. [Google Scholar] [CrossRef]
- Ugalde, C.; Hinttala, R.; Timal, S.; Smeets, R.; Rodenburg, R.J.T.; Uusimaa, J.; van Heuvel, L.P.; Nijtmans, L.G.J.; Majamaa, K.; Smeitink, J.A.M. Mutated ND2 impairs mitochondrial complex I assembly and leads to Leigh Syndrome. Mol. Genet. Metab. 2007, 90, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Rana, M.; De Coo, I.; Diaz, F.; Smeets, H.; Moraes, C.T. An out-of-frame cytochrome b gene deletion from a patient with parkinsonism is associated with impaired complex III assembly and an increase in free radical production. Ann. Neurol. 2000, 48, 774–781. [Google Scholar] [CrossRef]
- Protasoni, M.; Pérez-Pérez, R.; Lobo-Jarne, T.; Harbour, M.E.; Ding, S.; Peñas, A.; Diaz, F.; Moraes, C.T.; Fearnley, I.M.; Zeviani, M.; et al. Respiratory supercomplexes act as a platform for complex III -mediated maturation of human mitochondrial complexes I and IV. EMBO J. 2020, 39, e102817. [Google Scholar] [CrossRef] [PubMed]
- Bruno, C.; Martinuzzi, A.; Tang, Y.; Andreu, A.L.; Pallotti, F.; Bonilla, E.; Shanske, S.; Fu, J.; Sue, C.M.; Angelini, C.; et al. A stop-codon mutation in the human mtDNA cytochrome c oxidase I gene disrupts the functional structure of complex IV. Am. J. Hum. Genet. 1999, 65, 611–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campos, Y.; García-Redondo, A.; Fernández-Moreno, M.A.; Martínez-Pardo, M.; Goda, G.; Rubio, J.C.; Martín, M.A.; del Hoyo, P.; Cabello, A.; Bornstein, B.; et al. Early-onset multisystem mitochondrial disorder caused by a nonsense mutation in the mitochondrial DNA cytochrome C oxidase II gene. Ann. Neurol. 2001, 50, 409–413. [Google Scholar] [CrossRef]
- Lobo-Jarne, T.; Pérez-Pérez, R.; Fontanesi, F.; Timón-Gómez, A.; Wittig, I.; Peñas, A.; Serrano-Lorenzo, P.; García-Consuegra, I.; Arenas, J.; Martín, M.A.; et al. Multiple pathways coordinate assembly of human mitochondrial complex IV and stabilization of respiratory supercomplexes. EMBO J. 2020, 39, e103912. [Google Scholar] [CrossRef]
- Bourens, M.; Barrientos, A. Human mitochondrial cytochrome c oxidase assembly factor COX18 acts transiently as a membrane insertase within the subunit 2 maturation module. J. Biol. Chem. 2017, 292, 7774–7783. [Google Scholar] [CrossRef] [Green Version]
- Perez-Perez, R.; Lobo-Jarne, T.; Milenkovic, D.; Mourier, A.; Bratic, A.; Garcia-Bartolome, A.; Fernandez-Vizarra, E.; Cadenas, S.; Delmiro, A.; Garcia-Consuegra, I.; et al. COX7A2L Is a Mitochondrial Complex III Binding Protein that Stabilizes the III2+IV Supercomplex without Affecting Respirasome Formation. Cell Rep. 2016, 16, 2387–2398. [Google Scholar] [CrossRef] [Green Version]
- King, M.P.; Attardi, G. Isolation of human cell lines lacking mitochondrial DNA. Methods Enzymol. 1996, 264, 304–313. [Google Scholar]
- De Coo, I.F.; Renier, W.O.; Ruitenbeek, W.; Ter Laak, H.J.; Bakker, M.; Schägger, H.; Van Oost, B.A.; Smeets, H.J. A 4-base pair deletion in the mitochondrial cytochrome b gene associated with parkinsonism/MELAS overlap syndrome. Ann. Neurol. 1999, 45, 130–133. [Google Scholar] [CrossRef]
- Balsa, E.; Marco, R.; Perales-Clemente, E.; Szklarczyk, R.; Calvo, E.; Landázuri, M.O.; Enríquez, J.A. NDUFA4 is a subunit of complex IV of the mammalian electron transport chain. Cell Metab. 2012, 16, 378–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Aurelio, M.; Gajewski, C.D.; Lenaz, G.; Manfredi, G. Respiratory chain supercomplexes set the threshold for respiration defects in human mtDNA mutant cybrids. Hum. Mol. Genet. 2006, 15, 2157–2169. [Google Scholar] [CrossRef] [PubMed]
- Vives-Bauza, C.; Gonzalo, R.; Manfredi, G.; Garcia-Arumi, E.; Andreu, A.L. Enhanced ROS production and antioxidant defenses in cybrids harbouring mutations in mtDNA. Neurosci. Lett. 2006, 391, 136–141. [Google Scholar] [CrossRef]
- Gusdon, A.M.; Votyakova, T.V.; Mathews, C.E. mt-Nd2a Suppresses Reactive Oxygen Species Production by Mitochondrial Complexes I and III. J. Biol. Chem. 2008, 283, 10690–10697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lennicke, C.; Rahn, J.; Lichtenfels, R.; Wessjohann, L.A.; Seliger, B. Hydrogen peroxide - production, fate and role in redox signaling of tumor cells. Cell Commun. Signal. 2015, 13. [Google Scholar] [CrossRef] [Green Version]
- Suhler, E.; Lin, W.; Yin, H.L.; Lee, W.M. Decreased plasma gelsolin concentrations in acute liver failure, myocardial infarction, septic shock, and myonecrosis. Crit. Care Med. 1997, 25, 594–598. [Google Scholar] [CrossRef]
- Kulakowska, A.; Drozdowski, W.; Sadzynski, A.; Bucki, R.; Janmey, P.A. Gelsolin concentration in cerebrospinal fluid from patients with multiple sclerosis and other neurological disorders. Eur. J. Neurol. 2008, 15, 584–588. [Google Scholar] [CrossRef]
- Osborn, T.M.; Verdrengh, M.; Stossel, T.P.; Tarkowski, A.; Bokarewa, M. Decreased levels of the gelsolin plasma isoform in patients with rheumatoid arthritis. Arthritis Res. Ther. 2008, 10. [Google Scholar] [CrossRef] [Green Version]
- Mounzer, K.C.; Moncure, M.; Smith, Y.R.; Dinubile, M.J. Relationship of admission plasma gelsolin levels to clinical outcomes in patients after major trauma. Am. J. Respir. Crit. Care Med. 1999, 160, 1673–1681. [Google Scholar] [CrossRef]
- Smith, D.B.; Janmey, P.A.; Sherwood, J.A.; Howard, R.J.; Lind, S.E. Decreased plasma gelsolin levels in patients with Plasmodium falciparum malaria: A consequence of hemolysis? Blood 1988, 72, 214–218. [Google Scholar] [CrossRef] [Green Version]
- Maldonado, E.N. VDAC-tubulin, an anti-Warburg pro-oxidant switch. Front. Oncol. 2017, 7, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, E.L.; Klimova, T.A.; Eisenbart, J.; Moraes, C.T.; Murphy, M.P.; Budinger, G.R.S.; Chandel, N.S. The Qo site of the mitochondrial complex III is required for the transduction of hypoxic signaling via reactive oxygen species production. J. Cell Biol. 2007, 177, 1029–1036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blázquez, A.; Gil-Borlado, M.C.; Morán, M.; Verdú, A.; Cazorla-Calleja, M.R.; Martín, M.A.; Arenas, J.; Ugalde, C. Infantile mitochondrial encephalomyopathy with unusual phenotype caused by a novel BCS1L mutation in an isolated complex III-deficient patient. Neuromuscul. Disord. 2009, 19, 143–146. [Google Scholar] [CrossRef] [PubMed]
- Korzeniewski, B. Effects of OXPHOS complex deficiencies and ESA dysfunction in working intact skeletal muscle: Implications for mitochondrial myopathies. Biochim. Biophys. Acta 2015, 1847, 1310–1319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valsecchi, F.; Esseling, J.J.; Koopman, W.J.H.; Willems, P.H.G.M. Calcium and ATP handling in human NADH: Ubiquinone oxidoreductase deficiency. Biochim. Biophys. Acta 2009, 1792, 1130–1137. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nº | Age | Sex | Genetics | Clinical Phenotype | Enzyme Defect | mGSN | pGSN | mGSN/pGSN |
|---|---|---|---|---|---|---|---|---|
| P5 | 30 | M | Single deletion | CPEO | IV | 6.23 | 1.35 | 4.615 |
| P6 | 38 | F | Single deletion | CPEO + Myopathy | I, III, IV | 0.37 | 0.69 | 0.536 |
| P7 | 48 | M | OPA1 | Neuropathy | I | 2.93 | 0.62 | 4.726 |
| P8 | 53 | M | POLG | Exercise intolerance | I (mild) | 1.36 | 1.03 | 1.320 |
| P9 | 35 | M | POLG | Exercise intolerance | III (mild) | 2.53 | 1.09 | 2.321 |
| P10 | 58 | M | TK2 | Myopathy | Normal | 1.43 | 0.90 | 1.589 |
| P11 | 29 | F | TK2 | Myopathy | I, III, IV | 3.91 | 1.05 | 3.724 |
| P12 | 22 | M | MT-CO3 m.9319A>G | Myopathy | I, III | 1.70 | 0.92 | 1.848 |
| P13 | 45 | M | MT-TN m.5688T>C | Myopathy | Normal | 0.47 | 0.79 | 0.595 |
| Control | Age | Sex | mGSN | pGSN | mGSN/pGSN |
|---|---|---|---|---|---|
| C1 | 22 | M | 0.306 | 1.06 | 0.289 |
| C2 | 33 | M | 0.181 | 0.80 | 0.226 |
| C3 | 38 | M | 1.160 | 1.00 | 1.160 |
| C4 | 53 | M | 0.505 | 0.65 | 0.777 |
| C5 | 56 | M | 0.430 | 1.03 | 0.417 |
| C6 | 61 | M | 0.775 | 1.23 | 0.630 |
| C7 | 33 | F | 0.349 | 1.08 | 0.323 |
| C8 | 42 | F | 1.885 | 1.20 | 1.571 |
| C9 | 61 | F | 2.223 | 1.15 | 1.933 |
| Cell Type | Sample | mGSN | pGSN | mGSN/pGSN |
|---|---|---|---|---|
| Mutant Cybrids | CON | 1 ± 0.40 | 1 ± 0.20 | 1 |
| CI−KD | 4.66 ± 0.29 *** | 0.42 ± 0.23 *** | 11.10 | |
| CIII−KO | 3.70 ± 0.54 *** | 0.65 ± 0.09 *** | 5.69 | |
| CIV−KO1 | 5.11 ± 0.98 *** | 0.76 ± 0.28 * | 6.72 | |
| CIV−KO2 | 3.78 ± 1.44 *** | 0.56 ± 0.29 *** | 6.75 | |
| CON | 1 ± 0.25 | 1 ± 0.07 | 1 | |
| P1 | 2.40 ± 1.00 | 0.25 ± 0.08 | 9.60 | |
| BCSL1 Mutant | P2 | 2.27 ± 0.31 | 0.27 ± 0.17 | 8.41 |
| Fibroblasts ¶ | P3 | 2.60 ± 0.94 | 0.20 ± 0.09 | 13.00 |
| P4 | 2.23 ± 0.64 | 0.51 ± 0.15 | 4.37 | |
| CON | 1 ± 0.25 | 1 ± 0.33 | 1 | |
| 2 nM AA | 1.07 ± 0.25 | 0.81 ± 0.27 | 1.32 | |
| AA-treated | 20 nM AA | 1.16 ± 0.33 | 0.76 ± 0.38 | 1.53 |
| Fibroblasts ¶ | 200 nM AA | 1.35 ± 0.35 * | 0.67 ± 0.26 * | 2.02 |
| 500 nM AA | 1.39 ± 0.34 *** | 0.34 ± 0.16 * | 4.09 | |
| HEK 293T Cells | CON | 1 ± 0.17 | 1 ± 0.27 | 1 |
| COX18−KO | 1.32 ± 0.06 | 0.76 ± 0.13 | 1.75 | |
| COX18−KO+COX18 | 1.18 ± 0.15 | 1.01 ± 0.20 | 1.16 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-Bartolomé, A.; Peñas, A.; Illescas, M.; Bermejo, V.; López-Calcerrada, S.; Pérez-Pérez, R.; Marín-Buera, L.; Domínguez-González, C.; Arenas, J.; Martín, M.A.; et al. Altered Expression Ratio of Actin-Binding Gelsolin Isoforms Is a Novel Hallmark of Mitochondrial OXPHOS Dysfunction. Cells 2020, 9, 1922. https://doi.org/10.3390/cells9091922
García-Bartolomé A, Peñas A, Illescas M, Bermejo V, López-Calcerrada S, Pérez-Pérez R, Marín-Buera L, Domínguez-González C, Arenas J, Martín MA, et al. Altered Expression Ratio of Actin-Binding Gelsolin Isoforms Is a Novel Hallmark of Mitochondrial OXPHOS Dysfunction. Cells. 2020; 9(9):1922. https://doi.org/10.3390/cells9091922
Chicago/Turabian StyleGarcía-Bartolomé, Alberto, Ana Peñas, María Illescas, Verónica Bermejo, Sandra López-Calcerrada, Rafael Pérez-Pérez, Lorena Marín-Buera, Cristina Domínguez-González, Joaquín Arenas, Miguel A. Martín, and et al. 2020. "Altered Expression Ratio of Actin-Binding Gelsolin Isoforms Is a Novel Hallmark of Mitochondrial OXPHOS Dysfunction" Cells 9, no. 9: 1922. https://doi.org/10.3390/cells9091922