CDK7 Inhibition Is Effective in all the Subtypes of Breast Cancer: Determinants of Response and Synergy with EGFR Inhibition

Abstract
:1. Introduction
2. Materials and Methods
2.1. Kaplan–Meier Plotter Online Survival Analysis
2.2. Cell Lines and Reagents
2.3. Western Blotting
2.4. RNA Extraction, Reverse Transcription and q-PCR
2.5. Cell Proliferation Assays
2.6. Cell Cycle Analysis
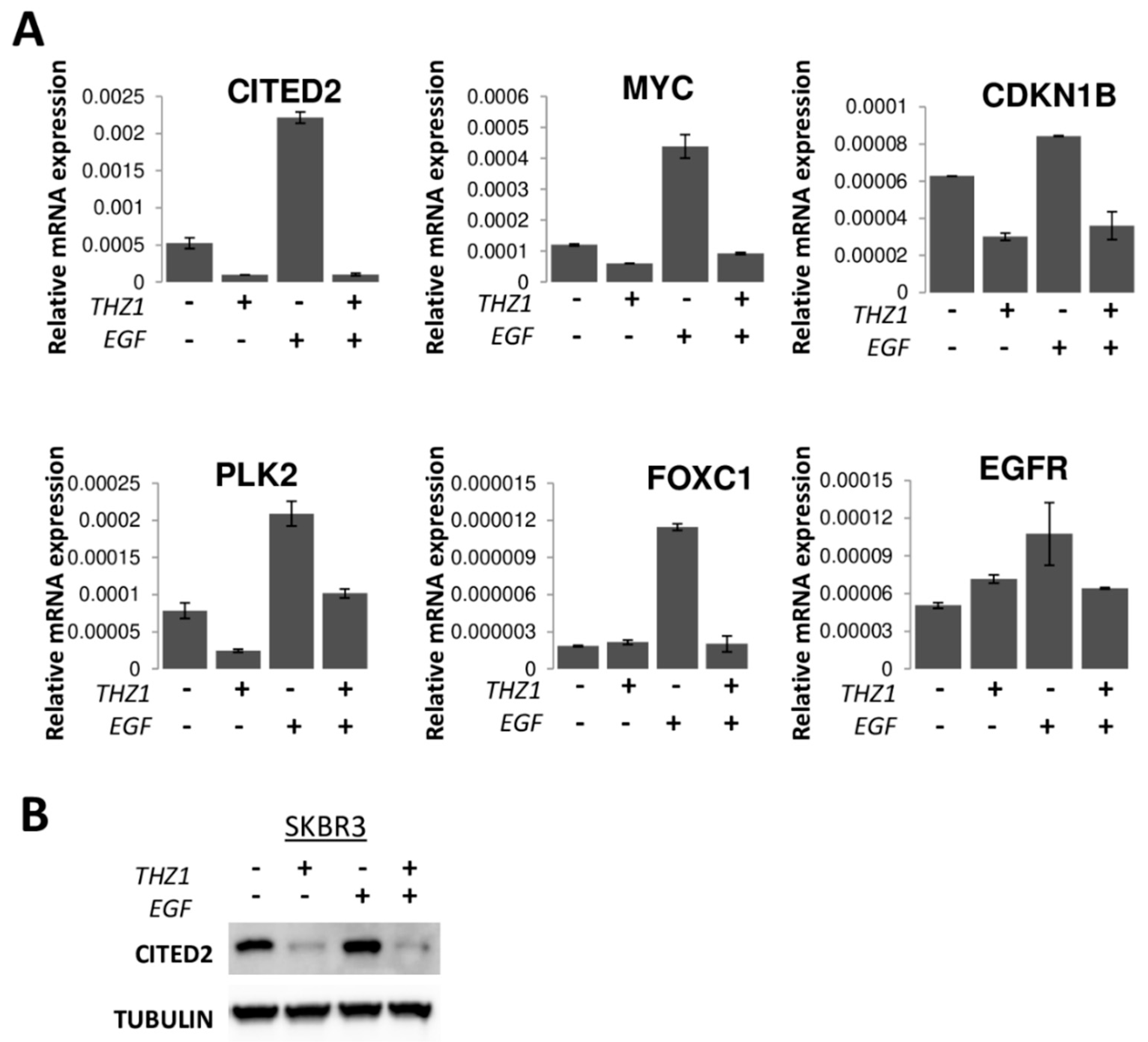
2.7. EGF Stimulation
2.8. Synergy Analysis
2.9. Statistical Analysis
3. Results
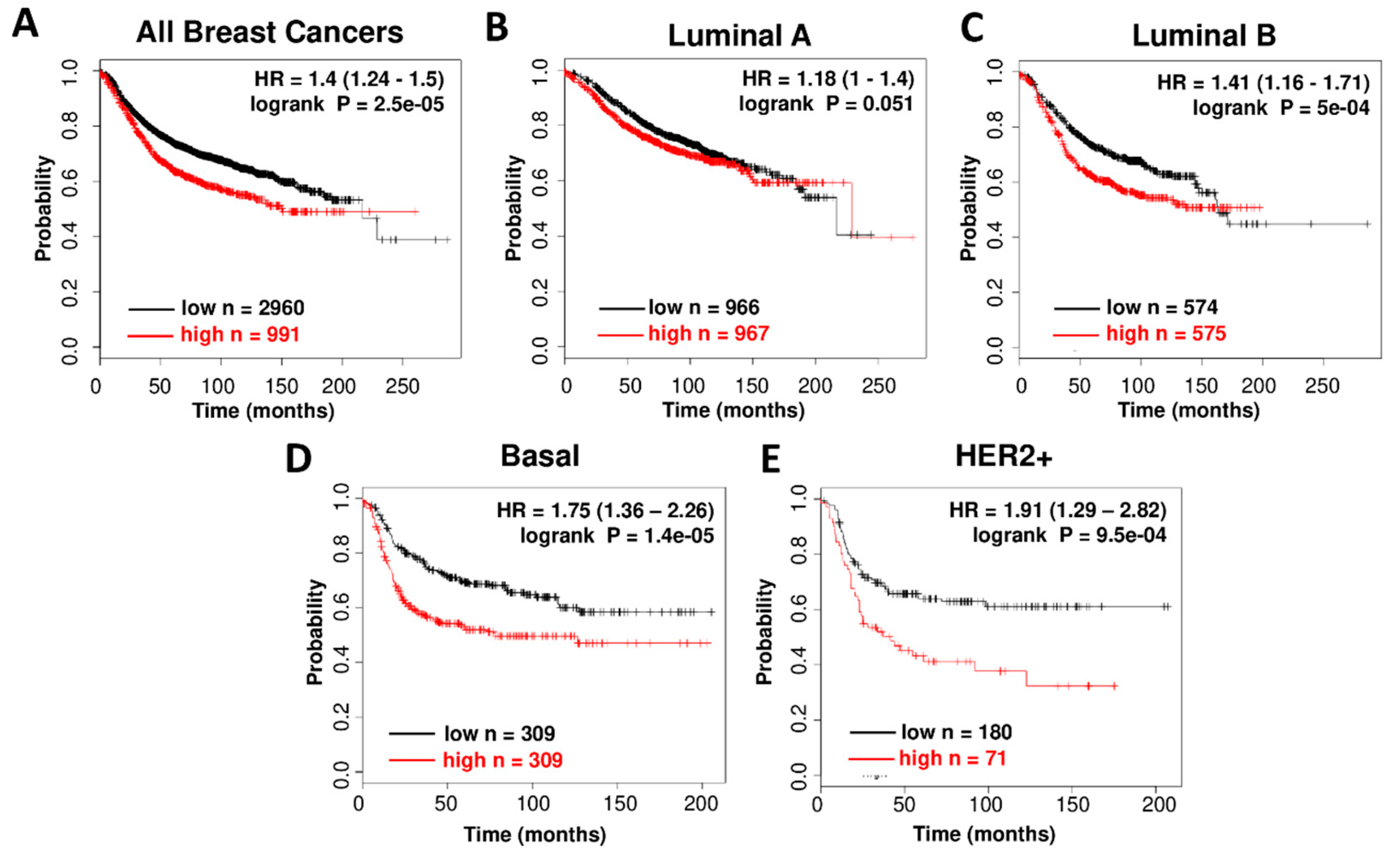
3.1. High CDK7 Expression is Associated with Worse Relapse Free Survival in Breast Cancer Subtypes
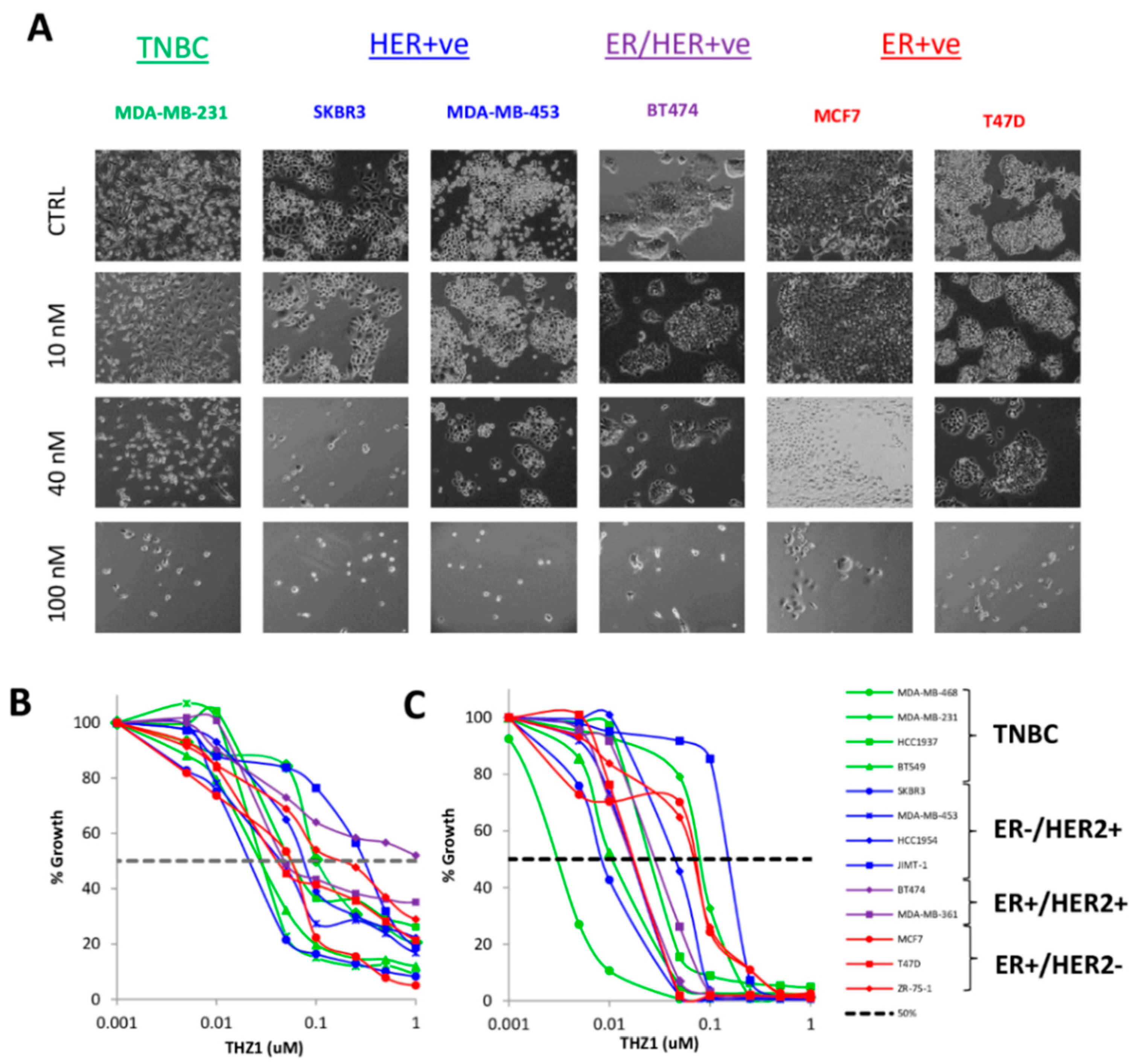
3.2. Breast Cancer Growth is Dependent on CDK7 Regardless of Subtype
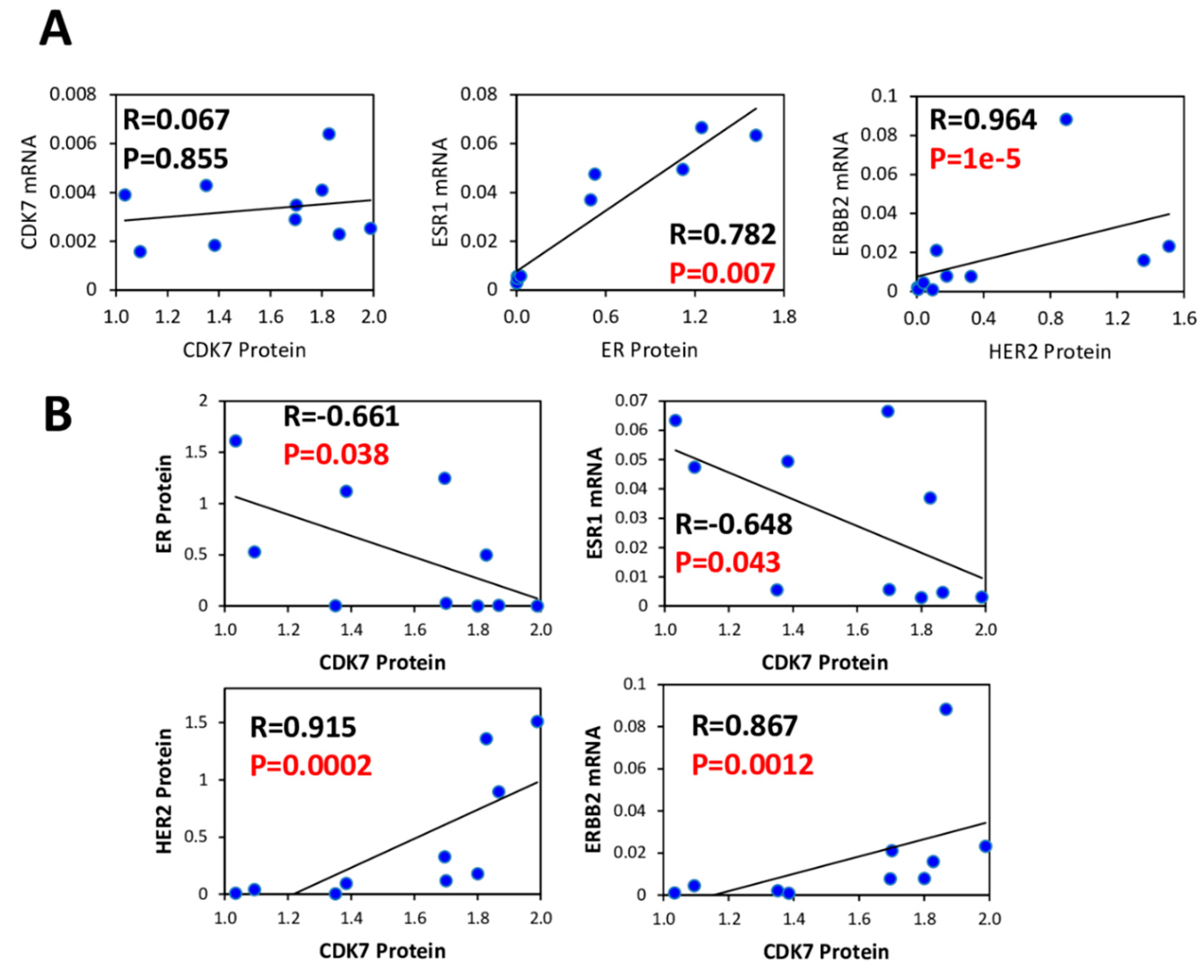
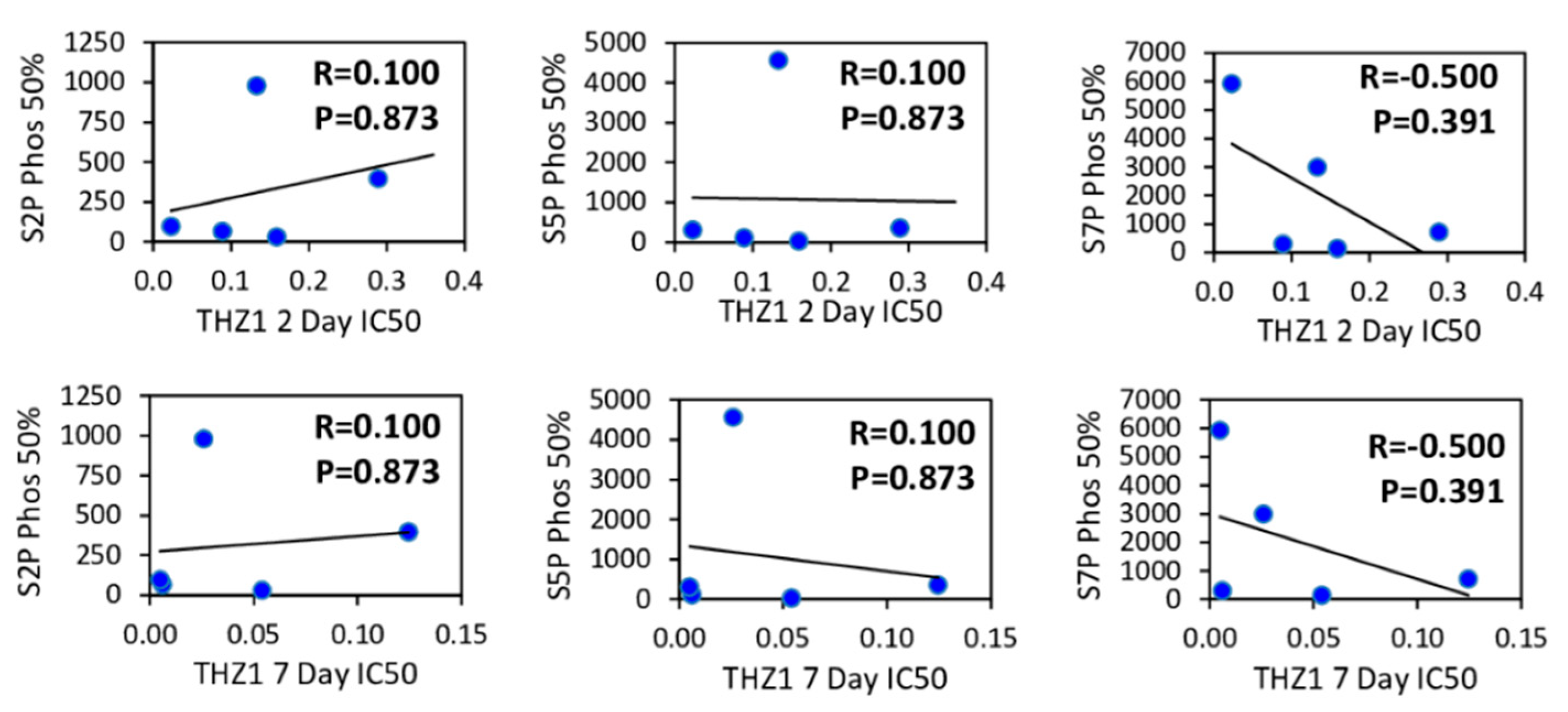
3.3. CDK7, ER and HER2 Expression are not Predictive Biomarkers of THZ1 Response
3.4. Phenotypic Responses to CDK7 Inhibition
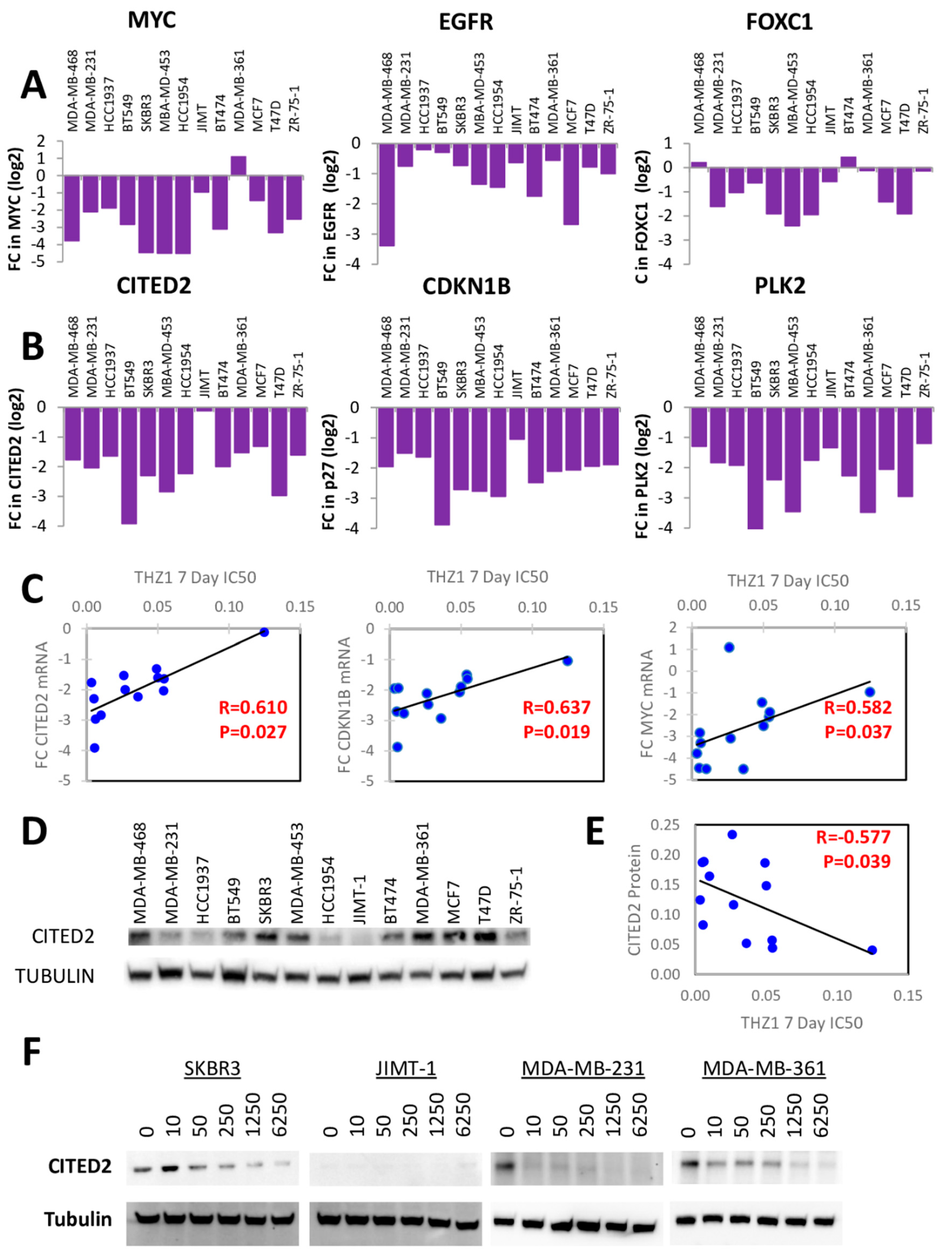
3.5. Transcriptional Regulation by THZ1 in Breast Cancer
3.6. CITED2 as a Determinant of CDK7 Dependence
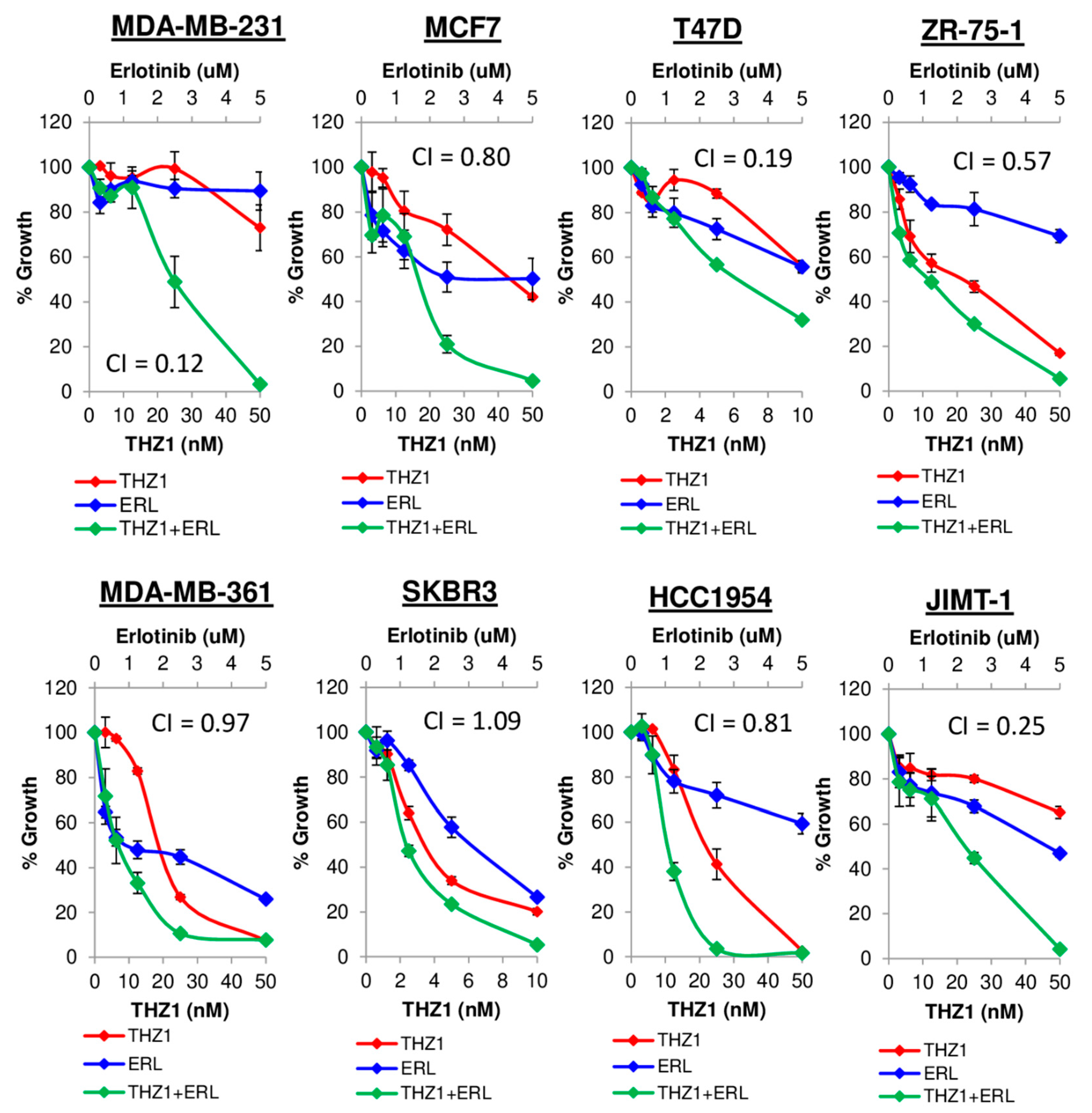
3.7. Synergy Between CDK7 and EGFR Inhibitors in Breast Cancer
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Whittaker, S.R.; Mallinger, A.; Workman, P.; Clarke, P.A. Inhibitors of cyclin-dependent kinases as cancer therapeutics. Pharmacol. Ther. 2017, 173, 83–105. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, T.; Kwiatkowski, N.; Abraham, B.J.; Lee, T.I.; Xie, S.; Yuzugullu, H.; Von, T.; Li, H.; Lin, Z.; et al. CDK7-dependent transcriptional addiction in triple-negative breast cancer. Cell 2015, 163, 174–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morales, F.; Giordano, A. Overview of CDK9 as a target in cancer research. Cell Cycle 2016, 15, 519–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDermott, M.S.; Chumanevich, A.A.; Lim, C.U.; Liang, J.; Chen, M.; Altilia, S.; Oliver, D.; Rae, J.M.; Shtutman, M.; Kiaris, H.; et al. Inhibition of CDK8 mediator kinase suppresses estrogen dependent transcription and the growth of estrogen receptor positive breast cancer. Oncotarget 2017, 8, 12558–12575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roeder, R.G. Transcriptional regulation and the role of diverse coactivators in animal cells. FEBS Lett. 2005, 579, 909–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larochelle, S.; Amat, R.; Glover-Cutter, K.; Sanso, M.; Zhang, C.; Allen, J.J.; Shokat, K.M.; Bentley, D.L.; Fisher, R.P. Cyclin-dependent kinase control of the initiation-to-elongation switch of RNA polymerase II. Nat. Struct. Mol. Biol. 2012, 19, 1108–1115. [Google Scholar] [CrossRef] [Green Version]
- Fisher, R.P. The CDK Network: Linking Cycles of Cell Division and Gene Expression. Genes Cancer 2012, 3, 731–738. [Google Scholar] [CrossRef] [Green Version]
- Schachter, M.M.; Merrick, K.A.; Larochelle, S.; Hirschi, A.; Zhang, C.; Shokat, K.M.; Rubin, S.M.; Fisher, R.P. A Cdk7-Cdk4 T-loop phosphorylation cascade promotes G1 progression. Mol. Cell 2013, 50, 250–260. [Google Scholar] [CrossRef] [Green Version]
- Kwiatkowski, N.; Zhang, T.; Rahl, P.B.; Abraham, B.J.; Reddy, J.; Ficarro, S.B.; Dastur, A.; Amzallag, A.; Ramaswamy, S.; Tesar, B.; et al. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature 2014, 511, 616–620. [Google Scholar] [CrossRef] [Green Version]
- Chipumuro, E.; Marco, E.; Christensen, C.L.; Kwiatkowski, N.; Zhang, T.; Hatheway, C.M.; Abraham, B.J.; Sharma, B.; Yeung, C.; Altabef, A.; et al. CDK7 inhibition suppresses super-enhancer-linked oncogenic transcription in MYCN-driven cancer. Cell 2014, 159, 1126–1139. [Google Scholar] [CrossRef] [Green Version]
- Christensen, C.L.; Kwiatkowski, N.; Abraham, B.J.; Carretero, J.; Al-Shahrour, F.; Zhang, T.; Chipumuro, E.; Herter-Sprie, G.S.; Akbay, E.A.; Altabef, A.; et al. Targeting transcriptional addictions in small cell lung cancer with a covalent CDK7 inhibitor. Cancer Cell 2014, 26, 909–922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenall, S.A.; Lim, Y.C.; Mitchell, C.B.; Ensbey, K.S.; Stringer, B.W.; Wilding, A.L.; O’Neill, G.M.; McDonald, K.L.; Gough, D.J.; Day, B.W.; et al. Cyclin-dependent kinase 7 is a therapeutic target in high-grade glioma. Oncogenesis 2017, 6, e336. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Peng, H.; Wang, X.; Yin, X.; Ma, P.; Jing, Y.; Cai, M.C.; Liu, J.; Zhang, M.; Zhang, S.; et al. Preclinical Efficacy and Molecular Mechanism of Targeting CDK7-Dependent Transcriptional Addiction in Ovarian Cancer. Mol. Cancer Ther. 2017, 16, 1739–1750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, H.; Abduljabbar, R.; Lai, C.F.; Periyasamy, M.; Harrod, A.; Gemma, C.; Steel, J.H.; Patel, N.; Busonero, C.; Jerjees, D.; et al. Expression of CDK7, Cyclin H, and MAT1 Is Elevated in Breast Cancer and Is Prognostic in Estrogen Receptor-Positive Breast Cancer. Clin. Cancer Res. 2016, 22, 5929–5938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.; Washbrook, E.; Sarwar, N.; Bates, G.J.; Pace, P.E.; Thirunuvakkarasu, V.; Taylor, J.; Epstein, R.J.; Fuller-Pace, F.V.; Egly, J.M.; et al. Phosphorylation of human estrogen receptor alpha at serine 118 by two distinct signal transduction pathways revealed by phosphorylation-specific antisera. Oncogene 2002, 21, 4921–4931. [Google Scholar] [CrossRef] [Green Version]
- Kavarthapu, R.; Dufau, M.L. Essential role of endogenous prolactin and CDK7 in estrogen-induced upregulation of the prolactin receptor in breast cancer cells. Oncotarget 2017, 8, 27353–27363. [Google Scholar] [CrossRef] [Green Version]
- Sun, B.; Mason, S.; Wilson, R.C.; Hazard, S.E.; Wang, Y.; Fang, R.; Wang, Q.; Yeh, E.S.; Yang, M.; Roberts, T.M.; et al. Inhibition of the transcriptional kinase CDK7 overcomes therapeutic resistance in HER2-positive breast cancers. Oncogene 2019. [Google Scholar] [CrossRef]
- Li, B.; Ni Chonghaile, T.; Fan, Y.; Madden, S.F.; Klinger, R.; O’Connor, A.E.; Walsh, L.; O’Hurley, G.; Mallya Udupi, G.; Joseph, J.; et al. Therapeutic Rationale to Target Highly Expressed CDK7 Conferring Poor Outcomes in Triple-Negative Breast Cancer. Cancer Res. 2017, 77, 3834–3845. [Google Scholar] [CrossRef] [Green Version]
- Gyorffy, B.; Lanczky, A.; Eklund, A.C.; Denkert, C.; Budczies, J.; Li, Q.; Szallasi, Z. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res. Treat. 2010, 123, 725–731. [Google Scholar] [CrossRef] [Green Version]
- Chou, T.C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [Green Version]
- Held, J.M.; Britton, D.J.; Scott, G.K.; Lee, E.L.; Schilling, B.; Baldwin, M.A.; Gibson, B.W.; Benz, C.C. Ligand binding promotes CDK-dependent phosphorylation of ER-alpha on hinge serine 294 but inhibits ligand-independent phosphorylation of serine 305. Mol. Cancer Res. 2012, 10, 1120–1132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayaraman, S.; Doucet, M.; Lau, W.M.; Kominsky, S.L. CITED2 Modulates Breast Cancer Metastatic Ability through Effects on IKKalpha. Mol. Cancer Res. 2016, 14, 730–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minemura, H.; Takagi, K.; Sato, A.; Takahashi, H.; Miki, Y.; Shibahara, Y.; Watanabe, M.; Ishida, T.; Sasano, H.; Suzuki, T. CITED2 in breast carcinoma as a potent prognostic predictor associated with proliferation, migration and chemoresistance. Cancer Sci. 2016, 107, 1898–1908. [Google Scholar] [CrossRef] [PubMed]
- Chou, Y.T.; Hsieh, C.H.; Chiou, S.H.; Hsu, C.F.; Kao, Y.R.; Lee, C.C.; Chung, C.H.; Wang, Y.H.; Hsu, H.S.; Pang, S.T.; et al. CITED2 functions as a molecular switch of cytokine-induced proliferation and quiescence. Cell Death Differ. 2012, 19, 2015–2028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levin, E.R. Bidirectional signaling between the estrogen receptor and the epidermal growth factor receptor. Mol. Endocrinol. 2003, 17, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Harrod, A.; Fulton, J.; Nguyen, V.T.M.; Periyasamy, M.; Ramos-Garcia, L.; Lai, C.F.; Metodieva, G.; de Giorgio, A.; Williams, R.L.; Santos, D.B.; et al. Genomic modelling of the ESR1 Y537S mutation for evaluating function and new therapeutic approaches for metastatic breast cancer. Oncogene 2017, 36, 2286–2296. [Google Scholar] [CrossRef] [Green Version]
- Lolli, G.; Johnson, L.N. CAK-Cyclin-dependent Activating Kinase: A key kinase in cell cycle control and a target for drugs? Cell Cycle 2005, 4, 572–577. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.Y.; Lin, D.C.; Mayakonda, A.; Hazawa, M.; Ding, L.W.; Chien, W.W.; Xu, L.; Chen, Y.; Xiao, J.F.; Senapedis, W.; et al. Targeting super-enhancer-associated oncogenes in oesophageal squamous cell carcinoma. Gut 2017, 66, 1358–1368. [Google Scholar] [CrossRef] [Green Version]
- Cayrol, F.; Praditsuktavorn, P.; Fernando, T.M.; Kwiatkowski, N.; Marullo, R.; Calvo-Vidal, M.N.; Phillip, J.; Pera, B.; Yang, S.N.; Takpradit, K.; et al. THZ1 targeting CDK7 suppresses STAT transcriptional activity and sensitizes T-cell lymphomas to BCL2 inhibitors. Nat. Commun. 2017, 8, 14290. [Google Scholar] [CrossRef] [Green Version]
- Kelso, T.W.; Baumgart, K.; Eickhoff, J.; Albert, T.; Antrecht, C.; Lemcke, S.; Klebl, B.; Meisterernst, M. Cyclin-dependent kinase 7 controls mRNA synthesis by affecting stability of preinitiation complexes, leading to altered gene expression, cell cycle progression, and survival of tumor cells. Mol. Cell Biol. 2014, 34, 3675–3688. [Google Scholar] [CrossRef] [Green Version]
- Obaya, A.J.; Kotenko, I.; Cole, M.D.; Sedivy, J.M. The proto-oncogene c-myc acts through the cyclin-dependent kinase (Cdk) inhibitor p27(Kip1) to facilitate the activation of Cdk4/6 and early G(1) phase progression. J. Biol. Chem. 2002, 277, 31263–31269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharya, S.; Michels, C.L.; Leung, M.K.; Arany, Z.P.; Kung, A.L.; Livingston, D.M. Functional role of p35srj, a novel p300/CBP binding protein, during transactivation by HIF-1. Genes Dev. 1999, 13, 64–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, Y.T.; Wang, H.; Chen, Y.; Danielpour, D.; Yang, Y.C. Cited2 modulates TGF-beta-mediated upregulation of MMP9. Oncogene 2006, 25, 5547–5560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yahata, T.; Shao, W.; Endoh, H.; Hur, J.; Coser, K.R.; Sun, H.; Ueda, Y.; Kato, S.; Isselbacher, K.J.; Brown, M.; et al. Selective coactivation of estrogen-dependent transcription by CITED1 CBP/p300-binding protein. Genes Dev. 2001, 15, 2598–2612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, W.M.; Weber, K.L.; Doucet, M.; Chou, Y.T.; Brady, K.; Kowalski, J.; Tsai, H.L.; Yang, J.; Kominsky, S.L. Identification of prospective factors promoting osteotropism in breast cancer: A potential role for CITED2. Int. J. Cancer 2010, 126, 876–884. [Google Scholar] [CrossRef] [PubMed]
- Lau, W.M.; Doucet, M.; Huang, D.; Weber, K.L.; Kominsky, S.L. CITED2 modulates estrogen receptor transcriptional activity in breast cancer cells. Biochem Biophys Res. Commun. 2013, 437, 261–266. [Google Scholar] [CrossRef] [Green Version]
- Berry, F.B.; Mirzayans, F.; Walter, M.A. Regulation of FOXC1 stability and transcriptional activity by an epidermal growth factor-activated mitogen-activated protein kinase signaling cascade. J. Biol. Chem. 2006, 281, 10098–10104. [Google Scholar] [CrossRef] [Green Version]
- Ran, W.; Dean, M.; Levine, R.A.; Henkle, C.; Campisi, J. Induction of c-fos and c-myc mRNA by epidermal growth factor or calcium ionophore is cAMP dependent. Proc. Natl. Acad. Sci. USA 1986, 83, 8216–8220. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Jo, M.; Curry, T.E., Jr.; Liu, J. Hormonal induction of polo-like kinases (Plks) and impact of Plk2 on cell cycle progression in the rat ovary. PLoS ONE 2012, 7, e41844. [Google Scholar] [CrossRef]
- Fang, Y.; Wang, Y.; Wang, Y.; Meng, Y.; Zhu, J.; Jin, H.; Li, J.; Zhang, D.; Yu, Y.; Wu, X.R.; et al. A new tumour suppression mechanism by p27Kip1: EGFR down-regulation mediated by JNK/c-Jun pathway inhibition. Biochem. J. 2014, 463, 383–392. [Google Scholar] [CrossRef] [Green Version]
- Masuda, H.; Zhang, D.; Bartholomeusz, C.; Doihara, H.; Hortobagyi, G.N.; Ueno, N.T. Role of epidermal growth factor receptor in breast cancer. Breast Cancer Res. Treat. 2012, 136, 331–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLaughlin, R.P.; He, J.; van der Noord, V.E.; Redel, J.; Foekens, J.A.; Martens, J.W.M.; Smid, M.; Zhang, Y.; van de Water, B. A kinase inhibitor screen identifies a dual cdc7/CDK9 inhibitor to sensitise triple-negative breast cancer to EGFR-targeted therapy. Breast Cancer Res. 2019, 21, 77. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | 2 Day IC50 (µM) | 7 Day IC50 (µM) |
|---|---|---|
| MDA-MB-468 | 0.1451 | 0.0034 |
| MDA-MB-231 | 0.1590 | 0.0540 |
| HCC1937 | 0.0847 | 0.0543 |
| BT549 | 0.0937 | 0.0054 |
| SKBR3 | 0.0230 | 0.0050 |
| MDA-MB-453 | 0.1330 | 0.0100 |
| HCC 1954 | 0.1580 | 0.0360 |
| JIMT-1 | 0.2890 | 0.1246 |
| BT474 | 0.3598 | 0.0270 |
| MDA-MB-361 | 0.1330 | 0.0260 |
| MCF7 | 0.0320 | 0.0490 |
| T47D | 0.0890 | 0.0061 |
| ZR-75-1 | 0.1288 | 0.0500 |
| Gene Name | 468 FC | 549 FC | T47D FC | ZR751 FC |
|---|---|---|---|---|
| CYR61 | –4.82 | –2.96 | –1.96 | –1.81 |
| DKK1 | –4.7 | –5.01 | –1.09 | –4.15 |
| MYC | –4.64 | –3.06 | –2.68 | –2.98 |
| CITED2 | –4.53 | –4.12 | –2.71 | –2.64 |
| FBXO5 | –4.19 | –4.09 | –2.65 | –3.01 |
| CDKN1B | –4.11 | –3.06 | –2.3 | –2.57 |
| PIM3 | –4.05 | –2.73 | –2.54 | –3.96 |
| MARS2 | –4.04 | –3.73 | –2.3 | –3.75 |
| NEDD9 | –4.03 | –1.53 | –3.32 | –2.68 |
| E2F8 | –4.01 | –3.09 | –4.01 | –4.01 |
| PLK2 | –3.99 | –3.7 | –2.48 | –3.04 |
| FADD | –3.93 | –3.1 | –3.29 | –4.12 |
| ELF3 | –3.88 | –1.11 | –1.22 | –1.65 |
| MOCS3 | –3.84 | –4.2 | –3.86 | –4.1 |
| BAMBI | –3.8 | –2.97 | –1.74 | –3.77 |
| ZNF217 | –3.77 | –3.66 | –2.77 | –4.13 |
| PHLDA1 | –3.71 | –2.72 | –3.81 | –2.55 |
| TRIB1 | –3.69 | –4.07 | –2.31 | –3.54 |
| WEE1 | –3.69 | –2.95 | –1.96 | –3.25 |
| ZNF627 | –3.68 | –3.39 | –2.75 | –3.38 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
McDermott, M.S.J.; Sharko, A.C.; Munie, J.; Kassler, S.; Melendez, T.; Lim, C.-u.; Broude, E.V. CDK7 Inhibition Is Effective in all the Subtypes of Breast Cancer: Determinants of Response and Synergy with EGFR Inhibition. Cells 2020, 9, 638. https://doi.org/10.3390/cells9030638
McDermott MSJ, Sharko AC, Munie J, Kassler S, Melendez T, Lim C-u, Broude EV. CDK7 Inhibition Is Effective in all the Subtypes of Breast Cancer: Determinants of Response and Synergy with EGFR Inhibition. Cells. 2020; 9(3):638. https://doi.org/10.3390/cells9030638
Chicago/Turabian StyleMcDermott, Martina S. J., Amanda C. Sharko, Jessica Munie, Susannah Kassler, Theresa Melendez, Chang-uk Lim, and Eugenia V. Broude. 2020. "CDK7 Inhibition Is Effective in all the Subtypes of Breast Cancer: Determinants of Response and Synergy with EGFR Inhibition" Cells 9, no. 3: 638. https://doi.org/10.3390/cells9030638