The Effects of Sodium Dichloroacetate on Mitochondrial Dysfunction and Neuronal Death Following Hypoglycemia-Induced Injury

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Experimental Animals
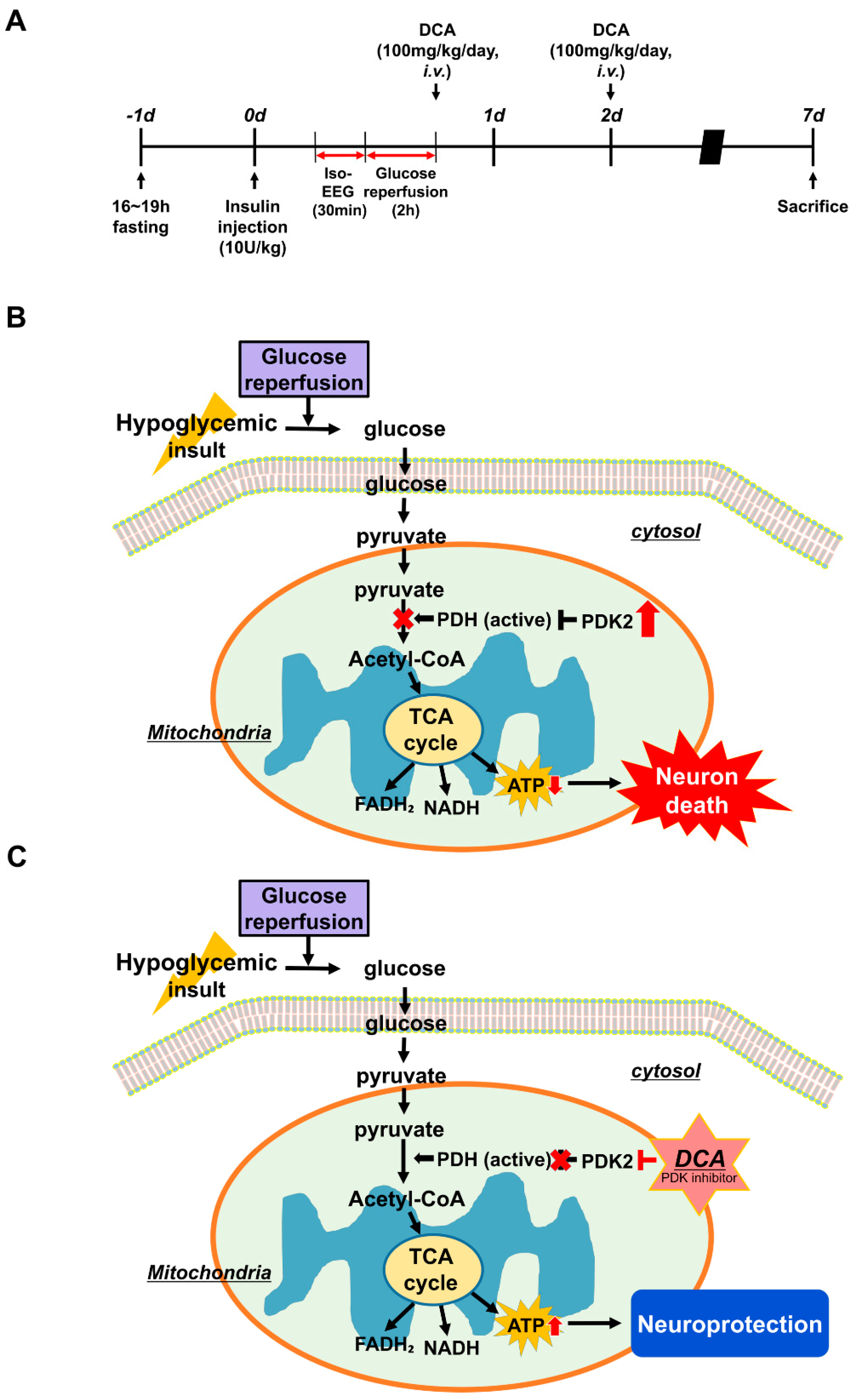
2.3. Animal Surgery and Severe Hypoglycemia Induction
2.4. Injection of DCA
2.5. Brain Sample Preparation
2.6. Detection of PDH and PDK2
2.7. Detection of Neuronal Death
2.8. Detection of Live Neurons
2.9. Detection of Oxidative Injury
2.10. Detection of Microglia and Astrocyte Activation
2.11. Detection of BBB Disruption and Neutrophils
2.12. Data Analysis
3. Results
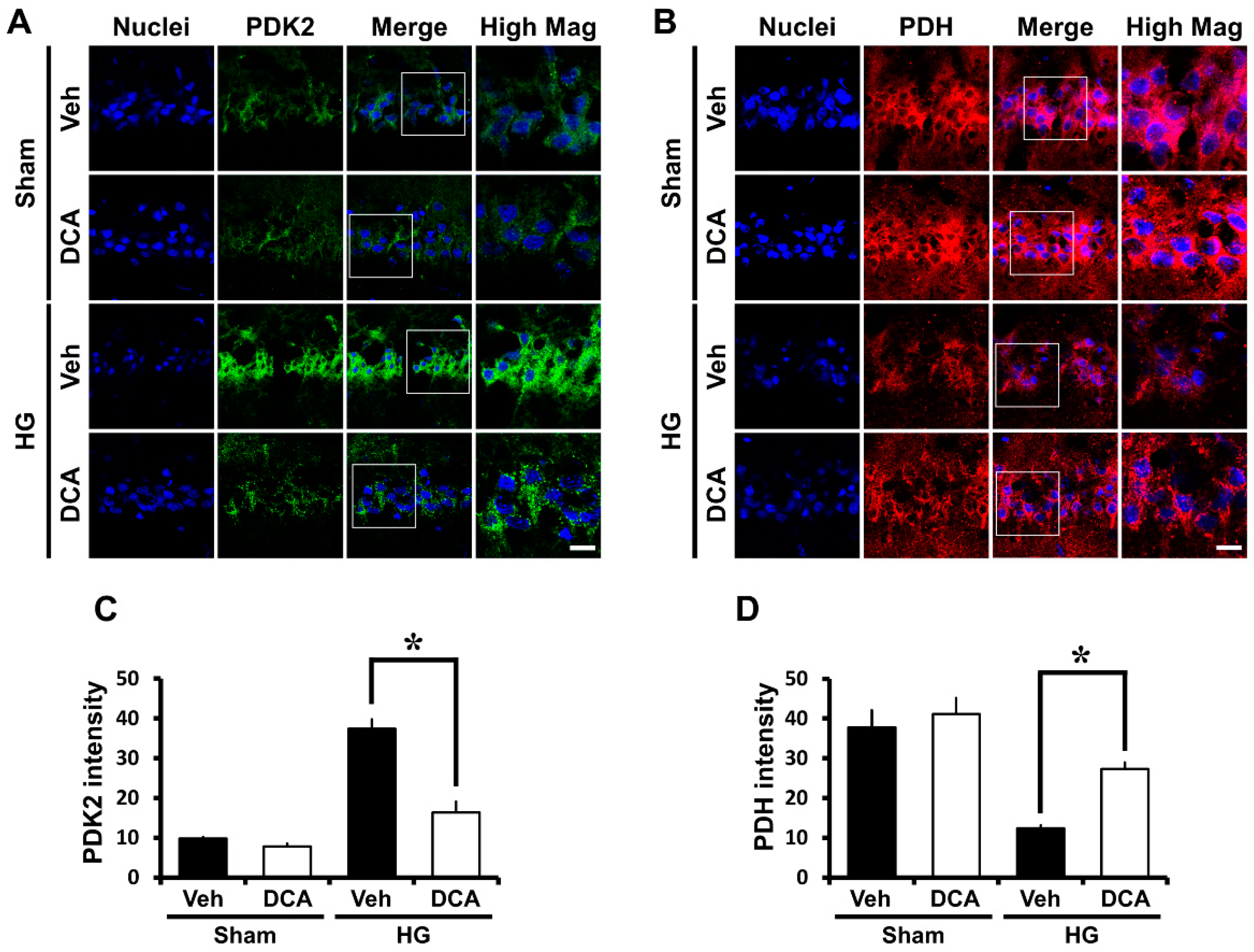
3.1. DCA Inhibits PDK2 after Hypoglycemia
3.2. DCA Increases PDH after Hypoglycemia
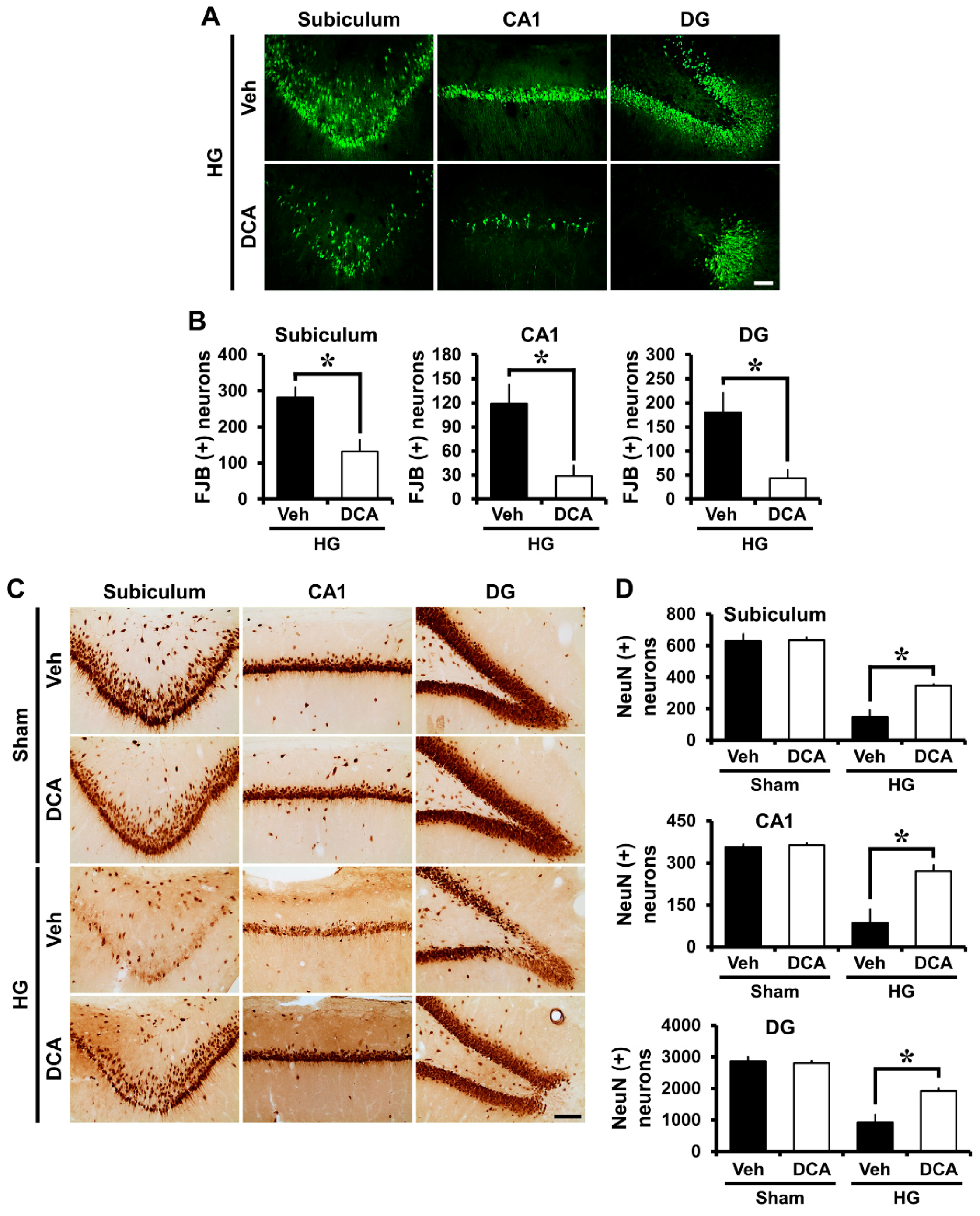
3.3. DCA Decreases Neuronal Death after Hypoglycemia
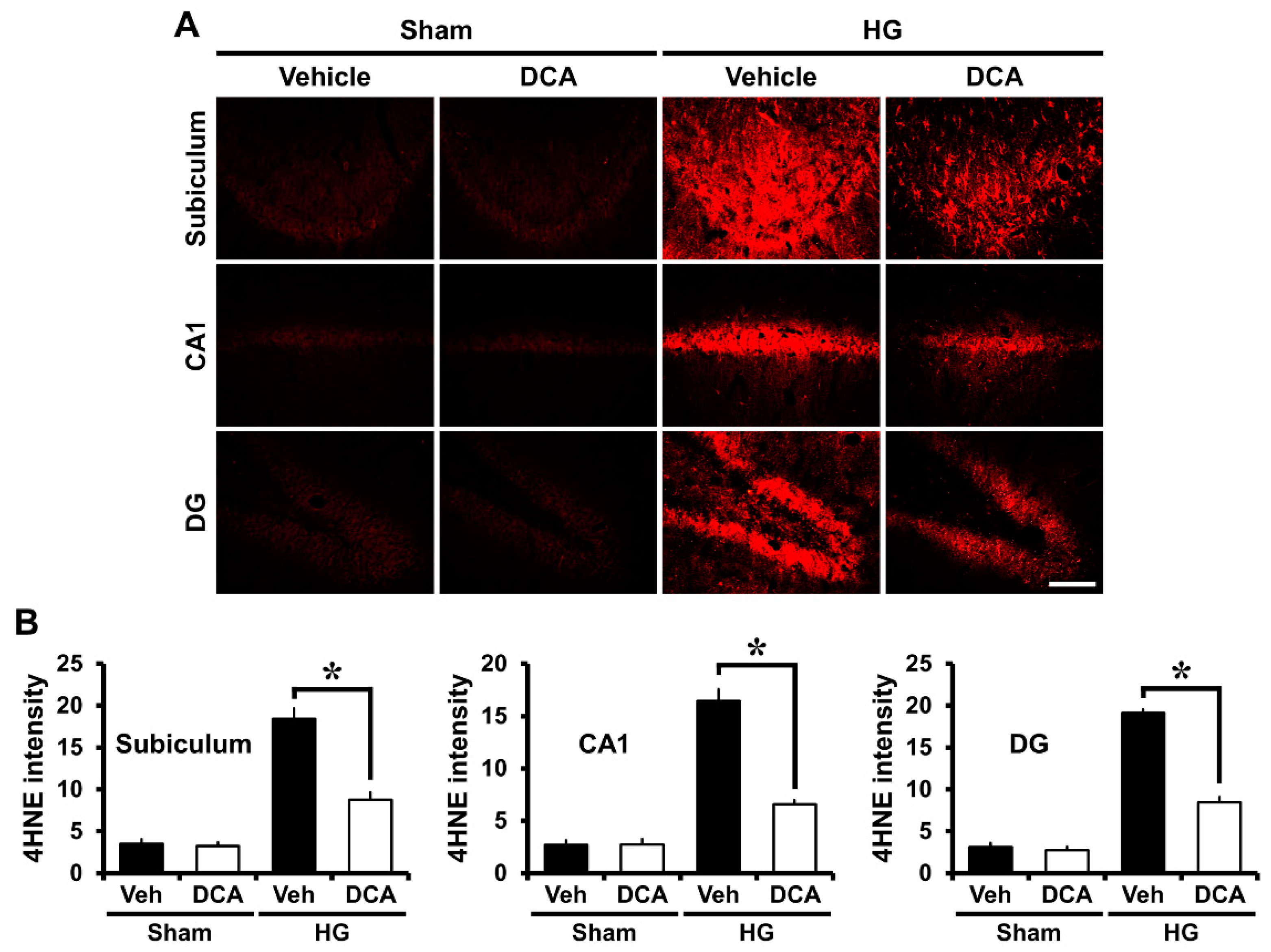
3.4. DCA Reduces Hypoglycemia-Induced Oxidative Injury
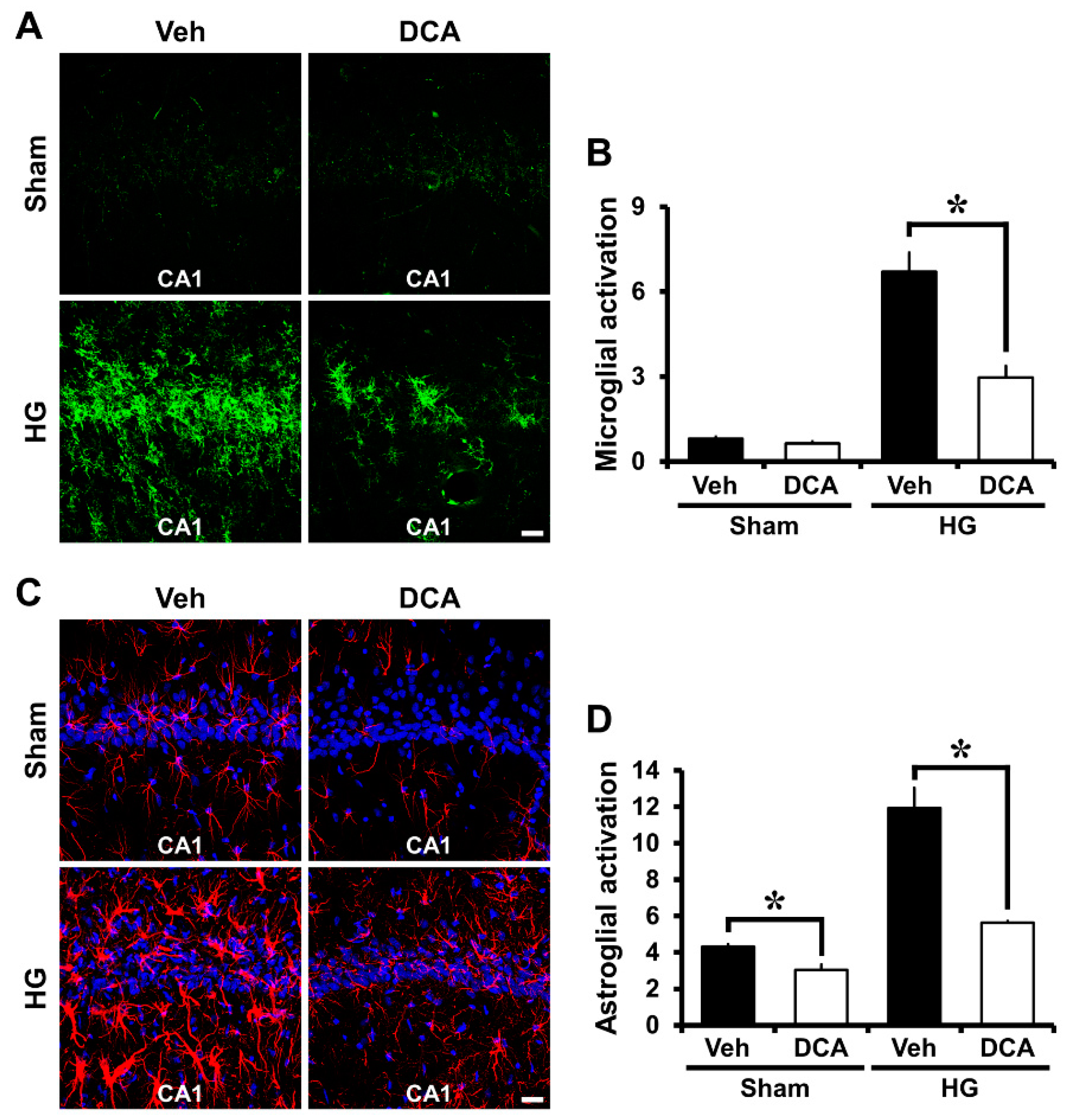
3.5. DCA Reduces Hypoglycemia-Induced Microglia and Astrocyte Activation
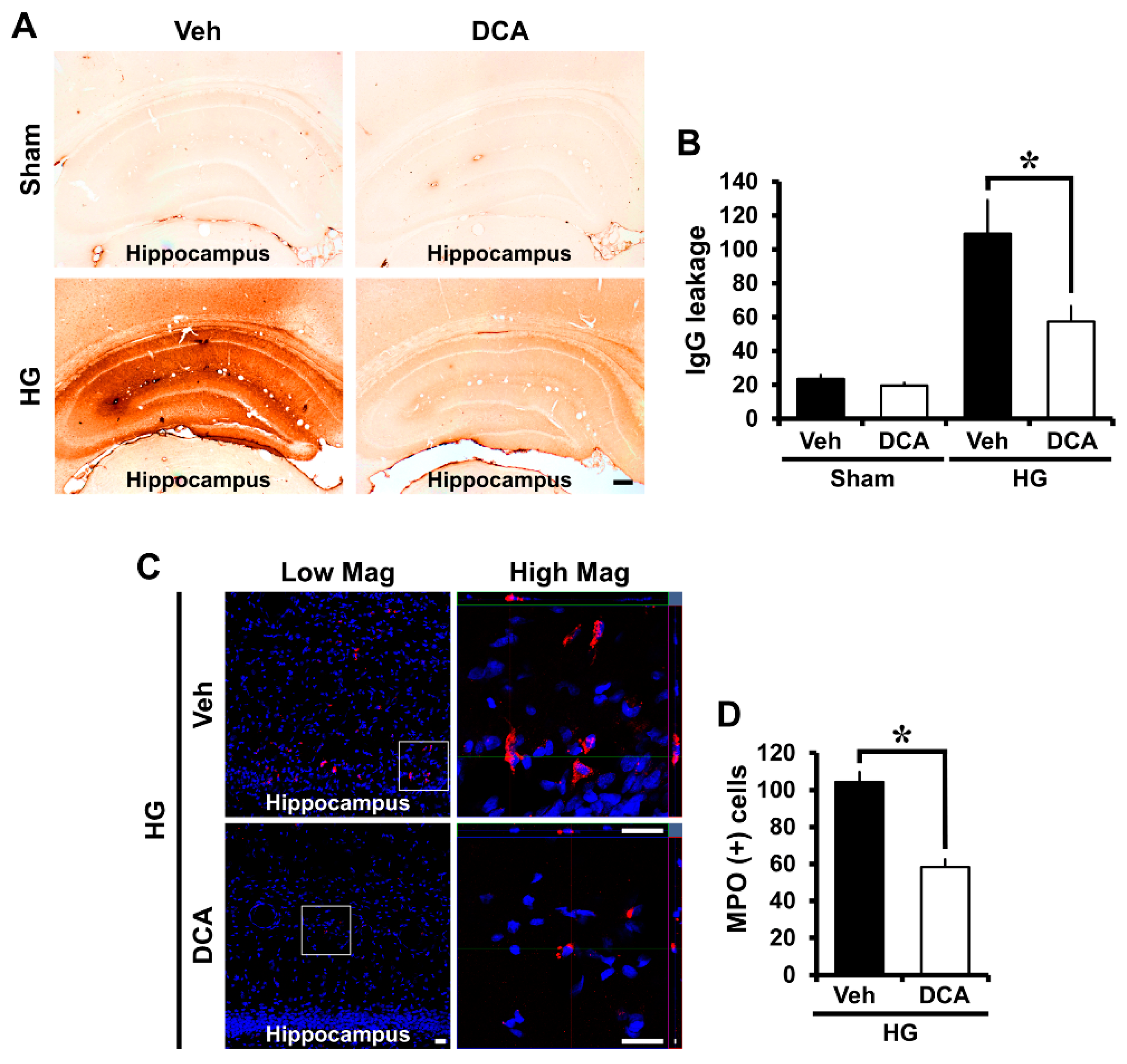
3.6. DCA Prevents Hypoglycemia-Induced Blood-Brain Barrier (BBB) Disruption
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Seltzer, H.S. Drug-induced hypoglycemia. A review of 1418 cases. Endocrinol. Metab. Clin. North. Am. 1989, 18, 163–183. [Google Scholar] [CrossRef]
- Tallroth, G.; Lindgren, M.; Stenberg, G.; Rosen, I.; Agardh, C.D. Neurophysiological changes during insulin-induced hypoglycaemia and in the recovery period following glucose infusion in type 1 (insulin-dependent) diabetes mellitus and in normal man. Diabetologia 1990, 33, 319–323. [Google Scholar] [CrossRef] [PubMed]
- Graveling, A.J.; Deary, I.J.; Frier, B.M. Acute hypoglycemia impairs executive cognitive function in adults with and without type 1 diabetes. Diabetes Care 2013, 36, 3240–3246. [Google Scholar] [CrossRef]
- Suh, S.W.; Gum, E.T.; Hamby, A.M.; Chan, P.H.; Swanson, R.A. Hypoglycemic neuronal death is triggered by glucose reperfusion and activation of neuronal NADPH oxidase. J. Clin. Invest. 2007, 117, 910–918. [Google Scholar] [CrossRef] [PubMed]
- Auer, R.N.; Olsson, Y.; Siesjo, B.K. Hypoglycemic brain injury in the rat. Correlation of density of brain damage with the EEG isoelectric time: A quantitative study. Diabetes 1984, 33, 1090–1098. [Google Scholar] [CrossRef]
- Auer, R.N.; Hall, P.; Ingvar, M.; Siesjo, B.K. Hypotension as a complication of hypoglycemia leads to enhanced energy failure but no increase in neuronal necrosis. Stroke 1986, 17, 442–449. [Google Scholar] [CrossRef] [PubMed]
- Suh, S.W.; Shin, B.S.; Ma, H.; Van Hoecke, M.; Brennan, A.M.; Yenari, M.A.; Swanson, R.A. Glucose and NADPH oxidase drive neuronal superoxide formation in stroke. Ann. Neurol. 2008, 64, 654–663. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Yoo, B.H.; Won, S.J.; Choi, B.Y.; Lee, B.E.; Kim, I.Y.; Kho, A.; Lee, S.H.; Sohn, M.; Suh, S.W. Melatonin Reduces Hypoglycemia-Induced Neuronal Death in Rats. Neuroendocrinology 2015, 102, 300–310. [Google Scholar] [CrossRef] [PubMed]
- Suh, S.W.; Garnier, P.; Aoyama, K.; Chen, Y.; Swanson, R.A. Zinc release contributes to hypoglycemia-induced neuronal death. Neurobiol. Dis. 2004, 16, 538–545. [Google Scholar] [CrossRef] [PubMed]
- Martin, E.; Rosenthal, R.E.; Fiskum, G. Pyruvate dehydrogenase complex: Metabolic link to ischemic brain injury and target of oxidative stress. J. Neurosci. Res. 2005, 79, 240–247. [Google Scholar] [CrossRef]
- Sun, W.; Liu, Q.; Leng, J.; Zheng, Y.; Li, J. The role of Pyruvate Dehydrogenase Complex in cardiovascular diseases. Life Sci. 2015, 121, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Sidhu, S.; Gangasani, A.; Korotchkina, L.G.; Suzuki, G.; Fallavollita, J.A.; Canty, J.M., Jr.; Patel, M.S. Tissue-specific pyruvate dehydrogenase complex deficiency causes cardiac hypertrophy and sudden death of weaned male mice. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H946–H952. [Google Scholar] [CrossRef]
- Holness, M.J.; Sugden, M.C. Regulation of pyruvate dehydrogenase complex activity by reversible phosphorylation. Biochem. Soc. Trans. 2003, 31, 1143–1151. [Google Scholar] [CrossRef]
- Hutson, N.J.; Randle, P.J. Enhanced activity of pyruvate dehydrogenase kinase in rat heart mitochondria in alloxan-diabetes or starvation. FEBS Lett. 1978, 92, 73–76. [Google Scholar] [CrossRef]
- Jha, M.K.; Lee, I.K.; Suk, K. Metabolic reprogramming by the pyruvate dehydrogenase kinase-lactic acid axis: Linking metabolism and diverse neuropathophysiologies. Neurosci. Biobehav. Rev. 2016, 68, 1–19. [Google Scholar] [CrossRef]
- Holness, M.J.; Kraus, A.; Harris, R.A.; Sugden, M.C. Targeted upregulation of pyruvate dehydrogenase kinase (PDK)-4 in slow-twitch skeletal muscle underlies the stable modification of the regulatory characteristics of PDK induced by high-fat feeding. Diabetes 2000, 49, 775–781. [Google Scholar] [CrossRef]
- Sugden, M.C.; Kraus, A.; Harris, R.A.; Holness, M.J. Fibre-type specific modification of the activity and regulation of skeletal muscle pyruvate dehydrogenase kinase (PDK) by prolonged starvation and refeeding is associated with targeted regulation of PDK isoenzyme 4 expression. Biochem. J. 2000, 346, 651–657. [Google Scholar] [CrossRef] [PubMed]
- Peters, S.J.; Harris, R.A.; Heigenhauser, G.J.; Spriet, L.L. Muscle fiber type comparison of PDH kinase activity and isoform expression in fed and fasted rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2001, 280, R661–R668. [Google Scholar] [CrossRef]
- Wu, P.; Blair, P.V.; Sato, J.; Jaskiewicz, J.; Popov, K.M.; Harris, R.A. Starvation increases the amount of pyruvate dehydrogenase kinase in several mammalian tissues. Arch. Biochem. Biophys. 2000, 381, 1–7. [Google Scholar] [CrossRef]
- Di, R.M.; Feng, Q.T.; Chang, Z.; Luan, Q.; Zhang, Y.Y.; Huang, J.; Li, X.L.; Yang, Z.Z. PDK1 plays a critical role in regulating cardiac function in mice and human. Chin. Med. J. 2010, 123, 2358–2363. [Google Scholar]
- Sugden, M.C.; Bulmer, K.; Augustine, D.; Holness, M.J. Selective modification of pyruvate dehydrogenase kinase isoform expression in rat pancreatic islets elicited by starvation and activation of peroxisome proliferator-activated receptor-alpha: Implications for glucose-stimulated insulin secretion. Diabetes 2001, 50, 2729–2736. [Google Scholar] [CrossRef] [PubMed]
- Spriet, L.L.; Tunstall, R.J.; Watt, M.J.; Mehan, K.A.; Hargreaves, M.; Cameron-Smith, D. Pyruvate dehydrogenase activation and kinase expression in human skeletal muscle during fasting. J. Appl. Physiol. (1985) 2004, 96, 2082–2087. [Google Scholar] [CrossRef] [PubMed]
- Bowker-Kinley, M.M.; Davis, W.I.; Wu, P.; Harris, R.A.; Popov, K.M. Evidence for existence of tissue-specific regulation of the mammalian pyruvate dehydrogenase complex. Biochem. J. 1998, 329, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Sato, J.; Zhao, Y.; Jaskiewicz, J.; Popov, K.M.; Harris, R.A. Starvation and diabetes increase the amount of pyruvate dehydrogenase kinase isoenzyme 4 in rat heart. Biochem. J. 1998, 329, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Jha, M.K.; Jeon, S.; Suk, K. Pyruvate Dehydrogenase Kinases in the Nervous System: Their Principal Functions in Neuronal-glial Metabolic Interaction and Neuro-metabolic Disorders. Curr. Neuropharmacol. 2012, 10, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.K.; Kho, A.R.; Choi, B.Y.; Lee, S.H.; Jeong, J.H.; Lee, S.H.; Park, K.H.; Park, J.B.; Suh, S.W. Combined Treatment With Dichloroacetic Acid and Pyruvate Reduces Hippocampal Neuronal Death After Transient Cerebral Ischemia. Front. Neurol. 2018, 9, 137. [Google Scholar] [CrossRef] [PubMed]
- McDonald, T.S.; Borges, K. Impaired hippocampal glucose metabolism during and after flurothyl-induced seizures in mice: Reduced phosphorylation coincides with reduced activity of pyruvate dehydrogenase. Epilepsia 2017, 58, 1172–1180. [Google Scholar] [CrossRef]
- Bonnet, S.; Archer, S.L.; Allalunis-Turner, J.; Haromy, A.; Beaulieu, C.; Thompson, R.; Lee, C.T.; Lopaschuk, G.D.; Puttagunta, L.; Bonnet, S.; et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 2007, 11, 37–51. [Google Scholar] [CrossRef]
- Lewandowski, E.D.; Johnston, D.L. Reduced substrate oxidation in postischemic myocardium: 13C and 31P NMR analyses. Am. J. Physiol. 1990, 258, H1357–H1365. [Google Scholar] [CrossRef]
- Saddik, M.; Gamble, J.; Witters, L.A.; Lopaschuk, G.D. Acetyl-CoA carboxylase regulation of fatty acid oxidation in the heart. J. Biol. Chem. 1993, 268, 25836–25845. [Google Scholar]
- Wahr, J.A.; Childs, K.F.; Bolling, S.F. Dichloroacetate enhances myocardial functional and metabolic recovery following global ischemia. J. Cardiothorac. Vasc. Anesth. 1994, 8, 192–197. [Google Scholar] [CrossRef]
- Lewandowski, E.D.; White, L.T. Pyruvate dehydrogenase influences postischemic heart function. Circulation 1995, 91, 2071–2079. [Google Scholar] [CrossRef] [PubMed]
- McVeigh, J.J.; Lopaschuk, G.D. Dichloroacetate stimulation of glucose oxidation improves recovery of ischemic rat hearts. Am. J. Physiol. 1990, 259, H1079–H1085. [Google Scholar] [CrossRef] [PubMed]
- Stacpoole, P.W.; Henderson, G.N.; Yan, Z.; James, M.O. Clinical pharmacology and toxicology of dichloroacetate. Environ. Health Perspect. 1998, 106 (Suppl. 4), 989–994. [Google Scholar]
- Yang, H.M.; Davis, M.E. Dichloroacetic acid pretreatment of male and female rats increases chloroform metabolism in vitro. Toxicology 1997, 124, 53–62. [Google Scholar] [CrossRef]
- Suh, S.W.; Aoyama, K.; Chen, Y.; Garnier, P.; Matsumori, Y.; Gum, E.; Liu, J.; Swanson, R.A. Hypoglycemic neuronal death and cognitive impairment are prevented by poly(ADP-ribose) polymerase inhibitors administered after hypoglycemia. J. Neurosci. 2003, 23, 10681–10690. [Google Scholar] [CrossRef] [PubMed]
- Suh, S.W.; Aoyama, K.; Matsumori, Y.; Liu, J.; Swanson, R.A. Pyruvate administered after severe hypoglycemia reduces neuronal death and cognitive impairment. Diabetes 2005, 54, 1452–1458. [Google Scholar] [CrossRef] [PubMed]
- Suh, S.W.; Hamby, A.M.; Gum, E.T.; Shin, B.S.; Won, S.J.; Sheline, C.T.; Chan, P.H.; Swanson, R.A. Sequential release of nitric oxide, zinc, and superoxide in hypoglycemic neuronal death. J. Cereb. Blood Flow Metab. 2008, 28, 1697–1706. [Google Scholar] [CrossRef]
- Kho, A.R.; Choi, B.Y.; Kim, J.H.; Lee, S.H.; Hong, D.K.; Lee, S.H.; Jeong, J.H.; Sohn, M.; Suh, S.W. Prevention of hypoglycemia-induced hippocampal neuronal death by N-acetyl-L-cysteine (NAC). Amino Acids 2017, 49, 367–378. [Google Scholar] [CrossRef]
- Schmued, L.C.; Hopkins, K.J. Fluoro-Jade B: A high affinity fluorescent marker for the localization of neuronal degeneration. Brain Res. 2000, 874, 123–130. [Google Scholar] [CrossRef]
- Kauppinen, T.M.; Swanson, R.A. Poly(ADP-ribose) polymerase-1 promotes microglial activation, proliferation, and matrix metalloproteinase-9-mediated neuron death. J. Immunol. 2005, 174, 2288–2296. [Google Scholar] [CrossRef] [PubMed]
- Kauppinen, T.M.; Higashi, Y.; Suh, S.W.; Escartin, C.; Nagasawa, K.; Swanson, R.A. Zinc triggers microglial activation. J. Neurosci. 2008, 28, 5827–5835. [Google Scholar] [CrossRef] [PubMed]
- Ruth, R.E.; Feinerman, G.S. Foreign and endogenous serum protein extravasation during harmaline tremors or kainic acid seizures in the rat: A comparison. Acta Neuropathol. 1988, 76, 380–387. [Google Scholar] [CrossRef]
- Hsu, S.M.; Raine, L.; Fanger, H. Use of avidin-biotin-peroxidase complex (ABC) in immunoperoxidase techniques: A comparison between ABC and unlabeled antibody (PAP) procedures. J. Histochem. Cytochem. 1981, 29, 577–580. [Google Scholar] [CrossRef]
- Xing, G.; Ren, M.; O’Neill, J.T.; Verma, A.; Watson, W.D. Controlled cortical impact injury and craniotomy result in divergent alterations of pyruvate metabolizing enzymes in rat brain. Exp. Neurol. 2012, 234, 31–38. [Google Scholar] [CrossRef]
- Jeong, J.Y.; Jeoung, N.H.; Park, K.G.; Lee, I.K. Transcriptional regulation of pyruvate dehydrogenase kinase. Diabetes Metab. J. 2012, 36, 328–335. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Hulver, M.W.; McMillan, R.P.; Cline, M.A.; Gilbert, E.R. The pivotal role of pyruvate dehydrogenase kinases in metabolic flexibility. Nutr. Metab. 2014, 11, 10. [Google Scholar] [CrossRef] [PubMed]
- Sorbi, S.; Bird, E.D.; Blass, J.P. Decreased pyruvate dehydrogenase complex activity in Huntington and Alzheimer brain. Ann. Neurol. 1983, 13, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Sheu, K.F.; Kim, Y.T.; Blass, J.P.; Weksler, M.E. An immunochemical study of the pyruvate dehydrogenase deficit in Alzheimer’s disease brain. Ann. Neurol. 1985, 17, 444–449. [Google Scholar] [CrossRef]
- Chinopoulos, C.; Adam-Vizi, V. Calcium, mitochondria and oxidative stress in neuronal pathology. Novel aspects of an enduring theme. FEBS J. 2006, 273, 433–450. [Google Scholar] [CrossRef] [PubMed]
- McGowan, J.E.; Chen, L.; Gao, D.; Trush, M.; Wei, C. Increased mitochondrial reactive oxygen species production in newborn brain during hypoglycemia. Neurosci. Lett. 2006, 399, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Won, S.J.; Yoo, B.H.; Kauppinen, T.M.; Choi, B.Y.; Kim, J.H.; Jang, B.G.; Lee, M.W.; Sohn, M.; Liu, J.; Swanson, R.A.; et al. Recurrent/moderate hypoglycemia induces hippocampal dendritic injury, microglial activation, and cognitive impairment in diabetic rats. J. Neuroinflammation 2012, 9, 182. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.M.; Dunn, E.; Oostveen, J.A.; Hall, E.D.; Carter, D.B. Induction of apolipoprotein E mRNA in the hippocampus of the gerbil after transient global ischemia. Brain Res. Mol. Brain Res. 1996, 38, 37–44. [Google Scholar] [CrossRef]
- Hirsch, E.C.; Hunot, S. Neuroinflammation in Parkinson’s disease: A target for neuroprotection? Lancet Neurol. 2009, 8, 382–397. [Google Scholar] [CrossRef]
- Kho, A.R.; Choi, B.Y.; Lee, S.H.; Hong, D.K.; Lee, S.H.; Jeong, J.H.; Park, K.H.; Song, H.K.; Choi, H.C.; Suh, S.W. Effects of Protocatechuic Acid (PCA) on Global Cerebral Ischemia-Induced Hippocampal Neuronal Death. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.Y.; Jang, B.G.; Kim, J.H.; Lee, B.E.; Sohn, M.; Song, H.K.; Suh, S.W. Prevention of traumatic brain injury-induced neuronal death by inhibition of NADPH oxidase activation. Brain Res. 2012, 1481, 49–58. [Google Scholar] [CrossRef]
- Kim, J.H.; Jang, B.G.; Choi, B.Y.; Kim, H.S.; Sohn, M.; Chung, T.N.; Choi, H.C.; Song, H.K.; Suh, S.W. Post-treatment of an NADPH oxidase inhibitor prevents seizure-induced neuronal death. Brain Res. 2013, 1499, 163–172. [Google Scholar] [CrossRef]
- Deng, J.; Zhao, F.; Yu, X.; Zhao, Y.; Li, D.; Shi, H.; Sun, Y. Expression of aquaporin 4 and breakdown of the blood-brain barrier after hypoglycemia-induced brain edema in rats. PLoS ONE 2014, 9, e107022. [Google Scholar] [CrossRef]
- Zhao, F.; Deng, J.; Yu, X.; Li, D.; Shi, H.; Zhao, Y. Protective effects of vascular endothelial growth factor in cultured brain endothelial cells against hypoglycemia. Metab. Brain Dis. 2015, 30, 999–1007. [Google Scholar] [CrossRef]
- Luo, S.; Li, S.; Zhu, L.; Fang, S.H.; Chen, J.L.; Xu, Q.Q.; Li, H.Y.; Luo, N.C.; Yang, C.; Luo, D.; et al. Effect of baicalin on oxygen-glucose deprivation-induced endothelial cell damage. Neuroreport 2017, 28, 299–306. [Google Scholar] [CrossRef]
- Murakami, K.; Yoshino, M. Zinc inhibition of pyruvate kinase of M-type isozyme. Biometals 2017, 30, 335–340. [Google Scholar] [CrossRef]
- Stanley, W.C.; Lopaschuk, G.D.; Hall, J.L.; McCormack, J.G. Regulation of myocardial carbohydrate metabolism under normal and ischaemic conditions. Potential for pharmacological interventions. Cardiovasc. Res. 1997, 33, 243–257. [Google Scholar] [CrossRef]
- Ussher, J.R.; Wang, W.; Gandhi, M.; Keung, W.; Samokhvalov, V.; Oka, T.; Wagg, C.S.; Jaswal, J.S.; Harris, R.A.; Clanachan, A.S.; et al. Stimulation of glucose oxidation protects against acute myocardial infarction and reperfusion injury. Cardiovasc. Res. 2012, 94, 359–369. [Google Scholar] [CrossRef]
- Pulsinelli, W.A.; Levy, D.E.; Duffy, T.E. Regional cerebral blood flow and glucose metabolism following transient forebrain ischemia. Ann. Neurol. 1982, 11, 499–502. [Google Scholar] [CrossRef] [PubMed]
- Yokobori, S.; Mazzeo, A.T.; Gajavelli, S.; Bullock, M.R. Mitochondrial neuroprotection in traumatic brain injury: Rationale and therapeutic strategies. CNS Neurol. Disord. Drug Targets 2014, 13, 606–619. [Google Scholar] [CrossRef]
- Fiskum, G.; Rosenthal, R.E.; Vereczki, V.; Martin, E.; Hoffman, G.E.; Chinopoulos, C.; Kowaltowski, A. Protection against ischemic brain injury by inhibition of mitochondrial oxidative stress. J. Bioenerg. Biomembr. 2004, 36, 347–352. [Google Scholar] [CrossRef] [PubMed]
- Opii, W.O.; Nukala, V.N.; Sultana, R.; Pandya, J.D.; Day, K.M.; Merchant, M.L.; Klein, J.B.; Sullivan, P.G.; Butterfield, D.A. Proteomic identification of oxidized mitochondrial proteins following experimental traumatic brain injury. J. Neurotrauma 2007, 24, 772–789. [Google Scholar] [CrossRef] [PubMed]
- Roche, T.E.; Hiromasa, Y. Pyruvate dehydrogenase kinase regulatory mechanisms and inhibition in treating diabetes, heart ischemia, and cancer. Cell. Mol. Life Sci. 2007, 64, 830–849. [Google Scholar] [CrossRef]
- Xing, G.; Ren, M.; Watson, W.D.; O’Neill, J.T.; Verma, A. Traumatic brain injury-induced expression and phosphorylation of pyruvate dehydrogenase: A mechanism of dysregulated glucose metabolism. Neurosci. Lett. 2009, 454, 38–42. [Google Scholar] [CrossRef]
- Leinninger, G.M.; Backus, C.; Sastry, A.M.; Yi, Y.B.; Wang, C.W.; Feldman, E.L. Mitochondria in DRG neurons undergo hyperglycemic mediated injury through Bim, Bax and the fission protein Drp1. Neurobiol. Dis. 2006, 23, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Katyare, S.S.; Patel, S.P. Insulin status differentially affects energy transduction in cerebral mitochondria from male and female rats. Brain Res. Bull. 2006, 69, 458–464. [Google Scholar] [CrossRef]
- Otera, H.; Ishihara, N.; Mihara, K. New insights into the function and regulation of mitochondrial fission. Biochim. Biophys. Acta 2013, 1833, 1256–1268. [Google Scholar] [CrossRef]
- Calcutt, N.A.; Lopez, V.L.; Bautista, A.D.; Mizisin, L.M.; Torres, B.R.; Shroads, A.L.; Mizisin, A.P.; Stacpoole, P.W. Peripheral neuropathy in rats exposed to dichloroacetate. J. Neuropathol. Exp. Neurol. 2009, 68, 985–993. [Google Scholar] [CrossRef]
- Moser, V.C.; Phillips, P.M.; McDaniel, K.L.; MacPhail, R.C. Behavioral evaluation of the neurotoxicity produced by dichloroacetic acid in rats. Neurotoxicol. Teratol. 1999, 21, 719–731. [Google Scholar] [CrossRef]
- Sun, X.Q.; Zhang, R.; Zhang, H.D.; Yuan, P.; Wang, X.J.; Zhao, Q.H.; Wang, L.; Jiang, R.; Jan Bogaard, H.; Jing, Z.C. Reversal of right ventricular remodeling by dichloroacetate is related to inhibition of mitochondria-dependent apoptosis. Hypertens Res. 2016, 39, 302–311. [Google Scholar] [CrossRef]
- Durie, D.; McDonald, T.S.; Borges, K. The effect of dichloroacetate in mouse models of epilepsy. Epilepsy Res. 2018, 145, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Staneviciute, J.; Jukneviciene, M.; Palubinskiene, J.; Balnyte, I.; Valanciute, A.; Vosyliute, R.; Suziedelis, K.; Lesauskaite, V.; Stakisaitis, D. Sodium Dichloroacetate Pharmacological Effect as Related to Na-K-2Cl Cotransporter Inhibition in Rats. Dose Response 2018, 16, 1559325818811522. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, J.; Hu, H.; Lu, S.; Lu, Q.; Quan, N.; Rousselle, T.; Patel, M.S.; Li, J. Dichloroacetate Ameliorates Cardiac Dysfunction Caused by Ischemic Insults Through AMPK Signal Pathway-Not Only Shifts Metabolism. Toxicol. Sci. 2019, 167, 604–617. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Palma, L.; Miquel, E.; Lagos-Rodriguez, V.; Barbeito, L.; Cassina, A.; Cassina, P. Mitochondrial Modulation by Dichloroacetate Reduces Toxicity of Aberrant Glial Cells and Gliosis in the SOD1G93A Rat Model of Amyotrophic Lateral Sclerosis. Neurotherapeutics 2019, 16, 203–215. [Google Scholar] [CrossRef] [PubMed]
- Stacpoole, P.W. The pharmacology of dichloroacetate. Metabolism 1989, 38, 1124–1144. [Google Scholar] [CrossRef]
- D’Alessandro, G.; Calcagno, E.; Tartari, S.; Rizzardini, M.; Invernizzi, R.W.; Cantoni, L. Glutamate and glutathione interplay in a motor neuronal model of amyotrophic lateral sclerosis reveals altered energy metabolism. Neurobiol. Dis. 2011, 43, 346–355. [Google Scholar] [CrossRef]
- Jozefczak, M.; Remans, T.; Vangronsveld, J.; Cuypers, A. Glutathione is a key player in metal-induced oxidative stress defenses. Int. J. Mol. Sci. 2012, 13, 3145–3175. [Google Scholar] [CrossRef]
- Choi, B.Y.; Kim, I.Y.; Kim, J.H.; Lee, B.E.; Lee, S.H.; Kho, A.R.; Jung, H.J.; Sohn, M.; Song, H.K.; Suh, S.W. Decreased cysteine uptake by EAAC1 gene deletion exacerbates neuronal oxidative stress and neuronal death after traumatic brain injury. Amino Acids 2016, 48, 1619–1629. [Google Scholar] [CrossRef] [PubMed]
- Jha, M.K.; Park, D.H.; Kook, H.; Lee, I.K.; Lee, W.H.; Suk, K. Metabolic Control of Glia-Mediated Neuroinflammation. Curr. Alzheimer Res. 2016, 13, 387–402. [Google Scholar]
- O’Neill, L.A.; Hardie, D.G. Metabolism of inflammation limited by AMPK and pseudo-starvation. Nature 2013, 493, 346–355. [Google Scholar] [CrossRef]
- Orihuela, R.; McPherson, C.A.; Harry, G.J. Microglial M1/M2 polarization and metabolic states. Br. J. Pharmacol. 2016, 173, 649–665. [Google Scholar] [CrossRef] [PubMed]
- Klimaszewska-Lata, J.; Gul-Hinc, S.; Bielarczyk, H.; Ronowska, A.; Zysk, M.; Gruzewska, K.; Pawelczyk, T.; Szutowicz, A. Differential effects of lipopolysaccharide on energy metabolism in murine microglial N9 and cholinergic SN56 neuronal cells. J. Neurochem. 2015, 133, 284–297. [Google Scholar] [CrossRef]
- Jha, M.K.; Song, G.J.; Lee, M.G.; Jeoung, N.H.; Go, Y.; Harris, R.A.; Park, D.H.; Kook, H.; Lee, I.K.; Suk, K. Metabolic Connection of Inflammatory Pain: Pivotal Role of a Pyruvate Dehydrogenase Kinase-Pyruvate Dehydrogenase-Lactic Acid Axis. J. Neurosci. 2015, 35, 14353–14369. [Google Scholar] [CrossRef]
- Liu, W.; Wang, X.; Yang, S.; Huang, J.; Xue, X.; Zheng, Y.; Shang, G.; Tao, J.; Chen, L. Electroacupunctre improves motor impairment via inhibition of microglia-mediated neuroinflammation in the sensorimotor cortex after ischemic stroke. Life Sci. 2016, 151, 313–322. [Google Scholar] [CrossRef]
- Ghaemi, A.; Alizadeh, L.; Babaei, S.; Jafarian, M.; Khaleghi Ghadiri, M.; Meuth, S.G.; Kovac, S.; Gorji, A. Astrocyte-mediated inflammation in cortical spreading depression. Cephalalgia 2018, 38, 626–638. [Google Scholar] [CrossRef]
- Dvoriantchikova, G.; Barakat, D.; Brambilla, R.; Agudelo, C.; Hernandez, E.; Bethea, J.R.; Shestopalov, V.I.; Ivanov, D. Inactivation of astroglial NF-kappa B promotes survival of retinal neurons following ischemic injury. Eur. J. Neurosci. 2009, 30, 175–185. [Google Scholar] [CrossRef]
- Kirkley, K.S.; Popichak, K.A.; Afzali, M.F.; Legare, M.E.; Tjalkens, R.B. Microglia amplify inflammatory activation of astrocytes in manganese neurotoxicity. J. Neuroinflammation 2017, 14, 99. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Munch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Blumcke, I.; Pauli, E.; Clusmann, H.; Schramm, J.; Becker, A.; Elger, C.; Merschhemke, M.; Meencke, H.J.; Lehmann, T.; von Deimling, A.; et al. A new clinico-pathological classification system for mesial temporal sclerosis. Acta Neuropathol. 2007, 113, 235–244. [Google Scholar] [CrossRef]
- Mueller, S.G.; Stables, L.; Du, A.T.; Schuff, N.; Truran, D.; Cashdollar, N.; Weiner, M.W. Measurement of hippocampal subfields and age-related changes with high resolution MRI at 4T. Neurobiol. Aging 2007, 28, 719–726. [Google Scholar] [CrossRef]
- West, M.J.; Kawas, C.H.; Stewart, W.F.; Rudow, G.L.; Troncoso, J.C. Hippocampal neurons in pre-clinical Alzheimer’s disease. Neurobiol. Aging 2004, 25, 1205–1212. [Google Scholar] [CrossRef]
- Chodobski, A.; Zink, B.J.; Szmydynger-Chodobska, J. Blood-brain barrier pathophysiology in traumatic brain injury. Transl. Stroke Res. 2011, 2, 492–516. [Google Scholar] [CrossRef]
- Tomkins, O.; Shelef, I.; Kaizerman, I.; Eliushin, A.; Afawi, Z.; Misk, A.; Gidon, M.; Cohen, A.; Zumsteg, D.; Friedman, A. Blood-brain barrier disruption in post-traumatic epilepsy. J. Neurol. Neurosurg. Psychiatry 2008, 79, 774–777. [Google Scholar] [CrossRef]
- Scholz, M.; Cinatl, J.; Schadel-Hopfner, M.; Windolf, J. Neutrophils and the blood-brain barrier dysfunction after trauma. Med. Res. Rev. 2007, 27, 401–416. [Google Scholar] [CrossRef]
- Alves, J.L. Blood-brain barrier and traumatic brain injury. J. Neurosci. Res. 2014, 92, 141–147. [Google Scholar] [CrossRef]
- Doll, D.N.; Hu, H.; Sun, J.; Lewis, S.E.; Simpkins, J.W.; Ren, X. Mitochondrial crisis in cerebrovascular endothelial cells opens the blood-brain barrier. Stroke 2015, 46, 1681–1689. [Google Scholar] [CrossRef]
- Hu, H.; Doll, D.N.; Sun, J.; Lewis, S.E.; Wimsatt, J.H.; Kessler, M.J.; Simpkins, J.W.; Ren, X. Mitochondrial Impairment in Cerebrovascular Endothelial Cells is Involved in the Correlation between Body Temperature and Stroke Severity. Aging Dis. 2016, 7, 14–27. [Google Scholar] [CrossRef]
- Salmina, A.B.; Kuvacheva, N.V.; Morgun, A.V.; Komleva, Y.K.; Pozhilenkova, E.A.; Lopatina, O.L.; Gorina, Y.V.; Taranushenko, T.E.; Petrova, L.L. Glycolysis-mediated control of blood-brain barrier development and function. Int. J. Biochem. Cell Biol. 2015, 64, 174–184. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kho, A.R.; Choi, B.Y.; Lee, S.H.; Hong, D.K.; Jeong, J.H.; Kang, B.S.; Kang, D.H.; Park, K.-H.; Park, J.B.; Suh, S.W. The Effects of Sodium Dichloroacetate on Mitochondrial Dysfunction and Neuronal Death Following Hypoglycemia-Induced Injury. Cells 2019, 8, 405. https://doi.org/10.3390/cells8050405
Kho AR, Choi BY, Lee SH, Hong DK, Jeong JH, Kang BS, Kang DH, Park K-H, Park JB, Suh SW. The Effects of Sodium Dichloroacetate on Mitochondrial Dysfunction and Neuronal Death Following Hypoglycemia-Induced Injury. Cells. 2019; 8(5):405. https://doi.org/10.3390/cells8050405
Chicago/Turabian StyleKho, A Ra, Bo Young Choi, Song Hee Lee, Dae Ki Hong, Jeong Hyun Jeong, Beom Seok Kang, Dong Hyeon Kang, Kyoung-Ha Park, Jae Bong Park, and Sang Won Suh. 2019. "The Effects of Sodium Dichloroacetate on Mitochondrial Dysfunction and Neuronal Death Following Hypoglycemia-Induced Injury" Cells 8, no. 5: 405. https://doi.org/10.3390/cells8050405