
A New Family of Vinyl Selenocyanates with the Amide Function Based on the Reaction of Potassium Selenocyanate with 3-Trimethylsilyl-2-Propynamides


Abstract
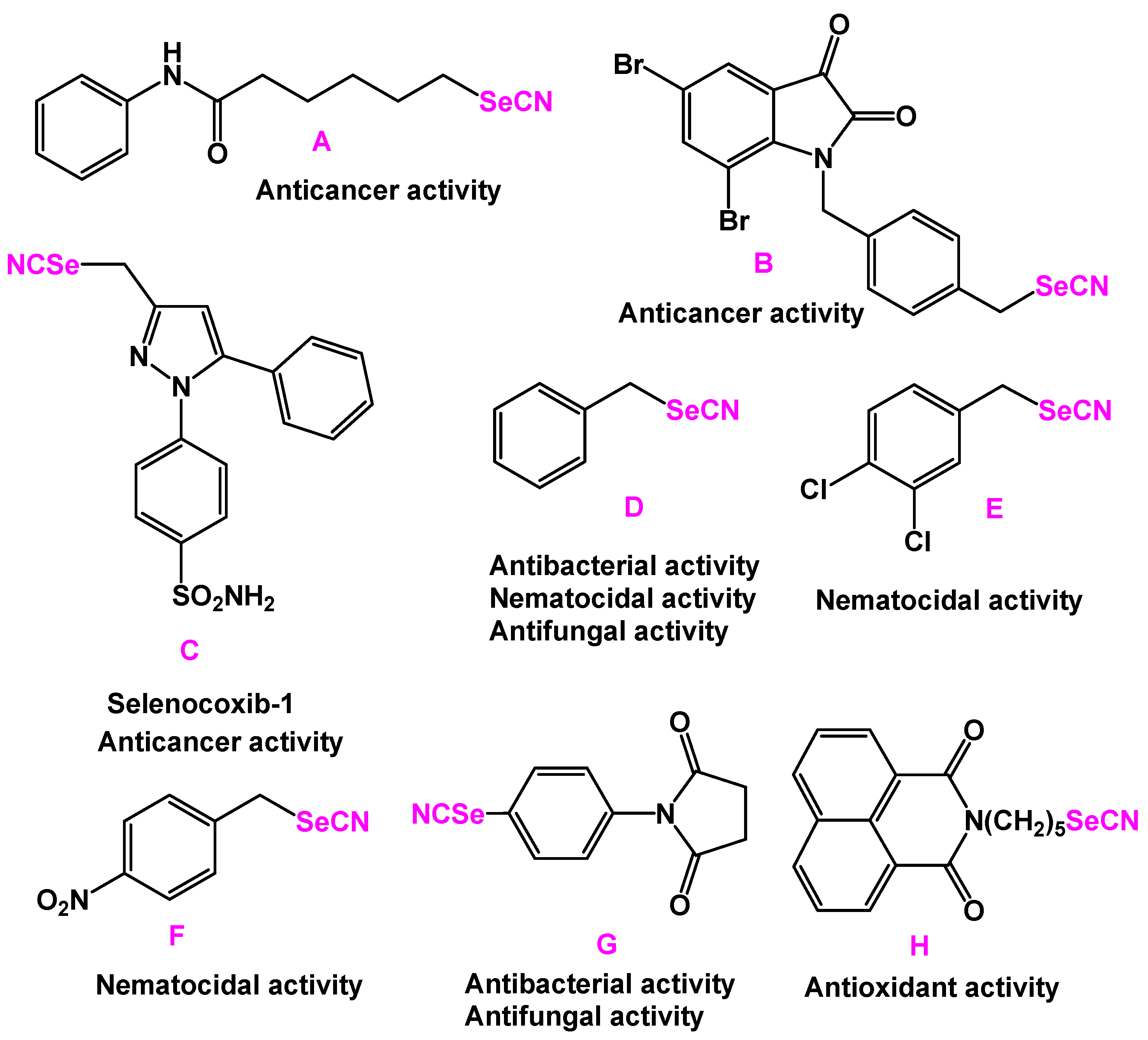
:1. Introduction
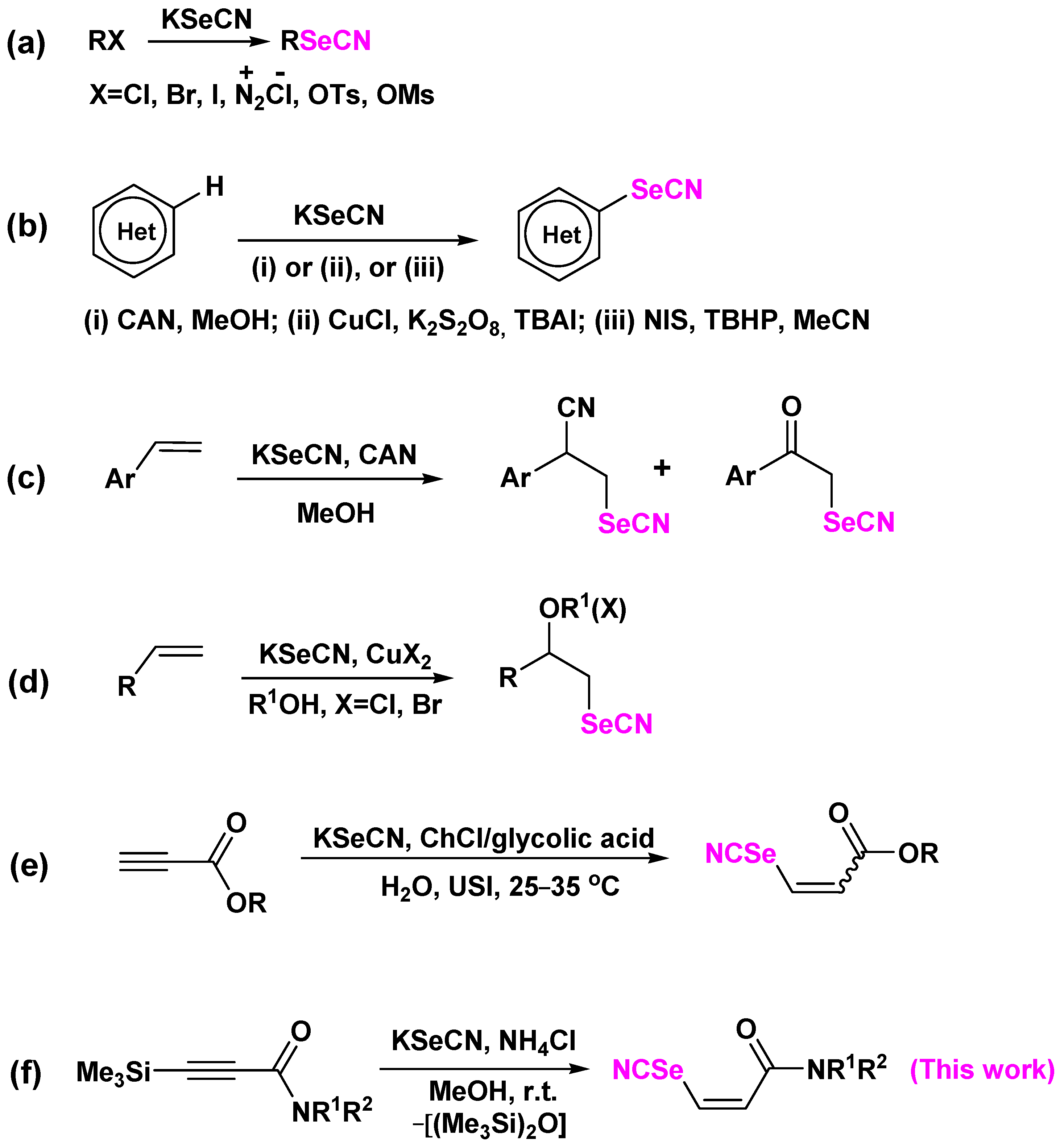
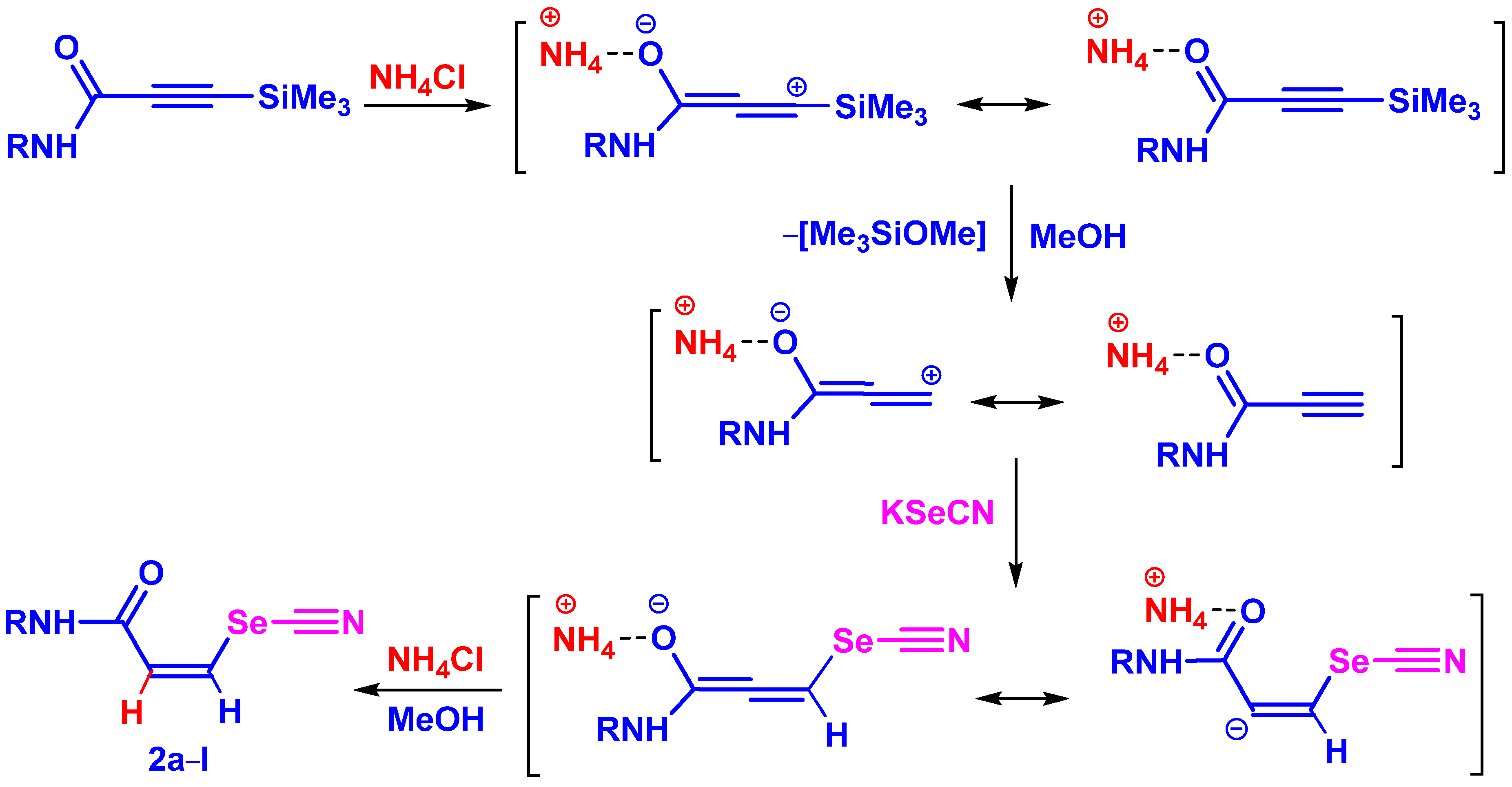
2. Results and Discussion
3. Materials and Methods
3.1. General Information
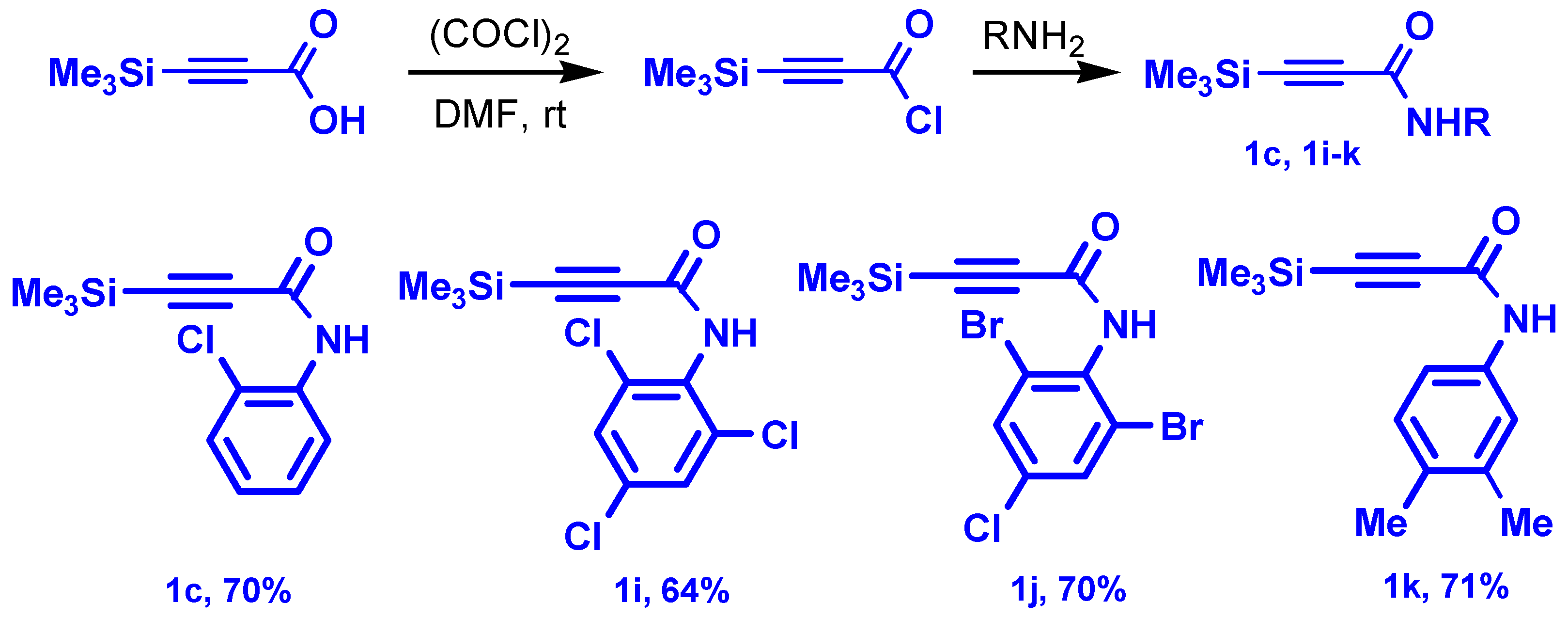
3.2. Synthesis of New 3-Trimethylsilyl-2-Propynamides
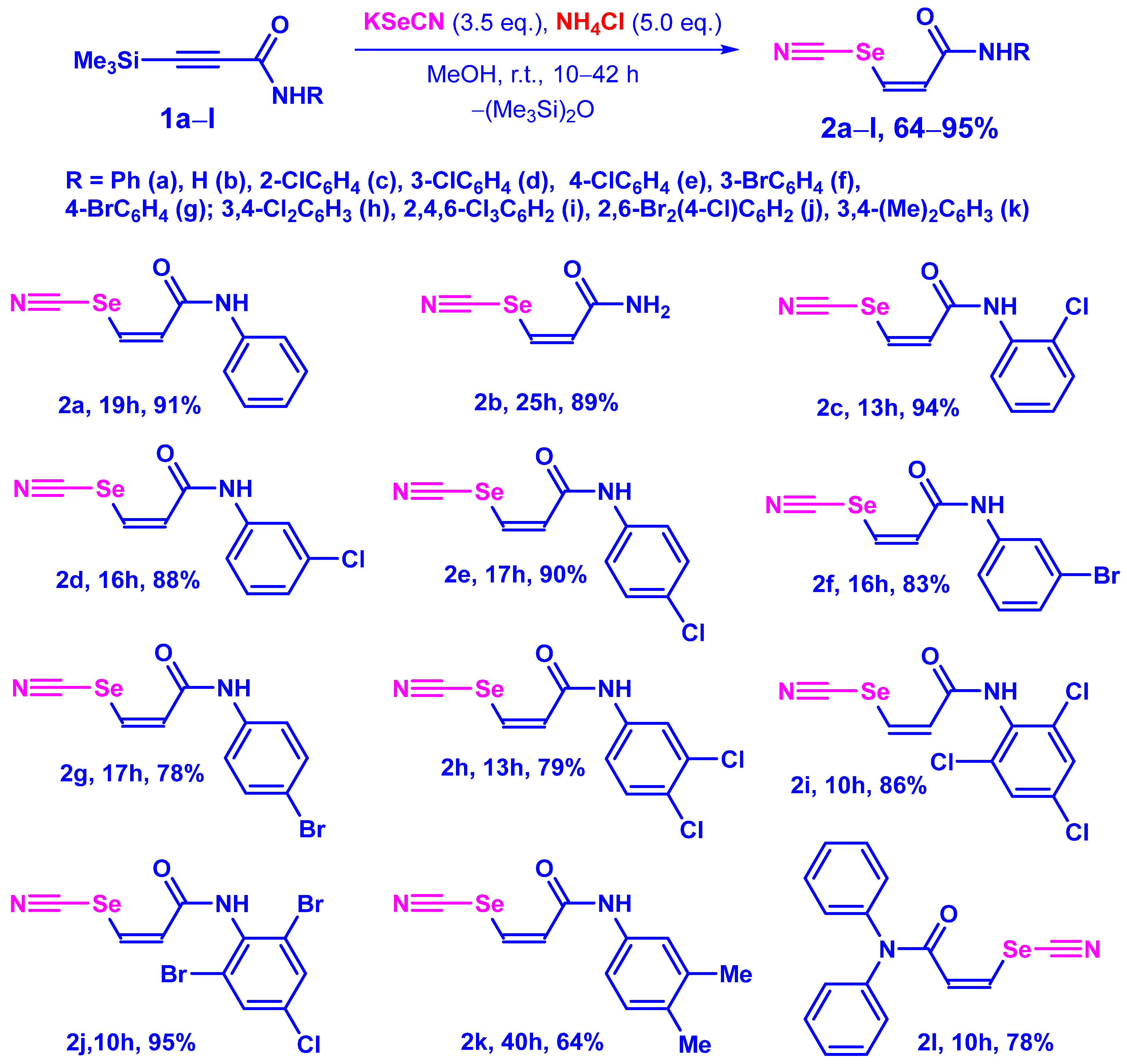
3.3. Synthesis of (Z)-3-Amino-3-Oxo-1-Propenyl Selenocyanates 2a–l
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gandhil, U.H.; Nagaraja, T.P.; Prabhu, K.S. Selenoproteins and their role in oxidative stress and inflammation. Curr. Chem. Biol. 2013, 7, 65–73. [Google Scholar] [CrossRef]
- Longtin, R. A forgotten debate: Is selenocysteine the 21st amino acid? J. Nat. Cancer Inst. 2004, 96, 504–505. [Google Scholar] [CrossRef] [PubMed]
- Tiekink, E.R.T. Therapeutic potential of selenium and tellurium compounds: Opportunities yet unrealized. Dalton Trans. 2012, 41, 6390–6395. [Google Scholar] [CrossRef] [PubMed]
- Młochowski, J.; Kloc, K.; Lisiak, R.; Potaczek, P.; Wójtowicz, H. Developments in the chemistry of selenaheterocyclic compounds of practical importance in synthesis and medicinal biology. Arkivoc 2007, 2007, 14–46. [Google Scholar] [CrossRef]
- Banerjee, B.; Koketsu, M. Recent developments in the synthesis of biologically relevant selenium-containing scaffolds. Coord. Chem. Rev. 2017, 339, 104–127. [Google Scholar] [CrossRef]
- Rafique, J.; Canto, R.F.S.; Saba, S.; Barbosa, F.A.R.; Braga, A.L. Recent Advances in the Synthesis of Biologically Relevant Selenium-containing 5-Membered Heterocycles. Curr. Org. Chem. 2016, 20, 166–188. [Google Scholar] [CrossRef]
- Woollins, J.D.; Laitinen, R.S. (Eds.) Selenium and Tellurium Chemistry. From Small Molecules to Biomolecules and Materials; Springer: Heidelberg, Germany, 2011; 334p. [Google Scholar]
- Santi, C. (Ed.) Organoselenium Chemistry: Between Synthesis and Biochemistry; Bentham Science: Sharjah, United Arab Emirates, 2014; 563p. [Google Scholar]
- Shaaban, S.; Ashmawy, A.M.; Negm, A.; Wessjohann, L.A. Synthesis and biochemical studies of novel organic selenides with increased selectivity for hepatocellular carcinoma and breast adenocarcinoma. Eur. J. Med. Chem. 2019, 179, 515–526. [Google Scholar] [CrossRef]
- Ramos-Inza, S.; Encío, I.; Raza, A.; Sharma, A.K.; Sanmartín, C.; Plano, D. Design, synthesis and anticancer evaluation of novel Se-NSAID hybrid molecules: Identification of a Se-indomethacin analog as a potential therapeutic for breast cancer. Eur. J. Med. Chem. 2022, 244, 114839. [Google Scholar] [CrossRef]
- Astrain-Redin, N.; Paoletti, N.; Plano, D.; Bonardi, A.; Gratteri, P.; Angeli, A.; Sanmartin, C.; Supuran, C.T. Selenium-analogs based on natural sources as cancer-associated carbonic anhydrase isoforms IX and XII inhibitors. J. Enzyme Inhib. Med. Chem. 2023, 38, 2191165. [Google Scholar] [CrossRef]
- Gouda, M.; Abbas, Y.J.; Abd El-Lateef, H.M.; Khalaf, M.M.; Shaaban, S. Design, Synthesis, and Biological Evaluations of Novel Naphthalene-based Organoselenocyanates. Biointerface Res. Appl. Chem. 2023, 13, 219. [Google Scholar]
- Di Leo, I.; Messina, F.; Nascimento, V.; Nacca, F.G.; Pietrella, D.; Lenardão, E.J.; Perin, G.; Sancineto, L. Synthetic Approaches to Organoselenium Derivatives with Antimicrobial and Anti-Biofilm Activity. Mini-Rev. Org. Chem. 2018, 16, 589–601. [Google Scholar] [CrossRef]
- Sarturi, J.M.; Dornelles, L.; Segatto, N.V.; Collares, T.; Seixas, F.K.; Piccoli, B.C.; D’Avila da Silva, F.; Omage, F.B.; Rocha, J.B.T.; Balaguez, R.A.; et al. Chalcogenium-AZT Derivatives: A Plausible Strategy to Tackle The RT-Inhibitors-Related Oxidative Stress While Maintaining Their Anti- HIV Properties. Curr. Med. Chem. 2023, 30, 2449–2462. [Google Scholar] [CrossRef] [PubMed]
- Omage, F.B.; Madabeni, A.; Tucci, A.R.; Nogara, P.A.; Bortoli, M.; dos Santos Rosa, A.; dos Santos Ferreira, V.N.; Rocha, J.B.T.; Miranda, M.D.; Orian, L. Diphenyl Diselenide and SARS-CoV-2: In silico Exploration of the Mechanisms of Inhibition of Main Protease (Mpro) and Papain-like Protease (PLpro). J. Chem. Inf. Model. 2023, 63, 2226–2239. [Google Scholar] [CrossRef] [PubMed]
- Refaay, D.A.; Ahmed, D.M.; Mowafy, A.M.; Shaaban, S. Evaluation of novel multifunctional organoselenium compounds as potential cholinesterase inhibitors against Alzheimer’s disease. Med. Chem. Res. 2022, 31, 894–904. [Google Scholar] [CrossRef]
- Mamgain, R.; Kostic, M.; Singh, F.V. Synthesis and Antioxidant Properties of Organoselenium Compounds. Curr. Med. Chem. 2023, 30, 2421–2448. [Google Scholar] [CrossRef] [PubMed]
- Elsherbini, M.; Hamama, W.S.; Zoorob, H.H. Recent advances in the chemistry of selenium-containing heterocycles: Five-membered ring systems. Coord. Chem. Rev. 2016, 312, 149–177. [Google Scholar] [CrossRef]
- Elsherbini, M.; Hamama, W.S.; Zoorob, H.H. Recent advances in the chemistry of selenium-containing heterocycles: Six-membered ring systems. Coord. Chem. Rev. 2017, 330, 110–126. [Google Scholar] [CrossRef]
- Azad, G.K.; Tomar, R.S. Ebselen, a promising antioxidant drug: Mechanisms of action and targets of biological pathways. Mol. Biol. Rep. 2014, 41, 4865–4879. [Google Scholar] [CrossRef]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef]
- Obieziurska-Fabisiak, M.; Pacuła-Miszewska, A.J.; Laskowska, A.; Ścianowski, J. Organoselenium compounds as antioxidants. Arkivoc 2023, v, 69–92. [Google Scholar] [CrossRef]
- Toshimitsu, A. Organic selenocyanates, tellurocyanates and related compounds. Synthesis and reactions. In Patai’s Chemistry of Functional Groups. Organic Selenium and Tellurium Compounds; Rappoport, Z., Ed.; John Wiley and Sons, Inc.: Chichester, UK, 2013; pp. 845–884. [Google Scholar]
- Guillemin, J.-C. Organic Selenocyanates: Synthesis, Characterization and Uses in Chemistry and Biology. Curr. Org. Chem. 2011, 15, 1670–1687. [Google Scholar] [CrossRef]
- Shaaban, S.; Arafat, M.A.; Hamama, W.S. Vistas in the domain of organoselenocyanates. ARKIVOC 2014, 1, 470–505. [Google Scholar] [CrossRef]
- Hassanpour, A.; Ghavidelaghdam, E.; Ebadi, A.G.; Heravi, M.R.P.; Vessally, E. Progress and recent trends in the direct selenocyanation of (hetero)aromatic C–H bonds. RSC Adv. 2021, 11, 22305–22316. [Google Scholar] [CrossRef] [PubMed]
- Salama, P.; Bernard, C. Chemoselective Synthesis of Functionalized Diselenides. Tetrahedron Lett. 1995, 36, 5711–5714. [Google Scholar] [CrossRef]
- Müller, J.; Terfort, A. Synthesis of pure aromatic, aliphatic, and araliphatic diselenides. Inorg. Chim. Acta 2006, 359, 4821–4827. [Google Scholar] [CrossRef]
- Plano, D.; Baquedano, Y.; Moreno-Mateos, D.; Font, M.; Jiménez-Ruiz, A.; Palop, J.A.; Sanmartín, C. Selenocyanates and diselenides: A new class of potent antileishmanial agents. Eur. J. Med. Chem. 2011, 46, 3315–3323. [Google Scholar] [CrossRef]
- Manna, T.; Misra, A.K. Aqueous Medium Preparation of Dialkyldiselenides. SynOpen 2018, 2, 229–233. [Google Scholar] [CrossRef]
- Heredia, A.A.; Peñéñory, A.B. Stereoselective synthesis of alkyl styryl selenides in one-pot: A straightforward approach by in situ dialkyl diselenide formation under transition metalfree conditions. RSC Adv. 2015, 5, 105699–105706. [Google Scholar] [CrossRef]
- Cooksey, J.P.; Kocieński, P.J.; Blacker, A.J. A Scalable Process for the Synthesis of 1,2-Dialkyldiselanes and 1-Alkaneselenols. Org. Process Res. Dev. 2019, 23, 2571–2575. [Google Scholar] [CrossRef]
- Potapov, V.A. Organic diselenides, ditellurides, polyselenides and polytellurides. Synthesis and reactions. In Patai’s Chemistry of Functional Groups. Organic Selenium and Tellurium Compounds; Rappoport, Z., Ed.; John Wiley and Sons, Inc.: Chichester, UK, 2013. [Google Scholar]
- Heredia, A.A.; Peñéñory, A.B. Transition-metal-free one-pot synthesis of alkynyl selenides from terminal alkynes under aerobic and sustainable conditions. Beilstein J. Org. Chem. 2017, 13, 910–918. [Google Scholar] [CrossRef] [PubMed]
- Wakamatsu, T.; Hara, H.; Ban, Y. A Formal Total Synthesis of (f)-Vernolepin and (f)-Vernomenin. J. Org. Chem. 1985, 50, 108–112. [Google Scholar] [CrossRef]
- Wojaczyńka, E.; Skarżewski, J. Novel C2-symmetric chiral ligands: Enantioselective transformation of cyclic 1,2-diols into 1,2-bis(phenylsulfenyl) and 1,2-bis(phenylselenyl) derivatives. Tetrahedron Asymmetry 2008, 19, 593–597. [Google Scholar] [CrossRef]
- Lari, A.; Bleiholder, C.; Rominger, F.; Gleiter, R. Intramolecular Nonbonded Interactions Between Divalent Selenium Centers with Donor and Acceptor Substituents. Eur. J. Org. Chem. 2009, 2765–2774. [Google Scholar] [CrossRef]
- Kumar, A.V.; Reddy, V.P.; Reddy, C.S.; Rao, K.R. Potassium selenocyanate as an efficient selenium source in C–Se cross-coupling catalyzed by copper iodide in water. Tetrahedron Lett. 2011, 52, 3978–3981. [Google Scholar] [CrossRef]
- Sureshbabu, V.V.; Vasantha, B.; Hemantha, H.P. Synthesis of N-Fmoc-Protected Amino Alkyl Thiocyanates/Selenocyanates and Their Application in the Preparation of 5-Substituted S/Se-Linked Tetrazoles. Synthesis 2011, 1447–1455. [Google Scholar] [CrossRef]
- Özkan, H.; Yavuz, S.; Dişli, A.; Yıldırır, Y.; Türker, L. Novel 5-Aryl-1H-Tetrazoles. Heteroatom Chem. 2007, 18, 255–258. [Google Scholar] [CrossRef]
- Dişli, A.; Salman, M. Synthesis of Some New 5-Substituted 1H-Tetrazoles. Rus. J. Org. Chem. 2009, 45, 151–153. [Google Scholar] [CrossRef]
- Kanakaraju, S.; Prasanna, B.; Chandramouli, G.V.P. Ionic liquid mediated a facile and convenient synthesis of new selanyl tetrazoles via one-pot three-component reaction. J. Chem. Pharm. Res. 2012, 4, 2994–2998. [Google Scholar]
- Maity, P.; Paroi, B.; Ranu, B.C. Transition-Metal-Free Iodine Catalyzed Selenocianation of Styrenyl Bromides and an Easy Access to Benzoselenophenes via Intermediacy of Styrenyl Selenocyanate. Org. Lett. 2017, 19, 5748–5751. [Google Scholar] [CrossRef]
- Cruz, A.; Macías-Mendoza, D.; Barragan-Rodríguez, E.; Tlahuext, H.; Nöth, H.; Contreras, R. Synthesis of optically active boroxazolidine, borathiazolidine and boraselenazolidine and their N-borane adducts from the corresponding 2-imino-heteroazolidines. Tetrahedron Asymmetry 1997, 8, 3903–3911. [Google Scholar] [CrossRef]
- Ueda, S.; Terauchi, H.; Suzuki, K.; Watanabe, N. Direct synthesis of novel 2-imino-1,3-selenazolidine derivatives from O-methanesulfonyl β-amino alcohol hydrochlorides. Tetrahedron Lett. 2005, 46, 233–236. [Google Scholar] [CrossRef]
- Cruz, A.; Padilla-Martínez, I.I.; García-Báez, E.V.; Contreras, R. Reactivity of chlorodeoxypseudoephedrines with oxo-, thio-, and selenocyanates. Tetrahedron Asymmetry 2007, 18, 123–130. [Google Scholar] [CrossRef]
- Castilla, J.; Marín, I.; Matheu, M.I.; Díaz, Y.; Castillón, S. Short and General Procedure for Synthesizing Cis-1,2-Fused 1,3-Oxathiolan-, 1,3-Oxaselenolan-, and 1,3-Oxazolidin-2-imine Carbohydrate Derivatives. J. Org. Chem. 2010, 75, 514–517. [Google Scholar] [CrossRef] [PubMed]
- Debnath, S.; Agarwal, A.; Kumar, N.R.; Bedi, A. Selenium-Based Drug Development for Antioxidant and Anticancer Activity. Future Pharmacol. 2022, 2, 595–607. [Google Scholar] [CrossRef]
- Gowda, R.; Madhunapantula, S.V.; Desai, D.; Amin, S.; Robertson, G.P. Selenium-containing histone deacetylase inhibitors for melanoma management. Cancer Biol. Ther. 2012, 13, 756–765. [Google Scholar] [CrossRef] [PubMed]
- Desai, D.; Salli, U.; Vrana, K.E.; Amin, S. SelSA, selenium analogs of SAHA as potent histone deacetylase inhibitors. Bioorg. Med. Chem. Lett. 2010, 20, 2044–2047. [Google Scholar] [CrossRef] [PubMed]
- Krishnegowda, G.; Gowda, A.P.; Tagaram, H.R.S.; Staveley-O’Carroll, K.F.; Irby, R.B.; Sharma, A.K.; Amin, S. Synthesis and biological evaluation of a novel class of isatin analogs as dual inhibitors of tubulin polymerization and Akt pathway. Bioorg. Med. Chem. 2011, 19, 6006–6014. [Google Scholar] [CrossRef]
- Desai, D.; Sinha, I.; Null, K.; Wolter, W.; Suckow, M.A.; King, T.; Amin, S.; Sinha, R. Synthesis and antitumor properties of selenocoxib-1 against rat prostate adenocarcinoma cells. Int. J. Cancer 2010, 127, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Nasim, M.J.; Witek, K.; Kincses, A.; Abdin, A.Y.; Żesławska, E.; Marć, M.A.; Gajdács, M.; Spengler, G.; Nitek, W.; Latacz, G.; et al. Pronounced activity of aromatic selenocyanates against multidrug resistant ESKAPE bacteria. New J. Chem. 2019, 43, 6021–6031. [Google Scholar] [CrossRef]
- Shaaban, S.; Negm, A.; Sobh, M.A.; Wessjohann, L.A. Organoselenocyanates and symmetrical diselenides redox modulators: Design, synthesis and biological evaluation. Eur. J. Med. Chem. 2015, 97, 190–201. [Google Scholar] [CrossRef]
- Sk, U.H.; Bhattacharya, S. Prevention of cadmium induced lipid peroxidation, depletion of some antioxidative enzymes and glutathione by a series of novel organoselenocyanates. Environ. Toxicol. Pharmacol. 2006, 22, 298–308. [Google Scholar] [CrossRef]
- Abdo, M.; Sun, Z.; Knapp, S. Biohybrid -Se-S- Coupling Reactions of an Amino Acid Derived Seleninate. Molecules 2013, 18, 1963–1972. [Google Scholar] [CrossRef] [PubMed]
- Krafft, G.A.; Meinke, P.T. Selenoaldehydes: Preparation and Dienophilic Reactivity. J. Am. Chem. Soc. 1986, 108, 1314–1315. [Google Scholar] [CrossRef]
- Guillemin, J.-C.; Bajor, G.; Riague El, H.; Khater, B.; Veszprémi, T. Allenyl and Alkynyl Selenols and Selenocyanates. Synthesis, Spectroscopic Characterization, and Quantum Chemical Study. Organomet 2007, 26, 2507–2518. [Google Scholar] [CrossRef]
- Nair, V.; Augustine, A.; George, T.G. A Facile CAN-Mediated Synthesis of Selenocyanates from Arylalkenes and Heteroarenes. Eur. J. Org. Chem. 2002, 2002, 2363–2366. [Google Scholar] [CrossRef]
- Chen, J.; Wang, T.; Wang, T.; Lin, A.; Yao, H.; Xu, J. Copper-catalyzed C5-selective thio/selenocyanation of 8-aminoquinolines. Org. Chem. Front. 2017, 4, 130–134. [Google Scholar] [CrossRef]
- Muniraj, N.; Dhineshkumar, J.; Prabhu, K.R. N-Iodosuccinimide Catalyzed Oxidative Selenocyanation and Thiocyanation of Electron Rich Arenes. ChemistrySelect 2016, 1, 1033–1038. [Google Scholar] [CrossRef]
- Bajor, G.; Veszprémi, T.; Riague, E.H.; Guillemin, J.-C. Alkenyl Selenols and Selenocyanates: Synthesis, Spectroscopic Characterization by Photoelectron Spectroscopy, and Quantum Chemical Study. Chem. Eur. J. 2004, 10, 3649–3656. [Google Scholar] [CrossRef] [PubMed]
- Toshimitsu, A.; Kosawa, Y.; Uemura, S.; Okano, M. Alkoxy- and Halogeno-selenocyanation of Olefins with Copper(II) Chloride or Bromide and Potassium Selenocyanate. J. Chem. Soc. Perkin Trans. 1 1978, 11, 1273–1278. [Google Scholar] [CrossRef]
- Wu, C.; Xiao, H.-J.; Wang, S.-W.; Tang, M.-S.; Tang, Z.-L.; Xia, W.; Li, W.-F.; Cao, Z.; He, W.-M. Natural Deep Eutectic Solvent-Catalyzed Selenocyanation of Activated Alkynes via an Intermolecular H-Bonding Activation Process. ACS Sustain. Chem. Eng. 2019, 7, 2169–2175. [Google Scholar] [CrossRef]
- Lu, L.-H.; Wang, Z.; Xia, W.; Cheng, P.; Zhang, B.; Cao, Z.; He, W.-M. Sustainable routes for quantitative green selenocyanation of activated alkynes. Chin. Chem. Lett. 2019, 30, 1237–1240. [Google Scholar] [CrossRef]
- Medvedeva, A.S.; Andreev, M.V.; Safronova, L.P. One-Pot Synthesis of 3-(Trimethylsilyl)propynamides. Russ. J. Org. Chem. 2010, 46, 1466–1470. [Google Scholar] [CrossRef]
- Hoberg, H.; Guhl, D. Ni0-induzierte CC-verknüpfung von fluorsubstituierten alkenen mit phenylisocyanat. J. Organomet. Chem. 1989, 378, 279–292. [Google Scholar] [CrossRef]
- Graziano, M.L.; Cimminiello, G. A Simple Route to 4,4-Dialkoxy-2-azetidinones: Useful Intermediates for Organic Synthesis. Synthesis 1989, 54–56. [Google Scholar] [CrossRef]
- Potapov, V.A.; Andreev, M.V.; Musalov, M.V.; Sterkhova, I.V.; Amosova, S.V.; Larina, L.I. Regio- and Stereoselective Synthesis of (Z,Z)-Bis(3-amino-3-oxo-1-propenyl) Selenides and Diselenides Based on 2-propynamides: A Novel Family of Diselenides with High Glutathione Peroxidase-Like Activity. Inorganics 2022, 10, 74. [Google Scholar] [CrossRef]
- Mareev, A.V.; Andreev, M.V.; Ushakov, I.A. Base-Catalyzed Hydration of Silicon-Containing Activated Alkynes: The Effect of Substituents at the Triple Bond. ChemistrySelect 2020, 5, 10736–10742. [Google Scholar] [CrossRef]
- Larina, L.I.; Albanov, A.I.; Zelinskiy, S.N.; Annenkov, V.V.; Rusakova, I.L. Acrylamide Derivatives: A Dynamic Nuclear Magnetic Resonance Spectroscopy. Magn. Reson. Chem. 2023, 61, 277–283. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | KSeCN a (Equiv) | MeOH (mL) | Conditions b | NH4Cl (Equiv) | Time (h) | Content (mol %) | Yield, 2a (%) | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 2a | 3 | 4 | 5 | 6 | |||||||
| 1 | 3.5 | 0.6 | r.t. b | – | 23 | – | 87 | 13 | – | – | – |
| 2 | 1.0 | 3.0 | reflux b | – | 3 | – | 35 | 65 | – | – | – |
| 3 | 3.5 | 0.6 | r.t. b | 1.0 | 23 | 63 | – | – | 31 | 6 | 65 |
| 4 | 3.5 | 0.6 | r.t. b | 1.0 | 68 | 38 | – | – | – | 62 | 36 |

| Entry | KSeCN a (eq) | Time (h) b | Content (mol %) | Yield of 2a c (%) | ||
|---|---|---|---|---|---|---|
| 2a | 5 | 6 | ||||
| 1 | 1.0 | 5 | 60 | 36 | 4 | 51 |
| 2 | 1.5 | 5 | 67 | 23 | 10 | 60 |
| 3 | 1.5 | 10 | 72 | 18 | 10 | 62 |
| 4 | 4.5 | 3 | 86 | – | 14 | 73 |

| Entry | Time (h) | NH4Cl (Equiv) | MeOH (mL) | Conc. 1a (mmol/L) | Conversion 2a (%) | Yield 2a (%) |
|---|---|---|---|---|---|---|
| 1 | 23 | 5 | 0.6 | 0.78 | 77 | 69 c |
| 2 | 23 | 10 | 0.6 | 0.78 | 83 | 78 c |
| 3 | 23 | 20 | 0.6 | 0.78 | 90 | 82 c |
| 4 | 23 | 30 | 0.6 | 0.78 | 92 | 82 c |
| 5 | 29 | 20 | 0.6 | 0.78 | 100 | 90 d |
| 6 | 19 | 5 | 0.4 | 1.18 | 100 | 91 d |
| 7 | 19 | 10 | 0.4 | 1.18 | 100 | 90 d |
| 8 | 19 | 3 | 0.4 | 1.18 | 89 | – |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andreev, M.V.; Potapov, V.A.; Musalov, M.V.; Larina, L.I. A New Family of Vinyl Selenocyanates with the Amide Function Based on the Reaction of Potassium Selenocyanate with 3-Trimethylsilyl-2-Propynamides. Catalysts 2023, 13, 1257. https://doi.org/10.3390/catal13091257
Andreev MV, Potapov VA, Musalov MV, Larina LI. A New Family of Vinyl Selenocyanates with the Amide Function Based on the Reaction of Potassium Selenocyanate with 3-Trimethylsilyl-2-Propynamides. Catalysts. 2023; 13(9):1257. https://doi.org/10.3390/catal13091257
Chicago/Turabian StyleAndreev, Mikhail V., Vladimir A. Potapov, Maxim V. Musalov, and Lyudmila I. Larina. 2023. "A New Family of Vinyl Selenocyanates with the Amide Function Based on the Reaction of Potassium Selenocyanate with 3-Trimethylsilyl-2-Propynamides" Catalysts 13, no. 9: 1257. https://doi.org/10.3390/catal13091257