
Microwave-Assisted Regioselective Suzuki Coupling of 2,4-Dichloropyrimidines with Aryl and Heteroaryl Boronic Acids



Faculty of Pharmacy, University of Ljubljana, Aškerčeva 7, 1000 Ljubljana, Slovenia
*
Author to whom correspondence should be addressed.
Catalysts 2021, 11(4), 439; https://doi.org/10.3390/catal11040439
Submission received: 5 March 2021
/
Revised: 24 March 2021
/
Accepted: 26 March 2021
/
Published: 30 March 2021
(This article belongs to the Special Issue Advances in Transition Metal Catalyzed Cross-Coupling)
Abstract
:Suzuki coupling reaction has been often used for the preparation of a diverse set of substituted pyrimidines. In this study, the Suzuki coupling of 2,4-dichloropyrimidines with aryl and heteroaryl boronic acids was investigated. A thorough screening of reaction conditions and the use of microwave irradiation led to a very efficient and straightforward synthetic procedure providing C4-substituted pyrimidines in good to excellent yields. Short reaction time (15 min) and extremely low catalyst loading (0.5 mol%) are the main advantages of our tetrakis(triphenylphosphine)palladium(0) catalyzed microwave-assisted procedure, which could be used for quick and low-cost regioselective preparation of substituted pyrimidine rings.
1. Introduction
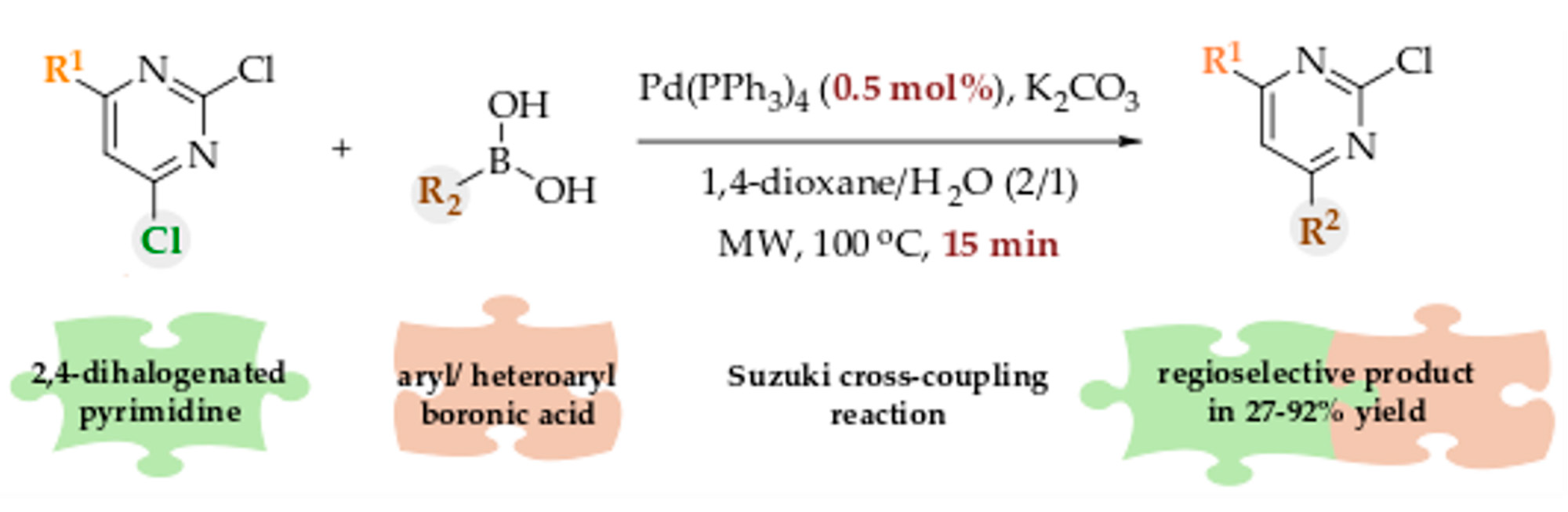
Substituted pyrimidine rings as scaffolds are of great interest for medicinal chemists being a part of many biologically active compounds [1,2]. The pyrimidine moiety is present in many natural compounds [3] (e.g., nucleic acids, alkaloids, folic acid, etc.) as well as synthetic analogs and also in approved drugs on the market, e.g., anticancer [4,5,6], antiviral [4,7], antibacterial [8], antilipidemic [9], anti-inflammatory [3,10], and antimalarial agents (Figure 1). Large number of synthetic methods have been described for the preparation of pyrimidine-based compounds [11,12,13,14,15,16]. Cyclocondensation between guanidine, amidine or thiourea derivatives and 1,3-diketones or 1,3-diesters is the most classical method for the synthesis of the main pyrimidine core [15,17], whereas one of the approaches to prepare substituted pyrimidine rings is via halogenated pyrimidines [18,19,20], which are greatly commercially available. The most common reactions involving various halogenated pyrimidines are cross-coupling reactions since pyrimidine ring is an electron-deficient aromatic system being far more reactive in comparison with analogous benzene halides [17,20]. Halogenated pyrimidines are thus very convenient substrates for substantial assortment of nucleophilic aromatic substitutions. Suzuki coupling of halogenated pyrimidines with boronic acids has been a commonly used approach for the preparation of a diverse set of substituted pyrimidines [20].
Polyhalogenated pyrimidines are commonly used substrates in cross-coupling reactions since each substituent could be added sequentially to a pyrimidine ring due to the intrinsic differences in reactivity at different positions [21,22,23,24]. In case of commonly used and commercially available 2,4-dichloropyrimidines, the regioselectivity preference in a Suzuki cross-coupling reaction was observed for C4-position due to the favored oxidative addition of palladium into the C4-chlorine bond [22,24,25]. Some anomalies have been observed, favoring C2 position over C4, mainly due to the steric reasons when additional substituents are present at other positions, i.e., C5 [24]. In the publication from Anderson and Handy in 2010, efficient synthesis of diarylated pyrimidines via regioselective double Suzuki coupling of 2,4-dichloropyrimidine was presented [21]. Similarly, the reaction occurred at the C4-position when 2,4-dichloropyrimidine or 2,4,6-trichloropyrimidine were coupled with alkenyl-boronic acids [26].
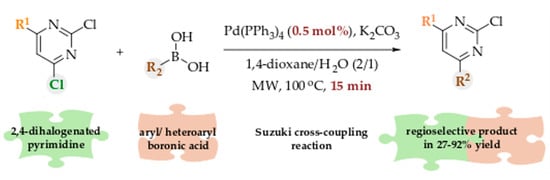
The aim of our study was to develop a quick, efficient, and regioselective synthetic procedure for substituted pyrimidines at position C4 from readily available 2,4-dichloropyrimidines via Suzuki coupling with a diverse set of aryl and heteroaryl boronic acids. The selection of appropriate solvents, catalysts, and reactions conditions (temperature, time) was systemically performed. Last, but not least, the optimal procedure was also transferred to a microwave reactor and further optimization (i.e., temperature, reaction time catalyst loading) was carried out.
2. Results
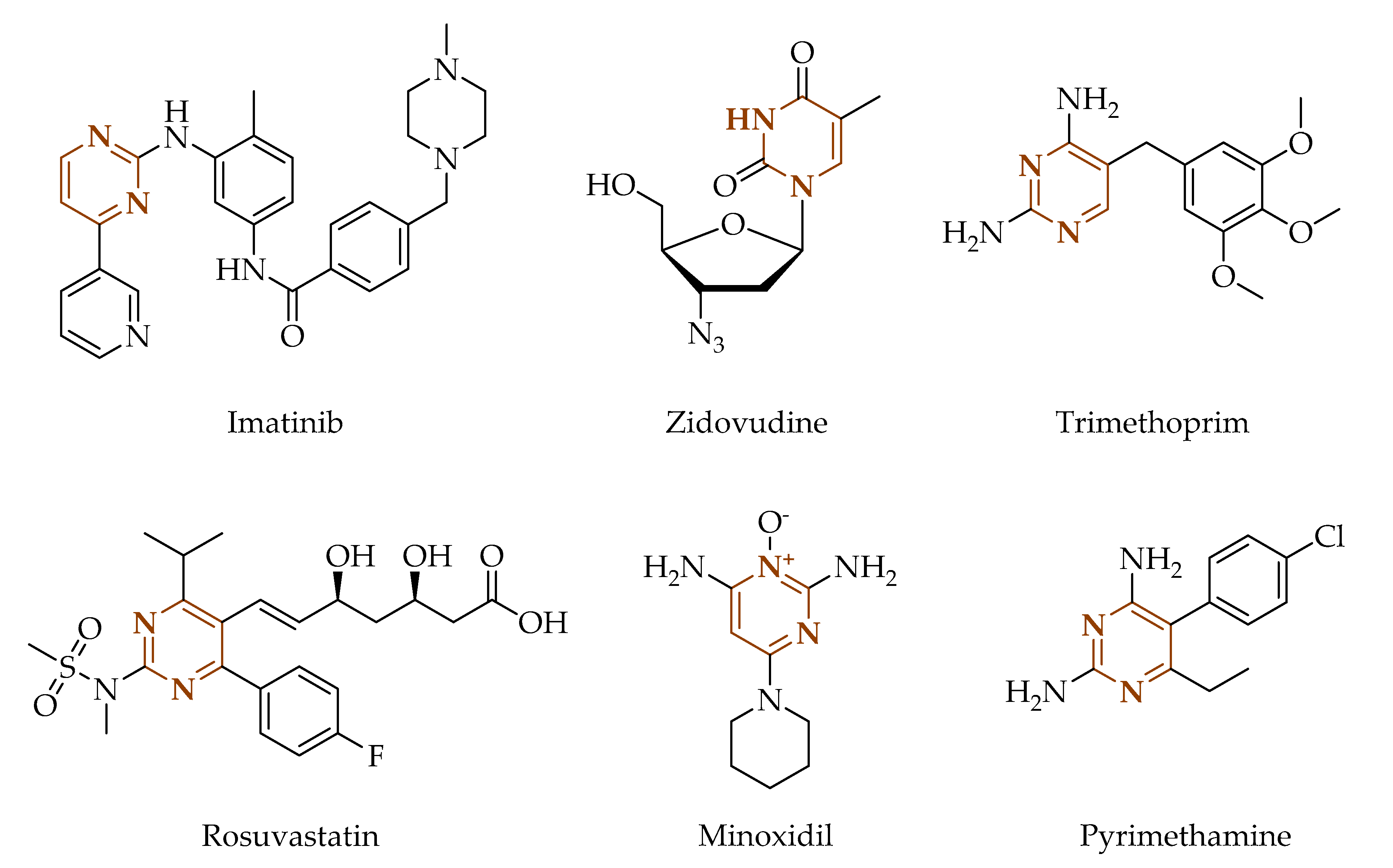
One of the most straightforward approaches to prepare substituted pyrimidines is via Suzuki coupling of halogenated pyrimidines with boronic acids [20,21]. There have been a variety of studies performed in order to propose the reaction mechanism underlaying the Suzuki-Miyaura coupling, consisting of several key steps. The initial pre-catalyst activation allows the formation of Pd(0) species. In the second step, during oxidative addition of aryl halide to the palladium center, the insertion of metal atom into the Cipso and halide (X) bond occurs and the complex [aryl-Pd(II)(Ln)-X] forms, where L represents the potentially bound ligand to the palladium centre, with n ranging from 1 to 4 [28]. Then the oxygen-containing nucleophile (either hydroxide from base followed by addition of boronic acid RB(OH)2, or boronate RB(OH)3–) replaces the halide group at palladium, which is later followed by transmetalation leading to the formation of aryl-Pd(II)(Ln)-R species. The final step includes reductive elimination of a product aryl-R with newly formed C-C bond, and regeneration of Pd(II) to Pd(0) [29]. In this study, Suzuki cross-coupling reaction between commercially available 2,4-dichloropyrimidine (1) and phenylboronic acid (2) was used as a model reaction to select appropriate catalyst, catalyst loading, solvent(s), and optimal reactions conditions (temperature, time). Our first attempt was to systemically screen the most common solvents used in Suzuki coupling reactions. Based on previous studies [21], tetrakis(triphenylphosphine)palladium(0) (Pd(PPh3)4) along with potassium carbonate K2CO3 as a base was selected as the starting catalytic system (Scheme 1).
A good selection of appropriate solvent for cross-coupling reactions is imperative. The screening of most common solvents for cross-coupling reactions (i.e., H2O, MeOH, THF, DMF, 1,4-dioxane, isopropanol, ethylene glycol) was performed at a room temperature (r.t.) and higher temperatures (60–100 °C), with yields determined by LC-MS being obtained after 1 h, 2 h, and 24 h (see Supporting Information, Table S1). The highest yields were obtained for less polar solvents, like isopropanol (64%) and 1,4-dioxane (72%), contrary to polar H2O, MeOH, DMF, and ethylene glycol, which corresponds to the observation made by Miyaura about Pd(PPh3)4 [30]. The choice of solvent (therefore its boiling point) defines the maximum operating temperature [31]. On the set of less polar solvents THF, isopropanol, and 1,4-dioxane with boiling points of 66 °C, 83 °C, and 101 °C, respectively, we confirmed that higher temperatures vastly improve the yield of reaction due to the high activation barriers reagents need to overcome for successful coupling. The C2 side product was formed in less than 8%, which is consistent with the literature reports [24].
We decided to repeat the reaction in four best solvents (i.e., THF, DMF, 1,4-dioxane and isopropanol) and isolate the pure product 3 by column chromatography. The isolated yields are presented in Table 1. The highest yield of 71% was obtained for non-polar 1,4-dioxane, which has been reported to complement Pd(PPh3)4 [30], thus we decided to proceed with further optimization in this solvent.
In a continuation of our study, we screened the most common commercially available palladium catalysts for this type of reaction. Tris(dibenzylideneacetone)palladium(0) (Pd2dba3) was not efficient for this type of reaction as has already been observed [32,33], whereas as an adduct with chloroform (Pd2dba3 · CHCl3) and together with tri-tert-butylphosphonium tetrafluoroborate (TTBP · HBF4) as a ligand it gave 35% and 23% yield, respectively (Table 2, entries 1-3). With bis(triphenylphosphine)palladium(II) chloride (PdCl2(PPh3)2) similarly low yield of 36% was achieved (Table 2, entry 4). Further we screened palladium(II) acetate (Pd(OAc)2) in combination with three different ligands, namely tricyclohexylphosphine (PCy3), triphenylphosphine (PPh3), and triphenylphosphine bound of divinylbenzene (PPh3 on DVB), with yields not significantly improved (Table 2, entries 5-7). According to the mechanism proposed, Pd(II) pre-catalysts have to be reduced to Pd(0) prior to their involvement in oxidative addition. It has been reported that trimeric Pd(OAc)2 is more susceptible to reduction in its monomeric form, whose formation has been shown to be proportional to the dipole moment of the solvent used [34]. In our screening, 1,4-dioxane with low dipole moment of only 0.45 D was used, which might be the reason for lower yields of Suzuki coupling with Pd(OAc)2. Solid-supported catalysts represent an attractive approach in the field of green organic chemistry, since they can often be recycled without loss of activity, in addition to the products and solution waste remaining free from metal contamination [35]. Despite several successful applications in Suzuki-Miyaura coupling [36,37,38], no product was formed when palladium on multiwall carbon nanotubes was used as a catalyst. Additionally, the screening was performed on a set of classic and solid-supported catalysts Pd(OAc)2 with PPh3 and Pd(OAc)2 with PPh3 on DVB, respectively, with the latter exhibiting inferior catalytic ability and yield in our model reaction. Only one catalyst ([1,1′-Bis(diphenylphosphino)ferrocene]dichloropalladium(II), in complex with dichloromethane (Pd(dppf)Cl2 · CH2Cl2) led to a higher yield of 70% (Table 2, entry 8), which could be due to its ability (i.e., wide bite-angle P-Pd-P) of driving very effective reductive elimination [39]. Since there was no improvement compared to Pd(PPh3)4 (Table 2, entry 9), all further reactions were performed with Pd(PPh3)4.
One parameter was screened additionally after selection of the appropriate catalyst, i.e., temperature (see Supporting Information, Table S2). Lowering the temperature significantly reduced the yield, thus all further reactions were performed at 100 °C.
After the first optimization process, we decided to further upgrade the reaction procedure with the aim to improve the yields and shorten the reaction time by performing it in a microwave reactor. We started with the solvent scan, where H2O due to favorable dielectric constant for microwave heating was added in different proportions to THF or 1,4-dioxane. With this experiment, where total solvent volume was 6 mL, we tried to find the optimal solvent ratio (v/v) to obtain the highest yield of 3 and as little as possible of any side products (Table 3). Generally, the yield of 3 was improved, when H2O was present in the reaction mixture, with the peak at solvent ratio 1:1 or 2:1 in the favor of non-polar solvent for THF and 1,4-dioxane, respectively (Table 3, entries 1 and 8). In addition to possessing dipole moment, the presence of water is also beneficial due to increased amount of hydrophilic boronates RB(OH)3– [31]. Overall, solvent mixture of 1,4-dioxane and H2O in ratio 2:1 appeared to be the most optimal, with 80% yield.
A scan of minimal solvent volume of 1,4-dioxane/ H2O (2:1) was performed (see Supporting Information, Table S3). Initial total volume of 6 mL in addition to 4.5 mL were found as the most suitable, while the yield significantly decreased when smaller solvent volumes were used. All further experiments were performed in total volume of 6 mL. Microwave-assisted reaction conditions, i.e., temperature and time, were further evaluated. First, the reaction mixture was subjected to temperature scan ranging from 60 °C to 140 °C with the interval of 20 °C (Table 4). The temperature of 100 °C appeared to be the most optimal leading to 80% yield. Higher temperatures (i.e., 120 °C and 140 °C, entries 7 and 10, respectively) did not improve the yield, since the side product 2,4-diphenylpyrimidine was formed. Additionally, time of the reaction was monitored for temperatures equal or above 100 °C. Reaction time of 15 min at 100 °C (yield 81%, entry 4) was found as equally efficient as 20 min. Therefore, these conditions were used for all further reactions.
To investigate the optimal amount of catalyst needed for the microwave-assisted Suzuki coupling, we performed the scan of catalyst loading, ranging from 5 mol% to 0.05 mol%. Contrary to the results from the experiments, performed in a flask, the minimal amount of catalyst needed for microwave-assisted reaction was only 0.5 mol% (Table 5), with lower amounts shown to be insufficient for all starting material to react.
A scale-up of the model reaction on proposed reaction conditions has further been performed. The reaction was performed on 4.0 mmol scale of starting reagents, with obtained yield of 3 being 53%, and 30% of 1 still remaining unreacted in the reaction mixture. Therefore, we prolonged the microwave reaction time for 5 min to 20 min and improved the yield of 3 to 74%, with only 19% of 1 still being present in the reaction mixture. Taken together, we can conclude that our microwave-assisted procedure is also suitable for production of aryl pyrimidines on larger scales.
With the optimal conditions being determined, the scope of several aryl and heteroaryl boronic acids was investigated (Table 6). On the series of methoxyphenyl boronic acids, different substituent positions on the aromatic ring were evaluated (products 26, 27, and 28); however, no significant effect on the yield and selectivity was found between ortho, meta or para position of a methoxy group. Therefore, we preferentially focused on meta-substituents in the following reactions. Overall, the main product was consistently the 4-substituted 2-chloropyrimidine, with little or usually none of 2-substituted 4-chloropyrimidine being formed during the reaction. Several different substituents were screened for and it was noticed that yields of phenylboronic acids with electron-withdrawing groups on meta position (30 to 33) were rather high and comparable to the model reaction (3). Other boronic acids with carboxylic and electron-donating groups (i.e., amino group) led to lower yields. As expected, the naphthyl boronic acid (product 37) displayed similar yield to our model reaction with phenylboronic acid 2 (3).
Some heteroaryl boronic acids were also screened, with the reaction being selective and successful for both furanylboronic acids (38 and 39) with comparable yield. However, the coupling of 2,4-dichloropyrimidine with thiophen-2-boronic acid failed due to the sulphur poisoning of the palladium catalyst [40]. Furthermore, in case of pyridinylboronic acids, the reaction was also unsuccessful.
The microwave-assisted procedure for formation of 4-phenylpyrimidines was further screened on the series of differently substituted 2,4-dihalogenopyrimidines (Table 7). Despite chloroarenes being generally inert to oxidative addition, some electron-deficient heteroaryl chlorides (i.e., 2,4-dichloropyrimidines) are enough reactive for successful cross-coupling reactions [30]. Interestingly, the coupling of 2 with 2,4-dibromopyridine (19) did not offer improved yield (product 40) and the formation of a side product 2-phenyl-4-bromopyrimidine was neither increased. The substitutions on the position 6 of 2,4-dichloropyrimidine (19–25) with electron-withdrawing and electron-donating groups did not significantly affect the yield of the reaction (e.g., 43 and 45 with electron donor substituent versus 42 with electron withdrawing group), with the exception of 41 and 46, which led to lower yields of 27% and 54%, respectively. Thus, we wanted to improve the yield of 41. The addition of 3 mol% of Pd(PPh3)4 significantly improved the formation of 41, leading to 45% yield. Interestingly, the reaction of 2 with 2,4-dichloro-6-methoxypyrimidine (23) led to the production of both isomers 2-chloro-4-methoxy-6-phenylpyrimidine (44A) and 4-chloro-6-methoxy-2-phenylpyrimidine (44B) with no selectivity between them.
Taken together, the reaction is less susceptible to different substitutions on aryl boronic acids, with most of them giving high yield and selectivity at coupling with 2,4-dichloropyrimidine. Since one of the major obstacles in coupling with heteroaryl chlorides is their potentially insufficient electron-deficiency, substitutions on the scaffold have immense effect on the outcome of Suzuki-Miyaura cross-coupling reactions.
3. Materials and Methods
3.1. Chemistry and Chemical Characterization of Compounds
The reagents and solvents were purchased from commercial sources (Sigma-Aldrich, St. Louis, MO, USA; Acros Organics, Antwerp, Belgium; Alfa Aesar, Haverhill, MA, USA; TCI, Tokio, Japan) and used without further purification. The microwave-assisted reactions were performed using an Anton Paar Monowave 200 microwave reactor (Anton Paar GmbH, Graz, Austria). Flash column chromatography was performed on Merck silica gel 60 (mesh size, 70–230), using the indicated solvents. Monitoring of the purification was done by thin-layer chromatography on silica-gel plates (Merck DC Fertig-platten Kieselgel 60 GF254, Merck, Darmstadt, Germany) and visualized under UV light. Yields refer to the purified products or were determined by LC-MS, as stated, and were not optimized. All of the melting points were determined on a Reichert hot-stage apparatus, and are uncorrected. 1H and 13C NMR spectra were recorded at 295 K in CDCl3 or DMSO-d6 on a Bruker Avance III NMR spectrometer equipped with a Broadband decoupling inverse 1H probe. The coupling constants (J) are in Hz, and the splitting patterns are designated as: s, singlet; d, doublet; dd, double doublet; td, triple doublet; t, triplet; dt, double triplet; ddd, double of doublet of doublet; and m, multiplet. High-resolution mass measurements were performed on ExativeTM Plus Orbitrap Mass Spectrometer (Thermo Fischer Scientific, Inc., Waltham, MA, USA). Monitoring of the reactions and determination of purities of the assayed compounds were done by Liquid Chromatography Mass Spectrometry (LC-MS) on Agilent 1260 Infinity II LC System (Agilent Technologies, Santa Clara, CA, USA) equipped with quaternary pump and a variable wavelength detector, coupled to ADVION Expression CMSL Mass Spectrometer (Advion Inc., Ithaca, NY, USA). A Waters xBridge BEH C18 column was used (4.6 mm × 150 mm, 3.5 µm) at 40 °C, with a flow rate of 1.5 mL/min, injection volume of 10 µL, detection at 254 nm and an eluent system of: A, 1% of CH3CN and 0.1% of HCOOH in ddH2O; B, CH3CN. The following gradients were applied: 0–1 min, 25% B; 1–6 min, 25% → 98% B; 6–6.5 min, 98% B; 6.5–7 min, 98% → 25% B; 7–10 min, 25% B.
1H and 13C NMR spectra are available in the Supplementary Material related to this manuscript.
3.2. General Procedure for Solvent Screening (Table 1)
2,4-dichloropyrimidine 1 (149 mg, 1.0 mmol) was dissolved/ suspended in the selected solvent (3.5 mL), where the air (oxygen) was displaced with argon before use. Then potassium carbonate K2CO3 (415 mg, 3.0 mmol), phenylboronic acid 2 (122 mg, 1.0 mmol), and 5 mol% of tetrakis(triphenylphosphine)palladium(0) Pd(PPh3)4 (58 mg, 0.05 mmol) were added and stirred under argon atmosphere at selected temperature (i.e., 60, 80, or 100 °C). After 24 h, the reaction mixture was extracted with EtOAc, brine, and dried over anhydrous Na2SO4. Solvents were removed under reduced pressure, the remaining solid was purified using flash column chromatography with EtOAc/n-Hex as an eluent.
3.3. General Procedure for Catalyst Screening (Table 2)
1 (75 mg, 0.5 mmol) was dissolved in 1,4-dioxane (3.5 mL), where the air (oxygen) was displaced with argon before use. Then K2CO3 (207 mg, 1.5 mmol), 2 (61 mg, 0.5 mmol), and 5 mol% of a selected catalyst (0.05 mmol) with 10 mol% of a ligand where applicable were added and stirred under argon atmosphere at 100 °C.
50 µL of reaction mixture was diluted with 950 µL of internal standard solution (prepared by 102 mg of acetanilide in 100 mL of CH3CN). The sample was further diluted (1/10) with CH3CN and filtered to obtain the final sample for LC-MS analysis.
In case of Pd(dppf)Cl2·CHCl2 and Pd(PPh3)4 as catalyst, after 24 h, the reaction mixture was extracted with EtOAc, brine and dried over anhydrous Na2SO4. Solvents were removed under reduced pressure, the remaining solid was purified using flash column chromatography with EtOAc/n-Hex as an eluent.
3.4. General Procedure for Catalyst Screening—Use of Palladium on Multiwall Carbon Nanotubes
1 (75 mg, 0.5 mmol) was dissolved in 1,4-dioxane (3.5 mL), where the air (oxygen) was displaced with argon before use. Then K2CO3 (207 mg, 1.5 mmol), 2 (61 mg, 0.5 mmol), and 0.2 mol% of palladium on multiwall carbon nanotubes (10.6 mg, 0.11 mg, 1.0 µM) was added and stirred under argon atmosphere at 100 °C.
50 µL of reaction mixture was diluted with 950 µL of internal standard solution (prepared by 102 mg of acetanilide in 100 mL of CH3CN). The sample was further diluted (1/10) with CH3CN and filtered to obtain the final sample for LC-MS analysis.
3.5. General Procedure for Solvent Mixture Screening (Table 3)
1 (75 mg, 0.5 mmol) was dissolved in a mixture of THF or 1,4-dioxane and H2O (total 6 mL), where the air (oxygen) was displaced with argon before use. Then K2CO3 (207 mg, 1.5 mmol), 2 (61 mg, 0.5 mmol), and 3 mol% of Pd(PPh3)4 (17.3 mg, 0.015 mmol) were added and stirred under argon atmosphere. The reaction mixture was then stirred for 20 min at 100 °C in a microwave reactor.
50 µL of reaction mixture was diluted with 950 µL of internal standard solution (prepared by 102 mg of acetanilide in 100 mL of CH3CN). The sample was further diluted (1/10) with CH3CN and filtered to obtain the final sample for LC-MS analysis.
3.6. General Procedure for Temperatuer and Time Screening (Table 4)
1 (75 mg, 0.5 mmol) was dissolved in a mixture of 1,4-dioxane (4 mL) and H2O (2 mL), where the air (oxygen) was displaced with argon before use. Then K2CO3 (207 mg, 1.5 mmol), 2 (61 mg, 0.5 mmol), and 3 mol% of Pd(PPh3)4 (17.3 mg, 0.015 mmol) were added and stirred under argon atmosphere. The reaction mixture was then stirred at several different temperatures (i.e., 60, 80, 100, 120, and 140 °C) in a microwave reactor for a specified amount of time (i.e., 5, 10, 15, and 20 min).
50 µL of reaction mixture was diluted with 950 µL of internal standard solution (prepared by 102 mg of acetanilide in 100 mL of CH3CN). The sample was further diluted (1/10) with CH3CN and filtered to obtain the final sample for LC-MS analysis.
3.7. General Procedure for Catalyst Loading Screening (Table 5)
1 (75 mg, 0.5 mmol) was dissolved in a mixture of 1,4-dioxane (4 mL) and H2O (2 mL), where the air (oxygen) was displaced with argon before use. Then K2CO3 (207 mg, 1.5 mmol), 2 (61 mg, 0.5 mmol), and appropriate amount of Pd(PPh3)4 (i.e., 0.05, 0.2, 0.5, 1, 2, 3, and 5 mol%) were added and stirred under argon atmosphere. The reaction mixture was then stirred for 15 min at 100 °C in a microwave reactor.
50 µL of reaction mixture was diluted with 950 µL of internal standard solution (prepared by 102 mg of acetanilide in 100 mL of CH3CN). The sample was further diluted (1/10) with CH3CN and filtered to obtain the final sample for LC-MS analysis.
3.8. General Procedure for Scale-Up
1 (600 mg, 4.0 mmol) was dissolved in a mixture of 1,4-dioxane (10 mL) and H2O (5 mL), where the air (oxygen) was displaced with argon before use. Then K2CO3 (1.656 g, 12.0 mmol), 2 (488 mg, 4.0 mmol), and 0.5 mol% of Pd(PPh3)4 (23.12 mg, 0.02 mmol) were added and stirred under argon atmosphere. The reaction mixture was then stirred for a specified amount of time (i.e., 15, 20 min) at 100 °C in a microwave reactor.
50 µL of reaction mixture was diluted with 950 µL of internal standard solution (prepared by 102 mg of acetanilide in 100 mL of CH3CN). The sample was further diluted (1/10) with CH3CN and filtered to obtain the final sample for LC-MS analysis.
3.9. General Procedure for Screening of Boronic Acids (Table 6)
1 (75 mg, 0.5 mmol) was dissolved in a mixture of 1,4-dioxane (4 mL) and H2O (2 mL), where the air (oxygen) was displaced with argon before use. Then K2CO3 (207 mg, 1.5 mmol), appropriate boronic acid (0.5 mmol), and 0.5 mol% of Pd(PPh3)4 (2.9 mg, 0.0025 mmol) were added and stirred under argon atmosphere. The reaction mixture was then stirred for 15 min at 100 °C in a microwave reactor. Afterwards, reaction mixture was extracted with EtOAc, brine, and dried over anhydrous Na2SO4. Solvents were removed under reduced pressure, the remaining solid was purified using flash column chromatography with EtOAc/n-Hex as an eluent.
3.10. General Procedure for Screening of 2,4-Dihalogenopyrimidines (Table 7)
Appropriate 2,4-dihalogenopyrimidine (0.5 mmol) was dissolved in a mixture of 1,4-dioxane (4 mL) and H2O (2 mL), where the air (oxygen) was displaced with argon before use. Then K2CO3 (207 mg, 1.5 mmol), 2 (61 mg, 0.5 mmol), and 0.5 mol% of Pd(PPh3)4 (2.9 mg, 0.0025 mmol) were added and stirred under argon atmosphere. The reaction mixture was then stirred for 15 min at 100°C in a microwave reactor. Afterwards, reaction mixture was extracted with EtOAc, brine, and dried over anhydrous Na2SO4. Solvents were removed under reduced pressure, the remaining solid was purified using flash column chromatography with EtOAc/n-Hex as an eluent.
3.11. Analytics of the Synthesized Compounds
2-chloro-4-phenylpyrimidine (3): white solid, Mp 86–87 °C (lit. 84–86 °C [41]); 1H NMR (400 MHz, CDCl3): δ (ppm) = 7.48–7.54 (m, 3H), 7.64 (d, J = 5.3 Hz, 1H), 8.07–8.10 (m, 2H), 8.63 (d, J = 5.3 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ (ppm) = 115.27, 127.55, 129.25, 132.04, 135.17, 159.95, 161.99, 167.30; HRMS (ESI+) m/z calc. for C10H8N2Cl [M + H]+ 191.0371, found 191.0369; HPLC tR = 6.41 min; Rf = 0.43 (EtOAc/n-Hex, 1:2, v/v).
4-chloro-2-phenylpyrimidine (4): white solid, Mp 72–73 °C (lit. 73–74 °C [42]); 1H NMR (400 MHz, CDCl3): δ (ppm) = 7.23 (d, J = 5.2 Hz, 1H), 7.48–7.52 (m, 3H), 8.43–8.46 (m, 2H), 8.67 (d, J = 5.2 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ (ppm) = 119.45, 128.66, 128.81, 131.68, 136.23, 158.39, 161.70, 165.71; HRMS (ESI+) m/z calc. for C10H8N2Cl [M + H]+ 191.0371, found 191.0371; HPLC tR = 7.09 min; Rf = 0.55 (EtOAc/n-Hex, 1:2, v/v).
2-chloro-4-(2-methoxyphenyl)pyrimidine (26): Yield: 76%; white solid; Mp 53–54 °C; 1H NMR (400 MHz, CDCl3): δ (ppm) = 3.92 (s, 3H), 7.00–7.04 (m, 1H), 7.11 (td, J1 = 1.1 Hz, J2 = 7.6 Hz, 1H), 7.48 (ddd, J1 = 1.8 Hz, J2 = 7.4 Hz, J3 = 8.4 Hz, 1H), 7.97 (d, J = 5.3 Hz, 1H), 8.09 (dd, J1 = 1.8 Hz, J2 = 7.8 Hz, 1H), 8.57 (d, J = 5.3 Hz, 1H) as reported [43]; 13C NMR (100 MHz, CDCl3): δ (ppm) = 55.76, 111.65, 120.21, 121.43, 124.53, 131.47, 13.86, 158.32, 159.05, 161.31, 165.82; HRMS (ESI+) m/z calc. for C10H10N2OCl [M + H]+ 221.0476, found 221.0476; HPLC tR = 6.41 min; Rf = 0.36 (EtOAc/n-Hex, 1:4, v/v).
2-chloro-4-(3-methoxyphenyl)pyrimidine (27): Yield: 72%; white solid; Mp 68–70 °C (lit. 83–86 °C [44]); 1H NMR (400 MHz, CDCl3): δ (ppm) = 3.90 (s, 3H), 7.09 (ddd, J1 = 0.9 Hz, J2 = 2.6 Hz, J3 = 8.2 Hz, 1H), 7.42 (t, J = 8.0 Hz, 1H), 7.63 (d, J = 5.3 Hz, 1H), 7.61–7.67 (m, 2H), 8.64 (d, J = 5.3 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ (ppm) = 55.67, 112.61, 115.48, 118.06, 119.92, 130.29, 136.64, 159.97, 160.41, 161.99, 167.17; HRMS (ESI+) m/z calc. for C10H10N2OCl [M + H]+ 221.0476, found 221.0476; HPLC tR = 6.34 min; Rf = 0.24 (EtOAc/n-Hex, 1:4, v/v).
2-chloro-4-(4-methoxyphenyl)pyrimidine (28): Yield: 77%; white solid; Mp 106–108 °C (lit. 137–140 °C [44]); 1H NMR (400 MHz, CDCl3): δ (ppm) = 3.89 (s, 3H), 6.99–7.03 (m, 2H), 7.57 (d, J = 5.3 Hz, 1H), 8.06–8.10 (m, 2H), 8.56 (d, J = 5.3 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ (ppm) = 55.65, 114.32, 114.63, 127.57, 129.30, 159.58, 161.90, 162.96, 166.79; HRMS (ESI+) m/z calc. for C11H10ON2Cl [M + H]+ 221.0476, found 221.0476; HPLC tR = 6.30 min; Rf = 0.18 (EtOAc/n-Hex, 1:4, v/v).
2-chloro-4-(m-tolyl)pyrimidine (29): Yield: 75%; white solid; Mp 74–75 °C; 1H NMR (400 MHz, CDCl3): δ (ppm) = 2.45 (s, 3H), 7.34–7.38 (m, 1H), 7.40 (t, J = 7.5 Hz, 1H), 7.64 (d, J = 5.3 Hz, 1H), 7.84–7.87 (m, 1H), 7.92–7.94 (m, 1H), 8.62 (d, J = 5.3 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ (ppm) = 21.59, 115.38, 124.72, 128.18, 129.16, 132.87, 135.18, 139.16, 159.88, 161.98, 167.56; HRMS (ESI+) m/z calc. for C11H10N2Cl [M + H]+ 205.0527, found 205.0529; HPLC tR = 6.87 min; Rf = 0.35 (EtOAc/n-Hex, 1:3, v/v).
2-chloro-4-(3-nitrophenyl)pyrimidine (30): Yield: 79%; yellow solid; Mp 123–125 °C; 1H NMR (400 MHz, CDCl3): δ (ppm) = 7.74 (t, J = 8.0 Hz, 1H), 7.76 (d, J = 5.2 Hz, 1H), 8.41 (ddd, J1 = 1.1 Hz, J2 = 2.2 Hz, J3 = 8.2 Hz, 1H), 8.48 (ddd, J1 = 1.1 Hz, J2 = 1.8 Hz, J3 = 7.8 Hz, 1H), 8.76 (d, J = 5.2 Hz, 1H), 8.93 (t, J = 1.9 Hz, 2H) as reported [45]; 13C NMR (100 MHz, CDCl3): δ (ppm) = 115.46, 122.46, 126.40, 130.48, 133.26, 136.96, 149.01, 160.77, 162.37, 164.64; HRMS (ESI+) m/z calc. for C10H7O2N3Cl [M + H]+ 236.0221, found 236.0221; HPLC tR = 6.33 min; Rf = 0.17 (EtOAc/n-Hex, 1:2, v/v).
3-(2-chloropyrimidin-4-yl)benzonitrile (31): Yield: 83%; pale pink solid; Mp 180–182 °C; 1H NMR (400 MHz, CDCl3): δ (ppm) = 7.64–7.68 (m, 1H), 7.68 (d, J = 5.2 Hz, 1H), 7.83 (dt, J1 = 1.4 Hz, J2 = 7.7 Hz, 1H), 8.33 (ddd, J1 = 1.2 Hz, J2 = 1.9 Hz, J3 = 8.0 Hz, 1H), 8.41 (t, J = 1.8 Hz, 1H), 8.74 (d, J = 5.2 Hz, 1H) as reported [46]; 13C NMR (100 MHz, CDCl3): δ (ppm) = 113.84, 115.31, 118.10, 130.23, 131.18, 131.56, 135.00, 136.47, 160.70, 162.34, 164.79; HRMS (ESI+) m/z calc. for C11H7N3Cl [M + H]+ 216.0323, found 216.0323; HPLC tR = 5.97 min; Rf = 0.17 (EtOAc/n-Hex, 1:2, v/v).
3-(2-chloropyrimidin-4-yl)benzaldehyde (32): Yield: 70%; white solid; Mp 120–122 °C; 1H NMR (400 MHz, CDCl3): δ (ppm) = 7.70 (t, J = 7.7 Hz, 1H), 7.74 (d, J = 5.3 Hz, 1H), 8.05 (dt, J1 = 1.4 Hz, J2 = 7.6 Hz, 1H), 8.38 (dt, J1 = 1.5 Hz, J2= 7.7 Hz, 1H), 8.58 (t, J = 1.8 Hz, 1H), 8.71 (d, J = 5.1 Hz, 1H), 10.12 (s, 1H); 13C NMR (100 MHz, CDCl3): δ (ppm) = 115.40, 128.63, 130.11, 132.71, 133.13, 136.22, 137.19, 160.45, 162.17, 165.78, 191.58; HRMS (ESI+) m/z calc. for C11H8ON2Cl [M + H]+ 219.0320, found 219.0322 as reported [47]; HPLC tR = 5.61 min; Rf = 0.32 (EtOAc/n-Hex, 1:1, v/v).
Methyl 3-(2-chloropyrimidin-4-yl)benzoate (33): Yield: 62%; white solid; Mp 108–110 °C; 1H NMR (400 MHz, CDCl3): δ (ppm) = 3.97 (s, 3H), 7.61 (t, J = 7.8 Hz, 1H), 7.72 (d, J = 5.3 Hz, 1H), 8.20 (dt, J1 = 1.4 Hz, J2 = 7.8 Hz, 1H), 8.34 (ddd, J1 = 1.2 Hz, J2 = 1.9 Hz, J3 = 7.9 Hz, 1H), 8.68 (d, J = 5.3 Hz, 1H), 8.69–8.70 (m, 1H) as reported [48]; 13C NMR (100 MHz, CDCl3): δ (ppm) = 52.58, 115.42, 128.57, 129.52, 131.30, 131.88, 132.86, 135.61, 160.28, 162.13, 166.24, 166.43; HRMS (ESI+) m/z calc. for C12H10O2N2Cl [M + H]+ 249.0425, found 249.0424; HPLC tR = 6.29 min; Rf = 0.25 (EtOAc/n-Hex, 1:2, v/v).
3-(2-chloropyrimidin-4-yl)benzoic acid (34): Yield: 55%; white solid; Mp 196–198 °C; 1H NMR (400 MHz, DMSO-d6): δ (ppm) = 7.49 (t, J = 7.7 Hz, 1H), 8.12–8.16 (m, 3H), 8.72–8.74 (m, 1H), 8.76 (d, J = 5.3 Hz, 1H), 1H from COOH is exchanged as reported [47]; 13C NMR (100 MHz, DMSO-d6): δ (ppm) = 116.07, 128.19, 128.28, 132.80, 133.70, 140.01, 160.51, 161.08, 166.51, 169.84; HRMS (ESI–) m/z calc. for C11H6O2N2Cl [M − H]– 233.0123, found 233.0122; HPLC tR = 4.96 min; Rf = 0.14 (DCM/MeOH, 9:1, v/v).
3-(2-chloropyrimidin-4-yl)aniline (35): Yield: 55%; yellow solid; Mp 120–122 °C (lit. 137–138 °C [49]); 1H NMR (400 MHz, CDCl3): δ (ppm) = 3.86 (s, 2H), 6.85 (ddd, J1 = 1.0 Hz, J2 = 2.4 Hz, J3 = 7.9 Hz, 1H), 7.28 (t, J = 7.9 Hz, 1H), 7.38 (ddd, J1 = 1.0 Hz, J2 = 1.8 Hz, J3 = 7.9 Hz, 1H), 7.48 (t, J = 2.0 Hz, 1H), 7.60 (d, J = 5.3 Hz, 1H), 8.61 (d, J = 5.3 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ (ppm) = 113.67, 115.41, 117.61, 118.60, 130.15, 136.21, 147.37, 159.83, 161.86, 167.51; HRMS (ESI+) m/z calc. for C10H9N3Cl [M + H]+ 206.0480, found 206.0478; HPLC tR = 3.35 min; Rf = 0.23 (EtOAc/n-Hex, 1:1, v/v).
2-chloro-4-(4-fluorophenyl)pyrimidine (36): Yield: 62%; white solid; Mp 101–103 °C; 1H NMR (400 MHz, CDCl3): δ (ppm) = 7.18–7.24 (m, 2H), 7.61 (d, J = 5.3 Hz, 1H), 8.10–8.15 (m, 2H), 8.64 (d, J = 5.3 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ (ppm) = 114.91, 116.47 (d, JC-F = 22.0 Hz), 129.82 (d, JC-F = 8.9 Hz), 131.37, 160.06, 162.07, 162.25 (d, JC-F = 253.3 Hz), 166.62; HRMS (ESI+) m/z calc. for C10H7N2ClF [M + H]+ 209.0276, found 209.0278; HPLC tR = 6.44 min; Rf = 0.31 (EtOAc/n-Hex, 1:3, v/v).
2-chloro-4-(naphthalen-2-yl)pyrimidine (37): Yield: 87%; white solid; Mp 114–116 °C; 1H NMR (400 MHz, CDCl3): δ (ppm) = 7.54–7.62 (m, 2H), 7.79 (d, J = 5.3 Hz, 1H), 7.88–7.92 (m, 1H), 7.95–8.01 (m, 2H), 8.15 (dd, J1 = 1.9 Hz, J2 = 8.6 Hz, 1H), 8.65–8.67 (m, 1H), 8.68 (d, J = 5.3 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ (ppm) = 115.49, 123.78, 127.08, 127.95, 128.19, 128.40, 129.17, 129.38, 132.42, 133.26, 135.17, 160.01, 162.10, 167.27; HRMS (ESI+) m/z calc. for C14H10N2Cl [M + H]+ 241.0527, found 241.0528 as reported [50]; HPLC tR = 7.31 min; Rf = 0.28 (EtOAc/n-Hex, 1:3, v/v).
2-chloro-4-(furan-2-yl)pyrimidine (38): Yield: 50%; white solid; Mp 67–69 °C; 1H NMR (400 MHz, CDCl3): δ (ppm) = 6.61 (dd, J1 = 1.7 Hz, J2 = 3.5 Hz, 1H), 7.39 (dd, J1 = 0.7 Hz, J2 = 3.5 Hz, 1H), 7.53 (d, J = 5.2 Hz, 1H), 7.63 (dd, J1 = 0.7 Hz, J2 = 1.7 Hz, 1H), 8.58 (d, J = 5.2 Hz, 1H) as reported [51]; 13C NMR (100 MHz, CDCl3): δ (ppm) = 113.10, 113.13, 114.58, 146.24, 150.52, 158.11, 159.92, 161.76; HRMS (ESI+) m/z calc. for C8H6ON2Cl [M + H]+ 181.0163, found 181.0163; HPLC tR = 5.03 min; Rf = 0.27 (EtOAc/n-Hex; 1:4, v/v).
2-chloro-4-(furan-3-yl)pyrimidine (39): Yield: 66%; grey solid; Mp 62–64 °C (lit. 87–89 °C [52]); 1H NMR (400 MHz, CDCl3): δ (ppm) = 6.90 (dd, J1 = 0.9 Hz, J2 = 1.9 Hz, 1H), 7.32 (d, J = 5.1 Hz, 1H), 7.52–7.55 (m, 1H), 8.21–8.23 (m, 1H), 8.55 (d, J = 5.2 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ (ppm) = 108.36, 115.06, 124.41, 144.77, 144.88, 159.67, 161.91, 161.98; HRMS (ESI+) m/z calc. for C8H6ON2Cl [M + H]+ 181.0163, found 181.0161; HPLC tR = 5.29 min; Rf = 0.18 (EtOAc/n-Hex, 1:4, v/v).
2-bromo-4-phenylpyrimidine (40): Yield: 48%; white solid; Mp 68–70 °C (lit. 85–87 °C [20]; 1H NMR (400 MHz, CDCl3): δ (ppm) = 7.49–7.55 (m, 3H), 7.69 (d, J = 5.3 Hz, 1H), 8.07–8.10 (m, 2H), 8.57 (d, J = 5.3 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ (ppm) = 115.67, 127.58, 129.28, 132.08, 135.13, 153.79, 159.73, 167.13; HRMS (ESI+) m/z calc. for C10H7BrN2 [M + H]+ 234.9865, found 234.9869; HPLC tR = 6.45 min; Rf = 0.38 (EtOAc/n-Hex, 1:2, v/v).
2-chloro-4-phenyl-6-(trifluoromethyl)pyrimidine (41): Yield: 27%; pale yellow solid; Mp 47–49 °C; 1H NMR (400 MHz, CDCl3): δ (ppm) = 7.54–7.63 (m, 3H), 7.96 (s, 1H), 8.15–8.18 (m, 2H); 13C NMR (100 MHz, CDCl3): δ (ppm) = 100.13, 111.06, 120.08 (q, JC-F = 274.1 Hz), 127.93, 129.55, 133.14, 133.78, 134.25, 169.84; HRMS (ESI+) m/z calc. for C11H7N2ClF3 [M + H]+ 259.0244, found 259.0250; HPLC tR = 7.51 min; Rf = 0.60 (EtOAc/n-Hex, 1:4, v/v).
2-chloro-6-phenylpyrimidine-4-carboxylic acid (42): Yield: 92%; appearance; Mp > 300 °C; 1H NMR (400 MHz, DMSO-d6): δ (ppm) = 7.51–7.58 (m, 3H), 8.13–8.16 (m, 2H), 8.29 (s, 1H), 1H from COOH is exchanged; 13C NMR (100 MHz, DMSO-d6): δ (ppm) = 114.06, 127.24, 129.17, 131.80, 134.90, 160.30, 165.40, 166.62, 168.53; HRMS (ESI–) m/z calc. for C11H6O2N2Cl [M − H ]– 233.0123, found 233.0121; HPLC tR = 5.29 min; Rf = 0.04 (DCM/MeOH, 9:1, v/v)
2-chloro-4-methyl-6-phenylpyrimidine (43): Yield: 78%; pale orange solid; Mp 42–44 °C; 1H NMR (400 MHz, CDCl3): δ (ppm) = 2.60 (s, 3H), 7.48–7.55 (m, 4H), 8.06–8.08 (m, 2H) as reported [53]; 13C NMR (100 MHz, CDCl3): δ (ppm) = 24.37, 100.13, 114.87, 127.55, 129.19, 131.75, 135.49, 166.96, 170.98; HRMS (ESI+) m/z calc. for C11H10N2Cl [M + H]+ 205.0527, found 205.0528; HPLC tR = 6.60 min; Rf = 0.41 (EtOAc/n-Hex, 1:2, v/v).
2-chloro-4-methoxy-6-phenylpyrimidine (44A): Yield: 34%; white solid; Mp 88–90 °C; 1H NMR (400 MHz, CDCl3): δ (ppm) = 4.05 (s, 3H), 7.03 (s, 1H), 7.47–7.51 (m, 3H), 8.00–8.03 (m, 2H); 13C NMR (100 MHz, CDCl3): δ (ppm) = 54.87, 102.05, 127.36, 129.06, 131.38, 135.62, 160.45, 167.10, 171.87; HRMS (ESI+) m/z calc. for C11H10ON2Cl [M + H]+ 221.0476, found 221.0477; HPLC tR = 7.41 min; Rf = 0.62 (EtOAc/n-Hex, 1:2, v/v).
4-chloro-6-methoxy-2-phenylpyrimidine (44B): Yield: 36%; white solid; Mp 49–51 °C; 1H NMR (400 MHz, CDCl3): δ (ppm) = 4.11 (s, 3H), 6.66 (s, 1H), 7.47–7.54 (m, 3H), 8.42–8.46 (m, 2H); 13C NMR (100 MHz, CDCl3): δ (ppm) = 54.44, 105.24, 128.62, 128.67, 131.59, 136.35, 161.10, 164.79, 170.77; HRMS (ESI+) m/z calc. for C11H10ON2Cl [M + H]+ 221.0476, found 221.0475; HPLC tR = 7.96 min; Rf = 0.65 (EtOAc/n-Hex, 1:2, v/v).
2-chloro-6-phenylpyrimidin-4-amine (45): Yield: 84%; pale yellow solid; Mp 160–162 °C; 1H NMR (400 MHz, CDCl3): δ (ppm) = 5.39 (s, 2H), 6.72 (s, 1H), 7.44–7.48 (m, 3H), 7.94–7.96 (m, 2H); 13C NMR (100 MHz, CDCl3): δ (ppm) = 98.87, 127.20, 128.94, 131.05, 136.12, 161.13, 165.10, 166.03; HRMS (ESI+) m/z calc. for C10H9N3Cl [M + H]+ 206.0480, found 206.0477; HPLC tR = 5.25 min; Rf = 0.43 (EtOAc/n-Hex, 1:1, v/v).
2-chloro-4-phenylquinazoline (46): Yield: 54%; white solid; Mp 91–92 °C (lit. 112–114 °C [41]); 1H NMR (400 MHz, DMSO-d6): δ (ppm) = 7.56–7.61 (m, 3H), 7.63 (ddd, J1 = 1.2 Hz, J2= 6.9 Hz, J3 = 8.3 Hz, 1H), 7.77–7.82 (m, 2H), 7.95 (ddd, J1 = 1.4 Hz, J2 = 6.9 Hz, J3 = 8.4 Hz, 1H), 8.06 (ddd, J1 = 0.7 Hz, J2 = 1.2 Hz, J3 = 8.5 Hz, 1H), 8.14 (ddd, J1 = 0.7 Hz, J2 = 1.4 Hz, J3 = 8.4 Hz, 1H); 13C NMR (100 MHz, DMSO-d6): δ (ppm) = 121.76, 127.64, 128.14, 128.26, 128.90, 130.28, 130.84, 135.02, 136.04, 153.16, 157.18, 171.84; HRMS (ESI+) m/z calc. for C14H10N2Cl [M + H]+ 241.0527, found 241.0523; HPLC tR = 7.08 min; Rf = 0.29 (EtOAc/n-Hex, 1:5, v/v).
4. Conclusions
Substituted pyrimidine rings represent an attractive scaffold, which has been often present in compounds with diverse biological activities. One of the most straightforward routes for preparation of substituted pyrimidines are cross-coupling reactions with Suzuki coupling being among them. In the present study we developed a quick, efficient, and regioselective procedure for preparation of C4-substituted pyrimidines from commercially available 2,4-dichloropyrimidines. A thorough screening of reaction conditions was performed, with the microwave-assisted 15-min protocol offering regioselective C4-pyrimidine substitution in good to high yields. Additionally, with only 0.5 mol% consumption of tetrakis(triphenylphosphine)palladium(0) catalyst, the environmental impact of the reaction is negligible. The scope of the reaction was investigated on several aryl and heteroaryl boronic acids and substituted 2,4-dihalogenopyrimidines, with most of them being readily amenable for Suzuki-Miyaura coupling. We believe that our microwave-assisted procedure bears significant potential for the rapid preparation of regioselective C4-substituted pyrimidines with high yields and most importantly extremely low catalyst loading.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/catal11040439/s1, Additional description of procedure optimization (Solvent screening, Temperature screening, Screening of the solvent volume for microwave-assisted Suzuki coupling between 1 and 2); 1H NMR and 13C NMR spectra of compounds.
Author Contributions
Conceptualization, A.D. and M.S.; methodology, A.D., K.M., and M.S.; validation, A.D. and K.M.; formal analysis, A.D. and K.M.; investigation, A.D., K.M., and M.S..; resources, M.S.; data curation, A.D. and M.S.; writing—original draft preparation, A.D. and M.S.; writing—review and editing, A.D. and M.S.; visualization, M.S.; supervision, M.S.; project administration, M.S.; funding acquisition, A.D. and M.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Slovenian Research Agency (research core funding No. P1-0208 and junior researcher’s programme for Ana Dolšak).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Chiacchio, M.A.; Iannazzo, D.; Romeo, R.; Giofrè, S.V.; Legnani, L. Pyridine and pyrimidine derivatives as privileged scaffolds in biologically active agents. Curr. Med. Chem. 2019, 26, 7166–7195. [Google Scholar] [CrossRef]
- Kumar, S.; Narasimhan, B. Therapeutic potential of heterocyclic pyrimidine scaffolds. Chem. Cent. J. 2018, 12, 38. [Google Scholar] [CrossRef]
- Lagoja, I.M. Pyrimidine as constituent of natural biologically active compounds. Chem. Biodivers. 2005, 2, 1–50. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Deep, A.; Narasimhan, B. A review on synthesis, anticancer and antiviral potentials of pyrimidine derivatives. Curr. Bioact. Compd. 2019, 15, 289–303. [Google Scholar] [CrossRef]
- MacCoss, M.; Robins, M.J. Anticancer pyrimidines, pyrimidine nucleosides and prodrugs. In The Chemistry of Antitumour Agents; Wilman, D.E.V., Ed.; Springer: Dordrecht, The Netherlands, 1990; pp. 261–298. ISBN 978-94-009-0397-5. [Google Scholar]
- Manley, P.W.; Cowan-Jacob, S.W.; Buchdunger, E.; Fabbro, D.; Fendrich, G.; Furet, P.; Meyer, T.; Zimmermann, J. Imatinib: A selective tyrosine kinase inhibitor. Eur. J. Cancer 2002, 38, S19–S27. [Google Scholar] [CrossRef]
- Langtry, H.D.; Campoli-Richards, D.M. Zidovudine. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic efficacy. Drugs 1989, 37, 408–450. [Google Scholar] [CrossRef] [PubMed]
- Brogden, R.N.; Carmine, A.A.; Heel, R.C.; Speight, T.M.; Avery, G.S. Trimethoprim: A review of its antibacterial activity, pharmacokinetics and therapeutic use in urinary tract infections. Drugs 1982, 23, 405–430. [Google Scholar] [CrossRef] [PubMed]
- White, C.M. A review of the pharmacologic and pharmacokinetic aspects of rosuvastatin. J. Clin. Pharmacol. 2002, 42, 963–970. [Google Scholar] [CrossRef]
- Sica, D.A. Minoxidil: An underused vasodilator for resistant or severe hypertension. J. Clin. Hypertens. 2004, 6, 283–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gore, R.P.; Rajput, A.P. A review on recent progress in multicomponent reactions of pyrimidine synthesis. Drug Invent Today 2013, 5, 148–152. [Google Scholar] [CrossRef]
- Mahfoudh, M.; Abderrahim, R.; Leclerc, E.; Campagne, J.-M. Recent approaches to the synthesis of pyrimidine derivatives. Eur. J. Org. Chem. 2017, 2017, 2856–2865. [Google Scholar] [CrossRef]
- Maji, P.K. Recent progress in the synthesis of pyrimidine heterocycles: A review. Curr. Org. Chem. 2020, 24, 1055–1096. [Google Scholar] [CrossRef]
- Roopan, S.M.; Sompalle, R. Synthetic chemistry of pyrimidines and fused pyrimidines: A review. Synth. Commun. 2016, 46, 645–672. [Google Scholar] [CrossRef]
- Eicher, T.; Hauptmann, S.; Speicher, A. Six-Membered Heterocycles: Sections 6.25–6.31. In The Chemistry of Heterocycles; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2003; pp. 381–416. ISBN 978-3-527-60183-7. [Google Scholar]
- Joule, J.A.; Mills, K. Heterocyclic Chemistry, 5th ed.; Wiley-Blackwell: Hoboken, NJ, USA, 2010; ISBN 978-1-4051-3300-5. [Google Scholar]
- Parks, E.L.; Sandford, G.; Christopher, J.A.; Miller, D.D. Perhalogenated pyrimidine scaffolds. Reactions of 5-chloro-2,4,6-trifluoropyrimidine with nitrogen centred nucleophiles. Beilstein J. Org. Chem. 2008, 4, 22. [Google Scholar] [CrossRef] [Green Version]
- Carver, D.R.; Komin, A.P.; Hubbard, J.S.; Wolfe, J.F. SRN1 mechanism in heteroaromatic nucleophilic substitution. reactions involving halogenated pyrimidines, pyridazines, and pyrazines. J. Org. Chem. 1981, 46, 294–299. [Google Scholar] [CrossRef]
- Delia, T.J. Grignard reactions involving halogenated pyrimidines. J. Heterocycl. Chem. 2013, 50, 735–745. [Google Scholar] [CrossRef]
- Schomaker, J.M.; Delia, T.J. Arylation of halogenated pyrimidines via a Suzuki coupling reaction. J. Org. Chem. 2001, 66, 7125–7128. [Google Scholar] [CrossRef]
- Anderson, S.C.; Handy, S.T. One-pot double Suzuki couplings of dichloropyrimidines. Synthesis 2010, 2010, 2721–2724. [Google Scholar] [CrossRef] [Green Version]
- Schröter, S.; Stock, C.; Bach, T. Regioselective cross-coupling reactions of multiple halogenated nitrogen-, oxygen-, and sulfur-containing heterocycles. Tetrahedron 2005, 61, 2245–2267. [Google Scholar] [CrossRef]
- Large, J.M.; Clarke, M.; Williamson, D.M.; McDonald, E.; Collins, I. Synthesis of trisubstituted pyrimidines by regioselective SNAr and Suzuki reactions of polyhalopyrimidines. Synlett 2006, 2006, 861–864. [Google Scholar] [CrossRef] [Green Version]
- Almond-Thynne, J.; Blakemore, D.C.; Pryde, D.C.; Spivey, A.C. Site-selective Suzuki–Miyaura coupling of heteroaryl halides—Understanding the trends for pharmaceutically important classes. Chem. Sci. 2016, 8, 40–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benneche, T.; Oscarson, S.; Kvarnström, I.; Niklasson, A.; Niklasson, G.; Svensson, S.C.T.; Edwards, J.V. Pyrimidinylpalladium(II) complexes in the synthesis of alkenylpyrimidines. Acta Chem. Scand. 1990, 44, 927–931. [Google Scholar] [CrossRef]
- Tan, J.; Chang, J.; Deng, M. The facile route to stereodefined alkenyl-substituted pyrimidines. Synth. Commun. 2004, 34, 3773–3783. [Google Scholar] [CrossRef]
- CRESTOR® (Rosuvastatin Calcium) Prescribing Information|AZ Medical Information. Available online: https://medicalinformation.astrazeneca-us.com/home/prescribing-information/crestor-pi.html (accessed on 1 December 2020).
- McMullin, C.L.; Fey, N.; Harvey, J.N. Computed ligand effects on the oxidative addition of phenyl halides to phosphine supported palladium(0) catalysts. Dalton Trans. 2014, 43, 13545–13556. [Google Scholar] [CrossRef] [PubMed]
- Yaman, T.; Harvey, J.N. Suzuki–Miyaura coupling revisited: An integrated computational study. Faraday Discuss. 2019, 220, 425–442. [Google Scholar] [CrossRef] [PubMed]
- Miyaura, N.; Suzuki, A. Palladium-catalyzed cross-coupling reactions of organoboron compounds. Chem. Rev. 1995, 95, 2457–2483. [Google Scholar] [CrossRef] [Green Version]
- Sherwood, J.; Clark, J.H.; Fairlamb, I.J.S.; Slattery, J.M. Solvent effects in palladium catalysed cross-coupling reactions. Green Chem. 2019, 21, 2164–2213. [Google Scholar] [CrossRef] [Green Version]
- Liu, N.; Liu, C.; Jin, Z. An efficient protocol for a Pd(OAc)2-catalyzed ligand-free Suzuki reaction in toluene. Chin. J. Catal. 2010, 31, 1316–1320. [Google Scholar] [CrossRef]
- Sun, J.; Jin, Z.; Xie, N.; Wang, H.; Gao, H. Studies on Suzuki coupling reactions of large sterically hindered substrates. Top. Chem. Mater. Eng. 2018, 1, 24–26. [Google Scholar] [CrossRef]
- Adrio, L.A.; Nguyen, B.N.; Guilera, G.; Livingston, A.G.; Hii, K.K. Speciation of Pd(OAac)2 in ligandless Suzuki–Miyaura reactions. Catal. Sci. Technol. 2012, 2, 316–323. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharyya, S.; Basu, B. Solid-Supported Catalysis. In Green Techniques for Organic Synthesis and Medicinal Chemistry; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2018; pp. 269–289. ISBN 978-1-119-28815-2. [Google Scholar]
- Yakukhnov, S.A.; Pentsak, E.O.; Galkin, K.I.; Mironenko, R.M.; Drozdov, V.A.; Likholobov, V.A.; Ananikov, V.P. Rapid “mix-and-stir” preparation of well-defined palladium on carbon catalysts for efficient practical use. ChemCatChem 2018, 10, 1869–1873. [Google Scholar] [CrossRef]
- Trzeciak, A.M.; Wojcik, P.; Lisiecki, R.; Gerasymchuk, Y.; Strek, W.; Legendziewicz, J. Palladium nanoparticles supported on graphene oxide as catalysts for the synthesis of diarylketones. Catalysts 2019, 9, 319. [Google Scholar] [CrossRef] [Green Version]
- Zheng, K.; Shen, C.; Qiao, J.; Tong, J.; Jin, J.; Zhang, P. Novel magnetically-recyclable, nitrogen-doped Fe3O4@Pd NPs for Suzuki–Miyaura coupling and their application in the synthesis of Crizotinib. Catalysts 2018, 8, 443. [Google Scholar] [CrossRef] [Green Version]
- Blakemore, D. Chapter 1: Suzuki–Miyaura coupling. In Synthetic Methods in Drug Discovery: Volume 1; The Royal Society of Chemistry: London, UK, 2015; pp. 1–69. [Google Scholar]
- Gravil, P.A.; Toulhoat, H. Hydrogen, sulphur and chlorine coadsorption on Pd(111): A theoretical study of poisoning and promotion. Surf. Sci. 1999, 430, 176–191. [Google Scholar] [CrossRef]
- Harden, D.B.; Mokrosz, M.J.; Strekowski, L. Addition and substitution reactions of chloropyrimidines with lithium reagents. J. Org. Chem. 1988, 53, 4137–4140. [Google Scholar] [CrossRef]
- Plas, H.C.V.D.; Haase, B.; Zuurdeeg, B.; Vollering, M.C. Ring transformations in reactions of heterocyclic halogeno compounds with nucleophiles (V): Conversion of some 2-alkyl-, 2-aryl- and 2,5-dialkyl-4-chloropyrimidines by potassium amide in liquid ammonia into 4-alkyl derivatives of 2-alkyl- or 2-aryl-s-triazines. Recl. Trav. Chim. Pays-Bas 1966, 85, 1101–1113. [Google Scholar] [CrossRef]
- Czudor, Z.; Balogh, M.; Bánhegyi, P.; Boros, S.; Breza, N.; Dobos, J.; Fábián, M.; Horváth, Z.; Illyés, E.; Markó, P.; et al. Novel compounds with potent CDK9 inhibitory activity for the treatment of myeloma. Bioorg. Med. Chem. Lett. 2018, 28, 769–773. [Google Scholar] [CrossRef]
- Cuccia, S.J.; Fleming, L.B.; France, D.J. A novel and efficient synthesis of 4-phenyl-2-chloropyrimidines from acetophenone cyanoimines. Synth. Commun. 2002, 32, 3011–3018. [Google Scholar] [CrossRef]
- Shu, L.; Chen, C.; Huan, X.; Huang, H.; Wang, M.; Zhang, J.; Yan, Y.; Liu, J.; Zhang, T.; Zhang, D. Design, synthesis, and pharmacological evaluation of 4- or 6-phenyl-pyrimidine derivatives as novel and selective Janus kinase 3 inhibitors. Eur. J. Med. Chem. 2020, 191, 112148. [Google Scholar] [CrossRef]
- Kemp, M.I.; Luckhurst, C.A.; Thompson, P.W. Substituted Cyanopyrrolidines with Activity as Usp30 Inhibitors. WO 2020/212351 A1, 14 April 2020. [Google Scholar]
- William, A.D.; Lee, A.C.-H.; Blanchard, S.; Poulsen, A.; Teo, E.L.; Nagaraj, H.; Tan, E.; Chen, D.; Williams, M.; Sun, E.T.; et al. Discovery of the macrocycle 11-(2-pyrrolidin-1-yl-ethoxy)-14,19-dioxa-5,7,26-triaza-tetracyclo[1 9.3.1.1(2,6).1(8,12)]heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene (SB1518), a potent Janus kinase 2/fms-like tyrosine kinase-3 (JAK2/FLT3) inhibitor for the treatment of myelofibrosis and lymphoma. J. Med. Chem. 2011, 54, 4638–4658. [Google Scholar] [CrossRef] [PubMed]
- Ganesamoorthy, S.; Muthu Tamizh, M.; Shanmugasundaram, K.; Karvembu, R. A sustainable heterogenized palladium catalyst for Suzuki-Miyaura cross coupling reaction of azaheteroaryl halides in aqueous media. J. Organomet. Chem. 2018, 862, 76–85. [Google Scholar] [CrossRef]
- Stanetty, P.; Röhrling, J.; Schnürch, M.; Mihovilovic, M.D. Synthesis of analogs of the phenylamino-pyrimidine type protein kinase C inhibitor CGP 60474 utilizing a Negishi cross-coupling strategy. Tetrahedron 2006, 62, 2380–2387. [Google Scholar] [CrossRef]
- Pelletier, J.C.; Lundquist, J.T.; Gilbert, A.M.; Alon, N.; Bex, F.J.; Bhat, B.M.; Bursavich, M.G.; Coleburn, V.E.; Felix, L.A.; Green, D.M.; et al. (1-(4-(Naphthalen-2-yl)pyrimidin-2-yl)piperidin-4-yl)methanamine: A wingless β-catenin agonist that increases bone formation rate. J. Med. Chem. 2009, 52, 6962–6965. [Google Scholar] [CrossRef] [PubMed]
- Bello, A.M.; Bende, T.; Wei, L.; Wang, X.; Majchrzak-Kita, B.; Fish, E.N.; Kotra, L.P. De novo design of nonpeptidic compounds targeting the interactions between interferon-α and its cognate cell surface receptor. J. Med. Chem. 2008, 51, 2734–2743. [Google Scholar] [CrossRef] [PubMed]
- Strekowski, L.; Harden, D.B.; Grubb, W.B.; Patterson, S.E.; Czarny, A.; Mokrosz, M.J.; Cegla, M.T.; Wydra, R.L. Synthesis of 2-chloro-4,6-di(heteroaryl)pyrimidines. J. Heterocycl. Chem. 1990, 27, 1393–1400. [Google Scholar] [CrossRef]
- Kamal, R.; Kumar, R.; Kumar, V.; Kumar, V.; Bansal, K.K.; Sharma, P.C. Synthesis, anthelmintic and antimicrobial evaluation of new 2-arylidene-1-(4-methyl-6-phenylpyrimidin-2-yl)hydrazines. ChemistrySelect 2019, 4, 713–717. [Google Scholar] [CrossRef]
Figure 1.
Representative drugs with pyrimidine moiety on the market: anticancer (e.g., imatinib [6]), antiviral (e.g., zidovudine [7]), antibacterial (e.g., trimethoprim [8]), antihyperlipidemic (e.g., rosuvastatin [9,27]), antihypertensive (e.g., minoxidil), and antimalarial (e.g., pyrimethamine).

Scheme 1.
Suzuki coupling of 2,4-dichloropyrimidine (1) with phenylboronic acid (2) as our model reaction.
Scheme 1.
Suzuki coupling of 2,4-dichloropyrimidine (1) with phenylboronic acid (2) as our model reaction.


{kind=link}
{kind=link}
{kind=link}
Table 1.
The isolated yield of product 3 after Suzuki coupling of 2,4-dichloropyrimidine (1) with phenylboronic acid (2) in four selected solvents.
Table 1.
The isolated yield of product 3 after Suzuki coupling of 2,4-dichloropyrimidine (1) with phenylboronic acid (2) in four selected solvents.
| Entry | Solvent | Temperature (°C) | Yield (%) 1,2 |
|---|---|---|---|
| 1 | THF | 60 | 38 |
| 2 | DMF | 100 | 31 |
| 3 | 1,4-dioxane | 100 | 71 |
| 4 | isopropanol | 80 | 52 |
1 Reaction conditions: 1 (149 mg, 1.0 mmol), solvent (7 mL), K2CO3 (415 mg, 3.0 mmol), 2 (122 mg, 1.0 mmol), Pd(PPh3)4 (58 mg, 0.05 mmol, 5 mol%), heating (60, 80 or 100 °C), 24 h. 2 Isolated yield of 3 after extraction and column chromatography.
Table 2.
Catalyst screening for Suzuki coupling of 1 with 2.
| Entry | Catalyst (ligand) | Yield (%) 1,2 | Selectivity Ratio 3:4 3 | ||
|---|---|---|---|---|---|
| 1 h | 2 h | 24 h | |||
| 1 | Pd2dba3 | 0 | 0 | 2 | / |
| 2 | Pd2dba3 · CHCl3 | 15 | 21 | 35 | 5 |
| 3 | Pd2dba3 (TTBP · HBF4) | 10 | 11 | 23 | 11.5 |
| 4 | PdCl2(PPh3)2 | 12 | 17 | 36 | 4.5 |
| 5 | Pd(OAc)2 (PCy3)2 | 10 | 12 | 26 | 13 |
| 6 | Pd(OAc)2 (PPh3)2 | 28 | 37 | 46 | 10 |
| 7 | Pd(OAc)2 (PPh3 on DVB)2 | 15 | 22 | 27 | 7 |
| 8 | Pd(dppf)Cl2 · CH2Cl2 | 26 | 38 | 70 (63) 4 | 10 |
| 9 | Pd(PPh3)4 | 29 | 41 | 72 (71) 4 | 9 |
1 Reaction conditions: 1 (75 mg, 0.50 mmol), 1,4-dioxane (3.5 mL), K2CO3 (207 mg, 1.50 mmol), 2 (61 mg, 0.50 mmol), 5 mol% of a catalyst (10 mol% of a ligand), 100 °C, 1–24 h. 2 Determined by LC-MS. 3 Determined by LC-MS after 24 h reaction time. 4 Isolated yield of 3 after extraction and column chromatography.
Table 3.
Solvent mixture screening for Suzuki coupling of 1 with 2.
| Entry | Non-Polar Solvent | Polar Solvent | Ratio Non-Polar vs. Polar Solvent (v/v) | Yield (%) 1,2 |
|---|---|---|---|---|
| 1 | THF | H2O | 1:1 | 53 |
| 2 | THF | H2O | 2:1 | 23 |
| 3 | THF | H2O | 3:1 | 20 |
| 4 | THF | H2O | 5:1 | 13 |
| 5 | THF | H2O | 9:1 | 14 |
| 6 | THF | / | / | 21 |
| 7 | 1,4-dioxane | H2O | 1:1 | 45 |
| 8 | 1,4-dioxane | H2O | 2:1 | 80 |
| 9 | 1,4-dioxane | H2O | 3:1 | 59 |
| 10 | 1,4-dioxane | H2O | 5:1 | 58 |
| 11 | 1,4-dioxane | H2O | 9:1 | 24 |
| 12 | 1,4-dioxane | / | / | 21 |
1 Reaction conditions: 1 (75 mg, 0.50 mmol), solvent(s) (6 mL), K2CO3 (207 mg, 1.50 mmol), 2 (61 mg, 0.50 mmol), Pd(PPh3)4 (17.3 mg, 0.015 mmol, 3 mol%), MW, 100 °C, 20 min. 2 Yield for 3, determined by LC-MS.
Table 4.
Temperature and time screening for Suzuki coupling of 1 with 2.
| Entry | Temperature [°C] | Time [min] | Yield (%) 1,2 |
|---|---|---|---|
| 1 | 60 | 20 | 12 |
| 2 | 80 | 20 | 60 |
| 3 | 100 | 20 | 80 |
| 4 | 100 | 15 | 81 |
| 5 | 100 | 10 | 59 |
| 6 | 100 | 5 | 46 |
| 7 | 120 | 20 | 60 |
| 8 | 120 | 10 | 61 |
| 9 | 120 | 5 | 66 |
| 10 | 140 | 20 | 56 |
| 11 | 140 | 5 | 61 |
1 Reaction conditions: 1 (75 mg, 0.50 mmol), 1,4-dioxane (4 mL), H2O (2 mL), K2CO3 (207 mg, 1.50 mmol), 2 (61 mg, 0.50 mmol), Pd(PPh3)4 (17.3 mg, 0.015 mmol, 3 mol%), MW, temperature, time. 2 Yield for 3, determined by LC-MS.
Table 5.
Screening of Pd(PPh3)4 loading for Suzuki coupling of 1 with 2.
| Entry | Catalyst loading (mol%) | Yield (%) 1,2 |
|---|---|---|
| 1 | 5 | 68 |
| 2 | 3 | 65 |
| 3 | 2 | 59 |
| 4 | 1 | 62 |
| 5 | 0.5 | 71 |
| 6 | 0.2 | 61 |
| 7 | 0.05 | 48 |
| 8 | without catalyst | 0 |
1 Reaction conditions: 1 (75 mg, 0.50 mmol), 1,4-dioxane (4 mL), H2O (2 mL), K2CO3 (207 mg, 1.50 mmol), 2 (61 mg, 0.50 mmol), Pd(PPh3)4 (0–5 mol%), MW, 100 °C, 15 min. 2 Determined by LC-MS.
Table 6.
Microwave-assisted Pd(PPh3)4-catalyzed Suzuki coupling of aryl and heteroaryl boronic acids with 1.
Table 6.
Microwave-assisted Pd(PPh3)4-catalyzed Suzuki coupling of aryl and heteroaryl boronic acids with 1.
 |
 |
 |
 |
Reaction conditions: 1 (75 mg, 0.50 mmol), 1,4-dioxane (4 mL), H2O (2 mL), K2CO3 (207 mg, 1.50 mmol), boronic acid (0.50 mmol), Pd(PPh3)4 (2.9 mg, 0.0025 mmol, 0.5 mol%), MW, 100 °C, 15 min. Isolated yields after extraction and column chromatography are presented.
Table 7.
Microwave-assisted Pd(PPh3)4-catalyzed Suzuki coupling of 2,4-dihalogenopyrimidines with 2.
Table 7.
Microwave-assisted Pd(PPh3)4-catalyzed Suzuki coupling of 2,4-dihalogenopyrimidines with 2.
 |
 |
 |
Reaction conditions: 2,4-dihalogenopyrimidine (0.50 mmol), 1,4-dioxane (4 mL), H2O (2 mL), K2CO3 (207 mg, 1.50 mmol), 2 (61 mg, 0.50 mmol), Pd(PPh3)4 (2.9 mg, 0.0025 mmol, 0.5 mol%), MW, 100 °C, 15 min. Isolated yields after extraction and column chromatography are presented.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Dolšak, A.; Mrgole, K.; Sova, M. Microwave-Assisted Regioselective Suzuki Coupling of 2,4-Dichloropyrimidines with Aryl and Heteroaryl Boronic Acids. Catalysts 2021, 11, 439. https://doi.org/10.3390/catal11040439
AMA Style
Dolšak A, Mrgole K, Sova M. Microwave-Assisted Regioselective Suzuki Coupling of 2,4-Dichloropyrimidines with Aryl and Heteroaryl Boronic Acids. Catalysts. 2021; 11(4):439. https://doi.org/10.3390/catal11040439
Chicago/Turabian StyleDolšak, Ana, Kristjan Mrgole, and Matej Sova. 2021. "Microwave-Assisted Regioselective Suzuki Coupling of 2,4-Dichloropyrimidines with Aryl and Heteroaryl Boronic Acids" Catalysts 11, no. 4: 439. https://doi.org/10.3390/catal11040439
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.