
Genetics of Heritable Thoracic Aortic Disease


1
Unit of Inherited and Rare Cardiovascular Diseases, Onassis Cardiac Surgery Center, Siggrou Avenue 356, Kallithea, 17674 Athens, Greece
2
Molecular and Clinical Sciences Research Institute, St George’s University of London, London SW17 0RE, UK
3
Greek National Network for Precision Medicine in Cardiology and Prevention of Sudden Cardiac Death in the Young, 17674 Kallithea, Greece
4
Molecular Immunopathology and Histocompatibility Unit, Division of Genetics, Onassis Cardiac Surgery Center, 17674 Athens, Greece
*
Author to whom correspondence should be addressed.
Cardiogenetics 2022, 12(1), 63-79; https://doi.org/10.3390/cardiogenetics12010006
Submission received: 27 December 2021
/
Revised: 24 January 2022
/
Accepted: 31 January 2022
/
Published: 4 February 2022
(This article belongs to the Special Issue Cardiogenetics: Feature Papers 2021)
Abstract
:Genetic testing plays an increasing diagnostic and prognostic role in the management of patients with heritable thoracic aortic disease (HTAD). The identification of a specific variant can establish or confirm the diagnosis of syndromic HTAD, dictate extensive evaluation of the arterial tree in HTAD with known distal vasculature involvement and justify closer follow-up and earlier surgical intervention in HTAD with high risk of dissection of minimal or normal aortic size. Evolving phenotype–genotype correlations lead us towards more precise and individualized management and treatment of patients with HTAD. In this review, we present the latest evidence regarding the role of genetics in patients with HTAD.
1. Introduction
Over the last two decades, genetic developments have significantly improved our understanding of heritable thoracic aortic disease (HTAD). The identification of new syndromes [1] and novel candidate genes [2] has changed the paradigm in the diagnostic evaluation of these patients. Specific genotype–phenotype correlations continue to emerge, promising a more precise and effective approach in the treatment of HTAD [3,4,5]. The goal of this review is to inform clinicians about the value and effect of genetics in the diagnosis, management, surveillance, risk stratification and familial evaluation of patients with HTAD.
1.1. Classification
The presence of syndromic systemic features and a positive family history of aortic aneurysm or dissection are the key elements that determine the classification and thus the management of patients with HTAD. Thoracic aortic disease at a younger age occurs more often in the context of a genetic syndrome.
Syndromic HTAD (sHTAD) typically exhibits a multiorgan phenotype and is caused by genetic variants that are involved in the transforming growth factor-β (TGF-β) pathway and genes encoding extracellular matrix proteins [2]. Nonsyndromic HTAD (nsHTAD) is typically characterized by isolated thoracic aortic aneurysm or dissection, without any recognizable systemic features, and can be familial in up to 20–25% of cases. A genetic defect, mainly in genes of the contractile apparatus, may be identified in up to 20% of familial nsHTAD [6].
1.2. Diagnostic Workup
Only 5% of patients present with alarming symptoms before an acute aortic event [7]. Most patients are usually diagnosed following a major complication, e.g., an aortic dissection, as part of familial evaluation, or based on characteristic physical findings suggestive of a specific syndrome.
Physical examination and history are vital in the assessment of patients with HTAD. The physician should be able to recognize any systemic features such as specific facial characteristics, skin lesions or skeletal manifestations, which suggest the presence of sHTAD. A detailed personal history should be obtained including history of recurrent pneumothorax, history of eye operations or ocular conditions. Ophthalmology evaluation, including slit-lamp examination, should be offered in all patients with a suspicion of Marfan syndrome (MFS). The reduced penetrance or incomplete expression and the phenotypic overlap and variability of hereditary aortopathy consist of major challenges, which make essential a multidisciplinary diagnostic approach.
2. Syndromic HTAD
2.1. Marfan Syndrome
Marfan syndrome (MFS; Online Mendelian Inheritance in Man, OMIM®. McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University, Baltimore, USA OMIM #154700, Orphanet rare disease nomenclature, French National Institute for Health and Medical Research, Paris, France ORPHA:558) is the most common syndromic aortopathy. It is characterized by aortic dilatation, ectopia lentis and skeletal abnormalities. MFS is associated with variants in the FBN1 gene, encoding the extracellular matrix protein called fibrillin-1 [8]. Other genes, typically not causing any ocular involvement, also can lead to a phenotype resembling MFS [2].
The diagnosis of MFS is based on the revised 2010 Ghent criteria [8]. A pathogenic FBN1 variant, along with ectopia lentis or enlarged aortic root (Z score ≥2 that corresponds to a diameter ≥2 standard deviations above normal, according to established aortic nomograms [9]), establishes the diagnosis. In the absence of a pathogenic FBN1 variant, the diagnosis can be made in patients with aortic dilatation (Z score ≥2) and ectopia lentis or a systemic score ≥7 (encompassing systemic features suggestive of MFS) [8]. In patients with a family history of MFS, the diagnosis can be made in the presence of ≥1 of the following criteria: (a) ectopia lentis, (b) systemic score ≥7, (c) aortic root enlargement with Z-score ≥2 in patients >20 years and (d) Z score ≥3 in patients <20 years [8].
Over half of MFS patients are diagnosed after adolescence (median age of diagnosis is 19 years) [10], and the first cardiovascular procedure occurs at average at the age of 36 years [11]. Aortic dilatation typically occurs at the level of the sinuses of Valsalva (SoV), but aortic dilatation or dissection can occur at every level of the aorta [12]. Most patients with MFS are diagnosed before severe cardiac complications occur. This is particularly relevant, as MFS patients appear to have better survival rates with appropriate medical management and when undergoing prophylactic elective surgery (complication rate of 1.5% vs. 11.7% of urgent procedures) [13]. Established clinical factors of high risk are: (a) aortic diameter at the SoV ≥5 cm, (b) rapid increase in aortic dilatation (≥3 mm per year), (c) family history of aortic dissection at a low aortic size, (d) progressive aortic regurgitation, (e) personal history of spontaneous vascular dissection and (f) desire for pregnancy [14,15]. Therapy is based on optimal blood pressure control, and medical management includes beta-blockers or angiotensin-1 antagonists (losartan) [14,16,17].
Most FBN1 variants are missense, having in most cases a dominant-negative effect (DN-FBN1), resulting in a disorganized extracellular matrix incorporating both mutated and nonmutated fibrillin-1 proteins. Haploinsufficient FBN1 variants (HI-FBN1), leading to reduced production of normal fibrillin-1 protein, are also well documented in up to 35% of MFS patients [5]. It has been reported that patients with HI-FBN1 are at increased risk of aortic dissection and death, have more rapid aortic root and ascending aorta dilation rates, manifest more severe cardiovascular complications and respond better to losartan therapy than patients with DN-FBN1 variants [5,18,19,20,21]. In a large cohort of MFS patients, premature termination codon variants leading to haploinsufficiency were associated with an 83% lifelong aortic event risk during life, shorter life expectancy, severe scoliosis and relatively lower rates of ectopia lentis surgery [22].
Specific DN-FBN1 variants have been linked with a similarly severe phenotype compared to HI-FBN1 variants. MFS patients with in-frame DN-FBN1 variants leading to a cysteine loss at the level of fibrillin-1 showed 73% lifelong risk of aortic dissection or surgery, high rates of severe scoliosis and ectopia lentis surgery. In-frame variants leading to a neutral cysteine effect were associated with an intermediate phenotype (61% of lifelong aortic event risk). In-frame variants leading to cysteine gain were associated with a better cardiovascular profile (29% of lifelong aortic event risk) and lower rates of severe scoliosis but high risk for ectopia lentis surgery [22]. Takeda et al. identified deleterious high-risk variants among DN-FBN1 Japanese patients, in-specific variants affecting or creating cysteine residues and in-frame deletion variants in exons 25–36 and 43–49 [19].
Furthermore, despite a greater frequency of surgery and type B aortic dissections in MFS patients harboring HI-FBN1 variants, all type A dissections that occurred at an aortic root diameter <50 mm were DN-FBN1 variants in a cohort of 954 MFS patients followed for a mean 9.1 years [23].
2.2. Loeys–Dietz Syndrome
Loeys–Dietz syndrome (LDS, ORPHA:60030) is characterized by the combination of arterial tortuosity with ascending aortic aneurysm/dissection, also involving the distal aorta and branching arteries, hypertelorism and bifid uvula or cleft palate. It was first described in 2005 as a novel autosomal dominant syndromic aortopathy [1]. Since 2005, loss-of-function variants in six genes have been linked to LDS (TGFBR1, TGFBR2, SMAD2, SMAD3, TGFB2 and TGFB3), all of which are involved in the TGF-β signaling pathway [4].
LDS has been originally associated with a very aggressive natural history, probably reflecting a selection bias in the first series of patients. Aortic dissection in young patients (mean age at death of 26 years), very high incidence of pregnancy-related complications and aortic dissections at only mildly increased or even much normal aortic dimensions have been reported [1]. Current American Heart Association/American College of Cardiology guidelines suggest an aggressive approach with prophylactic surgery in aortic size ≥42 mm in all patients with LDS [24]. Recent European Society of Cardiology (ESC) guidelines suggest surgery in patients with TGFBR1 or TGFBR2 pathogenic variants with maximal aortic sinus diameter ≥45 mm [15]. Although there is no scientific evidence published to date, drug treatment with beta-blockers and/or angiotensin blockade and optimal antihypertensive management is thought to improve prognosis in LDS patients in a similar fashion to MFS patients. Extensive and distal vascular involvement in LDS patients warrants regular and more extensive imaging of the arterial tree (from head to pelvis).
2.2.1. LDS Caused by Pathogenic Variants in TGFBR1 (OMIM #609192) and in TGFBR2 (OMIM #610168)
Most recent data from the biggest cohort to date, consisting of 441 patients with TGFBR1 and TGFBR2 variants from 228 families, showed a relatively more favorable overall clinical profile than previously reported and provided very important genotype–phenotype information [4]. No differences in survival or prevalence of syndromic characteristics between TGFBR1 and TGFBR2 carriers were identified.
The investigators identified a subgroup of female patients with a TGFBR2 variant, marked systemic features and low body surface area who exhibited aortic dissection in aortic sizes <45 mm. Hypertelorism, aortic tortuosity and wide scars were significantly associated with aortic dissection in this cohort and were present in all women with aortic dissection and minimal aortic enlargement or pregnancy-related dissections. In a considerable percentage of patients that had surgery for an aortic root aneurysm (10%), dissection of the ascending aorta occurred during follow-up. Therefore, in patients with TGFBR1 or TGBR2 variants, replacement of both the aortic root and the ascending aorta when an indication for surgery exists should be considered.
2.2.2. LDS Caused by Pathogenic Variants in SMAD3 (OMIM #613795)
Variants in SMAD3 cause a type of LDS also known as aneurysm–osteoarthritis syndrome, which is characterized by arterial tortuosity, aneurysms and dissection, as well as early-onset osteoarthritis [25,26,27]. Aside from osteoarthritis-related symptoms, which often may be missed if there is no high clinical suspicion [6], patients with SMAD3 variants present with fewer and milder syndromic features compared to the other types of LDS or MFS. This possibly leads to a belated diagnosis, with the majority of SMAD3 variant carriers presenting with type A dissections.
In contrast to TGFBR1 and TGFBR2, patients with SMAD3 variants show a relatively later onset of dilatation or dissection. Aortic events are extremely rare in children or adolescents [3,28]. A recent study indicated that missense variants in the region encoding the MAD homology 2 (MH2) domain may lead to a lower median age of the first aortic event compared with patients with haploinsufficient variants [3]. Aortic dissections typically occur with prior aortic root enlargement. In the largest series of patients with SMAD3 variants to date, dissections occurred at root diameter from 43 mm to 66 mm, with the majority of dissections or elective surgical repair happening in aortic root diameters of 50 mm or greater [3].
2.2.3. LDS Caused by Pathogenic Variants in TGFB2 (OMIM #614816) and SMAD2 or TGFB3 (OMIM #615582)
These variants are found in 10–15% of LDS cases and typically show milder phenotypes and reduced penetrance [27,29]. Aortic dissection in lower-than-standard surgical thresholds of 50 mm has been reported in LDS patients with TGFB2 variants [30], indicating a possible intermediate risk. A more conservative approach regarding aortic surgery following standard aortic size thresholds is reasonable in patients with SMAD2 or TGFB3 variants until new data emerge [2].
2.3. Rare Syndromic HTAD
2.3.1. Vascular Ehlers–Danlos Syndrome (vEDS; OMIM #130050)
It is a rare autosomal dominant syndromic HTAD caused by genetic defects in the COL3A1 gene. Rarely, vEDS can be caused by specific arginine-to-cysteine substitution variants in the COL1A1 gene. The syndrome is characterized by arterial, uterine or bowel ruptures, skin translucency with visible veins and easy bruising and characteristic facial features (thin pinched nose, prominent eyes and lobeless ears, lack of subcutaneous fat). Diagnosis is established using the 2017 International Classification of the Ehlers–Danlos syndrome [31], which updated the earlier nosology of Villefranche [32]. Men seem to have a poorer prognosis than women (median survival age of 46 ± 1.8 years vs. 54 ± 2.5 years) [33]. Surveillance may include periodic arterial screening.
Genetic testing is highly specific and sensitive for vEDS, revealing a genetic defect in 95% of cases [34]. Lethal arterial events in classic (nonvascular) Ehlers–Danlos syndrome (EDS) caused by COL5A1 or COL1A1 variants have also been reported [35]. Identification of a pathogenic variant establishes the diagnosis [32]. The clinical phenotype and prognosis of vEDS may be influenced by the type of COL3A1 variant. Patients heterozygous for “null” COL3A1 variants, leading to loss of the stable mRNA from one COL3A1 allele, show late-onset disease, reduced penetrance, solely vascular events and longest survival compared to missense and splicing variants [33,36,37,38]. Glycine substitutions, splice-site and in-frame insertions/deletions bear the poorer prognosis leading to earlier complications [39].
Although aortic dissection can occur at normal aortic sizes in up to 33% of patients [40], aortic surgery is not usually recommended due to the high rate of intraoperative mortality caused by extreme fragility of the vessel walls. Surgery is usually performed urgently to treat potentially life-threatening complications. Endovascular repair with coil embolization has shown promising results in selected cases of ruptured pseudoaneurysms, visceral aneurysms and carotid-cavernous fistulas. A multicenter, randomized and blinded open trial study showed significantly lower arterial events (rupture or dissection) in vEDS patients receiving celiprolol, a β(1)-adrenoceptor antagonist with a β(2)-adrenoceptor agonist action, compared to controls [41]. Encouraging reports from animal models and an observational study in favor of celiprolol have been published since; however, no randomized prospective trials exist to date [38,42].
2.3.2. Meester–Loeys Syndrome (MRLS; OMIM #300989)
Loss-of-function variants in the X-linked biglycan gene (BGN) have been described in five families with syndromic features overlapping with those of LDS and MFS patients. It is characterized by early-onset aneurysms of the aortic root or ascending aorta (as early as age 1) and aortic dissection (earliest at the age of 15 at an aortic size of 45 mm at the SoV) in male probands. Distal aneurysms in the brain have been detected in one patient. Female patients showed a relatively milder phenotype [43].
2.3.3. Filamin A-Related HTAD
Pathogenic variants in the X-linked filamin A (FLNA) gene, encoding an actin-binding protein that regulates the cytoskeleton and cell motility, cause the brain malformation periventricular heterotopia (PVNH; OMIM #300049, ORPHA:82004), which may also occur in association with EDS features [44]. Neurological symptoms include mainly seizures and dyslexia. Chen et al. reported on the largest series to date of 114 patients, with loss-of-function FLNA pathogenic variants and found aortic dilatation in 18.4% of the patients [45]. Aortic rupture occurred in a 41-year-old male patient at an aortic root size of 42 mm. Pulmonary artery dilatation and aneurysms of other vessels (in the subclavian, middle cerebral and internal carotid arteries, as well as in the abdominal aorta) were common.
2.3.4. LOX-Related HTAD
Loss-of-function variants in the LOX gene, encoding a lysyl oxidase involved in the remodeling of the extracellular matrix, have been shown to predispose to aortic root and fusiform aneurysms, involving both the aortic root and ascending aorta [46,47]. These patients exhibit some overlapping syndromic MFS features, without, however, fulfilling the Ghent criteria. No cases of aortic dissection in minimal aortic dimensions have been reported to date. Presence of a bicuspid aortic valve (BAV) has been found in up to 15% of LOX carriers [46].
2.3.5. Gorlin–Chaudhry–Moss Syndrome (GCMS; OMIM #612289, ORPHA:2095)
Recently, four case reports of aortic dilatation have been reported in patients with GCMS, including a 45-year-old female patient who presented with aortic dissection at an aortic size of 51 mm [48,49,50]. The syndrome is caused by variants in the SLC25A24 gene and is characterized by craniofacial dysostosis, hypertrichosis, underdeveloped genitalia, ocular and dental anomalies.
2.3.6. Shprintzen–Goldberg Craniosynostosis Syndrome (SGS; OMIM #182212, ORPHA:2462)
Pathogenic variants in the SKI gene, coding a protein that regulates the TGF-β signaling pathway, cause a hereditary syndromic aortopathy, phenotypically overlapping with MFS and LDS, characterized by craniosynostosis, marfanoid habitus, intellectual disability, camptodactyly and typical facial dysmorphism [51]. Infantile hypotonia, early developmental delay and intellectual disability are distinct features of the syndrome. Aortic dilatation is generally restricted to the aortic root, and a more benign course than MFS and LDS has been described [52].
2.3.7. Arterial Tortuosity Syndrome (ATORS; OMIM #208050, ORPHA:3342)
Pathogenic variants in the SLC2A10 gene, coding the protein GLUT10 that regulates the TGF-β signaling pathway, lead to ATORS. It is characterized by widespread arterial involvement with elongation, tortuosity and aneurysms of the large and middle-sized arteries along with craniofacial, skin and ocular manifestations [53]. Cardiovascular findings also include aortic coarctation, abnormal implantation of the aortic branches, pulmonary stenosis and aortic stenosis. Patients with ATORS are at higher risk of ischemic events. Aggressive aortic root aneurysm formation even in infancy and aortic dimensions up to 60 mm has been reported; however, there have been no reports of aortic dissection to date [54].
2.3.8. LTBP3-Related HTAD
The LTBP3 gene encodes an extracellular matrix protein regulating the TGF-β signaling pathway in a latent state. Homozygous loss-of-function pathogenic variants in the LTBP3 gene have been associated with dental anomalies and short stature (DASS) syndrome (OMIM #601216). Guo et al. reported segregation of compound heterozygous or homozygous LTBP3 variants in two families with DASS and thoracic aortic disease [55]. The affected individuals also manifested arterial involvement, including abdominal aortic aneurysms and multiple visceral and peripheral arterial aneurysms as well as mitral valve prolapse. Zhu et al. recently reported another compound heterozygous patient with DASS and aortic dissection at the age of 42 and an aortic sinus size of 53 mm [56].
Monoallelic LTBP3 variants appear to be involved in nsHTAD [55,56]. Guo et al. showed that relatives with heterozygous LTBP3 variants presented with late-onset aortic aneurysms and dissection without any systemic features. Additionally, heterozygous LTBP3 variants were identified in 9 out of 338 patients, with thoracic aortic dissection at <56 years of age and no family history or syndromic features [55]. Zhu et al., investigating a cohort of 266 Asian patients with thoracic aortic dissection and/or aneurysm, detected 4 patients with heterozygous LTBP3 variants who had experienced aortic dissections at 33–52 years of age.
2.3.9. ACTA2-Related sHTAD
Pathogenic variants in the ACTA2 gene, encoding the vascular isoform of the smooth muscle cell contractile protein alpha-actin, lead to diverse forms of syndromic and nonsyndromic HTAD [6,57,58]. Heterozygous missense ACTA2 variants cause a form of HTAD commonly associated with persistent livedo reticularis (a purplish skin discoloration caused by constriction or occlusion of deep dermal capillaries), iris floccule [58], premature onset of coronary artery disease, premature ischemic strokes due to Moyamoya disease (MYMY5; OMIM #614042, ORPHA:2573) (stenosis or occlusion of the terminal portion of the internal carotid artery with the formation of an abnormal vascular network in the vicinity of the arterial occlusion) or fusiform cerebral aneurysms [59], and BAV. The penetrance of thoracic aortic disease in these patients was estimated to be 50–70% and did not appear to be age dependent. Type A dissections were more common than type B dissections [58,60].
Pathogenic ACTA2 variants leading to arginine replacement by histidine, leucine or cysteine at position 179 (R179 variants) cause a distinct syndrome called multisystemic smooth muscle dysfunction syndrome (MSMDS, OMIM #613834, ORPHA:404463) characterized by congenital fixed pupils (mydriasis), large patent ductus arteriosus or aortopulmonary window, small vessel disease, urinary bladder dysfunction, intestinal malrotation, severe cerebrovascular disease and fully penetrant thoracic aortic disease by the age of 25 years [61,62,63,64,65].
The p.R179 and p.R258 ACTA2 variants are characterized by a significantly increased risk for aortic events, very early presentation typically in childhood warranting early repair, whereas the p.R185Q and p.R118Q variants seem to bare a more benign course [60]. Larger cohorts are required before implementation of these genotype–phenotype correlations into clinical practice. Dissections have been reported at aortic aneurysm sizes of as low as 40 mm [64,66]; therefore, earlier surgical intervention is advised in patients with established ACTA2 variants.
2.3.10. Importin-Related HTAD-VISS Syndrome (OMIM #619472)
Biallelic variants in the IPO8 gene, encoding the nuclear import protein importin 8, lead to a form of sHTAD presenting with a LDS/SGS-like phenotype with early-onset aortic aneurysms (before 1 year of age in the youngest), marked arterial tortuosity, structural heart disease in involving atrial or ventricular septal defects and patent ductus arteriosus, facial and skeletal anomalies, developmental delay, umbilical and/or inguinal hernias, immune dysregulation and allergic diseases [67,68,69]. No aortic dissection has been reported in a total of 28 patients who have been identified so far, despite a severe aneurysm phenotype in most affected individuals.
Turner syndrome (TS; OMIM #300082, ORPHA:881). TS is characterized by short stature, premature loss of ovarian function, webbing of the neck, lymphoedema, kidney and skeletal abnormalities in women and girls with complete or partial absence of the second X chromosome. Aortic coarctation (reported prevalence 7–18%) and BAV (reported prevalence 12–30%) are the most common congenital heart disease detected [70]. Aortic dilatation and dissection usually occur in TS patients with BAV or coarctation. However, the onset of aortic complications in TS occurs at a much younger age (20s and 30s) than nonsyndromic BAV or coarctation cases, aortic dilatation and dissection in the presence of apparently normal aortic valve has been reported, TS is an independent risk factor for aortic dilation and dissection and cystic medial necrosis has been found in a considerable portion of TS patients with aortic dissection during histology, indicating an inherent abnormality of the aortic tissue [70,71,72]. Aortic dissection in the absence of coarctation, BAV or hypertension has been reported in up to 11% of TS patients [73].
3. Thoracic Aortic Disease in Metabolic Storage Disease
Although metabolic storage diseases are not typically classified as syndromic HTAD, there is evidence of thoracic aortic disease in these conditions. El-Gharbawy et al. reported dilatation of the ascending aorta and/or aortic arch in five female patients (12.5% of the cohort) with late-onset Pompe disease (OMIM #232300, caused by variants in the GAA gene, leading to a deficiency in the acid α-glucosidase enzyme), including a 42-year-old patient with concomitant BAV who presented with aortic dissection of the ascending aorta at an aortic size >50 mm [74]. Aneurysms of the aortic root and ascending aorta, developing by the fifth decade of life, have been reported in 9.6% of male and 1.9% of female normotensive patients with Fabry disease (FD; OMIM #301500), an X-linked recessive disorder that is caused by deficiency of the lysosomal enzyme α-galactosidase A (GLA gene) [75,76,77]. Although vertebral artery and carotid artery dissection are relatively common and lead to stroke in FD, no cases of aortic dissection or rupture have been reported to date [76]. Aortic root dilatation (in 35–39% of the patients) has also been described in mucopolysaccharidoses, a group of 11 different lysosomal storage disorders, characterized by enzymatic deficiency leading to attenuated degradation and increased storage of glycosaminoglycans. No cases of aortic dissection have been published, and absolute aortic root dimensions greater than 45 mm are uncommon [78,79]. Hyperplasia and glycogen deposition vascular of smooth muscle cells, as indicated in animal models, may explain the presence of thoracic aortic disease in lysosomal storage disease [80].
4. Nonsyndromic HTAD
4.1. Bicuspid Aortic Valve Related HTAD
Bicuspid aortic valve (BAV; OMIM #109730, ORPHA:402075) is the most common congenital heart disease with an estimated prevalence of 0.5–0.8% [81]. Although BAV can be part of the phenotype in some cases of sHTAD such as MFS or LDS [82], there is also mounting evidence of familial clustering in up to 6–9% of first-degree relatives of nonsyndromic BAV [83,84]. An autosomal dominant inheritance pattern with variable expressivity and typically incomplete penetrance is recognized [82,84]. Up to 75% of patients with BAV might develop aortic dilatation [85], although this typically occurs later than other syndromic or nonsyndromic HTAD and at relatively slower growth rates (average of 0.19 cm/year) [86].
Variants in the NOTCH1 gene have been described in approximately 1% of sporadic BAV cases and in up to 7% of familial BAV cases [82,87,88] and are typically associated with prominent valve calcification [89]. Recently, loss-of-function SMAD6 variants have been found in up to 11% of nsHTAD patients with BAV [90,91]. ROBO4 variants, encoding a factor known to contribute to endothelial performance, and TBX20 variants, a transcription factor involved in the regulation of heart development, were shown to contribute to aortic aneurysm formation in families with nonsyndromic BAV [92,93].
Echocardiography screening of first-degree relatives of patients with BAV should be offered especially in boys, athletes and if hypertension is present [24]. Families with multiple affected relatives, a combination of other left-sided congenital abnormalities and a particularly malignant clinical profile should be offered genetic testing for at least ACTA2, SMAD6, TBX20, ROBO4 and NOTCH1 genes. Multiple gene panels should be considered in selected cases, taking into consideration the variable and incomplete penetrance of sHTAD that might lead to a mild phenotype with minimal or no systemic features in some patients. No specific genotype–phenotype correlations currently exist that could possibly guide surgical interventions or provide specific prognostic information. Recently, Pileggi et al. indicated that specific NOTCH1 variants could be associated with better prognosis and later-onset development of aortic stenosis [88].
4.2. Familial and Sporadic Nonsyndromic HTAD
Based on the presence of familial disease or not, nsHTAD is further categorized into familial and sporadic nsHTAD. A positive family history of thoracic aortic disease is associated with an increased aortic growth rate, a bigger chance of gene identification and earlier phenotypic manifestation [94]. The genetic etiology of familial nsHTAD is highly heterogeneous and usually involves genes that regulate the smooth muscle cell contractile apparatus. The genetic substrate of sporadic nsHTAD is largely unknown and seems to differ from familial nsHTAD cases.
To date, over 10 genes and 2 linked loci have been involved in the pathogenesis of nsHTAD, including genes involved in the (TGF-β) pathway and genes encoding extracellular matrix proteins that are typically associated with syndromic aortopathies [2,6,47]. Common single nucleotide polymorphisms at the 15q21.1 locus of the FBN1 gene have been shown to be associated with sporadic nsHTAD [95] without other systemic features of MFS. Arnaud et al. performed genetic screening in 226 consecutive nsHTAD, either sporadic in patients under 45 years of age or in documented familial cases, and identified an overall yield of pathogenic or likely pathogenic variants (SMAD3, FBN1, TGFBR1, TGFBR2, TGFB2, ACTA2, MYLK, FLNA, FBN2, LOX, MFAP5 genes) in 18% of the patients (11% in sporadic cases vs. 22% in familial cases), with almost two-thirds located in SMAD3 and FBN1 genes. Exclusively missense variants and no premature termination codon variants were identified in the FBN1 gene in this cohort. More careful clinical evaluation after the genetic result revealed clinical findings consistent with LDS in approximately half of the cases with SMAD3 variants and history of periventricular heterotopia in patients with the FLNA variant, reclassifying these cases as syndromic [6]. Weerakkody et al. investigated a cohort of 1025 unrelated HTAD cases, including many cases of sporadic HTAD, and reported a 4.9% yield of genetic testing for a 15-gene genetic panel. Patients with a family history of HTAD were four times more likely to carry a pathogenic or likely pathogenic variant than those without a family history (9.8 vs. 2.4%) [96]. Since clinical information (syndromic features or clinical diagnosis) was not available in a significant percentage of the cases, these cases cannot automatically be categorized as sporadic nsHTAD.
Overall, pathogenic ACTA2 variants are the most frequently encountered, as they are detected in 1–21% of nsHTAD [57,97,98] and are associated with a malignant aortic phenotype. Pathogenic variants in the MYLK gene [99,100,101], with missense pathogenic variants showing an earlier onset aortic event, and variants in the MYH11 [102] and PRKG1 genes [103] have also been recognized as relatively rare but aggressive causes of thoracic aortic dissection (~1% prevalence of each), in nsHTAD which are not always preceded by obvious aortic dilatation. There is no evidence to date that defects in the other genes identified (LOX, MFAP5, FOXE3, MAT2A, SMAD2, SMAD4, NOTCH1, PLOD1, TGFB2, TGFBR2, FBN1, FBN2) are linked to a more severe phenotype or earlier presentation of HTAD [6,57,58].
5. Genetic Testing and Surgical Intervention
Genetic testing of patients with established or suspected HTAD is an essential part of their assessment and should follow clinical evaluation and proper genetic counseling (Figure 1). Specific gene testing may be considered when the phenotype indicates a distinct syndromic aortopathy to aid clinical decisions and offer prognostic and diagnostic information. In most cases, however, a multigene panel should be used, consisting of at least of the 11 genes (MFS–FBN1, LDS–TGFBR1, TGFBR2, SMAD3, TGFB2, vEDS–COL3A1, ACTA2, MYLK, LOX, PRKG1, MYH11) that have been identified to have a “definitive” or “strong” association with HTAD [104]. Specific criteria for genetic testing have been proposed by the Rare Disease Group of VASCERN, based on expert opinion (Table 1) adopted in the most recent European guidelines of the ESC and the European Society of Human Genetics [105]. Exome sequencing may be performed in cases with equivocal diagnosis, nonsyndromic HTAD or for research purposes [106]. Cascade genetic screening should be offered in all family members of patients with well-established pathogenic or likely pathogenic variants. Surgery should be offered in patients with a malignant genetic and/or clinical profile and mildly dilated aortas (Table 2).
6. Conclusions
Genetic testing plays an increasing role in the management of patients with HTAD. The identification of a pathogenic variant can establish or confirm the diagnosis of syndromic HTAD, dictate extensive evaluation of the arterial tree in HTAD with known distal vasculature involvement and justify closer follow-up and earlier surgical intervention in HTAD with high risk of dissection of minimal or normal aortic size. Evolving phenotype–genotype correlations should soon allow for more precise and individualized management and treatment of patients with HTAD.
Author Contributions
Conceptualization, E.P. and A.A.; methodology, E.P., D.D. and A.A.; writing—original draft preparation, E.P.; writing—review and editing, E.P., D.D. and A.A.; supervision, E.P., D.D. and A.A.; All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Loeys, B.L.; Schwarze, U.; Holm, T.; Callewaert, B.L.; Thomas, G.H.; Pannu, H.; De Backer, J.F.; Oswald, G.L.; Symoens, S.; Manouvrier, S.; et al. Aneurysm Syndromes Caused by Mutations in the TGF-β Receptor. N. Engl. J. Med. 2006, 355, 788–798. [Google Scholar] [CrossRef] [PubMed]
- Verstraeten, A.; Luyckx, I.; Loeys, B. Aetiology and management of hereditary aortopathy. Nat. Rev. Cardiol. 2017, 14, 197–208. [Google Scholar] [CrossRef]
- Hostetler, E.M.; Regalado, E.S.; Guo, D.-C.; Hanna, N.; Arnaud, P.; Muiño-Mosquera, L.; Callewaert, B.L.; Lee, K.; Leal, S.M.; Wallace, S.E.; et al. SMAD3 pathogenic variants: Risk for thoracic aortic disease and associated complications from the Montalcino Aortic Consortium. J. Med. Genet. 2019, 56, 252–260. [Google Scholar] [CrossRef]
- Jondeau, G.; Ropers, J.; Regalado, E.; Braverman, A.; Evangelista, A.; Teixedo, G.; De Backer, J.; Muiño-Mosquera, L.; Naudion, S.; Zordan, C.; et al. International Registry of Patients Carrying TGFBR1 or TGFBR2 Mutations. Circ. Cardiovasc. Genet. 2016, 9, 548–558. [Google Scholar] [CrossRef] [Green Version]
- Franken, R.; Teixido-Tura, G.; Brion, M.; Forteza, A.; Palomares, J.F.R.; Gutierrez, L.; Dorado, D.G.; Pals, G.; Mulder, B.J.; Evangelista, A. Relationship between fibrillin-1 genotype and severity of cardiovascular involvement in Marfan syndrome. Heart 2017, 103, 1795–1799. [Google Scholar] [CrossRef]
- Arnaud, P.; Hanna, N.; Benarroch, L.; Aubart, M.; Bal, L.; Bouvagnet, P.; Busa, T.; Dulac, Y.; Dupuis-Girod, S.; Edouard, T.; et al. Genetic diversity and pathogenic variants as possible predictors of severity in a French sample of nonsyndromic heritable thoracic aortic aneurysms and dissections (nshTAAD). Genet. Med. 2019, 21, 2015–2024. [Google Scholar] [CrossRef]
- Elefteriades, J.A.; Farkas, E.A. Thoracic Aortic Aneurysm. Clinically Pertinent Controversies and Uncertainties. J. Am. Coll. Cardiol. 2010, 55, 841–857. [Google Scholar] [CrossRef] [PubMed]
- Loeys, B.L.; Dietz, H.C.; Braverman, A.C.; Callewaert, B.L.; De Backer, J.; Devereux, R.B.; Hilhorst-Hofstee, Y.; Jondeau, G.; Faivre, L.; Milewicz, D.M.; et al. The revised Ghent nosology for the Marfan syndrome. J. Med. Genet. 2010, 47, 476–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devereux, R.B.; De Simone, G.; Arnett, D.K.; Best, L.G.; Boerwinkle, E.; Howard, B.V.; Kitzman, D.; Lee, E.T.; Mosley, T.H. Weder, A. et al. Normal limits in relation to age, body size and gender of two-dimensional echocardiographic aortic root dimensions in persons ≥ 15 years of age. Am. J. Cardiol. 2012, 110, 1189–1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groth, K.A.; Hove, H.; Kyhl, K.; Folkestad, L.; Gaustadnes, M.; Vejlstrup, N.; Stochholm, K.; Østergaard, J.R.; Andersen, N.H.; Gravholt, C.H. Prevalence, incidence, and age at diagnosis in Marfan Syndrome. Orphanet J. Rare Dis. 2015, 10, 153. [Google Scholar] [CrossRef] [Green Version]
- Treasure, T.; Takkenberg, J.J.M.; Pepper, J. Surgical management of aortic root disease in Marfan syndrome and other congenital disorders associated with aortic root aneurysms. Heart 2014, 100, 1571–1576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Beaufort, H.W.L.; Trimarchi, S.; Korach, A.; Di Eusanio, M.; Gilon, D.; Montgomery, D.G.; Evangelista, A.; Braverman, A.C.; Chen, E.P.; Isselbacher, E.M.; et al. Aortic dissection in patients with Marfan syndrome based on the IRAD data. Ann. Cardiothorac. Surg. 2017, 6, 633–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gott, V.L.; Greene, P.S.; Alejo, D.E.; Cameron, D.E.; Naftel, D.C.; Miller, D.C.; Gillinov, A.M.; Laschinger, J.C.; Pyeritz, R.E.; Borst, H.G.; et al. Replacement of the aortic root in patients with Marfan’s syndrome. N. Engl. J. Med. 1999, 340, 1307–1313. [Google Scholar] [CrossRef]
- Erbel, R.; Aboyans, V.; Boileau, C.; Bossone, E.; Di Bartolomeo, R.; Eggebrecht, H.; Evangelista, A.; Falk, V.; Frank, H.; Gaemperli, O.; et al. 2014 ESC guidelines on the diagnosis and treatment of aortic diseases. Eur. Heart J. 2014, 35, 2873–2926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumgartner, H.; De Backer, J.; Babu-Narayan, S.V.; Budts, W.; Chessa, M.; Diller, G.-P.; Lung, B.; Kluin, J.; Lang, I.M.; Meijboom, F.; et al. 2020 ESC Guidelines for the management of adult congenital heart disease. Eur. Heart J. 2020, 42, 563–645. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Isselbacher, E.M.; Nienaber, C.A.; Pyeritz, R.E.; Eagle, K.A.; Tsai, T.T.; Cooper, J.V.; Januzzi, J.L.; Braverman, A.C.; Montgomery, D.G.; et al. Type-selective benefits of medications in treatment of acute aortic dissection (from the International Registry of Acute Aortic Dissection [IRAD]). Am. J. Cardiol. 2012, 109, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Update on Clinical Trials of Losartan with and Without β-Blockers to Block Aneurysm Growth in Patients With Marfan Syndrome: A Review. JAMA Cardiol. 2019, 4, 702–707. [CrossRef]
- Franken, R.; den Hartog, A.W.; Radonic, T.; Micha, D.; Maugeri, A.; van Dijk, F.S.; Meijers-Heijboer, H.E.; Timmermans, J.; Scholte, A.J.; van den Berg, M.P.; et al. Beneficial Outcome of Losartan Therapy Depends on Type of FBN1 Mutation in Marfan Syndrome. Circ. Cardiovasc. Genet. 2015, 8, 383–388. [Google Scholar] [CrossRef] [Green Version]
- Takeda, N.; Inuzuka, R.; Maemura, S.; Morita, H.; Nawata, K.; Fujita, D.; Taniguchi, Y.; Yamauchi, H.; Yagi, H.; Kato, M.; et al. Impact of Pathogenic FBN1 Variant Types on the Progression of Aortic Disease in Patients With Marfan Syndrome. Circ. Genom. Precis. Med. 2018, 11, e002058. [Google Scholar] [CrossRef] [Green Version]
- De Backer, J.; Campens, L.; Muiño Mosquera, L. Looking for the Missing Links: Challenges in the Search for Genotype-Phenotype Correlation in Marfan Syndrome. Circ. Genom. Precis. Med. 2018, 11, e002185. [Google Scholar] [CrossRef]
- Baudhuin, L.M.; Kotzer, K.E.; Lagerstedt, S.A. Increased frequency of FBN1 truncating and splicing variants in Marfan syndrome patients with aortic events. Genet. Med. 2015, 17, 177–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnaud, P.; Milleron, O.; Hanna, N.; Ropers, J.; Ould Ouali, N.; Affoune, A.; Langeois, M.; Eliahou, L.; Arnoult, F.; Renard, P.; et al. Clinical relevance of genotype–phenotype correlations beyond vascular events in a cohort study of 1500 Marfan syndrome patients with FBN1 pathogenic variants. Genet. Med. 2021, 23, 1296. [Google Scholar] [CrossRef]
- Milleron, O.; Arnoult, F.; Delorme, G.; Detaint, D.; Pellenc, Q.; Raffoul, R.; Tchitchinadze, M.; Langeois, M.; Guien, C.; Beroud, C.; et al. Pathogenic FBN1 Genetic Variation and Aortic Dissection in Patients With Marfan Syndrome. J. Am. Coll. Cardiol. 2020, 75, 843–853. [Google Scholar] [CrossRef] [PubMed]
- Hiratzka, L.F.; Bakris, G.L.; Beckman, J.A.; Bersin, R.M.; Carr, V.F.; Casey, D.E.; Eagle, K.A.; Hermann, L.K.; Isselbacher, E.M.; Kazerooni, E.A.; et al. 2010 ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM guidelines for the diagnosis and management of patients with thoracic aortic disease: Executive summary: A report of the American college of cardiology foundation/american heart association task force on practice guidelines, American association for thoracic surgery, American college of radiology, American stroke association. Circulation 2010, 121, 1544–1579. [Google Scholar]
- Aubart, M.; Gobert, D.; Aubart-Cohen, F.; Detaint, D.; Hanna, N.; d’Indya, H.; Lequintrec, J.-S.; Renard, P.; Vigneron, A.-M.; Dieudé, P.; et al. Early-Onset Osteoarthritis, Charcot-Marie-Tooth Like Neuropathy, Autoimmune Features, Multiple Arterial Aneurysms and Dissections: An Unrecognized and Life Threatening Condition. PLoS ONE 2014, 9, e96387. [Google Scholar] [CrossRef] [Green Version]
- Van de Laar, I.M.B.H.; Oldenburg, R.A.; Pals, G.; Roos-Hesselink, J.W.; de Graaf, B.M.; Verhagen, J.M.A.; Hoedemaekers, Y.M.; Willemsen, R.; Severijnen, L.-A.; Venselaar, H.; et al. Mutations in SMAD3 cause a syndromic form of aortic aneurysms and dissections with early-onset osteoarthritis. Nat. Genet. 2011, 43, 121–126. [Google Scholar] [CrossRef]
- Schepers, D.; Tortora, G.; Morisaki, H.; MacCarrick, G.; Lindsay, M.; Liang, D.; Mehta, S.G.; Hague, J.; Verhagen, J.; van de Laar, I.; et al. A mutation update on the LDS-associated genes TGFB2/3 and SMAD2/3. Hum. Mutat. 2018, 39, 621–634. [Google Scholar] [CrossRef] [Green Version]
- Wischmeijer, A.; Van Laer, L.; Tortora, G.; Bolar, N.A.; Van Camp, G.; Fransen, E.; Peeters, N.; di Bartolomeo, R.; Pacini, D.; Gargiulo, G.; et al. Thoracic Aortic Aneurysm in Infancy in Aneurysms-Osteoarthritis Syndrome Due to a Novel SMAD 3 Mutation: Further Delineation of the Phenotype. Am. J. Med. Genet. Part A 2013, 161, 1028–1035. [Google Scholar] [CrossRef]
- Boileau, C.; Guo, D.-C.; Hanna, N.; Regalado, E.S.; Detaint, D.; Gong, L.; Varret, M.; Prakash, S.K.; Li, A.H.; d’Indy, H.; et al. TGFB2 mutations cause familial thoracic aortic aneurysms and dissections associated with mild systemic features of Marfan syndrome. Nat. Genet. 2012, 44, 916–921. [Google Scholar] [CrossRef] [Green Version]
- Renard, M.; Callewaert, B.; Malfait, F.; Campens, L.; Sharif, S.; del Campo, M.; Valenzuela, I.; Mcwilliam, C.; Coucke, P.; De Paepe, A.; et al. Thoracic aortic-aneurysm and dissection in association with significant mitral valve disease caused by mutations in TGFB2. Int. J. Cardiol. 2013, 165, 584–587. [Google Scholar] [CrossRef]
- Malfait, F.; Francomano, C.; Byers, P.; Belmont, J.; Berglund, B.; Black, J.; Bloom, L.; Bowen, J.M.; Brady, A.F.; Burrows, N.P.; et al. The 2017 international classification of the Ehlers-Danlos syndromes; The 2017 international classification of the Ehlers-Danlos syndromes. Am. J. Med. Genet. Part C Semin. Med. Genet. 2017, 175, 8–26. [Google Scholar] [CrossRef] [Green Version]
- Beighton, P.; De Paepe, A.; Steinmann, B.; Tsipouras, P.; Wenstrup, R.J. Ehlers-Danlos syndromes: Revised nosology, Villefranche, 1997. Ehlers-Danlos National Foundation (USA) and Ehlers-Danlos Support Group (UK). Am. J. Med. Genet. 1998, 77, 31–37. [Google Scholar] [CrossRef]
- Pepin, M.; Schwarze, U.; Superti-Furga, A.; Byers, P.H. Clinical and Genetic Features of Ehlers–Danlos Syndrome Type IV, the Vascular Type. N. Engl. J. Med. 2000, 342, 673–680. [Google Scholar] [CrossRef]
- Schwarze, U.; Schievink, W.I.; Petty, E.; Jaff, M.R.; Babovic-Vuksanovic, D.; Cherry, K.J.; Pepin, M.; Byers, P.H. Haploinsufficiency for One COL3A1 Allele of Type III Procollagen Results in a Phenotype Similar to the Vascular Form of Ehlers-Danlos Syndrome, Ehlers-Danlos Syndrome Type IV. Am. J. Hum. Genet. 2001, 69, 989–1001. [Google Scholar] [CrossRef] [Green Version]
- Monroe, G.R.; Harakalova, M.; van der Crabben, S.N.; Majoor-Krakauer, D.; Bertoli-Avella, A.M.; Moll, F.L.; Oranen, B.I.; Dooijes, D.; Vink, A.; Knoers, N.V.; et al. Familial Ehlers-Danlos syndrome with lethal arterial events caused by a mutation in COL5A1. Am. J. Med. Genet. Part A 2015, 167, 1196–1203. [Google Scholar] [CrossRef] [PubMed]
- Leistritz, D.F.; Pepin, M.G.; Schwarze, U.; Byers, P.H. COL3A1 haploinsufficiency results in a variety of Ehlers-Danlos syndrome type IV with delayed onset of complications and longer life expectancy. Genet. Med. 2011, 13, 717–722. [Google Scholar] [CrossRef] [PubMed]
- Pepin, M.G.; Schwarze, U.; Rice, K.M.; Liu, M.; Leistritz, D.; Byers, P.H. Survival is affected by mutation type and molecular mechanism in vascular Ehlers–Danlos syndrome (EDS type IV). Genet. Med. 2014, 16, 881–888. [Google Scholar] [CrossRef] [Green Version]
- Frank, M.; Adham, S.; Seigle, S.; Legrand, A.; Mirault, T.; Henneton, P.; Albuisson, J.; Denarié, N.; Mazzella, J.M.; Mousseaux, E.; et al. Vascular Ehlers-Danlos Syndrome: Long-Term Observational Study. J. Am. Coll. Cardiol. 2019, 73, 1948–1957. [Google Scholar] [CrossRef]
- Frank, M.; Albuisson, J.; Ranque, B.; Golmard, L.; Mazzella, J.-M.; Bal-Theoleyre, L.; Fauret, A.-L.; Mirault, T.; Denarié, N.; Mousseaux, E.; et al. The type of variants at the COL3A1 gene associates with the phenotype and severity of vascular Ehlers–Danlos syndrome. Eur. J. Hum. Genet. 2015, 23, 1657–1664. [Google Scholar] [CrossRef] [PubMed]
- Bergqvist, D.; Björck, M.; Wanhainen, A. Treatment of Vascular Ehlers-Danlos Syndrome. Ann. Surg. 2013, 258, 257–261. [Google Scholar] [CrossRef]
- Ong, K.-T.; Perdu, J.; De Backer, J.; Bozec, E.; Collignon, P.; Emmerich, J.; Fauret, A.-L.; Fiessinger, J.-N.; Germain, D.P.; Georgesco, G.; et al. Effect of celiprolol on prevention of cardiovascular events in vascular Ehlers-Danlos syndrome: A prospective randomised, open, blinded-endpoints trial. Lancet 2010, 376, 1476–1484. [Google Scholar] [CrossRef]
- Dubacher, N.; Münger, J.; Gorosabel, M.C.; Crabb, J.; Ksiazek, A.A.; Caspar, S.M.; Bakker, E.N.T.P.; Van Bavel, E.; Ziegler, U.; Carrel, T.; et al. Celiprolol but not losartan improves the biomechanical integrity of the aorta in a mouse model of vascular Ehlers-Danlos syndrome. Cardiovasc. Res. 2020, 116, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Meester, J.A.N.; Vandeweyer, G.; Pintelon, I.; Lammens, M.; Van Hoorick, L.; De Belder, S.; Waitzman, K.; Young, L.; Markham, L.W.; Vogt, J.; et al. Loss-of-function mutations in the X-linked biglycan gene cause a severe syndromic form of thoracic aortic aneurysms and dissections. Genet. Med. 2017, 19, 386–395. [Google Scholar] [CrossRef] [Green Version]
- Sheen, V.L.; Jansen, A.; Chen, M.H.; Parrini, E.; Morgan, T.; Ravenscroft, R.; Ganesh, V.; Underwood, T.; Wiley, J.; Leventer, R.; et al. Filamin A mutations cause periventricular heterotopia with Ehlers-Danlos syndrome. Neurology 2005, 64, 254–262. [Google Scholar] [CrossRef]
- Chen, M.H.; Choudhury, S.; Hirata, M.; Khalsa, S.; Chang, B.; Walsh, C.A. Thoracic aortic aneurysm in patients with loss of function Filamin A mutations: Clinical characterization, genetics, and recommendations; Thoracic aortic aneurysm in patients with loss of function Filamin A mutations: Clinical characterization, genetics, and recommendations. Am. J. Med. Genet. A 2018, 176, 337–350. [Google Scholar] [PubMed]
- Guo, D.C.; Regalado, E.S.; Gong, L.; Duan, X.; Santos-Cortez, R.L.P.; Arnaud, P.; Ren, Z.; Cai, B.; Hostetler, E.M.; Moran, R.; et al. LOX mutations predispose to thoracic aortic aneurysms and dissections. Circ. Res. 2016, 118, 928–934. [Google Scholar] [CrossRef] [Green Version]
- Wolford, B.N.; Hornsby, W.E.; Guo, D.; Zhou, W.; Lin, M.; Farhat, L.; McNamara, J.; Driscoll, A.; Wu, X.; Schmidt, E.M.; et al. Clinical Implications of Identifying Pathogenic Variants in Individuals With Thoracic Aortic Dissection. Circ. Genom. Precis. Med. 2019, 12, e002476. [Google Scholar] [CrossRef] [Green Version]
- García, M.E.R.; Vinagre, F.J.C.; Cruz-Rojo, J.; Lorenzo, L.G.; Carnicero-Rodríguez, P.; Pozo, J.S.-D.; Martínez-Azorín, F. A rare male patient with Fontaine progeroid syndrome caused by p.R217H de novo mutation in SLC25A24. Am. J. Med. Genet. Part A 2018, 176, 2479–2486. [Google Scholar] [CrossRef] [PubMed]
- Ehmke, N.; Graul-Neumann, L.; Smorag, L.; Koenig, R.; Segebrecht, L.; Magoulas, P.; Scaglia, F.; Kilic, E.; Hennig, A.F.; Adolphs, N.; et al. De Novo Mutations in SLC25A24 Cause a Craniosynostosis Syndrome with Hypertrichosis, Progeroid Appearance, and Mitochondrial Dysfunction. Am. J. Hum. Genet. 2017, 101, 833–843. [Google Scholar] [CrossRef] [Green Version]
- Legué, J.; François, J.H.M.; van Rijswijk, C.S.P.; van Brakel, T.J. Is Gorlin–Chaudhry–Moss syndrome associated with aortopathy? Eur. J. Cardio-Thorac. Surg. 2020, 58, 654–655. [Google Scholar] [CrossRef]
- Schepers, D.; Doyle, A.J.; Oswald, G.; Sparks, E.; Myers, L.; Willems, P.J.; Mansour, S.; Simpson, M.A.; Frysira, H.; Maat-Kievit, A.; et al. The SMAD-binding domain of SKI: A hotspot for de novo mutations causing Shprintzen-Goldberg syndrome. Eur. J. Hum. Genet. 2015, 23, 224–228. [Google Scholar] [CrossRef] [PubMed]
- Doyle, A.J.; Doyle, J.J.; Bessling, S.L.; Maragh, S.; Lindsay, M.E.; Schepers, D.; Gillis, E.; Mortier, G.; Homfray, T.; Sauls, K.; et al. Mutations in the TGF-β repressor SKI cause Shprintzen-Goldberg syndrome with aortic aneurysm. Nat. Genet. 2012, 44, 1249–1254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callewaert, B.L.; Willaert, A.; Kerstjens-Frederikse, W.S.; De Backer, J.; Devriendt, K.; Albrecht, B.; Ramos-Arroyo, M.A.; Doco-Fenzy, M.; Hennekam, R.C.M.; Pyeritz, R.E.; et al. Arterial tortuosity syndrome: Clinical and molecular findings in 12 newly identified families. Hum. Mutat. 2008, 29, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Beyens, A.; Albuisson, J.; Boel, A.; Al-Essa, M.; Al-Manea, W.; Bonnet, D.; Bostan, O.; Boute, O.; Busa, T.; Canham, N.; et al. Arterial tortuosity syndrome: 40 new families and literature review. Genet. Med. 2018, 20, 1236–1245. [Google Scholar] [CrossRef] [Green Version]
- Guo, D.-C.; Regalado, E.S.; Pinard, A.; Chen, J.; Lee, K.; Rigelsky, C.; Zilberberg, L.; Hostetler, E.M.; Aldred, M.; Wallace, S.E.; et al. LTBP3 Pathogenic Variants Predispose Individuals to Thoracic Aortic Aneurysms and Dissections. Am. J. Hum. Genet. 2018, 102, 706–712. [Google Scholar] [CrossRef] [Green Version]
- Zhu, G.; Luo, M.; Chen, Q.; Zhang, Y.; Zhao, K.; Zhang, Y.; Shu, C.; Yang, H.; Zhou, Z. Novel LTBP3 mutations associated with thoracic aortic aneurysms and dissections. Orphanet J. Rare Dis. 2021, 16, 513. [Google Scholar] [CrossRef]
- Morisaki, H.; Akutsu, K.; Ogino, H.; Kondo, N.; Yamanaka, I.; Tsutsumi, Y.; Yoshimuta, T.; Okajima, T.; Matsuda, H.; Minatoya, K.; et al. Human Mutation RESEARCH ARTICLE Mutation of ACTA2 Gene as an Important Cause of Familial and Nonfamilial Nonsyndromatic Thoracic Aortic Aneurysm and/or Dissection (TAAD). Hum. Mutat. 2009, 30, 1406–1411. [Google Scholar] [CrossRef]
- Guo, D.-C.; Pannu, H.; Tran-Fadulu, V.; Papke, C.L.; Yu, R.K.; Avidan, N.; Bourgeois, S.; Estrera, A.L.; Safi, H.J.; Sparks, E.; et al. Mutations in smooth muscle α-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat. Genet. 2007, 39, 1488–1493. [Google Scholar] [CrossRef]
- Guo, D.C.; Papke, C.L.; Tran-Fadulu, V.; Regalado, E.S.; Avidan, N.; Johnson, R.J.; Kim, D.H.; Pannu, H.; Willing, M.C.; Sparks, E.; et al. Mutations in smooth muscle alpha-actin (ACTA2) cause coronary artery disease, stroke, and Moyamoya disease, along with thoracic aortic disease. Am. J. Hum. Genet. 2009, 84, 617–627. [Google Scholar] [CrossRef] [Green Version]
- Regalado, E.S.; Guo, D.; Prakash, S.; Bensend, T.A.; Flynn, K.; Estrera, A.; Safi, H.; Liang, D.; Hyland, J.; Child, A.; et al. Aortic Disease Presentation and Outcome Associated With ACTA2 Mutations. Circ. Cardiovasc. Genet. 2015, 8, 457–464. [Google Scholar] [CrossRef] [Green Version]
- Meuwissen, M.E.C.; Lequin, M.H.; Bindels-de Heus, K.; Bruggenwirth, H.T.; Knapen, M.F.C.M.; Dalinghaus, M.; de Coo, R.; van Bever, Y.; Winkelman, B.H.J.; Mancini, G.M.S. ACTA2 mutation with childhood cardiovascular, autonomic and brain anomalies and severe outcome. Am. J. Med. Genet. A 2013, 161A, 1376–1380. [Google Scholar] [CrossRef]
- Milewicz, D.M.; Østergaard, J.R.; Ala-Kokko, L.M.; Khan, N.; Grange, D.K.; Mendoza-Londono, R.; Bradley, T.J.; Olney, A.H.; Adès, L.; Maher, J.F.; et al. De novo ACTA2 mutation causes a novel syndrome of multisystemic smooth muscle dysfunction. Am. J. Med. Genet. A 2010, 152A, 2437–2443. [Google Scholar] [CrossRef] [Green Version]
- Munot, P.; Saunders, D.E.; Milewicz, D.M.; Regalado, E.S.; Ostergaard, J.R.; Braun, K.P.; Kerr, T.; Lichtenbelt, K.D.; Philip, S.; Rittey, C.; et al. A novel distinctive cerebrovascular phenotype is associated with heterozygous Arg179 ACTA2 mutations. Brain 2012, 135, 2506–2514. [Google Scholar] [CrossRef] [PubMed]
- Regalado, E.S.; Mellor-Crummey, L.; De Backer, J.; Braverman, A.C.; Ades, L.; Benedict, S.; Bradley, T.J.; Brickner, M.E.; Chatfield, K.C.; Child, A.; et al. Clinical History and Management Recommendations of the Smooth Muscle Dysfunction Syndrome Due to ACTA2 Arginine 179 Alterations. Genet. Med. 2018, 20, 1206–1215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yetman, A.T.; Starr, L.J.; Bleyl, S.B.; Meyers, L.; Delaney, J.W. Progressive Aortic Dilation Associated With ACTA2 Mutations Presenting in Infancy. Pediatrics 2015, 136, e262–e266. [Google Scholar] [CrossRef] [Green Version]
- Disabella, E.; Grasso, M.; Gambarin, F.I.; Narula, N.; Dore, R.; Favalli, V.; Serio, A.; Antoniazzi, E.; Mosconi, M.; Pasotti, M.; et al. Risk of dissection in thoracic aneurysms associated with mutations of smooth muscle alpha-actin 2 (ACTA2). Heart 2011, 97, 321–326. [Google Scholar] [CrossRef]
- Bertoli-Avella, A.M.; Kandaswamy, K.K.; Khan, S.; Ordonez-Herrera, N.; Tripolszki, K.; Beetz, C.; Rocha, M.E.; Urzi, A.; Hotakainen, R.; Leubauer, A.; et al. Combining exome/genome sequencing with data repository analysis reveals novel gene–disease associations for a wide range of genetic disorders. Genet. Med. 2021, 23, 1551–1568. [Google Scholar] [CrossRef] [PubMed]
- Van Gucht, I.; Meester, J.A.N.; Bento, J.R.; Bastiaansen, M.; Bastianen, J.; Luyckx, I.; Van Den Heuvel, L.; Neutel, C.H.G.; Guns, P.J.; Vermont, M.; et al. A human importin-β-related disorder: Syndromic thoracic aortic aneurysm caused by bi-allelic loss-of-function variants in IPO8. Am. J. Hum. Genet. 2021, 108, 1115–1125. [Google Scholar] [CrossRef]
- Ziegler, A.; Duclaux-Loras, R.; Revenu, C.; Charbit-Henrion, F.; Begue, B.; Duroure, K.; Grimaud, L.; Guihot, A.L.; Desquiret-Dumas, V.; Zarhrate, M.; et al. Bi-allelic variants in IPO8 cause a connective tissue disorder associated with cardiovascular defects, skeletal abnormalities, and immune dysregulation. Am. J. Hum. Genet. 2021, 108, 1126–1137. [Google Scholar] [CrossRef]
- Fuchs, M.M.; Attenhofer Jost, C.; Babovic-Vuksanovic, D.; Connolly, H.M.; Egbe, A. Long-Term Outcomes in Patients With Turner Syndrome: A 68-Year Follow-Up. J. Am. Heart Assoc. 2019, 8, e011501. [Google Scholar] [CrossRef] [Green Version]
- Thunström, S.; Krantz, E.; Thunström, E.; Hanson, C.; Bryman, I.; Landin-Wilhelmsen, K. Incidence of Aortic Dissection in Turner Syndrome: A 23-Year Prospective Cohort Study. Circulation 2019, 139, 2802–2804. [Google Scholar] [CrossRef] [PubMed]
- Lopez, L.; Arheart, K.L.; Colan, S.D.; Stein, N.S.; Lopez-Mitnik, G.; Lin, A.E.; Reller, M.D.; Ventura, R.; Silberbach, M. Turner syndrome is an independent risk factor for aortic dilation in the young. Pediatrics 2008, 121, e1622–e1627. [Google Scholar] [CrossRef] [PubMed]
- Carlson, M.; Silberbach, M. Dissection of the aorta in Turner syndrome: Two cases and review of 85 cases in the literature. Case Rep. 2009, 2009, bcr0620091998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Gharbawy, A.H.; Bhat, G.; Murillo, J.E.; Thurberg, B.L.; Kampmann, C.; Mengel, K.E.; Kishnani, P.S. Expanding the clinical spectrum of late-onset Pompe disease: Dilated arteriopathy involving the thoracic aorta, a novel vascular phenotype uncovered. Mol. Genet. Metab. 2011, 103, 362–366. [Google Scholar] [CrossRef]
- Barbey, F.; Qanadli, S.D.; Juli, C.; Brakch, N.; Palaek, T.; Rizzo, E.; Jeanrenaud, X.; Eckhardt, B.; Linhart, A. Aortic remodelling in Fabry disease. Eur. Heart J. 2010, 31, 347–353. [Google Scholar] [CrossRef] [Green Version]
- Anastasakis, A.; Papatheodorou, E.; Steriotis, A.K. Fabry disease and cardiovascular involvement. Curr. Pharm. Des. 2013, 19, 5997–6008. [Google Scholar] [CrossRef] [PubMed]
- Anastasakis, A.; Sevdalis, E.; Papatheodorou, E.; Stefanadis, C. Anderson-Fabry disease: A cardiomyopathy that can be cured. Hell. J. Cardiol. 2011, 52, 316–326. [Google Scholar]
- De Oliviera Poswar, F.; de Souza, C.F.M.; Giugliani, R.; Baldo, G. Aortic root dilatation in patients with mucopolysaccharidoses and the impact of enzyme replacement therapy. Heart Vessel. 2019, 34, 290–295. [Google Scholar] [CrossRef]
- Bolourchi, M.; Renella, P.; Wang, R. Aortic Root Dilatation in Mucopolysaccharidosis I–VII. Int. J. Mol. Sci. 2016, 17, 2004. [Google Scholar] [CrossRef] [Green Version]
- Belfiore, M.P.; Iacobellis, F.; Acampora, E.; Caiazza, M.; Rubino, M.; Monda, E.; Magaldi, M.R.; Tarallo, A.; Sasso, M.; De Pasquale, V.; et al. Aortopathies in mouse models of Pompe, Fabry and Mucopolysaccharidosis IIIB lysosomal storage diseases. PLoS ONE 2020, 15, e0233050. [Google Scholar] [CrossRef]
- Coffey, S.; Cairns, B.J.; Iung, B. The modern epidemiology of heart valve disease. Heart 2016, 102, 75–85. [Google Scholar] [CrossRef]
- Freeze, S.L.; Landis, B.J.; Ware, S.M.; Helm, B.M. Bicuspid Aortic Valve: A Review with Recommendations for Genetic Counseling. J. Genet. Couns. 2016, 25, 1171–1178. [Google Scholar] [CrossRef] [Green Version]
- Galian-Gay, L.; Carro Hevia, A.; Teixido-Turà, G.; Rodríguez Palomares, J.; Gutiérrez-Moreno, L.; Maldonado, G.; Gonzàlez-Alujas, M.T.; Sao-Aviles, A.; Gallego, P.; Calvo-Iglesias, F.; et al. Familial clustering of bicuspid aortic valve and its relationship with aortic dilation in first-degree relatives. Heart 2019, 105, 603–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huntington, K.; Hunter, A.G.; Chan, K.L. A prospective study to assess the frequency of familial clustering of congenital bicuspid aortic valve. J. Am. Coll. Cardiol. 1997, 30, 1809–1812. [Google Scholar] [CrossRef]
- Tadros, T.M.; Klein, M.D.; Shapira, O.M. Ascending Aortic Dilatation Associated With Bicuspid Aortic Valve. Circulation 2009, 119, 880–890. [Google Scholar] [CrossRef] [PubMed]
- Davies, R.R.; Kaple, R.K.; Mandapati, D.; Gallo, A.; Botta, D.M.; Elefteriades, J.A.; Coady, M.A. Natural History of Ascending Aortic Aneurysms in the Setting of an Unreplaced Bicuspid Aortic Valve. Ann. Thorac. Surg. 2007, 83, 1338–1344. [Google Scholar] [CrossRef] [PubMed]
- Prakash, S.K.; Bossé, Y.; Muehlschlegel, J.D.; Michelena, H.I.; Limongelli, G.; Della Corte, A.; Pluchinotta, F.R.; Russo, M.G.; Evangelista, A.; Benson, D.W.; et al. A roadmap to investigate the genetic basis of bicuspid aortic valve and its complications: Insights from the International BAVCon (Bicuspid Aortic Valve Consortium). J. Am. Coll. Cardiol. 2014, 64, 832–839. [Google Scholar] [CrossRef] [Green Version]
- Pileggi, S.; De Chiara, B.; Magnoli, M.; Franzosi, M.G.; Merlanti, B.; Bianchini, F.; Moreo, A.; Romeo, G.; Russo, C.F.; Rizzo, S.; et al. Sequencing of NOTCH1 gene in an Italian population with bicuspid aortic valve: Preliminary results from the GISSI OUTLIERS VAR study. Gene 2019, 715, 143970. [Google Scholar] [CrossRef]
- Kent, K.C.; Crenshaw, M.L.; Goh, D.L.; Dietz, H.C. Genotype–phenotype correlation in patients with bicuspid aortic valve and aneurysm. J. Thorac. Cardiovasc. Surg. 2013, 146, 158–165.e1. [Google Scholar] [CrossRef] [Green Version]
- Luyckx, I.; Maccarrick, G.; Kempers, M.; Meester, J.; Geryl, C.; Rombouts, O.; Peeters, N.; Claes, C.; Boeckx, N.; Sakalihasan, N.; et al. European Journal of Human Genetics Confirmation of the role of pathogenic SMAD6 variants in bicuspid aortic valve-related aortopathy. Eur. J. Hum. Genet. 2019, 5, 3552. [Google Scholar]
- Gillis, E.; Kumar, A.A.; Luyckx, I.; Preuss, C.; Cannaerts, E.; van de Beek, G.; Wieschendorf, B.; Alaerts, M.; Bolar, N.; Vandeweyer, G.; et al. Candidate Gene Resequencing in a Large Bicuspid Aortic Valve-Associated Thoracic Aortic Aneurysm Cohort: SMAD6 as an Important Contributor. Front. Physiol. 2017, 8, 400. [Google Scholar] [CrossRef] [PubMed]
- Gould, R.A.; Aziz, H.; Woods, C.E.; Seman-Senderos, M.A.; Sparks, E.; Preuss, C.; Wünnemann, F.; Bedja, D.; Moats, C.R.; McClymont, S.A.; et al. ROBO4 variants predispose individuals to bicuspid aortic valve and thoracic aortic aneurysm. Nat. Genet. 2019, 51, 42–50. [Google Scholar] [CrossRef]
- Luyckx, I.; Kumar, A.A.; Reyniers, E.; Dekeyser, E.; Vanderstraeten, K.; Vandeweyer, G.; Wünnemann, F.; Preuss, C.; Mazzella, J.-M.; Goudot, G.; et al. Copy number variation analysis in bicuspid aortic valve-related aortopathy identifies TBX20 as a contributing gene. Eur. J. Hum. Genet. 2019, 27, 1033–1043. [Google Scholar] [CrossRef] [PubMed]
- Coady, M.A.; Davies, R.R.; Roberts, M.; Goldstein, L.J.; Rogalski, M.J.; Rizzo, J.A.; Hammond, G.L.; Kopf, G.S.; Elefteriades, J.A. Familial Patterns of Thoracic Aortic Aneurysms. Arch. Surg. 1999, 134, 361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LeMaire, S.A.; McDonald, M.-L.N.; Guo, D.; Russell, L.; Miller, C.C.; Johnson, R.J.; Bekheirnia, M.R.; Franco, L.M.; Nguyen, M.; Pyeritz, R.E.; et al. Genome-wide association study identifies a susceptibility locus for thoracic aortic aneurysms and aortic dissections spanning FBN1 at 15q21.1. Nat. Genet. 2011, 43, 996–1000. [Google Scholar] [CrossRef]
- Weerakkody, R.; Ross, D.; Parry, D.A.; Ziganshin, B.; Vandrovcova, J.; Gampawar, P.; Abdullah, A.; Biggs, J.; Dumfarth, J.; Ibrahim, Y.; et al. Targeted genetic analysis in a large cohort of familial and sporadic cases of aneurysm or dissection of the thoracic aorta. Genet. Med. 2018, 20, 1414–1422. [Google Scholar] [CrossRef]
- Ke, T.; Han, M.; Zhao, M.; Wang, Q.K.; Zhang, H.; Zhao, Y.; Ruan, X.; Li, H.; Xu, C.; Sun, T. Alpha-actin-2 mutations in Chinese patients with a non-syndromatic thoracic aortic aneurysm. BMC Med. Genet. 2016, 17, 45. [Google Scholar] [CrossRef] [Green Version]
- Van De Laar, I.M.B.H.; Arbustini, E.; Loeys, B.; Björck, E.; Murphy, L.; Groenink, M.; Kempers, M.; Timmermans, J.; Roos-Hesselink, J.; Benke, K.; et al. European reference network for rare vascular diseases (VASCERN) consensus statement for the screening and management of patients with pathogenic ACTA2 variants. Orphanet J. Rare Dis. 2019, 14, 264. [Google Scholar] [CrossRef]
- Hannuksela, M.; Stattin, E.-L.; Klar, J.; Ameur, A.; Johansson, B.; Sörensen, K.; Carlberg, B. A novel variant in MYLK causes thoracic aortic dissections: Genotypic and phenotypic description. BMC Med. Genet. 2016, 17, 61. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Guo, D.-C.; Cao, J.; Gong, L.; Kamm, K.E.; Regalado, E.; Li, L.; Shete, S.; He, W.-Q.; Zhu, M.-S.; et al. Mutations in myosin light chain kinase cause familial aortic dissections. Am. J. Hum. Genet. 2010, 87, 701–707. [Google Scholar] [CrossRef] [Green Version]
- Wallace, S.E.; Regalado, E.S.; Gong, L.; Janda, A.L.; Guo, D.-C.; Russo, C.F.; Kulmacz, R.J.; Hanna, N.; Jondeau, G.; Boileau, C.; et al. MYLK pathogenic variants aortic disease presentation, pregnancy risk, and characterization of pathogenic missense variants. Genet. Med. 2019, 21, 144–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, L.; Vranckx, R.; Van Kien, P.K.; Lalande, A.; Boisset, N.; Mathieu, F.; Wegman, M.; Glancy, L.; Gasc, J.-M.; Brunotte, F.; et al. Mutations in myosin heavy chain 11 cause a syndrome associating thoracic aortic aneurysm/aortic dissection and patent ductus arteriosus. Nat. Genet. 2006, 38, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Ziganshin, B.A.; Bailey, A.E.; Coons, C.; Dykas, D.; Charilaou, P.; Tanriverdi, L.H.; Liu, L.; Tranquilli, M.; Bale, A.E.; Elefteriades, J.A. Routine Genetic Testing for Thoracic Aortic Aneurysm and Dissection in a Clinical Setting. Ann. Thorac. Surg. 2015, 100, 1604–1611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renard, M.; Francis, C.; Ghosh, R.; Scott, A.F.; Witmer, P.D.; Adès, L.C.; Andelfinger, G.U.; Arnaud, P.; Boileau, C.; Callewaert, B.L.; et al. Clinical Validity of Genes for Heritable Thoracic Aortic Aneurysm and Dissection. J. Am. Coll. Cardiol. 2018, 72, 605–615. [Google Scholar] [CrossRef]
- De Backer, J.; Bondue, A.; Budts, W.; Evangelista, A.; Gallego, P.; Jondeau, G.; Loeys, B.; Peña, M.L.; Teixido-Tura, G.; van de Laar, I.; et al. Genetic counselling and testing in adults with congenital heart disease: A consensus document of the ESC Working Group of Grown-Up Congenital Heart Disease, the ESC Working Group on Aorta and Peripheral Vascular Disease and the European Society of Human G. Eur. J. Prev. Cardiol. 2019, 27, 1423–1435. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Luo, M.; Fu, Y.; Cao, Y.; Yin, K.; Li, W.; Meng, C.; Ma, Y.; Zhang, J.; Fan, Y.; et al. Genetic testing of 248 Chinese aortopathy patients using a panel assay. Sci. Rep. 2016, 6, 33002. [Google Scholar] [CrossRef] [Green Version]
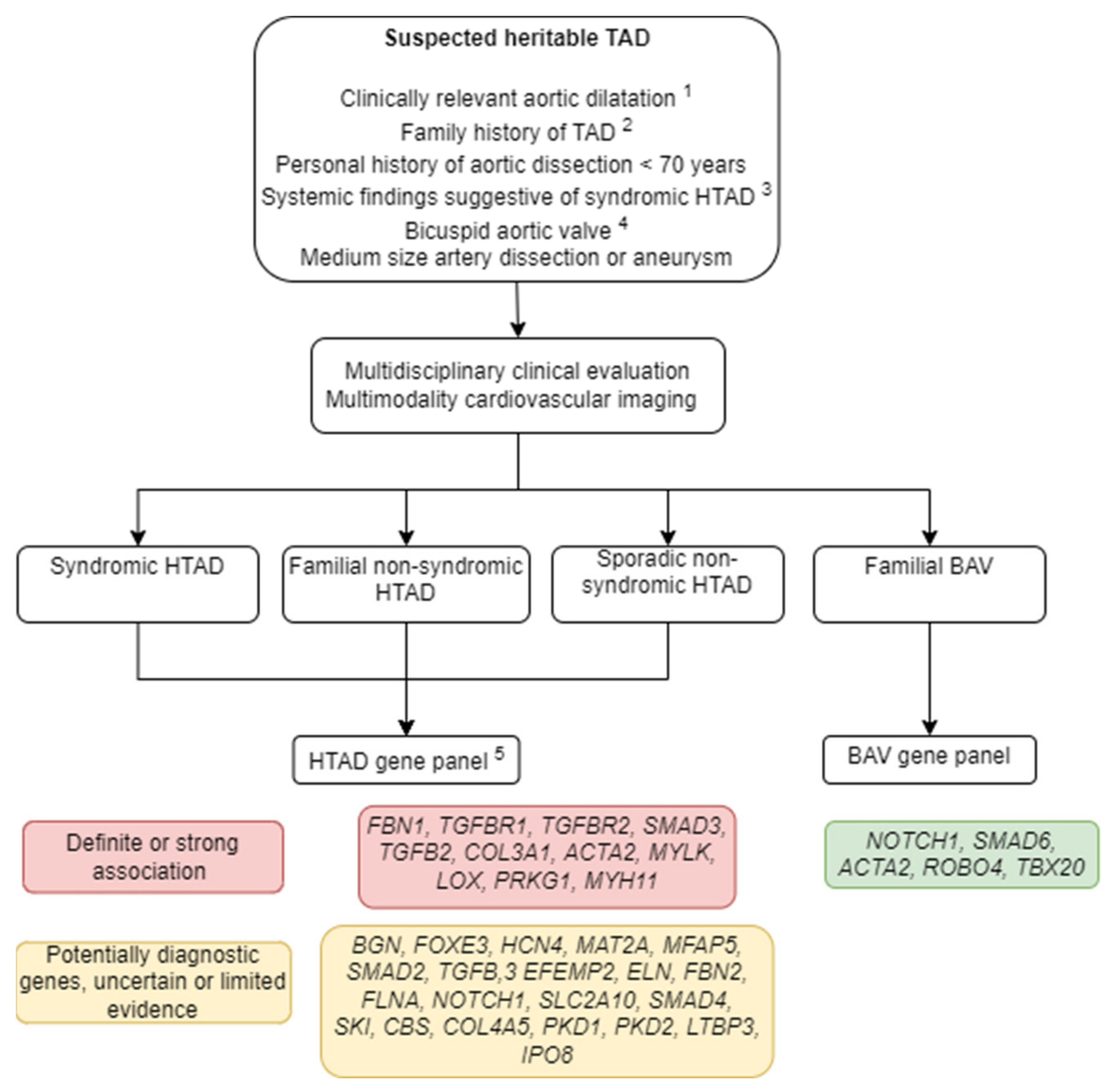
Figure 1.
Clinical and genetic evaluation of suspected heritable thoracic aortic disease. HTAD: Heritable thoracic aortic disease; TAD: thoracic aortic disease, BAV: bicuspid aortic valve. 1 This includes one of the following: (a) Aortic root diameter Z-score >3.5 (b) Aortic root diameter Z-score ≥3 in patients <18 years (c) Aortic root diameter Z-score 2.5–3.5 in patients 18–60 years (d) Aortic root diameter Z-score 2.5–3.5 without hypertension in patients >60 years. 2 1st- or 2nd-degree relative with aortic dissection or aneurysm aged <60 years or sudden cardiac death <45 years. 3 Ectopia lentis, hypertelorism, bifid uvula, premature and extensive osteoarthritis, club feet, cleft palate and other systemic features. 4 Familial cases, syndromic or extra phenotypic characteristics, young patients with severe disease. 5 In familial and/or syndromic cases consider exome sequencing, copy number variant testing if next-generation sequencing is negative and there is high clinical suspicion of HTAD or for research purposes.
Figure 1.
Clinical and genetic evaluation of suspected heritable thoracic aortic disease. HTAD: Heritable thoracic aortic disease; TAD: thoracic aortic disease, BAV: bicuspid aortic valve. 1 This includes one of the following: (a) Aortic root diameter Z-score >3.5 (b) Aortic root diameter Z-score ≥3 in patients <18 years (c) Aortic root diameter Z-score 2.5–3.5 in patients 18–60 years (d) Aortic root diameter Z-score 2.5–3.5 without hypertension in patients >60 years. 2 1st- or 2nd-degree relative with aortic dissection or aneurysm aged <60 years or sudden cardiac death <45 years. 3 Ectopia lentis, hypertelorism, bifid uvula, premature and extensive osteoarthritis, club feet, cleft palate and other systemic features. 4 Familial cases, syndromic or extra phenotypic characteristics, young patients with severe disease. 5 In familial and/or syndromic cases consider exome sequencing, copy number variant testing if next-generation sequencing is negative and there is high clinical suspicion of HTAD or for research purposes.


{kind=link}
Table 1.
Criteria for genetic testing in patients with suspected heritable thoracic aortic disease.
| Patient’s Age | Criteria for Genetic Testing a |
|---|---|
| All ages | Familial HTAD (≥2 relatives identified) |
| Personal history of aortic dissection | |
| Aortic root diameter Z-score >3.5 | |
| <18 years old | Aortic root diameter Z-score ≥3 |
| 18–60 years old | Aortic root diameter Z-score 2.5–3.5 |
| >60 | Aortic root diameter Z-score 2.5–3.5 without hypertension |
HTAD: Heritable thoracic aortic disease, a Based on expert opinion [105].
Table 2.
Genotype–phenotype correlations to be considered before aortic surgery in patients with heritable thoracic aortic disease.
Table 2.
Genotype–phenotype correlations to be considered before aortic surgery in patients with heritable thoracic aortic disease.
| Aortopathy | Genes | Aortic Size to Consider Surgical Operation a | |||
|---|---|---|---|---|---|
| ≥40–42 mm | ≥45 mm | ≥50 mm | ≥55 mm | ||
| Marfan syndrome | FBN1 | ≥1 high-risk factor b | No high-risk factors b | ||
| Loeys–Dietz syndrome | SMAD2, SMAD3, TGFB2, TGFB3, TGFBR1, TGFBR2 | TGFBR2 (females, severe phenotypic features, low body surface area) | SMAD3, TGFB2, TGFBR1, TGFBR2 | SMAD2, TGFB3 | |
| Vascular Ehlers–Danlos syndrome | COL3A1, COL1A1 | Surgical intervention not recommended due to high surgical risk; only as emergency treatment | |||
| Meester–Loeys syndrome | BGN (X--linked) | Aortic dissection in a patient with aortic size of 45 mm has been reported; individualized expert assessment is needed | |||
| Filamin-related HTAD | FLNA (X-linked) | Aortic rupture at aortic size of 42 mm has been reported; individualized expert assessment is needed | |||
| LOX-related HTAD | LOX | Aortic dissection at aortic size >50 mm has been reported; individualized expert assessment is needed | |||
| Gorlin–Chaudhry–Moss syndrome | SLC25A24 | Aortic dissection at aortic size of 51 mm has been reported; individualized expert assessment is needed | |||
| Shprintzen–Goldberg syndrome | SKI | Milder phenotype than Marfan syndrome or Loeys–Dietz syndrome; individualized expert assessment is needed | |||
| Importin-related HTAD | IPO8 (biallelic) | No aortic dissections have been reported | |||
| LTBP3-related HTAD | LTBP3 (biallelic) | Aortic dissection at aortic size >50 mm has been reported; individualized expert assessment is needed | |||
| ACTA2-related HTAD | ACTA2 (especially p.R179, p.R258) | Dissections have been reported at aortic aneurysm sizes of as low as 40 mm. More aggressive phenotype with p.R179 (multisystemic smooth muscle dysfunction syndrome) and p.R258 variants | |||
| Arterial tortuosity syndrome | SLC2A10 | No dissections reported; individualized expert assessment is needed | |||
| Turner syndrome | Complete or partial X chromosome monosomy | ASI >25 mm/m2, especially if ≥1 high-risk factor present c | |||
| Pompe disease | GAA (X-linked) | Aortic dissection at aortic size >50 mm has been reported; individualized expert assessment is needed | |||
| Fabry disease | GLA (X-linked) | No aortic dissections have been reported | |||
| Mucopolysaccharidoses | IDUA, IDS, SGSH, NAGLU, HGSNAT, GNS, GALNS, GLB1, ARSB, GUSB, HYAL1 | No aortic dissections have been reported | |||
| Bicuspid aortic valve-related aortopathy | NOTCH1, ACTA2, SMAD6, ROBO4, TBX20 | ≥1 high-risk factor present d | No high-risk factors present d | ||
| Nonsyndromic HTAD (familial or sporadic) | MYLK, MYH11, PRKG1, ACTA2, LOX, MFAP5, FOXE3, MAT2A, SMAD2, SMAD4, NOTCH1, PLOD1, TGFB2, TGFBR2, FBN1, FBN2, LTBP3 | MYLK, MYH11, PRKG1, ACTA2 (especially p.R179, p.R258) | All other genes; individualized expert assessment is needed | ||
HTAD: heritable thoracic aortic disease. a A lower threshold may be considered if there is a family history of aortic dissection, pregnancy or rapid aortic size increase on an individual basis; b High-risk factors for Marfan syndrome patients are: (a) aortic diameter at the sinuses of Valsalva ≥5 cm, (b) rapid increase in aortic dilatation (≥3 mm per year), (c) family history of aortic dissection at a low aortic size, (d) progressive aortic regurgitation, (e) personal history of spontaneous vascular dissection and (f) desire for pregnancy. c High-risk factors for Turner syndrome: (a) Bicuspid aortic valve, (b) elongation of the transverse aorta, (c) Aortic coarctation or (d) hypertension. d High-risk factors for bicuspid aortic valve: (a) Family history of dissection at a low diameter, (b) desire for pregnancy, (c) systemic hypertension, (d) increase >3 mm/year.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Papatheodorou, E.; Degiannis, D.; Anastasakis, A. Genetics of Heritable Thoracic Aortic Disease. Cardiogenetics 2022, 12, 63-79. https://doi.org/10.3390/cardiogenetics12010006
AMA Style
Papatheodorou E, Degiannis D, Anastasakis A. Genetics of Heritable Thoracic Aortic Disease. Cardiogenetics. 2022; 12(1):63-79. https://doi.org/10.3390/cardiogenetics12010006
Chicago/Turabian StylePapatheodorou, Efstathios, Dimitrios Degiannis, and Aris Anastasakis. 2022. "Genetics of Heritable Thoracic Aortic Disease" Cardiogenetics 12, no. 1: 63-79. https://doi.org/10.3390/cardiogenetics12010006