
KIT Mutations Correlate with Higher Galectin Levels and Brain Metastasis in Breast and Non-Small Cell Lung Cancer

 ,
,  , ,
, ,
Abstract
:Simple Summary
Abstract
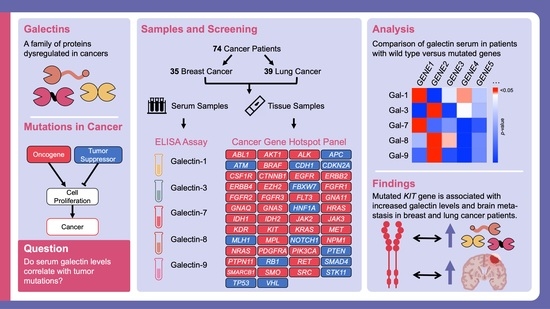
1. Introduction
1.1. Cancer-Critical Genes
1.2. Galectins and Their Role in Cancer
2. Materials and Methods
2.1. Patient Samples
2.2. Galectin Profiling
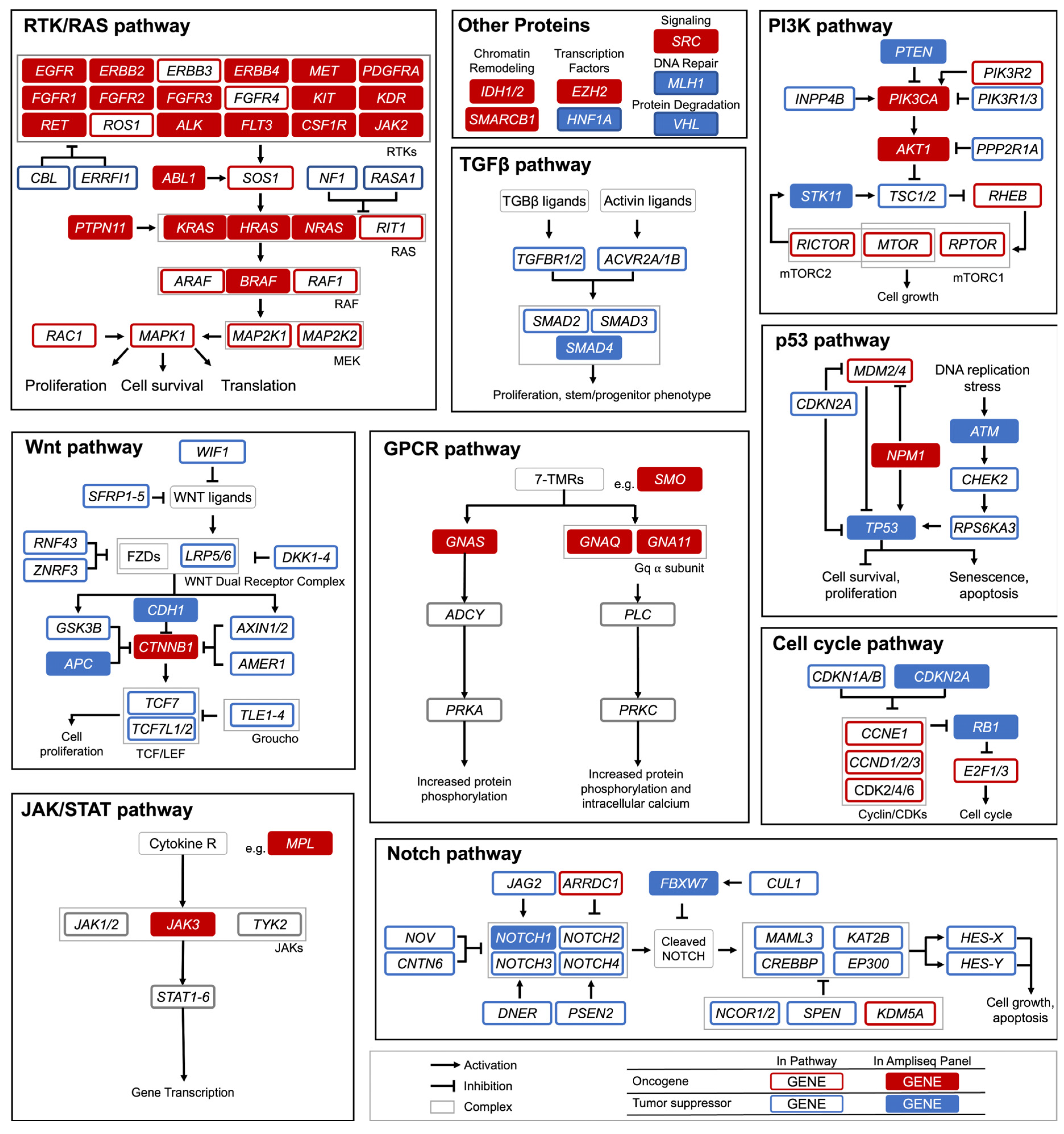
2.3. Cancer HotSpot Panel
2.4. Data Analysis
3. Results
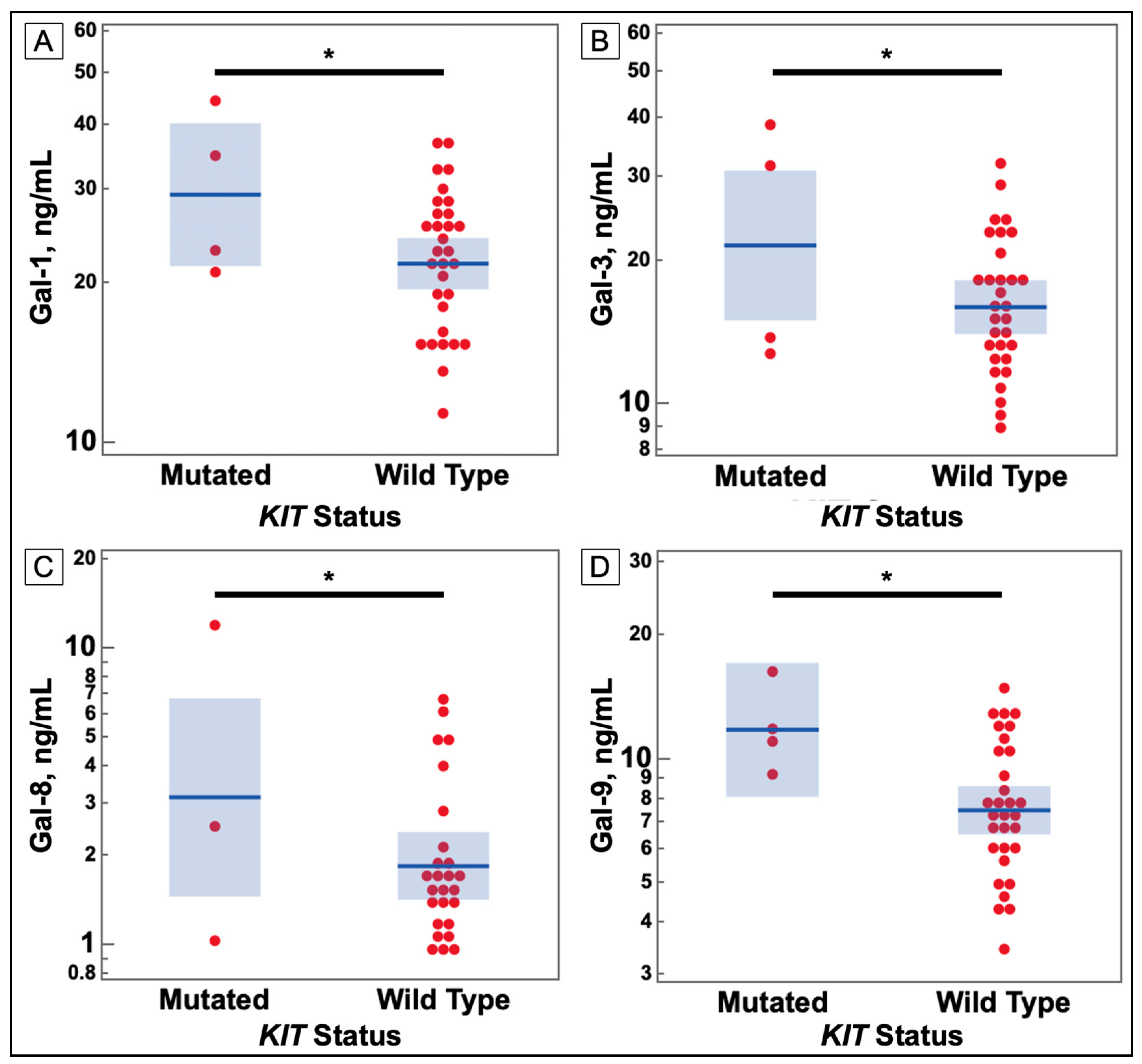
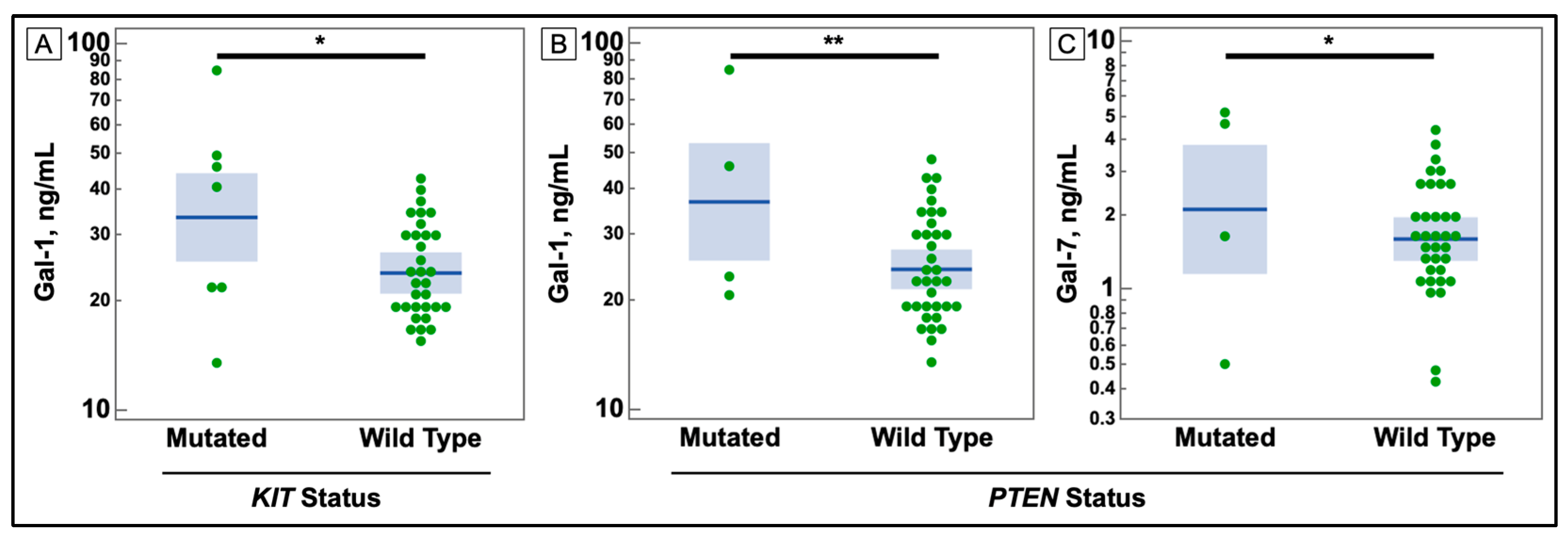
3.1. Serum Galectin Levels
3.2. Mutation Frequency
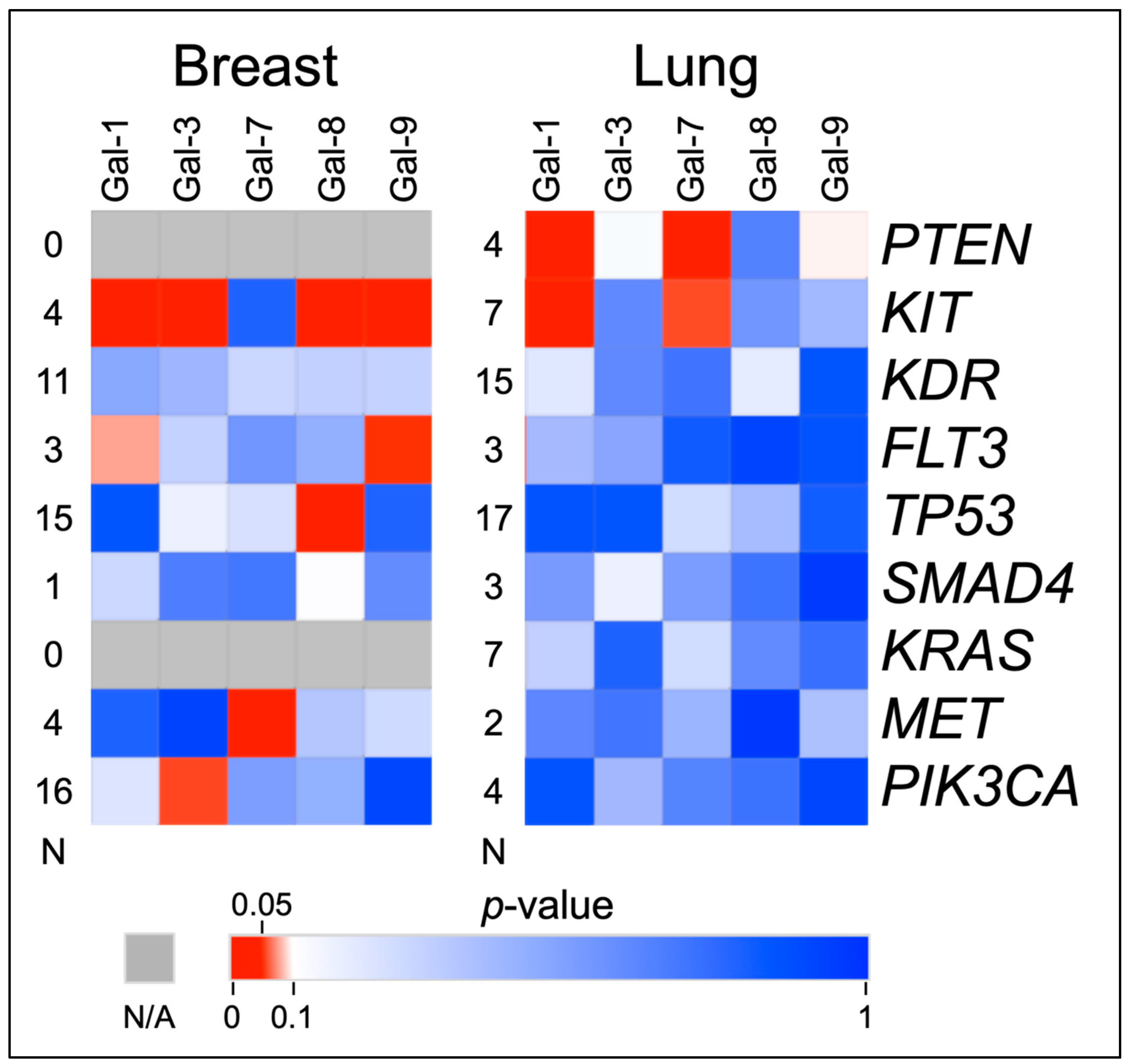
3.3. Associations with Galectins
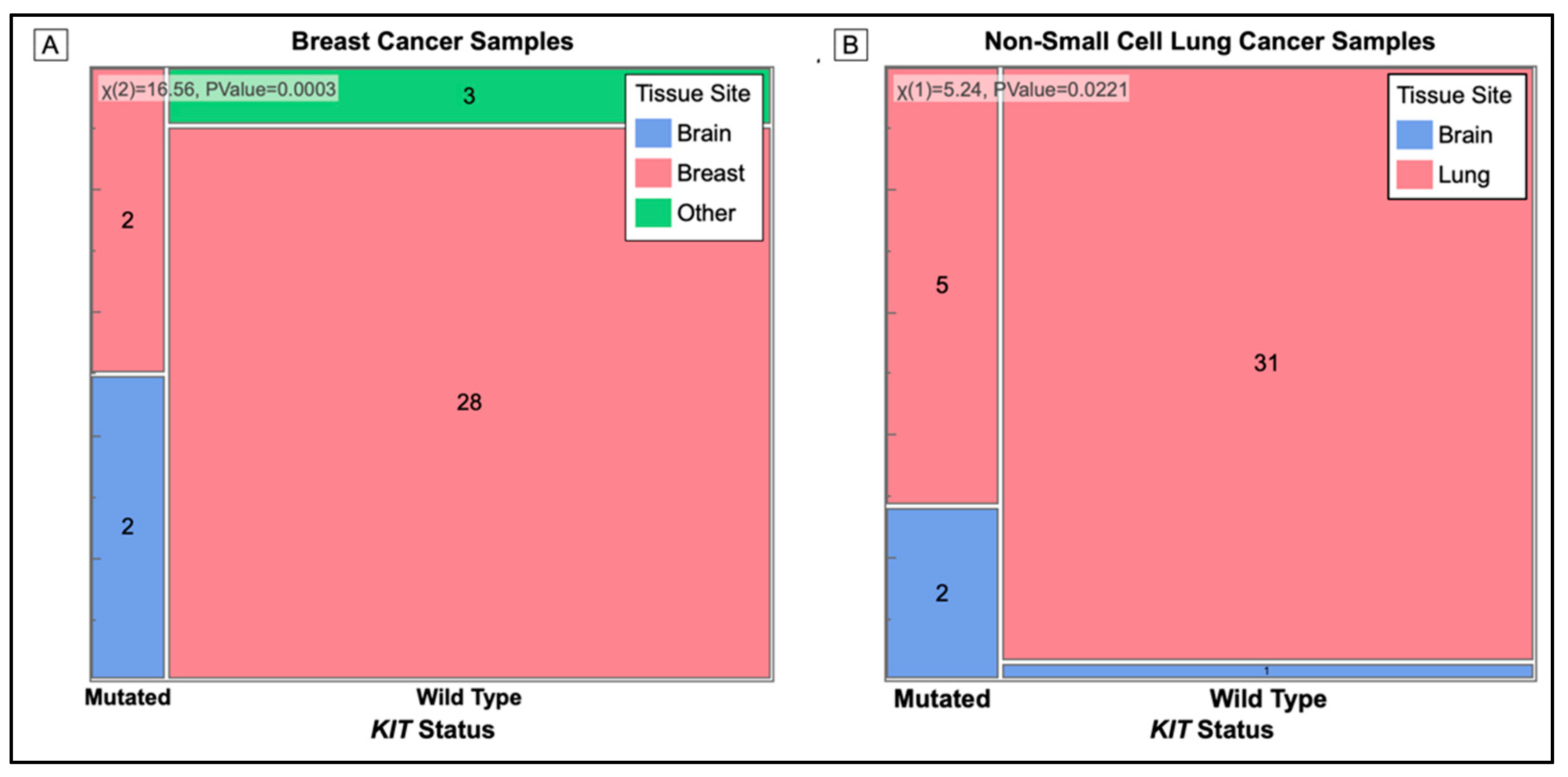
3.4. Associations with Brain Metastases
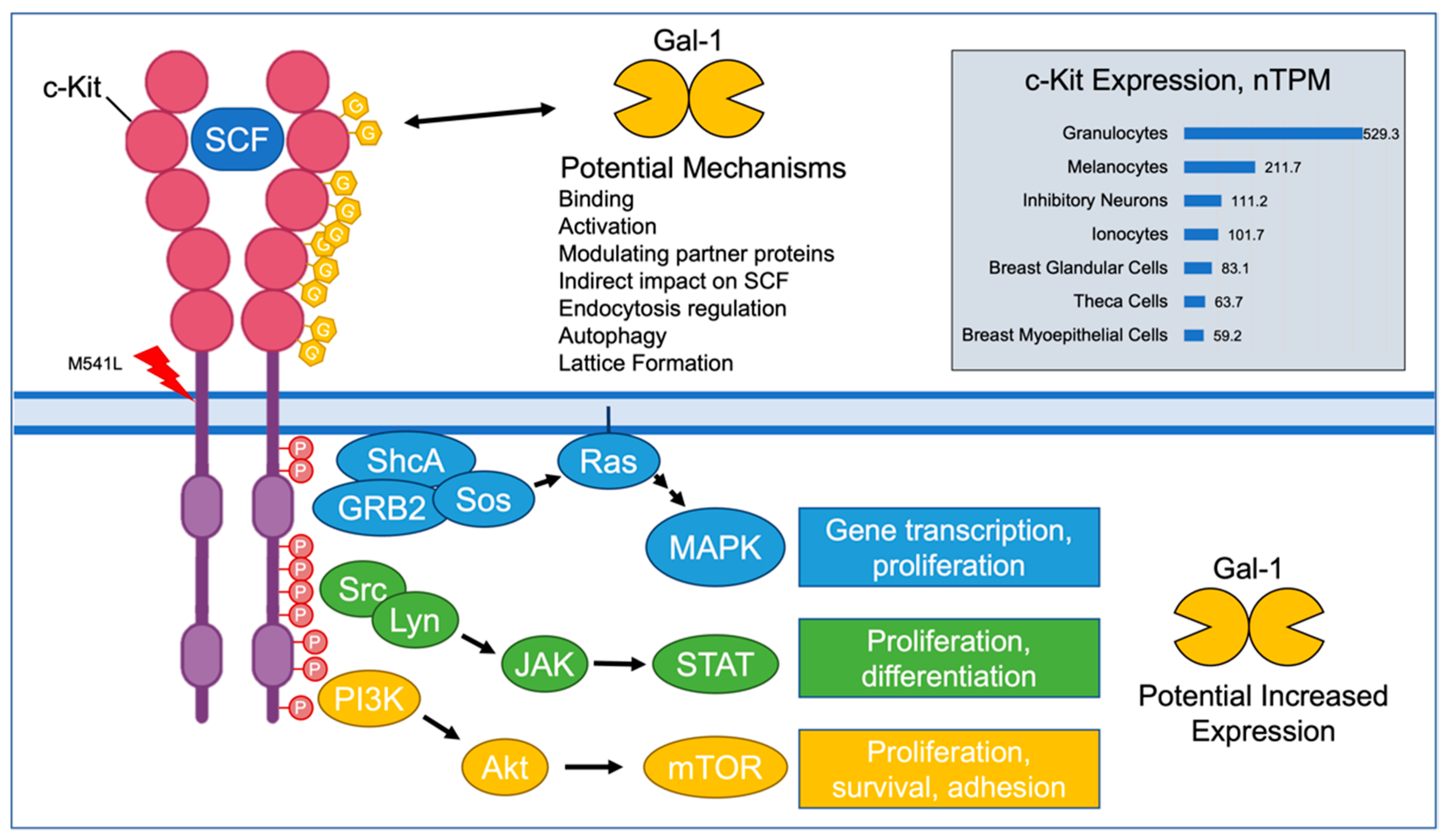
4. Discussion
4.1. Impact of Findings
4.2. Study Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Dillekås, H.; Rogers, M.S.; Straume, O. Are 90% of deaths from cancer caused by metastases? Cancer Med. 2019, 8, 5574–5576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schouten, L.J.; Rutten, J.; Huveneers, H.A.M.; Twijnstra, A. Incidence of brain metastases in a cohort of patients with carcinoma of the breast, colon, kidney, and lung and melanoma. Cancer 2002, 94, 2698–2705. [Google Scholar] [CrossRef] [PubMed]
- Barnholtz-Sloan, J.S.; Sloan, A.E.; Davis, F.G.; Vigneau, F.D.; Lai, P.; Sawaya, R.E. Incidence Proportions of Brain Metastases in Patients Diagnosed (1973 to 2001) in the Metropolitan Detroit Cancer Surveillance System. J. Clin. Oncol. 2004, 22, 2865–2872. [Google Scholar] [CrossRef]
- Bailey, M.H.; Tokheim, C.; Porta-Pardo, E.; Sengupta, S.; Bertrand, D.; Weerasinghe, A.; Colaprico, A.; Wendl, M.C.; Kim, J.; Reardon, B.; et al. Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 2018, 173, 371–385.E18. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018, 173, 321–337.e10. [Google Scholar] [CrossRef] [Green Version]
- Cardoso, H.; Figueira, M.; Socorro, S. The stem cell factor (SCF)/c-KIT signalling in testis and prostate cancer. J. Cell Commun. Signal. 2017, 11, 297–307. [Google Scholar] [CrossRef] [Green Version]
- Wandzioch, E.; Edling, C.E.; Palmer, R.H.; Carlsson, L.; Hallberg, B. Activation of the MAP kinase pathway by c-Kit is PI-3 kinase dependent in hematopoietic progenitor/stem cell lines. Blood 2004, 104, 51–57. [Google Scholar] [CrossRef]
- Linnekin, D.; Mou, S.; DeBerry, C.S.; Weiler, S.R.; Keller, J.R.; Ruscetti, F.W.; Longo, D.L. Stem Cell Factor, the JAK-STAT Pathway and Signal Transduction. Leuk. Lymphoma 1997, 27, 439–444. [Google Scholar] [CrossRef]
- Karlsson, M.; Zhang, C.; Méar, L.; Zhong, W.; Digre, A.; Katona, B.; Sjöstedt, E.; Butler, L.; Odeberg, J.; Dusart, P.; et al. A single–cell type transcriptomics map of human tissues. Sci. Adv. 2021, 7, eabh2169. [Google Scholar] [CrossRef]
- Ashman, L.K. The biology of stem cell factor and its receptor C-kit. Int. J. Biochem. Cell Biol. 1999, 31, 1037–1051. [Google Scholar] [CrossRef]
- Ashman, L.K.; Griffith, R. Therapeutic targeting of c-KIT in cancer. Expert Opin. Investig. Drugs 2012, 22, 103–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woodman, S.E.; Trent, J.C.; Stemke-Hale, K.; Lazar, A.J.; Pricl, S.; Pavan, G.M.; Fermeglia, M.; Gopal, Y.N.V.; Yang, D.; Podoloff, D.A.; et al. Activity of dasatinib against L576P KIT mutant melanoma: Molecular, cellular, and clinical correlates. Mol. Cancer Ther. 2009, 8, 2079–2085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bodemer, C.; Hermine, O.; Palmérini, F.; Yang, Y.; Grandpeix-Guyodo, C.; Leventhal, P.S.; Hadj-Rabia, S.; Nasca, L.; Georgin-Lavialle, S.; Cohen-Akenine, A.; et al. Pediatric Mastocytosis Is a Clonal Disease Associated with D816V and Other Activating c-KIT Mutations. J. Investig. Dermatol. 2010, 130, 804–815. [Google Scholar] [CrossRef] [Green Version]
- Pathania, S.; Pentikäinen, O.T.; Singh, P.K. A holistic view on c-Kit in cancer: Structure, signaling, pathophysiology and its inhibitors. Biochim. et Biophys. Acta 2021, 1876, 188631. [Google Scholar] [CrossRef]
- Slimane, K.; Andre, F.; Delaloge, S.; Dunant, A.; Perez, A.; Grenier, J.; Massard, C.; Spielmann, M. Risk factors for brain relapse in patients with metastatic breast cancer. Ann. Oncol. 2004, 15, 1640–1644. [Google Scholar] [CrossRef]
- Crivellari, D.; Pagani, O.; Veronesi, A.; Lombardi, D.; Nolè, F.; Thürlimann, B.; Hess, D.; Borner, M.; Bauer, J.; Martinelli, G.; et al. High incidence of central nervous system involvement in patients with metastatic or locally advanced breast cancer treated with epirubicin and docetaxel. Ann. Oncol. 2001, 12, 353–356. [Google Scholar] [CrossRef]
- Miller, K.D.; Weathers, T.; Haney, L.G.; Timmerman, R.; Dickler, M.; Shen, J.; Jr, G.W.S. Occult central nervous system involvement in patients with metastatic breast cancer: Prevalence, predictive factors and impact on overall survival. Ann. Oncol. 2003, 14, 1072–1077. [Google Scholar] [CrossRef]
- Tham, Y.-L.; Sexton, K.; Kramer, R.; Hilsenbeck, S.; Elledge, R. Primary breast cancer phenotypes associated with propensity for central nervous system metastases. Cancer 2006, 107, 696–704. [Google Scholar] [CrossRef]
- Lin, N.U.; Winer, E.P. Brain Metastases: The HER2 Paradigm. Clin. Cancer Res. 2007, 13, 1648–1655. [Google Scholar] [CrossRef]
- Gori, S.; Rimondini, S.; De Angelis, V.; Colozza, M.; Bisagni, G.; Moretti, G.; Sidoni, A.; Basurto, C.; Aristei, C.; Anastasi, P.; et al. Central Nervous System Metastases in HER-2–Positive Metastatic Breast Cancer Patients Treated with Trastuzumab: Incidence, Survival, and Risk Factors. Oncol. 2007, 12, 766–773. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.-Y.; Na, I.I.; Kim, C.H.; Park, S.; Baek, H.; Yang, S.H. EGFR Mutation and Brain Metastasis in Pulmonary Adenocarcinomas. J. Thorac. Oncol. 2014, 9, 195–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, I.; Zaorsky, N.; Palmer, J.D.; Mehra, R.; Lu, B. Targeting brain metastases in ALK-rearranged non-small-cell lung cancer. Lancet Oncol. 2015, 16, e510–e521. [Google Scholar] [CrossRef]
- Johung, K.L.; Yeh, N.; Desai, N.B.; Williams, T.M.; Lautenschlaeger, T.; Arvold, N.D.; Ning, M.S.; Attia, A.; Lovly, C.M.; Goldberg, S.; et al. Extended Survival and Prognostic Factors for Patients With ALK-Rearranged Non–Small-Cell Lung Cancer and Brain Metastasis. J. Clin. Oncol. 2016, 34, 123–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawyers, C. Targeted cancer therapy. Nature 2004, 432, 294–297. [Google Scholar] [CrossRef] [PubMed]
- Hanna, N.; Johnson, D.; Temin, S.; Baker, S.; Brahmer, J.; Ellis, P.M.; Giaccone, G.; Hesketh, P.J.; Jaiyesimi, I.; Leighl, N.B.; et al. Systemic Therapy for Stage IV Non–Small-Cell Lung Cancer: American Society of Clinical Oncology Clinical Practice Guideline Update. J. Clin. Oncol. 2017, 35, 3484–3515. [Google Scholar] [CrossRef]
- Planchard, D.; Popat, S.; Kerr, K.; Novello, S.; Smit, E.F.; Faivre-Finn, C.; Mok, T.S.; Reck, M.; Van Schil, P.E.; Hellmann, M.D.; et al. Metastatic non-small cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2018, 29 (Suppl. 4), iv192–iv237. [Google Scholar] [CrossRef]
- Wu, Y.-L.; Planchard, D.; Lu, S.; Sun, H.; Yamamoto, N.; Kim, D.-W.; Tan, D.; Yang, J.-H.; Azrif, M.; Mitsudomi, T.; et al. Pan-Asian adapted Clinical Practice Guidelines for the management of patients with metastatic non-small-cell lung cancer: A CSCO–ESMO initiative endorsed by JSMO, KSMO, MOS, SSO and TOS. Ann. Oncol. 2019, 30, 171–210. [Google Scholar] [CrossRef] [Green Version]
- Cross, D.A.E.; Ashton, S.E.; Ghiorghiu, S.; Eberlein, C.; Nebhan, C.A.; Spitzler, P.J.; Orme, J.P.; Finlay, M.R.V.; Ward, R.A.; Mellor, M.J.; et al. AZD9291, an Irreversible EGFR TKI, Overcomes T790M-Mediated Resistance to EGFR Inhibitors in Lung Cancer. Cancer Discov. 2014, 4, 1046–1061. [Google Scholar] [CrossRef] [Green Version]
- Mok, T.S.; Wu, Y.-L.; Ahn, M.-J.; Garassino, M.C.; Kim, H.R.; Ramalingam, S.S.; Shepherd, F.A.; He, Y.; Akamatsu, H.; Theelen, W.S.M.E.; et al. Osimertinib or Platinum–Pemetrexed in EGFR T790M–Positive Lung Cancer. N. Engl. J. Med. 2017, 376, 629–640. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.-L.; Ahn, M.-J.; Garassino, M.C.; Han, J.-Y.; Katakami, N.; Kim, H.R.; Hodge, R.; Kaur, P.; Brown, A.P.; Ghiorghiu, D.; et al. CNS Efficacy of Osimertinib in Patients With T790M-Positive Advanced Non–Small-Cell Lung Cancer: Data From a Randomized Phase III Trial (AURA3). J. Clin. Oncol. 2018, 36, 2702–2709. [Google Scholar] [CrossRef] [PubMed]
- Reungwetwattana, T.; Nakagawa, K.; Cho, B.C.; Cobo, M.; Cho, E.K.; Bertolini, A.; Bohnet, S.; Zhou, C.; Lee, K.H.; Nogami, N.; et al. CNS Response to Osimertinib Versus Standard Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors in Patients With Untreated EGFR-Mutated Advanced Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2018, 36, 3290–3297. [Google Scholar] [CrossRef] [PubMed]
- Raposo, C.; Canelas, A.; Barros, M. Human Lectins, Their Carbohydrate Affinities and Where to Find Them. Biomolecules 2021, 11, 188. [Google Scholar] [CrossRef] [PubMed]
- Di Lella, S.; Sundblad, V.; Cerliani, J.P.; Guardia, C.M.; Estrin, D.A.; Vasta, G.R.; Rabinovich, G.A. When Galectins Recognize Glycans: From Biochemistry to Physiology and Back Again. Biochemistry 2011, 50, 7842–7857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miko, E.; Meggyes, M.; Doba, K.; Barakonyi, A.; Szereday, L. Immune Checkpoint Molecules in Reproductive Immunology. Front. Immunol. 2019, 10, 846. [Google Scholar] [CrossRef] [PubMed]
- Méndez-Huergo, S.P.; Blidner, A.G.; A Rabinovich, G. Galectins: Emerging regulatory checkpoints linking tumor immunity and angiogenesis. Curr. Opin. Immunol. 2017, 45, 8–15. [Google Scholar] [CrossRef]
- Rabinovich, G.A.; Toscano, M.A. Turning ’sweet’ on immunity: Galectin–glycan interactions in immune tolerance and inflammation. Nat. Rev. Immunol. 2009, 9, 338–352. [Google Scholar] [CrossRef]
- Lau, K.; Partridge, E.A.; Grigorian, A.; Silvescu, C.I.; Reinhold, V.N.; Demetriou, M.; Dennis, J.W. Complex N-Glycan Number and Degree of Branching Cooperate to Regulate Cell Proliferation and Differentiation. Cell 2007, 129, 123–134. [Google Scholar] [CrossRef] [Green Version]
- Raz, A. Biological Modulation by Lectins and Their Ligands in Tumor Progression and Metastasis. Anti-Cancer Agents Med. Chem. 2008, 8, 22–36. [Google Scholar] [CrossRef] [Green Version]
- Markowska, A.I.; Liu, F.-T.; Panjwani, N. Galectin-3 is an important mediator of VEGF- and bFGF-mediated angiogenic response. J. Exp. Med. 2010, 207, 1981–1993. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.-T.; Rabinovich, G.A. Galectins as modulators of tumour progression. Nat. Rev. Cancer 2005, 5, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Mayoral, M.A.; Mayoral, C.; Meneses, A.; Villalvazo, L.; Guzmán, A.; Espinosa, B.; Ochoa, J.L.; Zenteno, E.; Guevara, J. Identification of Galectin-3 and Mucin-Type O-Glycans in Breast Cancer and Its Metastasis to Brain. Cancer Investig. 2008, 26, 615–623. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.-W.; Chang, H.-T.; Chen, C.-H.; Lin, S.-W.; Hsu, T.-L.; Wong, C.-H. Galectin-3 Binding Protein and Galectin-1 Interaction in Breast Cancer Cell Aggregation and Metastasis. J. Am. Chem. Soc. 2015, 137, 9685–9693. [Google Scholar] [CrossRef] [PubMed]
- Kocoglu, H.; Topcu, T.O.; Kavgaci, H.; Gunaldi, M.; Akyol, M.; Mentese, A.; Yaman, S.O.; Orem, A.; Ozdemir, F.; Aydin, F. The clinical importance of serum galectin-3 levels in breast cancer patients with and without metastasis. J. Cancer Res. Ther. 2018, 14, S583–S586. [Google Scholar] [CrossRef] [PubMed]
- Campion, C.G.; Labrie, M.; Lavoie, G.; St-Pierre, Y. Expression of Galectin-7 Is Induced in Breast Cancer Cells by Mutant p53. PLoS ONE 2013, 8, e72468. [Google Scholar] [CrossRef] [PubMed]
- Hadari, Y.; Arbel-Goren, R.; Levy, Y.; Amsterdam, A.; Alon, R.; Zakut, R.; Zick, Y. Galectin-8 binding to integrins inhibits cell adhesion and induces apoptosis. J. Cell Sci. 2000, 113, 2385–2397. [Google Scholar] [CrossRef] [PubMed]
- André, S.; Kojima, S.; Yamazaki, N.; Fink, C.; Kaltner, H.; Kayser, K.; Gabius, H.-J. Galectins-1 and -3 and their ligands in tumor biology: Non-uniform properties in cell-surface presentation and modulation of adhesion to matrix glycoproteins for various tumor cell lines, in biodistribution of free and liposome-bound galectins and in thei. J. Cancer Res. Clin. Oncol. 1999, 125, 461–474. [Google Scholar] [CrossRef]
- Grosset, A.-A.; Labrie, M.; Gagné, D.; Vladoiu, M.-C.; Gaboury, L.; Doucet, N.; St-Pierre, Y. Cytosolic galectin-7 impairs p53 functions and induces chemoresistance in breast cancer cells. BMC Cancer 2014, 14, 1–10. [Google Scholar] [CrossRef]
- Trebo, A.; Ditsch, N.; Kuhn, C.; Heidegger, H.H.; Zeder-Goess, C.; Kolben, T.; Czogalla, B.; Schmoeckel, E.; Mahner, S.; Jeschke, U.; et al. High Galectin-7 and Low Galectin-8 Expression and the Combination of both are Negative Prognosticators for Breast Cancer Patients. Cancers 2020, 12, 953. [Google Scholar] [CrossRef] [Green Version]
- Yang, R.; Sun, L.; Li, C.-F.; Wang, Y.-H.; Yao, J.; Li, H.; Yan, M.; Chang, W.-C.; Hsu, J.-M.; Cha, J.-H.; et al. Galectin-9 interacts with PD-1 and TIM-3 to regulate T cell death and is a target for cancer immunotherapy. Nat. Commun. 2021, 12, 1–17. [Google Scholar] [CrossRef]
- Yasinska, I.M.; Sakhnevych, S.S.; Pavlova, L.; Teo Hansen Selnø, A.; Teuscher Abeleira, A.M.; Benlaouer, O.; Gonçalves Silva, I.; Mosimann, M.; Varani, L.; Bardelli, M.; et al. The Tim-3-Galectin-9 Pathway and Its Regulatory Mechanisms in Human Breast Cancer. Front. Immunol. 2019, 10, 1594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, P.-L.; Hung, J.-Y.; Huang, S.-K.; Chou, S.-H.; Cheng, D.-E.; Jong, Y.-J.; Hung, C.-H.; Yang, C.-J.; Tsai, Y.-M.; Hsu, Y.-L.; et al. Lung Cancer-Derived Galectin-1 Mediates Dendritic Cell Anergy through Inhibitor of DNA Binding 3/IL-10 Signaling Pathway. J. Immunol. 2010, 186, 1521–1530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, L.-Y.; Tang, S.-J.; Sun, G.-H.; Chou, T.-Y.; Yeh, T.-S.; Yu, S.-L.; Sun, K.-H. Galectin-1 Promotes Lung Cancer Progression and Chemoresistance by Upregulating p38 MAPK, ERK, and Cyclooxygenase-2. Clin. Cancer Res. 2012, 18, 4037–4047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, X.-N.; Dang, Y.-F.; Gong, F.-L.; Guo, X.-L. Role and regulation mechanism of Gal-3 in non-small cell lung cancer and its potential clinical therapeutic significance. Chem. Interactions 2019, 309, 108724. [Google Scholar] [CrossRef]
- Schulkens, I.A.; Heusschen, R.; Boogaart, V.V.D.; Van Suylen, R.-J.; Dingemans, A.-M.C.; Griffioen, A.W.; Thijssen, V.L. Galectin Expression Profiling Identifies Galectin-1 and Galectin-9Δ5 as Prognostic Factors in Stage I/II Non-Small Cell Lung Cancer. PLoS ONE 2014, 9, e107988. [Google Scholar] [CrossRef]
- He, Y.; Jia, K.; Dziadziuszko, R.; Zhao, S.; Zhang, X.; Deng, J.; Wang, H.; Hirsch, F.R.; Zhou, C. Galectin-9 in non-small cell lung cancer. Lung Cancer 2019, 136, 80–85. [Google Scholar] [CrossRef]
- Dalotto-Moreno, T.; Croci, D.O.; Cerliani, J.P.; Martinez-Allo, V.C.; Dergan-Dylon, S.; Méndez-Huergo, S.P.; Stupirski, J.C.; Mazal, D.; Osinaga, E.; Toscano, M.; et al. Targeting Galectin-1 Overcomes Breast Cancer-Associated Immunosuppression and Prevents Metastatic Disease. Cancer Res. 2012, 73, 1107–1117. [Google Scholar] [CrossRef] [Green Version]
- Jung, E.-J.; Moon, H.-G.; Cho, B.I.; Jeong, C.-Y.; Joo, Y.-T.; Lee, Y.-J.; Hong, S.-C.; Choi, S.-K.; Ha, W.-S.; Kim, J.W.; et al. Galectin-1 expression in cancer-associated stromal cells correlates tumor invasiveness and tumor progression in breast cancer. Int. J. Cancer 2007, 120, 2331–2338. [Google Scholar] [CrossRef]
- Demers, M.; Rose, A.A.; Grosset, A.-A.; Biron-Pain, K.; Gaboury, L.; Siegel, P.M.; St-Pierre, Y. Overexpression of Galectin-7, A Myoepithelial Cell Marker, Enhances Spontaneous Metastasis of Breast Cancer Cells. Am. J. Pathol. 2010, 176, 3023–3031. [Google Scholar] [CrossRef]
- Danguy, A.; Rorive, S.; Decaestecker, C.; Bronckart, Y.; Kaltner, H.; Hadari, Y.R.; Goren, R.; Zich, Y.; Petein, M.; Salmon, I.; et al. Immunohistochemical profile of galectin-8 expression in benign and malignant tumors of epithelial, mesenchymatous and adipous origins, and of the nervous system. Histol. Histopathol. 2001, 16, 861–868. [Google Scholar] [CrossRef]
- Szöke, T.; Kayser, K.; Baumhäkel, J.-D.; Trojan, I.; Furak, J.; Tiszlavicz, L.; Horvath, A.; Szluha, K.; Gabius, H.-J.; Andre, S. Prognostic Significance of Endogenous Adhesion/Growth-Regulatory Lectins in Lung Cancer. Oncology 2005, 69, 167–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bassen, R.; Brichory, F.; Caulet-Maugendre, S.; Bidon, N.; Delaval, P.; Desrues, B.; Dazord, L. Expression of Po66-CBP, a type-8 galectin, in different healthy, tumoral and peritumoral tissues. Anticancer Res. 1999, 19, 5429–5433. [Google Scholar]
- Barrow, H.; Guo, X.; Wandall, H.H.; Pedersen, J.W.; Fu, B.; Zhao, Q.; Chen, C.; Rhodes, J.M.; Yu, L.-G. Serum Galectin-2, -4, and -8 Are Greatly Increased in Colon and Breast Cancer Patients and Promote Cancer Cell Adhesion to Blood Vascular Endothelium. Clin. Cancer Res. 2011, 17, 7035–7046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iurisci, I.; Tinari, N.; Natoli, C.; Angelucci, D.; Cianchetti, E.; Iacobelli, S. Concentrations of galectin-3 in the sera of normal controls and cancer patients. Clin. Cancer Res. 2000, 6, 1389–1393. [Google Scholar]
- Blair, B.B.; Funkhouser, A.T.; Goodwin, J.L.; Strigenz, A.M.; Chaballout, B.H.; Martin, J.C.; Arthur, C.M.; Funk, C.R.; Edenfield, W.J.; Blenda, A.V. Increased Circulating Levels of Galectin Proteins in Patients with Breast, Colon, and Lung Cancer. Cancers 2021, 13, 4819. [Google Scholar] [CrossRef]
- Gong, H.C.; Honjo, Y.; Nangia-Makker, P.; Hogan, V.; Mazurak, N.; Bresalier, R.; Raz, A. The NH2 terminus of galectin-3 governs cellular compartmentalization and functions in cancer cells. Cancer Res. 1999, 59, 6239–6245. [Google Scholar]
- Lipkowitz, M.S.; Leal-Pinto, E.; Cohen, B.E.; Abramson, R.G. Galectin 9 is the sugar-regulated urate transporter/channel UAT. Glycoconj. J. 2002, 19, 491–498. [Google Scholar] [CrossRef]
- Menon, S.; Kang, C.-M.; Beningo, K.A. Galectin-3 secretion and tyrosine phosphorylation is dependent on the calpain small subunit, Calpain 4. Biochem. Biophys. Res. Commun. 2011, 410, 91–96. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Liu, D.; Fan, Y.; Li, X.; Xue, H.; Ma, Y.; Zhou, Y.; Tai, G. The Two Endocytic Pathways Mediated by the Carbohydrate Recognition Domain and Regulated by the Collagen-like Domain of Galectin-3 in Vascular Endothelial Cells. PLoS ONE 2012, 7, e52430. [Google Scholar] [CrossRef] [Green Version]
- Ideo, H.; Hoshi, I.; Yamashita, K.; Sakamoto, M. Phosphorylation and externalization of galectin-4 is controlled by Src family kinases. Glycobiology 2013, 23, 1452–1462. [Google Scholar] [CrossRef] [Green Version]
- Ohtsubo, K.; Marth, J.D. Glycosylation in Cellular Mechanisms of Health and Disease. Cell 2006, 126, 855–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dube, D.H.; Bertozzi, C.R. Glycans in Cancer and Inflammation — Potential for Therapeutics and Diagnostics. Nat. Rev. Drug Discov. 2005, 4, 477–488. [Google Scholar] [CrossRef] [PubMed]
- Adamczyk, B.; Tharmalingam, T.; Rudd, P.M. Glycans as cancer biomarkers. Biochim. et Biophys. Acta Gen. Subj. 2011, 1820, 1347–1353. [Google Scholar] [CrossRef] [PubMed]
- Bresalier, R.S.; Byrd, J.C.; Tessler, D.; Lebel, J.; Koomen, J.; Hawke, D.; Half, E.; Liu, K.-F.; Mazurek, N. A circulating ligand for galectin-3 is a haptoglobin-related glycoprotein elevated in individuals with colon cancer. Gastroenterology 2004, 127, 741–748. [Google Scholar] [CrossRef] [PubMed]
- Endo, K.; Kohnoe, S.; Tsujita, E.; Watanabe, A.; Nakashima, H.; Baba, H.; Maehara, Y. Galectin-3 expression is a potent prognostic marker in colorectal cancer. Anticancer Res. 2005, 25, 3117–3121. [Google Scholar]
- Barrow, H.; Rhodes, J.M.; Yu, L.-G. The role of galectins in colorectal cancer progression. Int. J. Cancer 2011, 129, 1–8. [Google Scholar] [CrossRef]
- Sakaki, M.; Oka, N.; Nakanishi, R.; Yamaguchi, K.; Fukumori, T.; Kanayama, H.-O. Serum level of galectin-3 in human bladder cancer. J. Med Investig. 2008, 55, 127–132. [Google Scholar] [CrossRef] [Green Version]
- Gheysen, L.; Soumoy, L.; Trelcat, A.; Verset, L.; Journe, F.; Saussez, S. New Treatment Strategy Targeting Galectin-1 against Thyroid Cancer. Cells 2021, 10, 1112. [Google Scholar] [CrossRef]
- Shimura, T.; Shibata, M.; Gonda, K.; Kofunato, Y.; Okada, R.; Ishigame, T.; Kimura, T.; Kenjo, A.; Kono, K.; Marubashi, S. Significance of Circulating Galectin-3 in Patients with Pancreatobiliary Cancer. Anticancer Res. 2017, 37, 4979–4986. [Google Scholar] [CrossRef] [Green Version]
- Masoodi, M.; Shah, Z.A.; Beigh, A.H.; Ahmad, S.Z.; Mir, A.W.; Yasin, B.; Rasool, R.; Masoodi, K.Z.; Bhat, G.M. Galectin-1 as a predictive biomarker in ovarian cancer. J. Ovarian Res. 2021, 14, 1–9. [Google Scholar] [CrossRef]
- Girotti, M.R.; Salatino, M.; Dalotto-Moreno, T.; Rabinovich, G.A. Sweetening the hallmarks of cancer: Galectins as multifunctional mediators of tumor progression. J. Exp. Med. 2019, 217, e20182041. [Google Scholar] [CrossRef] [PubMed]
- Wdowiak, K.; Francuz, T.; Gallego-Colon, E.; Ruiz-Agamez, N.; Kubeczko, M.; Grochoła, I.; Wojnar, J. Galectin Targeted Therapy in Oncology: Current Knowledge and Perspectives. Int. J. Mol. Sci. 2018, 19, 210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paz, A.; Haklai, R.; Elad-Sfadia, G.; Ballan, E.; Kloog, Y. Galectin-1 binds oncogenic H-Ras to mediate Ras membrane anchorage and cell transformation. Oncogene 2001, 20, 7486–7493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elad-Sfadia, G.; Haklai, R.; Balan, E.; Kloog, Y. Galectin-3 Augments K-Ras Activation and Triggers a Ras Signal That Attenuates ERK but Not Phosphoinositide 3-Kinase Activity. J. Biol. Chem. 2004, 279, 34922–34930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, S.; Ji, B.; Ramachandran, V.; Wang, H.; Hafley, M.; Logsdon, C.; Bresalier, R. Overexpressed Galectin-3 in Pancreatic Cancer Induces Cell Proliferation and Invasion by Binding Ras and Activating Ras Signaling. PLoS ONE 2012, 7, e42699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shalom-Feuerstein, R.; Cooks, T.; Raz, A.; Kloog, Y. Galectin-3 Regulates a Molecular Switch from N-Ras to K-Ras Usage in Human Breast Carcinoma Cells. Cancer Res. 2005, 65, 7292–7300. [Google Scholar] [CrossRef] [Green Version]
- Partridge, E.A.; Le Roy, C.; Di Guglielmo, G.M.; Pawling, J.; Cheung, P.; Granovsky, M.; Nabi, I.R.; Wrana, J.L.; Dennis, J.W. Regulation of Cytokine Receptors by Golgi N-Glycan Processing and Endocytosis. Science 2004, 306, 120–124. [Google Scholar] [CrossRef] [Green Version]
- Bresalier, R.S.; Mazurek, N.; Sternberg, L.R.; Byrd, J.C.; Yunker, C.K.; Nangia-Makker, P.; Raz, A. Metastasis of human colon cancer is altered by modifying expression of the β-galactoside-binding protein galectin 3. Gastroenterology 1998, 115, 287–296. [Google Scholar] [CrossRef]
- Folgueira, M.A.A.K.; Maistro, S.; Katayama, M.L.H.; Roela, R.A.; Mundim, F.G.L.; Nanogaki, S.; de Bock, G.H.; Brentani, M.M. Markers of breast cancer stromal fibroblasts in the primary tumour site associated with lymph node metastasis: A systematic review including our case series. Biosci. Rep. 2013, 33, 921–929. [Google Scholar] [CrossRef]
- Lotan, R.; Belloni, P.N.; Tressler, R.J.; Lotan, D.; Xu, X.-C.; Nicolson, G.L. Expression of galectins on microvessel endothelial cells and their involvement in tumour cell adhesion. Glycoconj. J. 1994, 11, 462–468. [Google Scholar] [CrossRef]
- Brûle, F.V.D.; Califice, S.; Garnier, F.; Fernandez, P.L.; Berchuck, A.; Castronovo, V. Galectin-1 Accumulation in the Ovary Carcinoma Peritumoral Stroma Is Induced by Ovary Carcinoma Cells and Affects Both Cancer Cell Proliferation and Adhesion to Laminin-1 and Fibronectin. Lab. Investig. 2003, 83, 377–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, M.H.; Hong, T.M.; Cheng, H.W.; Pan, S.H.; Liang, Y.R.; Hong, H.C.; Chiang, W.F.; Wong, T.Y.; Shieh, D.B.; Shiau, A.L.; et al. Galectin-1-Mediated tumor invasion and metastasis, Up-Regulated matrix metalloproteinase expression, and reorganized actin cytoskeletons. Mol. Cancer Res. 2009, 7, 311–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shih, T.-C.; Liu, R.; Wu, C.-T.; Li, X.; Xiao, W.; Deng, X.; Kiss, S.; Wang, T.; Chen, X.-J.; Carney, R.P.; et al. Targeting Galectin-1 Impairs Castration-Resistant Prostate Cancer Progression and Invasion. Clin. Cancer Res. 2018, 24, 4319–4331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, M.; Kaur, T.; Kamboj, S.S.; Singh, J. Roles of Galectin-7 in Cancer. Asian Pac. J. Cancer Prev. 2016, 17, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thijssen, V.L.; Heusschen, R.; Caers, J.; Griffioen, A.W. Galectin expression in cancer diagnosis and prognosis: A systematic review. Biochim. et Biophys. Acta 2015, 1855, 235–247. [Google Scholar] [CrossRef] [PubMed]
- Gluck, W.L.; Callahan, S.P.; Brevetta, R.A.; Stenbit, A.E.; Smith, W.M.; Martin, J.C.; Blenda, A.V.; Arce, S.; Edenfield, W.J. Efficacy of therapeutic plasma exchange in the treatment of penn class 3 and 4 cytokine release syndrome complicating COVID-19. Respir. Med. 2020, 175, 106188. [Google Scholar] [CrossRef] [PubMed]
- Shuford, S.; Lipinski, L.; Abad, A.; Smith, A.M.; Rayner, M.; O’Donnell, L.; Stuart, J.; Mechtler, L.L.; Fabiano, A.J.; Edenfield, J.; et al. Prospective prediction of clinical drug response in high-grade gliomas using an ex vivo 3D cell culture assay. Neuro-Oncology Adv. 2021, 3, vdab065. [Google Scholar] [CrossRef]
- rs3822214 RefSNP Report-dbSNP-NCBI. Available online: https://www.ncbi.nlm.nih.gov/snp/rs3822214#clinical_significance (accessed on 5 January 2022).
- Murakami, T.; Fukai, K.; Oiso, N.; Hosomi, N.; Kato, A.; Garganta, C.; Barnicoat, A.; Poppelaars, F.; Aquaron, R.; Paller, A.S.; et al. New KIT mutations in patients with piebaldism. J. Dermatol. Sci. 2004, 35, 29–33. [Google Scholar] [CrossRef]
- Foster, R.; Byrnes, E.; Meldrum, C.; Griffith, R.; Ross, G.; Upjohn, E.; Braue, A.; Scott, R.; Varigos, G.; Ferrao, P.; et al. Association of paediatric mastocytosis with a polymorphism resulting in an amino acid substitution (M541L) in the transmembrane domain of c-KIT. Br. J. Dermatol. 2008, 159, 1160–1169. [Google Scholar] [CrossRef]
- Inokuchi, K.; Yamaguchi, H.; Tarusawa, M.; Futaki, M.; Hanawa, H.; Tanosaki, S.; Dan, K. Abnormality of c-kit oncoprotein in certain patients with chronic myelogenous leukemia–potential clinical significance. Leukemia 2002, 16, 170–177. [Google Scholar] [CrossRef] [Green Version]
- Lobl, M.B.; Hass, B.; Clarey, D.; Higgins, S.; Wysong, A. Next-generation sequencing identifies novel single nucleotide polymorphisms in high-risk cutaneous squamous cell carcinoma: A pilot study. Exp. Dermatol. 2020, 29, 667–671. [Google Scholar] [CrossRef] [PubMed]
- Kruger, S.; Emig, M.; Lohse, P.; Ehninger, G.; Hochhaus, A.; Schackert, H.K. The c-kit (CD117) sequence variation M541L, but not N564K, is frequent in the general population, and is not associated with CML in Caucasians. Leukemia 2005, 20, 354–355. [Google Scholar] [CrossRef] [PubMed]
- Porębska, N.; Poźniak, M.; Matynia, A.; Żukowska, D.; Zakrzewska, M.; Otlewski, J.; Opaliński, Ł. Galectins as modulators of receptor tyrosine kinases signaling in health and disease. Cytokine Growth Factor Rev. 2021, 60, 89–106. [Google Scholar] [CrossRef] [PubMed]
- Obermann, J.; Priglinger, C.S.; Merl-Pham, J.; Geerlof, A.; Priglinger, S.; Götz, M.; Hauck, S.M. Proteome-wide Identification of Glycosylation-dependent Interactors of Galectin-1 and Galectin-3 on Mesenchymal Retinal Pigment Epithelial (RPE) Cells. Mol. Cell. Proteom. 2017, 16, 1528–1546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jastrzebski, K.; Zdżalik-Bielecka, D.; Mamińska, A.; Kalaidzidis, Y.; Hellberg, C.; Miaczynska, M. Multiple routes of endocytic internalization of PDGFRβ contribute to PDGF-induced STAT3 signaling. J. Cell Sci. 2016, 130, 577–589. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.-H.; Chen, Y.-L.; Lee, K.-H.; Chang, C.-C.; Cheng, T.-M.; Wu, S.-Y.; Tu, C.-C.; Tsui, W.-L. Glycosylation-dependent galectin-1/neuropilin-1 interactions promote liver fibrosis through activation of TGF-β- and PDGF-like signals in hepatic stellate cells. Sci. Rep. 2017, 7, 1–16. [Google Scholar] [CrossRef]
- Feng, Z.-C.; Riopel, M.; Popell, A.; Wang, R. A survival Kit for pancreatic beta cells: Stem cell factor and c-Kit receptor tyrosine kinase. Diabetologia 2015, 58, 654–665. [Google Scholar] [CrossRef] [Green Version]
- Shi, X.; Sousa, L.P.; Mandel-Bausch, E.M.; Tome, F.; Reshetnyak, A.V.; Hadari, Y.; Schlessinger, J.; Lax, I. Distinct cellular properties of oncogenic KIT receptor tyrosine kinase mutants enable alternative courses of cancer cell inhibition. Proc. Natl. Acad. Sci. 2016, 113, E4784–E4793. [Google Scholar] [CrossRef] [Green Version]
- Chandrashekar, D.S.; Bashel, B.; Balasubramanya, S.A.H.; Creighton, C.J.; Ponce-Rodriguez, I.; Chakravarthi, B.V.S.K.; Varambally, S. UALCAN: A portal for facilitating tumor subgroup gene expression and survival analyses. Neoplasia 2017, 19, 649–658. [Google Scholar] [CrossRef]
- Tsutsui, S.; Yasuda, K.; Suzuki, K.; Takeuchi, H.; Nishizaki, T.; Higashi, H.; Era, S. A loss of c-kit expression is associated with an advanced stage and poor prognosis in breast cancer. Br. J. Cancer 2006, 94, 1874–1878. [Google Scholar] [CrossRef]
- Foster, B.M.; Zaidi, D.; Young, T.R.; Mobley, M.E.; Kerr, B.A. CD117/c-kit in Cancer Stem Cell-Mediated Progression and Therapeutic Resistance. Biomedicines 2018, 6, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potti, A.; Moazzam, N.; Ramar, K.; Hanekom, D.S.; Kargas, S.; Koch, M. CD117 (c-KIT) overexpression in patients with extensive-stage small-cell lung carcinoma. Ann. Oncol. 2003, 14, 894–897. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Galectin | n | Mean, ng/mL | SD, ng/mL | Min-Max, ng/mL |
|---|---|---|---|---|
| Gal-1 | 35 | 23.50 | 7.48 | 11.30–43.92 |
| Gal-3 | 35 | 17.52 | 6.94 | 8.85–38.20 |
| Gal-7 | 35 | 1.77 | 1.85 | 0.58–11.69 |
| Gal-8 | 29 | 2.50 | 2.35 | 0.95–11.84 |
| Gal-9 | 35 | 8.40 | 3.21 | 3.42–16.12 |
| Galectin | n | Mean, ng/mL | SD, ng/mL | Min-Max, ng/mL |
|---|---|---|---|---|
| Gal-1 | 39 | 27.12 | 12.84 | 13.41–83.69 |
| Gal-3 | 38 | 17.77 | 6.98 | 4.52–37.82 |
| Gal-7 | 39 | 1.91 | 1.12 | 0.42–5.07 |
| Gal-8 | 31 | 2.73 | 8.02 | 0.56–45.71 |
| Gal-9 | 39 | 10.15 | 4.87 | 3.79–26.64 |
| Gene | Mutation | Count | Percent |
|---|---|---|---|
| PIK3CA | p.His1047Arg | 7 | 20.0% |
| p.Ile391Met | 5 | 14.3% | |
| p.Glu545Lys | 4 | 10.3% | |
| Other | 2 | 5.7% | |
| Total | 18 | 51.4% | |
| TP53 | p.Pro72Arg | 5 | 14.3% |
| p.Arg273His | 2 | 5.7% | |
| Other | 10 | 28.6% | |
| Total | 17 | 48.6% | |
| KDR | p.Gln472His | 11 | 31.4% |
| Total | 11 | 31.4% | |
| KIT | p.Met541Leu | 3 | 8.6% |
| p.Val530Ile | 1 | 2.9% | |
| Total | 4 | 11.4% | |
| MET | p.Asn375Ser | 2 | 5.7% |
| p.Met362Thr | 2 | 5.7% | |
| Total | 4 | 11.4% | |
| Other | Other | 14 | 40.0% |
| Gene | Mutation | Count | Percent |
|---|---|---|---|
| TP53 | p.Pro72Arg | 4 | 10.2% |
| Other | 14 | 35.9% | |
| Total | 18 | 46.2% | |
| KDR | p.Gln472His | 15 | 38.5% |
| p.Val1356Ala | 1 | 2.6% | |
| Total | 16 | 41.0% | |
| KIT | p.Met541Leu | 6 | 15.4% |
| p.Glu849Gln | 1 | 2.6% | |
| Total | 7 | 18.0% | |
| KRAS | p.Gly12Asp | 2 | 5.1% |
| p.Gly12Cys | 2 | 5.1% | |
| p.Gly13Cys | 2 | 5.1% | |
| p.Gly13Asp | 1 | 2.6% | |
| Total | 7 | 18.0% | |
| PIK3CA | p.Ile391Met | 3 | 7.7% |
| p.His1047Arg | 1 | 2.6% | |
| Total | 4 | 10.3% | |
| PTEN | p.Arg173fs | 1 | 2.6% |
| p.Gly165Ter | 1 | 2.6% | |
| p.Gly244Cys | 1 | 2.6% | |
| p.Met1Ile | 1 | 2.6% | |
| Total | 4 | 10.3% | |
| Other | Other | 29 | 74.4% |
| Cancer | Mutated KIT | Wild-Type KIT | |||
|---|---|---|---|---|---|
| Brain Met | No Brain Met | Brain Met | No Brain Met | Odds Ratio | |
| Breast | 2 | 2 | 0 | 31 | - |
| Lung | 2 | 5 | 1 | 31 | 12.4 |
| Total | 4 | 7 | 1 | 62 | 35.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Funkhouser, A.T.; Strigenz, A.M.; Blair, B.B.; Miller, A.P.; Shealy, J.C.; Ewing, J.A.; Martin, J.C.; Funk, C.R.; Edenfield, W.J.; Blenda, A.V. KIT Mutations Correlate with Higher Galectin Levels and Brain Metastasis in Breast and Non-Small Cell Lung Cancer. Cancers 2022, 14, 2781. https://doi.org/10.3390/cancers14112781
Funkhouser AT, Strigenz AM, Blair BB, Miller AP, Shealy JC, Ewing JA, Martin JC, Funk CR, Edenfield WJ, Blenda AV. KIT Mutations Correlate with Higher Galectin Levels and Brain Metastasis in Breast and Non-Small Cell Lung Cancer. Cancers. 2022; 14(11):2781. https://doi.org/10.3390/cancers14112781
Chicago/Turabian StyleFunkhouser, Avery T., Alexander M. Strigenz, Bailey B. Blair, Andrew P. Miller, Jonah C. Shealy, Joseph A. Ewing, Julie C. Martin, Christopher R. Funk, William J. Edenfield, and Anna V. Blenda. 2022. "KIT Mutations Correlate with Higher Galectin Levels and Brain Metastasis in Breast and Non-Small Cell Lung Cancer" Cancers 14, no. 11: 2781. https://doi.org/10.3390/cancers14112781