
The Cellular Prion Protein and the Hallmarks of Cancer


1
Centre de Recherche des Cordeliers, Université de Paris, INSERM, Sorbonne Université, F-75006 Paris, France
2
Department of Biology, Institut du Cancer Paris CARPEM, APHP, Hôpital Européen Georges Pompidou, F-75015 Paris, France
*
Author to whom correspondence should be addressed.
Cancers 2021, 13(19), 5032; https://doi.org/10.3390/cancers13195032
Submission received: 8 September 2021
/
Revised: 30 September 2021
/
Accepted: 5 October 2021
/
Published: 8 October 2021
(This article belongs to the Special Issue Feature Paper from Journal Reviewers)
Abstract
:Simple Summary
The cellular prion protein PrPC is best known for its involvement, under its pathogenic isoform, in a group of neurodegenerative diseases. Notwithstanding, an emerging role for PrPC in various cancer-associated processes has attracted increasing attention over recent years. PrPC is overexpressed in diverse types of solid cancers and has been incriminated in various aspects of cancer biology, most notably proliferation, migration, invasion and metastasis, as well as resistance to cytotoxic agents. This article aims to provide a comprehensive overview of the current knowledge of PrPC with respect to the hallmarks of cancer, a reference framework encompassing the major characteristics of cancer cells.
Abstract
Beyond its causal involvement in a group of neurodegenerative diseases known as Transmissible Spongiform Encephalopathies, the cellular prion protein PrPC is now taking centre stage as an important contributor to cancer progression in various types of solid tumours. The prion cancer research field has progressively expanded in the last few years and has yielded consistent evidence for an involvement of PrPC in cancer cell proliferation, migration and invasion, therapeutic resistance and cancer stem cell properties. Most recent data have uncovered new facets of the biology of PrPC in cancer, ranging from its control on enzymes involved in immune tolerance to its radio-protective activity, by way of promoting angiogenesis. In the present review, we aim to summarise the body of literature dedicated to the study of PrPC in relation to cancer from the perspective of the hallmarks of cancer, the reference framework defined by Hanahan and Weinberg.
1. Introduction
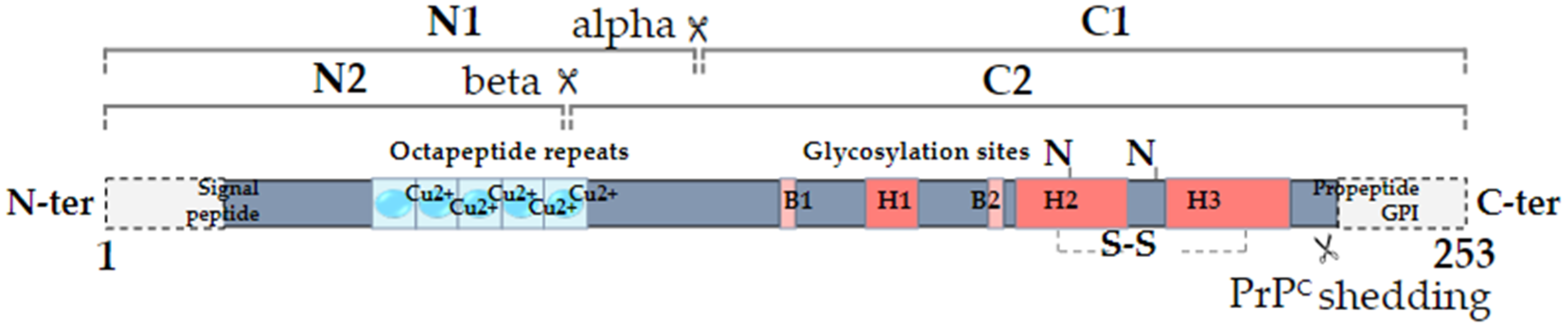
The cellular prion protein PrPC was discovered in the mid-1980s as the normal counterpart of the scrapie prion protein, denoted PrPSc, itself responsible for the development of a group of fatal neurodegenerative diseases known as Transmissible Spongiform Encephalopathies or prion diseases [1]. PrPC, encoded by the PRNP gene located on chromosome 20 in humans, is a ubiquitous protein that is highly conserved from fish to mammals [2]. It is a small glycoprotein of 253 amino acids subject to various types of post-translational modifications: removal of a N-terminal signal peptide responsible for the trafficking of the protein to the endoplasmic reticulum for subsequent maturation, replacement of the C-terminus with a glycosyl-phosphatidylinositol (GPI) moiety that allows PrPC anchoring at the extracellular plasma membrane, formation of a disulfide bridge and potential N-glycosylation on two Asparagine residues (reviewed in [3]). Beyond being majorly GPI-anchored at the cell membrane, PrPC may additionally exist as two topological transmembrane variants with either the N-terminus (CtmPrP) or C-terminus (NtmPrP) portion in the cytosol, possibly accounting for the interaction with cytosolic partners [3]. From a structure–function point of view, PrPC is composed of a N-terminal, intrinsically disordered domain, also referred to as “flexible tail”, a central hydrophobic domain and a C-terminal globular domain (Figure 1). A notable feature within the N-terminal domain is the presence of four histidine-containing octapeptide tandem repeats, which are involved in the binding of divalent ions such as copper or zinc, themselves promoting the endocytosis of PrPC [4]. In addition to full-length isoforms, several proteolytic processes can generate various truncated or soluble forms of PrPC (reviewed in [5]). First, the so-called alpha-cleavage, occurring at position 111/112, generates a N-terminal fragment termed N1 and a C-terminal, GPI-anchored fragment termed C1 (Figure 1). This alpha-cleavage occurs under physiological conditions and influences the endocytosis of PrPC as well as its interaction with diverse partners [5]. The beta-cleavage generating N2 and C2 fragments takes place within the octarepeat region (Figure 1). It is mostly described as a reactive oxygen species-dependent reaction sustaining the protective role of PrPC against oxidative stress [5]. Finally, a far C-terminal cleavage is responsible for the production of “shed PrPC”, which nearly encompasses the full sequence of PrPC (Figure 1). Shed PrPC is, however, distinct from soluble PrPC as it results from the phospholipase-C mediated hydrolysis of the GPI anchor [5]. Altogether, miscellaneous glycosylation and proteolytic processes in fine generate a variety of PrPC isoforms that may underlie its wide range of functions [6]. One should further bear in mind that the various soluble isoforms, N1, N2, shed PrPC as well as phospholipase-C-released PrPC, have the capacity to signal to neighbouring cells (see [5] for review). This also holds true for exosomal PrPC, as will be discussed below.
The variety of PrPC isoforms may explain why PrPC has been ascribed a plethora of functions, ranging from broad roles in the physiology of the central nervous system, resistance to various types of stresses, cell fate and differentiation, cell adhesion and cell signalling [6]. Unravelling the physiological roles exerted by PrPC has long emerged as a powerful strategy to understand how the corruption of these functions may contribute to pathological contexts, not only prion diseases [7] but also other disorders, including Alzheimer’s disease, immune disorders or cancer [8]. Obviously, the research dedicated to prion and cancer has lagged behind that of neurodegeneration, but this field is now becoming the focus of growing interest. We here chose to provide a comprehensive review of the current knowledge relating to PrPC in cancer through the lens of the hallmarks of cancer. As such, the contribution of PrPC to the emergence and/or maintenance of cancer stem cell properties or the potential therapeutic strategies to target this protein in cancer will not be discussed here and have been covered by several reviews [9,10,11,12]. The hallmarks of cancer are a reference framework introduced by Hanahan and Weinberg over 20 years ago [13] and further refined in 2011 [14]. It summarises the fundamental capacities endowing cancer cells with the ability to develop and escape control by the organism. In this review, we will summarise the overall data relating to the biology of PrPC in cancer according to each hallmark, except the enabling replicative immortality hallmark due to a lack of data on this axis. We further highlight some findings pertaining to the physiological function of PrPC and discuss their potential implications in the field of cancer.
2. Sustaining Proliferative Signalling
Since sustained proliferative capacity arguably represents one of the most fundamental traits of cancer cells, the contribution of PrPC to cancer cell proliferation has been extensively studied. The first demonstration that PrPC drives the proliferation of cancer cells was brought by the team of Daiming Fan using the SGC7901 and AGS gastric cancer cell lines [15]. In the low PrPC-expressing SGC7901 cells, PrP overexpression promoted an increase in cell proliferation in vitro, as well as tumour growth in xenografted nude mice [15]. To the opposite, PrPC silencing in the high PrPC-expressing AGS cell line triggered a reduction in their proliferative index [15]. At a mechanistic level, PrPC was shown to foster the transition from the G0/G1 to the S phase and to transcriptionally control the expression of Cyclin D1 via the PI3/AKT signalling pathway. From a structure-function point of view, it is interesting to note that the action of PrPC requires the presence of the N-terminal domain of the protein [15], in line with the importance of this region in the coupling of PrPC to PI3K/AKT signalling in non-tumoral cells [16].
Similarly, Li and colleagues documented a reduction in cell proliferation and in vivo tumour growth upon PrPC silencing in the pancreatic cancer cell lines BxPC3 and Pan 02.03 [17]. In a follow-up study based on the same cellular models together with the Capan-1 pancreatic cell line, PrPC was found to control the levels of Ki67 [18], a key marker of cell proliferation [19]. Interestingly, the decrease in cell proliferation observed after PrPC silencing in Capan-1 cells was abrogated upon a concomitant overexpression of the activated form of NOTCH1 [18]. This observation has to be brought together with the PrPC-dependent control on the Notch pathway that we documented in neural stem and progenitor cells [20]. A correlation between PrPC and Ki67 levels was also reported by Lopes et al. in a large cohort of patients with glioblastoma [21]. As with gastric and pancreatic cell lines, the latter study demonstrated a pro-proliferative action of PrPC in the U87 glioma cell line and corresponding xenografts, which was dependent upon the interaction of PrPC with its ligand STI1 [21]. The proliferative role of PrPC in glioblastoma was also confirmed in U87 cells grown as spheres to mimic glioblastoma stem cells [22] as well as in primary tumour cells [9]. In addition, PrPC expression was reported to vary according to the cell cycle in U87 glioma cells with significantly higher levels in the G2/M versus G1/S phase [23]. The PrPC-dependent control of proliferation was also exemplified in schwannoma [24] and colorectal cancer [25,26,27,28,29,30]. In the context of colorectal cancer, we notably brought to light a PrPC-dependent activation of the integrin linked kinase (ILK) that relays its control on cell proliferation [26]. Adding another layer of complexity to the picture, Yun et al. recently reported that the proliferation of various colorectal cancer cells can be sustained by exosomes derived from the same cells grown under hypoxia in a PrPC-dependent manner [31]. In this setting, two non-mutually exclusive mechanisms may be at play: the proliferation of recipient cells may be directly regulated by exosomal PrPC, the level of which is increased following hypoxia [31], or it may additionally depend upon other exosomal proteins whose abundance in exosomes is influenced by the expression of PrPC in cancer cells. In this respect, it is also worth noting that PrPC regulates the balance between exosome biogenesis and autophagy [32]. Thus, we may surmise that high PrPC-expressing cancer cells produce abundant levels of exosomes, enriched in PrPC, that may sustain their proliferation in an autocrine and paracrine manner, especially in a hypoxic environment.
We many finally note that the PrPC-dependent regulation of proliferation in the context of cancer may be viewed as a gain of its normal physiological function resulting from its overexpression. Indeed, the contribution of PrPC to normal cell proliferation has been extensively documented (reviewed in [6]), most notably in the context of stem cells (reviewed in [11]). The physiological PrPC-dependent regulation of proliferation may involve a modulation of the Epidermal Growth Factor Receptor (EGFR) activity as described by Llorens et al. [33]. Incidentally, PrPC and EGFR were shown to co-localise and to interact, as inferred by co-immunoprecipitation experiments, in the HT29 colorectal cell line [34]. These overall observations warrant investigating the signalling pathways through which PrPC sustains the proliferation of cancer cells and its potential functional interactions with growth factor receptors.
3. Evading Growth Suppressors
This hallmark corresponds to the ability of cancer cells to circumvent anti-growth signals [14]. They may do so by bypassing the activity of suppressors of proliferation such as TP53 and RB, encoding p53 and the retinoblastoma-associated protein, respectively, evading mechanisms of contact inhibition and/or corrupting anti-growth signalling circuitries such as the Transforming Growth Factor β (TGFβ) pathway. Although a direct contribution of PrPC to growth suppressors evasion has yet to be fully investigated, some observations are worth considering.
First, several studies have uncovered a modulation of p53 expression and activity by PrPC and its proteolytic fragments (reviewed in [35]). On the one hand, the full length PrPC was shown to up-regulate p53 activity and mRNA levels in neuronal cells upon exposure to the apoptotic inducer staurosporine, thereby sensitising cells to cell death [36]. This also holds true for the cleaved C1 fragment of PrPC, as shown in HEK293 cells [37]. On the other hand, the N1 soluble fragment of PrPC was found to exert an opposite effect on p53 and to protect cells from the full length and C1 PrPC-mediated potentiation of cell death [38]. Importantly, this set of studies, as well as others [39,40], indicated that overexpressed PrPC has no impact on the p53 pathway in basal conditions, i.e., in the absence of pro-apoptotic signals.
Nevertheless, an activation of p53 signalling was reported upon PrP overexpression in skeletal muscle [41]. In a more cancer-relevant context, Liang et al. documented that PrPC silencing promotes an increase in the expression levels of p53 in the gastric cancer cell line AGS, while an opposite effect was obtained upon PrPC overexpression [42]. Thus, until now, these scarce studies have provided only a glimpse of the potential regulation of p53 by PrPC, which may notably depend upon the relative abundance of its different isoforms.
Regarding contact inhibition, an interesting observation is the upregulation of PrPC in various types of Merlin-deficient tumours, including schwannoma and mesothelioma [24]. Merlin, encoded by the neurofibromatosis type 2 (NF2) gene, is a well-described regulator of cell–cell attachment, whose loss of function allows cells to evade contact inhibition [43]. Of note, we recently demonstrated that PrPC levels positively control the phosphorylation of NF2 on serine 518, itself negatively regulating NF2 activity, in colorectal cancer [26]. Mechanistically, PrPC operates via ILK [26], previously described as an upstream regulator of the NF2-Hippo pathway in various types of cancer cells [44]. Accordingly, we documented that the PrPC-ILK module promotes the activation of YAP/TAZ [26], the two transcriptional effectors of the Hippo pathway, which play a key role in promoting the poor-prognosis mesenchymal subtype of colorectal cancer [28,45]. Since YAP/TAZ are major orchestrators of organ growth and contact inhibition [46], their upstream regulation by PrPC clearly delineates a link between PrPC and contact inhibition. Furthermore, because NF2 controls the cell surface availability of various growth factor receptors [43], it will be interesting for future studies to evaluate the impact of its negative regulation by PrPC on growth factor receptor signalling.
Finally, regarding TGFβ, we recently demonstrated that PrPC controls the soluble levels of TGFβ in the supernatant of colorectal cancer cells [28]. Conversely, PrPC levels are increased in response to TGFβ [28]. Although the mechanisms involved still require further investigation, we were able to show that the PrPC-TGFβ axis contributes to the expression of several markers that specify the mesenchymal subtype of colorectal cancer, including that of ZEB1, a master regulator of Epithelial to Mesenchymal Transition (EMT) [28]. These observations call for a better understanding of the interplay between PrPC and TGFβ, considering the major role played by TGFβ not only in the poor prognosis subgroup of colorectal cancer [47] but more widely in various aspects of high-grade malignancy across cancer [48].
4. Resisting Cell Death
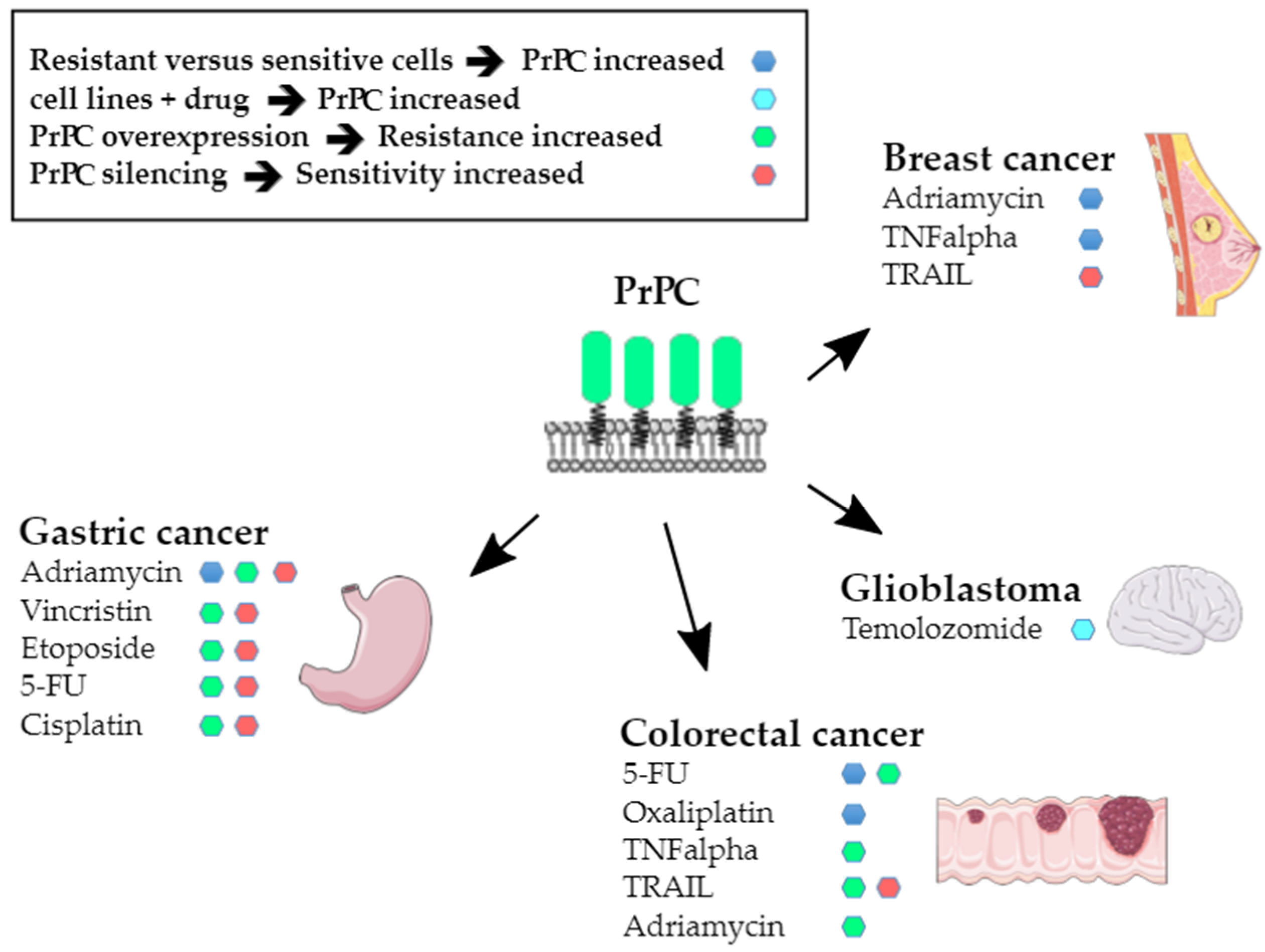
Resistance to apoptosis was the first hallmark to be connected with PrPC nearly 20 years ago, as PRNP transcripts were found to be upregulated in adriamycin-resistant SGC7901 gastric cancer cells as compared to the parental cell line [49]. That elevated PrPC may confer resistance to anticancer agents was soon confirmed by the demonstration of a causal relationship between increased PrPC expression and resistance to tumour necrosis factor-α (TNFα) in the MCF7 breast cancer cell line [50]. The involvement of PrPC in the resistance of cancer cells to cell death-inducing signals has been extensively studied by employing diverse experimental paradigms. The first set of data provided evidence for an upregulation of PrPC expression in drug-resistant contexts, as in a seminal Zhao study [49]. For instance, PRNP gene expression [51] and PrPC protein levels [52] were found to be upregulated in adriamycin-resistant MCF7 breast cancer cells, as well as in SNU-5C colorectal cancer cells resistant to 5-fluorouracil (5-FU) or oxaliplatin [53]. Zhuang et al. further showed a dose-dependent increase in PrPC expression in U87 and U251 glioblastoma cells in response to temolozomide [23]. Moreover, we reported that high expression levels of PrPC are associated with 5-FU resistance in a panel of colorectal cancer cell lines [28]. On the other hand, PrPC overexpression was reported to induce resistance to adriamycin, vincristine, etoposide, 5-FU and cisplatin in SGC7901 gastric cancer cells [54] and resistance to TNFα [30] or adriamycin [25] in LS174 colorectal cancer cells. Conversely, silencing of PrPC increased the sensitivity of MKN28 gastric cancer cells to adriamycin, vincristin, etoposide, 5-FU and cisplatin [55] and that of 5-FU-resistant SNU-5C/FUR [56] or MDST8 [28] colorectal cancer cells to 5-FU. It also cancelled the protection of hypoxia against TRAIL-mediated cell death in HCT116 colorectal cancer cells, while PrPC overexpression conferred resistance to TRAIL-induced cytotoxicity in normoxic conditions [57]. These observations are summarised in Figure 2.
From a translational perspective, it is interesting to note that the relationship between PrPC levels and chemoresistance was confirmed in patients. Indeed, an increased PrPC expression was reported in recurrent versus primary lesions of patients with glioblastoma, following combined temolozomide and radiation therapy [23]. In the same line, Yun et al. reported that plasma levels of PrPC are higher in colorectal cancer patients having received chemotherapy versus untreated patients [31]. Moreover, high PrPC levels in gastric cancer patients were found to be associated with a poor response to chemotherapy [58].
At a molecular level, PrPC may promote chemoresistance through the upregulation of MDR1, which encodes the P-glycoprotein, a transporter responsible for the efflux of anti-cancer drugs, as shown in SGC7901 gastric cancer cells [54]. The PrPC-dependent control on MDR1 appears to be mediated by the PI3K-Akt pathway [59] and to necessitate the octarepeat-rich N-terminal domain of PrPC [60], although the deletion of a single octarepeat appears without effect [61]. The recruitment of the PI3K-Akt cascade downstream from PrPC may rely upon its interaction with the 37kD laminin receptor precursor protein (37LRP), as suggested by Luo et al. [62]. A different, non-mutually exclusive mechanism is the positive regulation of the anti-apoptotic effector Bcl2, which may not only occur in gastric [42,54,58], breast [51] and glioma [63] cancer cells but is also a well-described pathway relaying the cell-survival physiological activity of PrPC (reviewed in [64]) and was even recently documented in the context of liver metabolism [65]. Other effects of PrPC on apoptotic effectors include the upregulation of survivin, cIAP-1 and XIAP levels in colorectal cancer cells [25] or the sequestration of the pro-apoptotic factor Par4 in glioblastoma cells [23]. Finally, Wiegmans et al. described a novel mechanism whereby soluble PrPC promotes the resistance of breast cancer cells to adriamycin through the direct binding and sequestration of the drug [66].
Beyond conferring resistance to anticancer drugs, PrPC was recently reported to protect breast and colorectal cancer cells from irradiation-induced toxicity [67]. Accordingly, PRNP expression levels were found to be increased following radiation treatment of breast or rectal tumours [67]. As with other PrPC-related processes, the radioprotective function of PrPC is not restricted to cancer cells but was also exemplified in hematopoietic progenitor cells [68]. PrPC actually appears to confer a broad resistance to genotoxic stress, in part by potentiating the activity of the APE1 endonuclease, a major player in DNA repair [69].
Overall, the relationship between PrPC and resistance to cell death in cancer is multifaceted and can be viewed as an exacerbation of one of its diverse physiological functions.
5. Inducing Angiogenesis
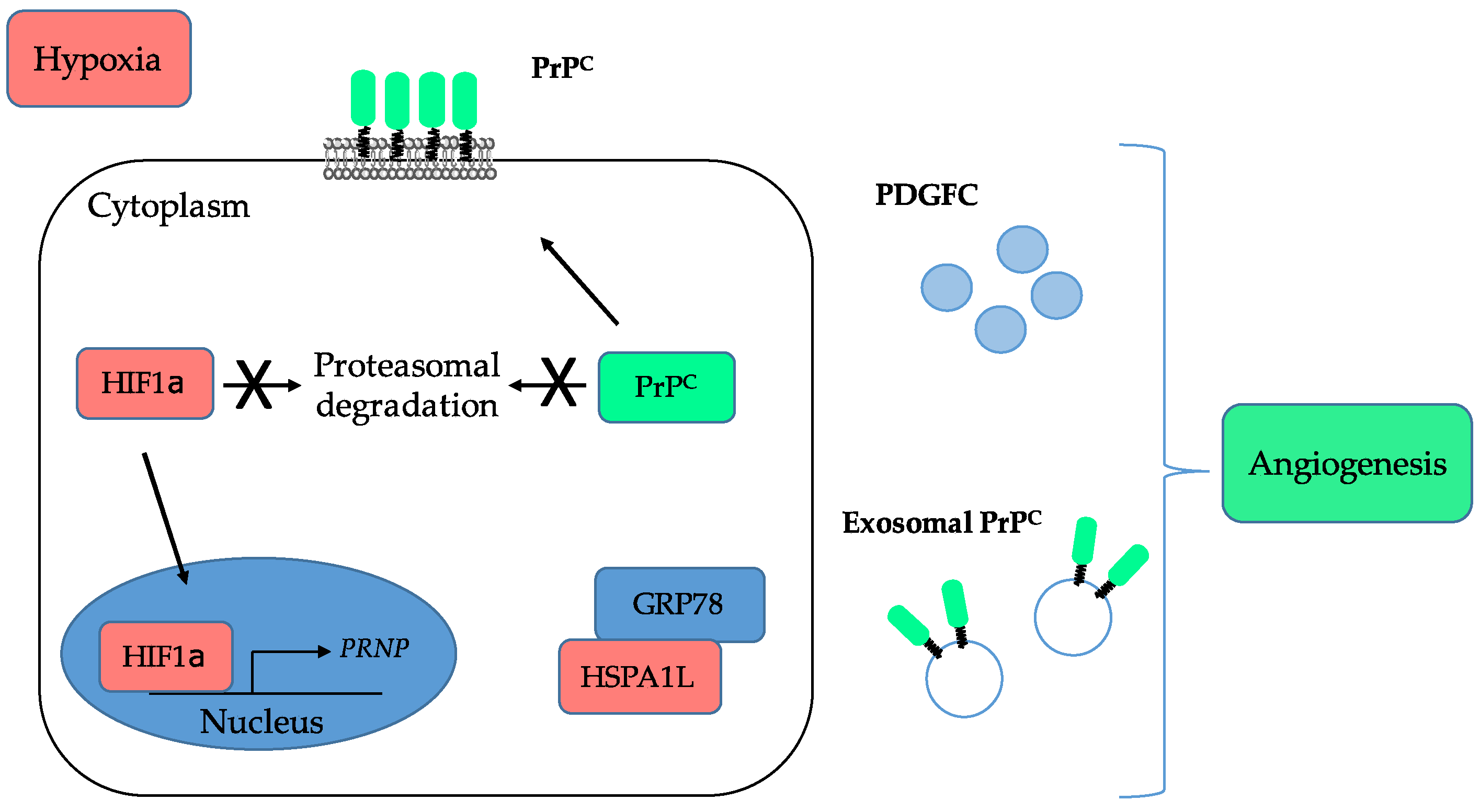
The link between PrPC and angiogenesis emerged almost two decades ago with several studies showing that PrPC is expressed and released by endothelial cells and that its levels are increased after ischemic injury (reviewed in [70]). These observations were subsequently refined with the demonstration that PrPC-deficient mice submitted to cerebral ischemia exhibit poorer recovery as compared with wild-type mice, including reduced neo-angiogenesis [71]. In that study, the authors suggested that PrPC operates, at least in part, by preventing the degradation of HIF1α by the proteasome (see Figure 3) [71].
In line with this, Alfaidy et al. suggested that PrPC is involved in placental angiogenesis by controlling the proliferation, migration and tube-like organisation of trophoblastic cells [72]. This study further showed an upregulation of PrPC in response to hypoxia [72], which is now quite well established under various paradigms (reviewed in [73]). The HIF1α-dependent control on PrPC expression actually extends to the stabilisation of the protein through the sequestration of the E3 ligase GP78 by hypoxia-induced HSPA1L (Figure 3), as shown by Lee et al. in colorectal cancer cells [56]. Very recently, these data were expanded with the demonstration that the levels of PrPC released in exosomes of colorectal cancer cells are increased under hypoxia [31]. In accordance with the broad role of hypoxia in tumour-associated angiogenesis [74] and the contribution of exosomes to this process [75], Yun et al. found that exosomes derived from colorectal cancer cells grown under hypoxia promote the proliferation, migration, invasion and permeability of human umbilical vein endothelial cells (HUVECs) (Figure 3) [31]. This seminal study provides the first direct evidence for a contribution of PrPC to cancer-associated angiogenesis. Other indirect support is brought by the downstream effectors of PrPC in cancer cells. For instance, we showed that, in colorectal cancer, PrPC controls the expression of Platelet-Derived Growth Factor C (PDGFC) (Figure 3) [28], a stimulator of angiogenesis [76]. It also activates YAP and TAZ [28], the two main effectors of the Hippo pathway, which have been shown to promote vascular mimicry, a process whereby cancer cells themselves, instead of endothelial cells, form angiogenic tubules to supply blood to the tumour [77].
Finally, we may note that the PrPC paralogue Doppel has been incriminated in both developmental [78] and cancer-associated angiogenesis [79]. As Doppel resembles the C-terminal moiety of PrPC [80], whether PrPC or its cleaved fragments recapitulate the functional interaction with VEGFR2 reported for Doppel in tumour endothelial cells [79] seems worth investigating.
Altogether, the contribution of PrPC to cancer-associated angiogenesis is only beginning to be unveiled and will certainly be an important axis for future research.
6. Activating Invasion and Metastasis
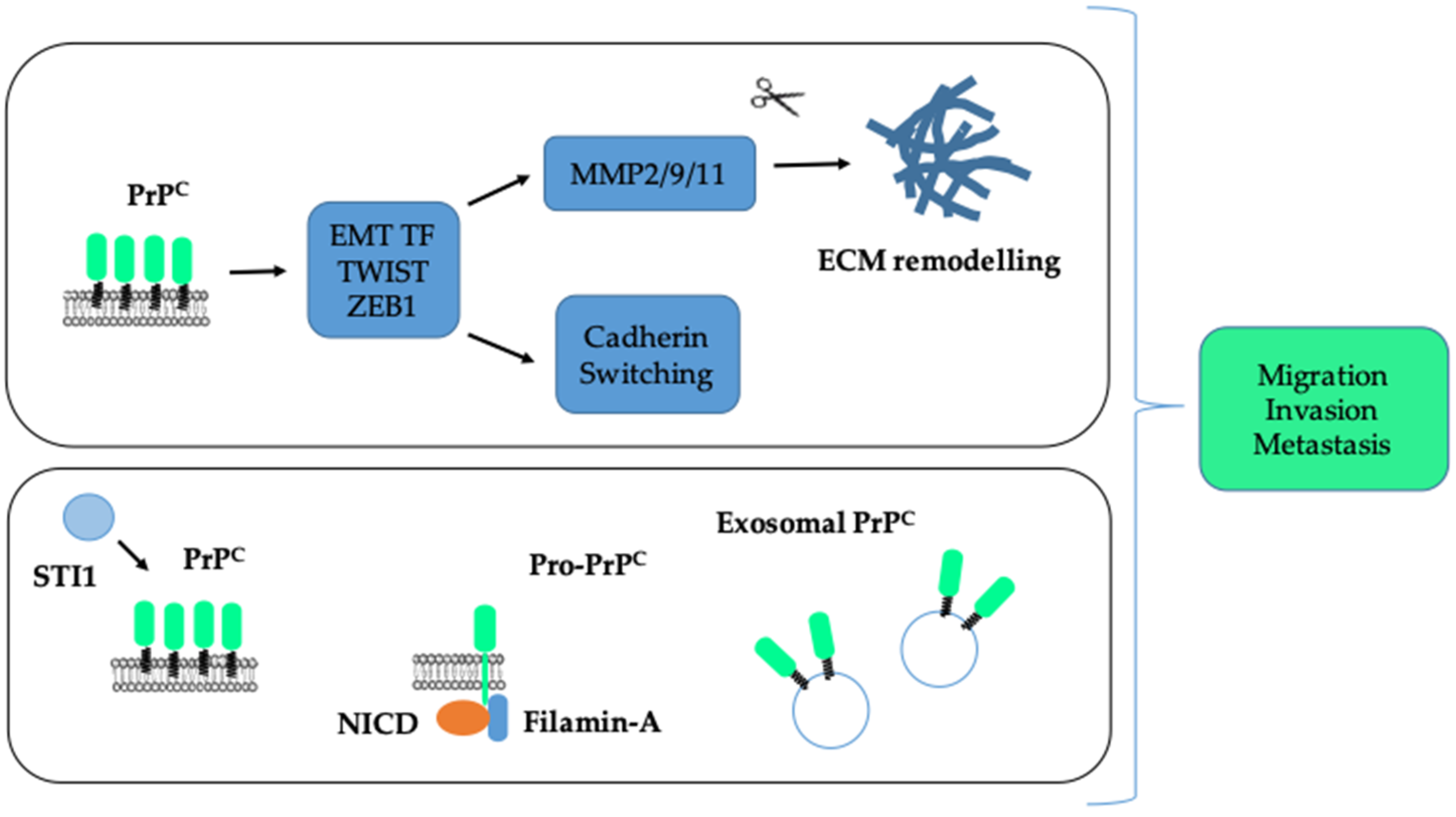
A most notable hallmark of cancer cells is their ability to disseminate to distant organs. One major biological process sustaining invasion and metastasis is a transcriptional program referred to as EMT, whereby cells lose cell–cell contacts and acquire the capacity to migrate and degrade the surrounding matrix for dissemination [81]. The EMT program is orchestrated by major transcription factors, such as SNAIL, SLUG, TWIST, ZEB1 or ZEB2 [81]. There are multiple lines of evidence indicating that PrPC promotes the invasion and migration of cancer cells and controls some EMT-associated features. Indeed, silencing of PrPC in adryamicin-resistant MCF7 breast cancer cells reduces their invasion and migration, as well as the expression of two metalloproteases MMP2 and MMP9 [52]. Conversely, PrPC overexpression in MCF7 cells enhances their invasion and migration as well as the expression and activity of MMP9 [82]. Similarly, depleting PrPC in U87 glioma cells reduces their migration on laminin [22]. In the Capan-1 pancreatic cell line, Wang et al. found that the reduction in cell migration induced by the depletion of PrPC was rescued upon overexpression of the activated form of NOTCH1 [18]. In addition, PrPC-silencing in SGC7901 or MKN45 gastric cancer cells caused a reduction in their invasion, their expression of MMP11, as well as their ability to metastasise to the liver after tail vein injection [83]. In the context of lung cancer, higher PrPC levels were measured in a panel of invasive versus non-invasive lung cancer cell lines [84]. Furthermore, PrPC was shown to control the invasive and migratory properties of CL1-5 cells via a JNK pathway and the knock-down of PrPC reduced their metastatic potential in vivo [84]. Likewise, various studies based on gain and loss of function experiments in colorectal cancer cell lines confirmed the pro-invasive and pro-migratory action of PrPC [25,26,85]. In line with this, Go et al. depicted an increased invasion and migration in the PrPC-positive versus the PrPC-negative fraction of 5-FU resistant SNU-5C/FUR cells [27]. Finally, the presence of PrPC together with that of the cancer stem cell marker CD44 at the cell surface of primary colorectal cancer cells was found to control their migration in vitro as well as their metastatic potential after injection in the cecal wall [86]. Several studies have further documented a link between PrPC and EMT. Thus, in primary colorectal cancer cells, PrPC was found to control the expression of the EMT transcription factor TWIST and that of N-cadherin while repressing that of E-cadherin [86]. Similar findings were obtained with HT29 colorectal cancer cells grown under hypoxia [56]. Likewise, we documented that PrPC controls the expression of the EMT transcription factor ZEB1 in colorectal cancer cell lines and that PRNP gene expression is significantly correlated with an EMT signature in both colorectal cancer patients and cell panels [28]. These overall findings are summarised in Figure 4 (top panel).
At a mechanistic level, several findings are worth noting. Lacerda et al. found that the PrPC pro-invasive action in colorectal cancer cells depends upon its interaction with its ligand STI1 [87]. Moreover, we recently highlighted the importance of the PrPC-ILK coupling in the invasive and migratory properties of the MDST8 colorectal cancer cell line [26]. On the other hand, in melanoma cells, where PrPC is mainly found as a pro-PrP isoform retaining its C-terminus instead of a GPI anchor, invasion and migration depends upon the interaction of pro-PrP with Filamin-A [88], which was shown to form a complex with pro-PrP and NOTCH1 in pancreatic cancer cells [18]. Yun et al. further demonstrated that exosomes derived from various colorectal cancer cells grown under hypoxia can sustain their own invasion and migration in a PrPC-dependent manner (see Figure 4, bottom panel) [31].
From a clinical point of view, several studies have depicted a correlation between high levels of tumour PrPC—as inferred through immuno-histochemistry—and metastasis in patients with breast [89], gastric [83,90] and colorectal [31,56,86] cancer. In the same line, Lin et al. depicted increased levels of tumour PrPC in patients with invasive versus in situ lung adenocarcinoma [84]. Moreover, in colorectal cancer, we reported an enrichment in the expression of the PRNP gene in the mesenchymal subtype [28], itself associated with increased progression to advanced stages [91]. We further documented that plasma levels of PrPC have prognostic value in terms of disease control in metastatic colorectal cancer patients [28]. Thus, PrPC is unambiguously associated with this hallmark of cancer.
7. Reprogramming of Energy Metabolism (Emerging Hallmark)
Metabolic reprogramming features as one of the emerging hallmarks of cancer [14], which has taken centre stage over the past decade [92]. It is clear that studies addressing the question as to whether PrPC may influence the metabolism of cancer cells are very scarce. Most data come from the work by Li et al. who found a regulation of the expression of GLUT1, encoding the glucose transporter 1, downstream from PrPC in the DLD-1 colorectal cancer cell line [29]. Accordingly, the authors demonstrated that PrPC depletion reduces glucose uptake and the glycolytic rate of colorectal cancer cells [29]. From a translational point of view, it is worth noting that the expression levels of PrPC and GLUT1 were correlated in colorectal cancer patients [29]. These findings actually recall the identification of both PrPC and GLUT1 as specific cell-surface biomarkers of the adenoma-to-carcinoma transition in colorectal cancer [93]. Aside from cancer, the link between PrPC and glucose uptake is further strengthened by the work of Ashok et al. based upon a comparison of various tissues of mice deficient in PrPC versus their wild-type counterparts for the expression of glucose transporters [94]. However, in apparent contradiction with the data obtained in cancer cells, the absence of PrPC was associated with increased GLUT1, GLUT2 or GLUT3 levels according to the tissue—brain, retina or liver—considered [94]. Changes in the expression of the mono-carboxylate transporters MCT1 and MCT4, involved in the transport of lactate and pyruvate, were also observed in the brains of mice lacking PrPC, with positive or negative regulations depending on the cerebral region considered [95]. In this respect, it is interesting to note that PrPC was found to control the uptake of lactate by astrocytes through the interaction with Na+-K+ ATPase, the driving force for MCT1 activity [96]. Other points of interest include the interaction between PrPC and the lactate dehydrogenase isoforms LDH-A and LDH-B, which were uncovered through systematic proteomic assays for PrPC partners [97,98,99]. PrPC was later shown to potentiate the activity of LDH in the hypoxic brain, which may contribute to the protective role for PrPC against stress [100]. Other PrPC interactors involved in glycolysis include aldolase C (ALDOC) [101] and as well as aldolase A (ALDOA) [99], both catalysing the conversion of fructose-1,6-bisphosphate into glyceraldehyde 3-phosphate (G3P), alpha- and gamma- enolase (ENO1 and ENO2), two isoenzymes that converts 2-phosphoglycerate into phosphoenolpyruvate [97,99], the well-known glyceraldehyde-3-phosphate dehydrogenase (GAPDH) [97,98,99], the triose-phosphate isomerase TPI [98,99] as well as the pyruvate kinases PKM1/PKM2 that catalyse the de-phosphorylation of phosphoenolpyruvate into pyruvate [97,102]. PrPC was further shown to interact with both cytoplasmic malate dehydrogenase [97], which converts oxaloacetate into malate, itself being imported in the mitochondrial matrix, and mitochondrial malate dehydrogenase [99], which catalyses the oxidation of malate into oxaloacetate within the Krebs cycle. It is of note that these overall interactions have been recapitulated in several studies, suggesting functional implications. However, apart from LDH, how PrPC may influence the activity of these diverse enzymes remains to be explored.
On the other hand, several studies have brought to light links between PrPC and mitochondria. First, beyond its main location at the cell surface, PrPC was also found to locate in the mitochondria of healthy mice [103]. PrPC was further shown to co-localise with COX4 [103], one of the nuclear-encoded subunits of complex IV of the respiratory chain. In addition, PrPC-deficient mice were reported to have reduced numbers of mitochondria, which have a larger morphology, and an enhanced maximal respiratory capacity, presumably to compensate for low mitochondrial numbers [104,105]. A proteomic study comparing WT and PrPC-deficient neurons also exemplified reduced levels of the mitochondrial proteins COX2 in the absence of PrPC [106]. Finally, coming back to PrPC partners, two independent studies have identified citrate synthase (CS) as a PrPC interactor [97,98]. Again, whether PrPC modulates the activity of CS has yet to be investigated.
As a whole, despite the scarcity of data relating to PrPC and cancer cell metabolism, the findings obtained with respect to PrPC physiological role and, more importantly, to PrPC binding partners offer many avenues for future investigation.
8. Evading Immune Destruction (Emerging Hallmark)
The second emerging hallmark emphasised by Hanahan and Weinberg refers to the ability of cancer cells to thwart the immune system [14]. A first hint at a link between PrPC and immune-evasion can be inferred from the enrichment of PRNP gene expression in the mesenchymal subtype of colorectal cancer [28], itself associated with an immune-suppressive signature [107]. Secondly, a more general role for PrPC in immunological quiescence has been proposed, based on its pattern of expression in immune privilege organs as well as its cytoprotective and immune-regulatory function [108]. Mechanistically, PrPC may, by itself, induce cell signalling events sustaining immunomodulation, and thereby temper inflammation [8,108]. On the other hand, tumour associated PrPC controls the levels of several effectors known to promote immune evasion. One such effector is TGFβ, whose soluble levels are regulated by PrPC through a yet-to-be-uncovered mechanism [28]. It is indeed now well-acknowledged that TGFβ inhibits anti-tumour immunity through multiple mechanisms (see [109] for review). Another major player in immune tolerance is IDO (indoleamine 2,2 dioxygenase), an enzyme of the kynurenine pathway [110], which we recently identified as a molecular target downstream from PrPC signalling in colorectal cancer [26]. Thus, it appears that PrPC from cancer cells orchestrates diverse pathways that altogether tone down the anti-tumour immune response by favouring an immune-suppressive contexture.
9. Genome Instability and Mutation
Genomic alterations represent a key characteristic enabling cancer cells to acquire their diverse hallmarks [14]. The question as to whether PrPC may be linked to this enabling characteristic is twofold. First, we may ask whether genome instability and mutation foster the expression of PrPC. A second question is whether the expression of PrPC may afford protection against DNA damage. A major observation regarding the first point is the identification of cell surface PrPC as a marker of aneuploidy in a pan-cancer screening study [111]. The authors further reported an increase in PrPC levels in parental or aneuploid HCT116 colorectal cancer cells upon serum-deprivation, which they linked to oxidative stress, and showed that PrPC is protective against serum-deprivation-induced necrotic death [111]. On this basis, the authors proposed that the upregulation of PrPC in aneuploid cells is a consequence of the oxidative stress associated with this genomic alteration. This notion actually fully fits in with the physiological role described for PrPC in the protection against oxidative stress (reviewed in [6]). According to Qin et al. the induction of PRNP gene expression in response to oxidative stress involves the Ataxia Telangiectasia Mutated (ATM) kinase [112]. More recently, ATM was further shown to mediate the upregulation of PRNP transcription in response to irradiation [67]. In line with the Domingues study [111], PrPC expression was found to confer a protective role against irradiation [67]. This radioprotective action fits in with the protective role against other types of genotoxic stresses [69], as mentioned above.
Thus, since PrPC is induced in response to DNA injury and supports protection against DNA damage, we may propose that PrPC takes part in the balance between DNA damage and repair, a trade-off for cancer cell survival and growth [113].
10. Tumour-Promoting Inflammation
Chronic inflammation is a well-acknowledged driver of cancer development [114]. Several pieces of evidence indicate that inflammatory conditions may promote an increase in PrPC expression. In the context of cancer, the upregulation of PrPC in Merlin-deficient tumours was found to depend on the NFκB transcription factor [24], which embodies a major link between inflammation and cancer (reviewed in [115]). Whether NFκB positively regulates the expression of PrPC in other types of tumours obviously deserves further investigation. Conversely, PrPC was shown to activate NFκB-dependent transcription in breast cancer cells [82], or to be necessary for the TNFα-dependent activation of NFκB in melanoma M2 cells [116], thereby delineating a bidirectional link between PrPC and NFκB. An alternative yet still hypothetical mechanism leading to an upregulation of PrPC would be via ILK, which we showed to be both a downstream target and an upstream regulator of PrPC [26] and has been reported to be induced in colonic cells in response to inflammation [117]. Another observation worth noting is the induction of PrPC in the mucosa of patients with Helicobacter Pylori gastritis [118], a condition well-known to predispose patients to gastric cancer.
Paradoxically, Prnp-deficient mice were reported to be more sensitive than wild-type mice to dextran sulfate-induced colitis [119]. This is, however, reminiscent of the observations obtained in mice deficient for Yap [120], itself a downstream target of PrPC [28]. In the case of Yap, results were interpreted as Yap being necessary for tissue repair after injury, a function whose over-activation supports cancer progression [121]. Likewise, we may surmise that PrPC is mandatory for tissue regeneration, in accordance with its function in stem cell self-renewal [11], and that its upregulation under inflammatory conditions contributes to cancer development.
Altogether, the interplay between PrPC and tumour-promoting inflammation is currently supported by few studies and could be worthy of further exploration.
11. Conclusions/Future Prospects
In summary, we have enlightened the involvement of PrPC in cancer biology from the standpoint of the hallmarks of cancer (Figure 5).
While the participation of PrPC to some of those hallmarks—most notably proliferation, survival, invasion and metastasis—is substantiated by multiple cell-based, pre-clinical or clinical studies across cancer types, its links to other hallmarks, such as reprogramming of energy metabolism or evading immune destruction, are only beginning to be explored or even merely suggested from observations outside the field of cancer. These underappreciated roles of PrPC in cancer-related processes represent important areas for future research. Regarding the binding partners involved in the contribution of PrPC to each hallmark (Table 1), data remain scarce at present and are likely to expand in the near future.
Although some of the signalling pathways through which PrPC operates have been elucidated (Table 2), the picture is far from complete and also requires further investigation.
Casting further light on these specific points will undoubtedly help reach an integrated view of the multifaceted contribution of PrPC to cancer initiation, promotion and progression.
Author Contributions
S.M.-R. wrote the review. A.G. prepared the figures. P.L.-P. reviewed and edited the draft. All authors have read and agreed to the published version of the manuscript.
Funding
Institut National de la Santé et de la Recherche Médicale; Association pour la Recherche sur le Cancer (grant number PJA 20171206220); Cancéropôle Ile de France (grant number 2016-1-EMERG-36-UP 5-1); SIRIC CARPEM (Cancer Research for Personalized Medicine, INCa-DGOS Inserm_12561).
Acknowledgments
The authors wish to thank Fatima Djouadi for her critical reading of the review. We are grateful to D. Le Corre who produced some of the results summarised in the present review. We apologise to those whose work has not been cited owing to space limitations.
Conflicts of Interest
The authors declare no conflict of interest in the context of the present review.
Abbreviations
| 5-FU | 5-fluorouracil |
| APE1 | apurinic/apyrimidinic endonuclease 1 |
| ATM | ataxia telangiectasia mutated |
| EMT | epithelial to mesenchymal transition |
| EGFR | epidermal growth factor receptor |
| GPI | glycosyl-phosphatidylinositol |
| HIF-1α | hypoxia-inducible factor 1 alpha |
| HSPA1L | heat shock protein 70 member 1-like |
| HUVEC | human umbilical vein endothelial cell |
| ILK | integrin linked kinase |
| NF2 | neurofibromatosis type 2 |
| PDGFC | platelet-derived growth factor C |
| PrPC | cellular prion protein |
| PrPSc | scrapie prion protein |
| SNU-C5/FUR | 5-FU resistant SNU-C5 cells |
| TGFβ | transforming growth factor β |
| TNFα | tumour necrosis factor-α |
References
- Oesch, B.; Westaway, D.; Walchli, M.; McKinley, M.P.; Kent, S.B.; Aebersold, R.; Barry, R.A.; Tempst, P.; Teplow, D.B.; Hood, L.E.; et al. A Cellular Gene Encodes Scrapie PrP 27-30 Protein. Cell 1985, 40, 735–746. [Google Scholar] [CrossRef]
- Aguzzi, A.; Baumann, F.; Bremer, J. The Prion’s Elusive Reason for Being. Annu. Rev. Neurosci. 2008, 31, 439–477. [Google Scholar] [CrossRef] [PubMed]
- Sarnataro, D.; Pepe, A.; Zurzolo, C. Chapter Three—Cell Biology of Prion Protein. In Progress in Molecular Biology and Translational Science; Legname, G., Vanni, S., Eds.; Prion Protein; Academic Press: Cambridge, MA, USA, 2017; Volume 150, pp. 57–82. [Google Scholar]
- Evans, E.G.B.; Millhauser, G.L. Copper- and Zinc-Promoted Interdomain Structure in the Prion Protein: A Mechanism for Autoinhibition of the Neurotoxic N-Terminus. Prog. Mol. Biol. Transl. Sci. 2017, 150, 35–56. [Google Scholar] [CrossRef] [PubMed]
- Linsenmeier, L.; Altmeppen, H.C.; Wetzel, S.; Mohammadi, B.; Saftig, P.; Glatzel, M. Diverse Functions of the Prion Protein—Does Proteolytic Processing Hold the Key? Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 2128–2137. [Google Scholar] [CrossRef]
- Hirsch, T.Z.; Martin-Lannerée, S.; Mouillet-Richard, S. Functions of the Prion Protein. Prog. Mol. Biol. Transl. Sci. 2017, 150, 1–34. [Google Scholar] [CrossRef]
- Hirsch, T.Z.; Hernandez-Rapp, J.; Martin-Lanneree, S.; Launay, J.M.; Mouillet-Richard, S. PrPC Signalling in Neurons: From Basics to Clinical Challenges. Biochimie 2014, 104C, 2–11. [Google Scholar] [CrossRef]
- Manni, G.; Lewis, V.; Senesi, M.; Spagnolli, G.; Fallarino, F.; Collins, S.J.; Mouillet-Richard, S.; Biasini, E. The Cellular Prion Protein beyond Prion Diseases. Swiss Med. Wkly. 2020, 150, w20222. [Google Scholar] [CrossRef] [PubMed]
- Corsaro, A.; Bajetto, A.; Thellung, S.; Begani, G.; Villa, V.; Nizzari, M.; Pattarozzi, A.; Solari, A.; Gatti, M.; Pagano, A.; et al. Cellular Prion Protein Controls Stem Cell-like Properties of Human Glioblastoma Tumor-Initiating Cells. Oncotarget 2016, 7, 38638. [Google Scholar] [CrossRef]
- Go, G.; Lee, S.H. The Cellular Prion Protein: A Promising Therapeutic Target for Cancer. Int. J. Mol. Sci. 2020, 21, 9208. [Google Scholar] [CrossRef]
- Martin-Lannerée, S.; Hirsch, T.Z.; Hernandez-Rapp, J.; Halliez, S.; Vilotte, J.L.; Launay, J.M.; Mouillet-Richard, S. PrPC from Stem Cells to Cancer. Front. Cell Dev. Biol. 2014, 2, 55. [Google Scholar] [PubMed] [Green Version]
- Ryskalin, L.; Biagioni, F.; Busceti, C.L.; Giambelluca, M.A.; Morelli, L.; Frati, A.; Fornai, F. The Role of Cellular Prion Protein in Promoting Stemness and Differentiation in Cancer. Cancers 2021, 13, 170. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Liang, J.; Pan, Y.; Zhang, D.; Guo, C.; Shi, Y.; Wang, J.; Chen, Y.; Wang, X.; Liu, J.; Guo, X.; et al. Cellular Prion Protein Promotes Proliferation and G1/S Transition of Human Gastric Cancer Cells SGC7901 and AGS. FASEB J. 2007, 21, 2247–2256. [Google Scholar] [CrossRef]
- Vassallo, N.; Herms, J.; Behrens, C.; Krebs, B.; Saeki, K.; Onodera, T.; Windl, O.; Kretzschmar, H.A. Activation of Phosphatidylinositol 3-Kinase by Cellular Prion Protein and Its Role in Cell Survival. Biochem. Biophys. Res. Commun. 2005, 332, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Yu, S.; Nakamura, F.; Yin, S.; Xu, J.; Petrolla, A.A.; Singh, N.; Tartakoff, A.; Abbott, D.W.; Xin, W.; et al. Binding of Pro-Prion to Filamin a Disrupts Cytoskeleton and Correlates with Poor Prognosis in Pancreatic Cancer. J. Clin. Invest. 2009, 119, 2725–2736. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yu, S.; Huang, D.; Cui, M.; Hu, H.; Zhang, L.; Wang, W.; Parameswaran, N.; Jackson, M.; Osborne, B.; et al. Cellular Prion Protein Mediates Pancreatic Cancer Cell Survival and Invasion through Association with and Enhanced Signaling of Notch1. Am. J. Pathol. 2016, 186, 2945–2956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.T.; Jiang, G.; Chen, Q.; Zheng, J.N. Ki67 Is a Promising Molecular Target in the Diagnosis of Cancer (Review). Mol. Med. Rep. 2015, 11, 1566–1572. [Google Scholar] [CrossRef] [Green Version]
- Martin-Lannerée, S.; Halliez, S.; Hirsch, T.Z.; Hernandez-Rapp, J.; Passet, B.; Tomkiewicz, C.; Villa-Diaz, A.; Torres, J.-M.; Launay, J.-M.; Béringue, V.; et al. The Cellular Prion Protein Controls Notch Signaling in Neural Stem/Progenitor Cells. Stem Cells 2017, 35, 754–765. [Google Scholar] [CrossRef] [PubMed]
- Lopes, M.H.; Santos, T.G.; Rodrigues, B.R.; Queiroz-Hazarbassanov, N.; Cunha, I.W.; Wasilewska-Sampaio, A.P.; Costa-Silva, B.; Marchi, F.A.; Bleggi-Torres, L.F.; Sanematsu, P.I.; et al. Disruption of Prion Protein-HOP Engagement Impairs Glioblastoma Growth and Cognitive Decline and Improves Overall Survival. Oncogene 2014, 34, 3305–3314. [Google Scholar] [CrossRef]
- Iglesia, R.P.; Prado, M.B.; Cruz, L.; Martins, V.R.; Santos, T.G.; Lopes, M.H. Engagement of Cellular Prion Protein with the Co-Chaperone Hsp70/90 Organizing Protein Regulates the Proliferation of Glioblastoma Stem-like Cells. Stem Cell Res. Ther. 2017, 8, 76. [Google Scholar] [CrossRef]
- Zhuang, D.; Liu, Y.; Mao, Y.; Gao, L.; Zhang, H.; Luan, S.; Huang, F.; Li, Q. TMZ-Induced PrPc/Par-4 Interaction Promotes the Survival of Human Glioma Cells. Int. J. Cancer 2012, 130, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Provenzano, L.; Ryan, Y.; Hilton, D.A.; Lyons-Rimmer, J.; Dave, F.; Maze, E.A.; Adams, C.L.; Rigby-Jones, R.; Ammoun, S.; Hanemann, C.O. Cellular Prion Protein (PrPC) in the Development of Merlin-Deficient Tumours. Oncogene 2017, 36, 6132–6142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chieng, C.K.-L.; Say, Y.-H. Cellular Prion Protein Contributes to LS 174T Colon Cancer Cell Carcinogenesis by Increasing Invasiveness and Resistance against Doxorubicin-Induced Apoptosis. Tumour Biol. 2015, 36, 8107–8120. [Google Scholar] [CrossRef] [PubMed]
- Ghazi, A.; Le Corre, D.; Pilati, C.; Taieb, J.; Aparicio, T.; Didelot, A.; Dedhar, S.; Mulot, C.; Le Malicot, K.; Djouadi, F.; et al. Prognostic Value of the PrPC-ILK-IDO1 Axis in the Mesenchymal Colorectal Cancer Subtype. Oncoimmunology 2021, 10, 1940674. [Google Scholar] [CrossRef]
- Go, G.; Yun, C.W.; Yoon, Y.M.; Lim, J.H.; Lee, J.H.; Lee, S.H. Role of PrPC in Cancer Stem Cell Characteristics and Drug Resistance in Colon Cancer Cells. Anticancer Res. 2020, 40, 5611–5620. [Google Scholar] [CrossRef]
- Le Corre, D.; Ghazi, A.; Balogoun, R.; Pilati, C.; Aparicio, T.; Martin-Lannerée, S.; Marisa, L.; Djouadi, F.; Poindessous, V.; Crozet, C.; et al. The Cellular Prion Protein Controls the Mesenchymal-like Molecular Subtype and Predicts Disease Outcome in Colorectal Cancer. EBioMedicine 2019, 46, 94–104. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.Q.; Sun, Y.P.; Ruan, C.P.; Xu, X.Y.; Ge, J.H.; He, J.; Xu, Z.D.; Wang, Q.; Gao, W.C. Cellular Prion Protein Promotes Glucose Uptake through the Fyn-HIF-2α-Glut1 Pathway to Support Colorectal Cancer Cell Survival. Cancer Sci. 2011, 102, 400–406. [Google Scholar] [CrossRef]
- Yap, Y.H.-Y.; Say, Y.-H. Resistance against Tumour Necrosis Factor α Apoptosis by the Cellular Prion Protein Is Cell-Specific for Oral, Colon and Kidney Cancer Cell Lines. Cell Biol. Int. 2012, 36, 273–277. [Google Scholar] [CrossRef]
- Yun, C.-W.; Lee, J.-H.; Go, G.; Jeon, J.; Yoon, S.; Lee, S.-H. Prion Protein of Extracellular Vesicle Regulates the Progression of Colorectal Cancer. Cancers 2021, 13, 2144. [Google Scholar] [CrossRef]
- Dias, M.V.S.; Teixeira, B.L.; Rodrigues, B.R.; Sinigaglia-Coimbra, R.; Porto-Carreiro, I.; Roffé, M.; Hajj, G.N.M.; Martins, V.R. PRNP/Prion Protein Regulates the Secretion of Exosomes Modulating CAV1/Caveolin-1-Suppressed Autophagy. Autophagy 2016, 12, 2113–2128. [Google Scholar] [CrossRef] [Green Version]
- Llorens, F.; Carulla, P.; Villa, A.; Torres, J.M.; Fortes, P.; Ferrer, I.; del Rio, J.A. PrP(C) Regulates Epidermal Growth Factor Receptor Function and Cell Shape Dynamics in Neuro2a Cells. J. Neurochem. 2013, 127, 124–138. [Google Scholar] [CrossRef]
- Atkinson, C.J.; Kawamata, F.; Liu, C.; Ham, S.; Győrffy, B.; Munn, A.L.; Wei, M.Q.; Möller, A.; Whitehall, V.; Wiegmans, A.P. EGFR and Prion Protein Promote Signaling via FOXO3a-KLF5 Resulting in Clinical Resistance to Platinum Agents in Colorectal Cancer. Mol. Oncol. 2019, 13, 725–737. [Google Scholar] [CrossRef] [Green Version]
- Checler, F.; Alves da Costa, C. P53 in Neurodegenerative Diseases and Brain Cancers. Pharmacol. Ther. 2014, 142, 99–113. [Google Scholar] [CrossRef]
- Paitel, E.; Fahraeus, R.; Checler, F. Cellular Prion Protein Sensitizes Neurons to Apoptotic Stimuli through Mdm2-Regulated and P53-Dependent Caspase 3-like Activation. J. Biol. Chem. 2003, 278, 10061–10066. [Google Scholar] [CrossRef] [Green Version]
- Sunyach, C.; Cisse, M.A.; da Costa, C.A.; Vincent, B.; Checler, F. The C-Terminal Products of Cellular Prion Protein Processing, C1 and C2, Exert Distinct Influence on P53-Dependent Staurosporine-Induced Caspase-3 Activation. J. Biol. Chem. 2007, 282, 1956–1963. [Google Scholar] [CrossRef] [Green Version]
- Guillot-Sestier, M.V.; Sunyach, C.; Druon, C.; Scarzello, S.; Checler, F. The α-Secretase-Derived N-Terminal Product of Cellular Prion, N1, Displays Neuroprotective Function In Vitro and In Vivo. J. Biol. Chem. 2009, 284, 35973–35986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altmeppen, H.C.; Prox, J.; Puig, B.; Kluth, M.A.; Bernreuther, C.; Thurm, D.; Jorissen, E.; Petrowitz, B.; Bartsch, U.; De Strooper, B.; et al. Lack of A-Disintegrin-and-Metalloproteinase ADAM10 Leads to Intracellular Accumulation and Loss of Shedding of the Cellular Prion Protein In Vivo. Mol. Neurodegener. 2011, 6, 36. [Google Scholar] [CrossRef] [Green Version]
- Weiss, E.; Ramljak, S.; Asif, A.R.; Ciesielczyk, B.; Schmitz, M.; Gawinecka, J.; Schulz-Schaeffer, W.; Behrens, C.; Zerr, I. Cellular Prion Protein Overexpression Disturbs Cellular Homeostasis in SH-SY5Y Neuroblastoma Cells but Does Not Alter P53 Expression: A Proteomic Study. Neuroscience 2010, 169, 1640–1650. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Parchaliuk, D.; Medina, S.; Sorensen, G.; Landry, L.; Huang, S.; Wang, M.; Kong, Q.; Booth, S.A. Activation of P53-Regulated pro-Apoptotic Signaling Pathways in PrP-Mediated Myopathy. BMC Genomics 2009, 10, 201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, J.; Pan, Y.L.; Ning, X.X.; Sun, L.J.; Lan, M.; Hong, L.; Du, J.P.; Liu, N.; Liu, C.J.; Qiao, T.D.; et al. Overexpression of PrPC and Its Antiapoptosis Function in Gastric Cancer. Tumour Biol. 2006, 27, 84–91. [Google Scholar] [CrossRef]
- Curto, M.; McClatchey, A.I. Nf2/Merlin: A Coordinator of Receptor Signalling and Intercellular Contact. Br. J. Cancer 2008, 98, 256–262. [Google Scholar] [CrossRef] [Green Version]
- Serrano, I.; McDonald, P.C.; Lock, F.; Muller, W.J.; Dedhar, S. Inactivation of the Hippo Tumour Suppressor Pathway by Integrin-Linked Kinase. Nat. Commun. 2013, 4, 2976. [Google Scholar] [CrossRef]
- Mouillet-Richard, S.; Laurent-Puig, P. YAP/TAZ Signalling in Colorectal Cancer: Lessons from Consensus Molecular Subtypes. Cancers 2020, 12, 3160. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Tumaneng, K.; Guan, K.-L. The Hippo Pathway in Organ Size Control, Tissue Regeneration and Stem Cell Self-Renewal. Nat. Cell Biol. 2011, 13, 877–883. [Google Scholar] [CrossRef] [PubMed]
- Tauriello, D.V.F.; Batlle, E. Targeting the Microenvironment in Advanced Colorectal Cancer. Trends Cancer 2016, 2, 495–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batlle, E.; Massagué, J. Transforming Growth Factor-β Signaling in Immunity and Cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; You, H.; Liu, F.; An, H.; Shi, Y.; Yu, Q.; Fan, D. Differentially Expressed Gene Profiles between Multidrug Resistant Gastric Adenocarcinoma Cells and Their Parental Cells. Cancer Lett. 2002, 185, 211–218. [Google Scholar] [CrossRef]
- Diarra-Mehrpour, M.; Arrabal, S.; Jalil, A.; Pinson, X.; Gaudin, C.; Pietu, G.; Pitaval, A.; Ripoche, H.; Eloit, M.; Dormont, D.; et al. Prion Protein Prevents Human Breast Carcinoma Cell Line from Tumor Necrosis Factor α-Induced Cell Death. Cancer Res. 2004, 64, 719–727. [Google Scholar] [CrossRef] [Green Version]
- Meslin, F.; Hamai, A.; Gao, P.; Jalil, A.; Cahuzac, N.; Chouaib, S.; Mehrpour, M. Silencing of Prion Protein Sensitizes Breast Adriamycin-Resistant Carcinoma Cells to TRAIL-Mediated Cell Death. Cancer Res. 2007, 67, 10910–10919. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Tao, L.; Xu, J.; Li, Q.; Yu, J.; Jin, Y.; Chen, Q.; Xu, Z.; Zou, Q.; Liu, X. CD44/Cellular Prion Protein Interact in Multidrug Resistant Breast Cancer Cells and Correlate with Responses to Neoadjuvant Chemotherapy in Breast Cancer Patients. Mol. Carcinog. 2013, 53, 686–697. [Google Scholar] [CrossRef]
- Lee, J.H.; Yun, C.W.; Lee, S.H. Cellular Prion Protein Enhances Drug Resistance of Colorectal Cancer Cells via Regulation of a Survival Signal Pathway. Biomol. Ther. 2018, 26, 313–321. [Google Scholar] [CrossRef]
- Du, J.; Pan, Y.; Shi, Y.; Guo, C.; Jin, X.; Sun, L.; Liu, N.; Qiao, T.; Fan, D. Overexpression and Significance of Prion Protein in Gastric Cancer and Multidrug-Resistant Gastric Carcinoma Cell Line SGC7901/ADR. Int. J. Cancer 2005, 113, 213–220. [Google Scholar] [CrossRef]
- Liang, J.; Bai, F.; Luo, G.; Wang, J.; Liu, J.; Ge, F.; Pan, Y.; Yao, L.; Du, R.; Li, X.; et al. Hypoxia Induced Overexpression of PrP(C) in Gastric Cancer Cell Lines. Cancer Biol. Ther. 2007, 6, 769–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.H.; Han, Y.-S.; Yoon, Y.M.; Yun, C.W.; Yun, S.P.; Kim, S.M.; Kwon, H.Y.; Jeong, D.; Baek, M.J.; Lee, H.J.; et al. Role of HSPA1L as a Cellular Prion Protein Stabilizer in Tumor Progression via HIF-1α/GP78 Axis. Oncogene 2017, 36, 6555–6567. [Google Scholar] [CrossRef]
- Park, J.-Y.; Jeong, J.-K.; Lee, J.-H.; Moon, J.-H.; Kim, S.-W.; Lee, Y.-J.; Park, S.-Y. Induction of Cellular Prion Protein (PrPc) under Hypoxia Inhibits Apoptosis Caused by TRAIL Treatment. Oncotarget 2015, 6, 5342–5353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.-H.; Du, J.-P.; Zhang, Y.-H.; Zhao, X.-J.; Fan, R.-Y.; Wang, Z.-H.; Wu, Z.-T.; Han, Y. Dynamic Changes and Surveillance Function of Prion Protein Expression in Gastric Cancer Drug Resistance. World J. Gastroenterol. 2011, 17, 3986–3993. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Ge, F.; Guo, C.; Luo, G.; Wang, X.; Han, G.; Zhang, D.; Wang, J.; Li, K.; Pan, Y.; et al. Inhibition of PI3K/Akt Partially Leads to the Inhibition of PrP(C)-Induced Drug Resistance in Gastric Cancer Cells. FEBS J. 2009, 276, 685–694. [Google Scholar] [CrossRef]
- Wang, J.H.; Du, J.P.; Li, S.J.; Zhai, L.P.; Yang, X.Y.; Wang, Z.H.; Wu, Z.T.; Han, Y. Octarepeat Peptides of Prion Are Essential for Multidrug Resistance in Gastric Cancer Cells. J. Dig. Dis. 2012, 13, 143–152. [Google Scholar] [CrossRef]
- Liang, J.; Wang, J.; Luo, G.; Pan, Y.; Wang, X.; Guo, C.; Zhang, D.; Yin, F.; Zhang, X.; Liu, J.; et al. Function of PrPC (1-OPRD) in Biological Activities of Gastric Cancer Cell Lines. J. Cell. Mol. Med. 2009, 13, 4453–4464. [Google Scholar] [CrossRef] [Green Version]
- Luo, G.; Wang, W.; Wu, Q.; Lu, Y.; Su, T.; Gu, N.; Li, K.; Wang, J.; Du, R.; Zhao, X.; et al. MGr1-Antigen/37 KDa Laminin Receptor Precursor Promotes Cellular Prion Protein Induced Multi-Drug-Resistance of Gastric Cancer. Oncotarget 2017, 8, 71630–71641. [Google Scholar] [CrossRef] [Green Version]
- Barbieri, G.; Palumbo, S.; Gabrusiewicz, K.; Azzalin, A.; Marchesi, N.; Spedito, A.; Biggiogera, M.; Sbalchiero, E.; Mazzini, G.; Miracco, C.; et al. Silencing of Cellular Prion Protein (PrPC) Expression by DNA-Antisense Oligonucleotides Induces Autophagy-Dependent Cell Death in Glioma Cells. Autophagy 2011, 7, 840–853. [Google Scholar] [CrossRef] [Green Version]
- Roucou, X.; Gains, M.; LeBlanc, A.C. Neuroprotective Functions of Prion Protein. J. Neurosci. Res. 2004, 75, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Arora, A.S.; Zafar, S.; Latif, U.; Llorens, F.; Sabine, M.; Kumar, P.; Tahir, W.; Thüne, K.; Shafiq, M.; Schmitz, M.; et al. The Role of Cellular Prion Protein in Lipid Metabolism in the Liver. Prion 2020, 14, 95–108. [Google Scholar] [CrossRef] [Green Version]
- Wiegmans, A.P.; Saunus, J.M.; Ham, S.; Lobb, R.; Kutasovic, J.R.; Dalley, A.J.; Miranda, M.; Atkinson, C.; Foliaki, S.T.; Ferguson, K.; et al. Secreted Cellular Prion Protein Binds Doxorubicin and Correlates with Anthracycline Resistance in Breast Cancer. JCI Insight 2019, 5, e124092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernardino-Sgherri, J.; Siberchicot, C.; Auvré, F.; Busso, D.; Brocas, C.; El Masri, G.; Lioutsko, A.; Ferri, F.; Radicella, J.P.; Romeo, P.-H.; et al. Tumor Resistance to Radiotherapy Is Triggered by an ATM/TAK1-Dependent-Increased Expression of the Cellular Prion Protein. Oncogene 2021, 40, 3460–3469. [Google Scholar] [CrossRef] [PubMed]
- Siberchicot, C.; Gault, N.; Déchamps, N.; Barroca, V.; Aguzzi, A.; Roméo, P.-H.; Radicella, J.P.; Bravard, A.; Bernardino-Sgherri, J. Prion Protein Deficiency Impairs Hematopoietic Stem Cell Determination and Sensitizes Myeloid Progenitors to Irradiation. Haematologica 2020, 105, 1216–1222. [Google Scholar] [CrossRef] [Green Version]
- Bravard, A.; Auvré, F.; Fantini, D.; Bernardino-Sgherri, J.; Sissoëff, L.; Daynac, M.; Xu, Z.; Etienne, O.; Dehen, C.; Comoy, E.; et al. The Prion Protein Is Critical for DNA Repair and Cell Survival after Genotoxic Stress. Nucleic Acids Res. 2015, 43, 904–916. [Google Scholar] [CrossRef] [Green Version]
- Turu, M.; Slevin, M.; Ethirajan, P.; Luque, A.; Elasbali, A.; Font, A.; Gaffney, J.; Cairols, M.; Kumar, P.; Kumar, S.; et al. The Normal Cellular Prion Protein and Its Possible Role in Angiogenesis. Front. Biosci. J. Virtual Libr. 2008, 13, 6491–6500. [Google Scholar] [CrossRef] [Green Version]
- Doeppner, T.R.; Kaltwasser, B.; Schlechter, J.; Jaschke, J.; Kilic, E.; Bähr, M.; Hermann, D.M.; Weise, J. Cellular Prion Protein Promotes Post-Ischemic Neuronal Survival, Angioneurogenesis and Enhances Neural Progenitor Cell Homing via Proteasome Inhibition. Cell Death Dis. 2015, 6, e2024. [Google Scholar] [CrossRef] [Green Version]
- Alfaidy, N.; Chauvet, S.; Andrei, S.; Salomon, A.; Saoudi, Y.; Richaud, P.; Aude-Garcia, C.; Hoffmann, P.; Andrieux, A.; Moulis, J.M.; et al. Prion Protein Expression and Functional Importance in Developmental Angiogenesis: Role in Oxidative Stress and Copper Homeostasis. Antioxid. Redox Signal. 2012, 18, 400–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramljak, S.; Herlyn, H.; Zerr, I. Cellular Prion Protein (PrPc) and Hypoxia: True to Each Other in Good Times and in Bad, in Sickness, and in Health. Front. Cell. Neurosci. 2016, 10, 292. [Google Scholar] [CrossRef] [Green Version]
- Singleton, D.C.; Macann, A.; Wilson, W.R. Therapeutic Targeting of the Hypoxic Tumour Microenvironment. Nat. Rev. Clin. Oncol. 2021. [Google Scholar] [CrossRef]
- Möller, A.; Lobb, R.J. The Evolving Translational Potential of Small Extracellular Vesicles in Cancer. Nat. Rev. Cancer 2020, 20, 697–709. [Google Scholar] [CrossRef]
- Cao, R.; Bråkenhielm, E.; Li, X.; Pietras, K.; Widenfalk, J.; Ostman, A.; Eriksson, U.; Cao, Y. Angiogenesis Stimulated by PDGF-CC, a Novel Member in the PDGF Family, Involves Activation of PDGFR-αα and-αβ Receptors. FASEB J. 2002, 16, 1575–1583. [Google Scholar] [CrossRef]
- Azad, T.; Ghahremani, M.; Yang, X. The Role of YAP and TAZ in Angiogenesis and Vascular Mimicry. Cells 2019, 8, 407. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Morales, J.E.; Avci, N.; Guerrero, P.A.; Rao, G.; Seo, J.H.; McCarty, J.H. The Vascular Endothelial Cell-Expressed Prion Protein Doppel Promotes Angiogenesis and Blood-Brain Barrier Development. Development 2020, 147, dev193094. [Google Scholar] [CrossRef]
- Al-Hilal, T.A.; Chung, S.W.; Choi, J.U.; Alam, F.; Park, J.; Kim, S.W.; Kim, S.Y.; Ahsan, F.; Kim, I.-S.; Byun, Y. Targeting Prion-like Protein Doppel Selectively Suppresses Tumor Angiogenesis. J. Clin. Invest. 2016, 126, 1251–1266. [Google Scholar] [CrossRef]
- Ciric, D.; Rezaei, H. Biochemical Insight into the Prion Protein Family. Front. Cell Dev. Biol. 2015, 3, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, W.; Kang, Y. Epithelial-Mesenchymal Plasticity in Cancer Progression and Metastasis. Dev. Cell 2019, 49, 361–374. [Google Scholar] [CrossRef] [PubMed]
- Gil, M.; Kim, Y.K.; Kim, K.-E.; Kim, W.; Park, C.-S.; Lee, K.J. Cellular Prion Protein Regulates Invasion and Migration of Breast Cancer Cells through MMP-9 Activity. Biochem. Biophys. Res. Commun. 2016, 470, 213–219. [Google Scholar] [CrossRef]
- Pan, Y.; Zhao, L.; Liang, J.; Liu, J.; Shi, Y.; Liu, N.; Zhang, G.; Jin, H.; Gao, J.; Xie, H.; et al. Cellular Prion Protein Promotes Invasion and Metastasis of Gastric Cancer. FASEB J. 2006, 20, 1886–1888. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.-C.; Lin, C.-H.; Shih, N.-C.; Liu, H.-L.; Wang, W.-C.; Lin, K.-Y.; Liu, Z.-Y.; Tseng, Y.-J.; Chang, H.-K.; Lin, Y.-C.; et al. Cellular Prion Protein Transcriptionally Regulated by NFIL3 Enhances Lung Cancer Cell Lamellipodium Formation and Migration through JNK Signaling. Oncogene 2020, 39, 385–398. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Qian, J.; Wang, F.; Ma, Z. Cellular Prion Protein Accelerates Colorectal Cancer Metastasis via the Fyn-SP1-SATB1 Axis. Oncol. Rep. 2012, 28, 2029–2034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, L.; Rao, G.; Wang, H.; Li, B.; Tian, W.; Cui, J.; He, L.; Laffin, B.; Tian, X.; Hao, C.; et al. CD44-Positive Cancer Stem Cells Expressing Cellular Prion Protein Contribute to Metastatic Capacity in Colorectal Cancer. Cancer Res. 2013, 73, 2682–2694. [Google Scholar] [CrossRef] [Green Version]
- De Lacerda, T.C.S.; Costa-Silva, B.; Giudice, F.S.; Dias, M.V.S.; de Oliveira, G.P.; Teixeira, B.L.; Dos Santos, T.G.; Martins, V.R. Prion Protein Binding to HOP Modulates the Migration and Invasion of Colorectal Cancer Cells. Clin. Exp. Metastasis 2016, 33, 441–451. [Google Scholar] [CrossRef]
- Li, C.; Yu, S.; Nakamura, F.; Pentikäinen, O.T.; Singh, N.; Yin, S.; Xin, W.; Sy, M.-S. Pro-Prion Binds Filamin A, Facilitating Its Interaction with Integrin β1, and Contributes to Melanomagenesis. J. Biol. Chem. 2010, 285, 30328–30339. [Google Scholar] [CrossRef] [Green Version]
- Dery, M.A.; Jodoin, J.; Ursini-Siegel, J.; Aleynikova, O.; Ferrario, C.; Hassan, S.; Basik, M.; Leblanc, A.C. Endoplasmic Reticulum Stress Induces PRNP Prion Protein Gene Expression in Breast Cancer. Breast Cancer Res. 2013, 15, R22. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Shang, Y.; Liu, C.; Li, J.; Hu, H.; Liang, C.; Han, Y.; Zhang, W.; Liang, J.; Wu, K. Overexpression of PrPc, Combined with MGr1-Ag/37LRP, Is Predictive of Poor Prognosis in Gastric Cancer. Int. J. Cancer 2014, 135, 2329–2337. [Google Scholar] [CrossRef]
- Coebergh van den Braak, R.R.J.; Ten Hoorn, S.; Sieuwerts, A.M.; Tuynman, J.B.; Smid, M.; Wilting, S.M.; Martens, J.W.M.; Punt, C.J.A.; Foekens, J.A.; Medema, J.P.; et al. Interconnectivity between Molecular Subtypes and Tumor Stage in Colorectal Cancer. BMC Cancer 2020, 20, 850. [Google Scholar] [CrossRef]
- Faubert, B.; Solmonson, A.; DeBerardinis, R.J. Metabolic Reprogramming and Cancer Progression. Science 2020, 368, eaaw5473. [Google Scholar] [CrossRef] [PubMed]
- De Wit, M.; Jimenez, C.R.; Carvalho, B.; Belien, J.A.M.; Delis-van Diemen, P.M.; Mongera, S.; Piersma, S.R.; Vikas, M.; Navani, S.; Pontén, F.; et al. Cell Surface Proteomics Identifies Glucose Transporter Type 1 and Prion Protein as Candidate Biomarkers for Colorectal Adenoma-to-Carcinoma Progression. Gut 2012, 61, 855–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashok, A.; Singh, N. Prion Protein Modulates Glucose Homeostasis by Altering Intracellular Iron. Sci. Rep. 2018, 8, 6556. [Google Scholar] [CrossRef] [PubMed]
- Ramljak, S.; Schmitz, M.; Repond, C.; Zerr, I.; Pellerin, L. Altered MRNA and Protein Expression of Monocarboxylate Transporter MCT1 in the Cerebral Cortex and Cerebellum of Prion Protein Knockout Mice. Int. J. Mol. Sci. 2021, 22, 1566. [Google Scholar] [CrossRef] [PubMed]
- Kleene, R.; Loers, G.; Langer, J.; Frobert, Y.; Buck, F.; Schachner, M. Prion Protein Regulates Glutamate-Dependent Lactate Transport of Astrocytes. J. Neurosci. Off. J. Soc. Neurosci. 2007, 27, 12331–12340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rutishauser, D.; Mertz, K.D.; Moos, R.; Brunner, E.; Rulicke, T.; Calella, A.M.; Aguzzi, A. The Comprehensive Native Interactome of a Fully Functional Tagged Prion Protein. PLoS ONE 2009, 4, e4446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watts, J.C.; Huo, H.; Bai, Y.; Ehsani, S.; Jeon, A.H.; Shi, T.; Daude, N.; Lau, A.; Young, R.; Xu, L.; et al. Interactome Analyses Identify Ties of PrP and Its Mammalian Paralogs to Oligomannosidic N-Glycans and Endoplasmic Reticulum-Derived Chaperones. PLoS Pathog. 2009, 5, e1000608. [Google Scholar] [CrossRef]
- Zafar, S.; von Ahsen, N.; Oellerich, M.; Zerr, I.; Schulz-Schaeffer, W.J.; Armstrong, V.W.; Asif, A.R. Proteomics Approach to Identify the Interacting Partners of Cellular Prion Protein and Characterization of Rab7a Interaction in Neuronal Cells. J. Proteome Res. 2011, 10, 3123–3135. [Google Scholar] [CrossRef] [PubMed]
- Ramljak, S.; Schmitz, M.; Zafar, S.; Wrede, A.; Schenkel, S.; Asif, A.R.; Carimalo, J.; Doeppner, T.R.; Schulz-Schaeffer, W.J.; Weise, J.; et al. Cellular Prion Protein Directly Interacts with and Enhances Lactate Dehydrogenase Expression under Hypoxic Conditions. Exp. Neurol. 2015, 271, 155–167. [Google Scholar] [CrossRef]
- Strom, A.; Diecke, S.; Hunsmann, G.; Stuke, A.W. Identification of Prion Protein Binding Proteins by Combined Use of Far-Western Immunoblotting, Two Dimensional Gel Electrophoresis and Mass Spectrometry. Proteomics 2006, 6, 26–34. [Google Scholar] [CrossRef]
- Zafar, S.; Asif, A.R.; Ramljak, S.; Tahir, W.; Schmitz, M.; Zerr, I. Anchorless 23-230 PrPC Interactomics for Elucidation of PrPC Protective Role. Mol. Neurobiol. 2014, 49, 1385–1399. [Google Scholar] [CrossRef] [PubMed]
- Faris, R.; Moore, R.A.; Ward, A.; Race, B.; Dorward, D.W.; Hollister, J.R.; Fischer, E.R.; Priola, S.A. Cellular Prion Protein Is Present in Mitochondria of Healthy Mice. Sci. Rep. 2017, 7, 41556. [Google Scholar] [CrossRef] [PubMed]
- Miele, G.; Jeffrey, M.; Turnbull, D.; Manson, J.; Clinton, M. Ablation of Cellular Prion Protein Expression Affects Mitochondrial Numbers and Morphology. Biochem. Biophys. Res. Commun. 2002, 291, 372–377. [Google Scholar] [CrossRef]
- Paterson, A.W.J.; Curtis, J.C.; Macleod, N.K. Complex I Specific Increase in Superoxide Formation and Respiration Rate by PrP-Null Mouse Brain Mitochondria. J. Neurochem. 2008, 105, 177–191. [Google Scholar] [CrossRef]
- Stella, R.; Cifani, P.; Peggion, C.; Hansson, K.; Lazzari, C.; Bendz, M.; Levander, F.; Sorgato, M.C.; Bertoli, A.; James, P. Relative Quantification of Membrane Proteins in Wild-Type and Prion Protein (PrP)-Knockout Cerebellar Granule Neurons. J. Proteome Res. 2012, 11, 523–536. [Google Scholar] [CrossRef] [PubMed]
- Becht, E.; de Reyniès, A.; Giraldo, N.A.; Pilati, C.; Buttard, B.; Lacroix, L.; Selves, J.; Sautès-Fridman, C.; Laurent-Puig, P.; Fridman, W.H. Immune and Stromal Classification of Colorectal Cancer Is Associated with Molecular Subtypes and Relevant for Precision Immunotherapy. Clin. Cancer Res. 2016, 22, 4057–4066. [Google Scholar] [CrossRef] [Green Version]
- Bakkebø, M.K.; Mouillet-Richard, S.; Espenes, A.; Goldmann, W.; Tatzelt, J.; Tranulis, M.A. The Cellular Prion Protein: A Player in Immunological Quiescence. Front. Immunol. 2015, 6, 450. [Google Scholar] [CrossRef] [Green Version]
- Angioni, R.; Sánchez-Rodríguez, R.; Viola, A.; Molon, B. TGF-β in Cancer: Metabolic Driver of the Tolerogenic Crosstalk in the Tumor Microenvironment. Cancers 2021, 13, 401. [Google Scholar] [CrossRef]
- Munn, D.H.; Mellor, A.L. IDO in the Tumor Microenvironment: Inflammation, Counter-Regulation, and Tolerance. Trends Immunol. 2016, 37, 193–207. [Google Scholar] [CrossRef] [Green Version]
- Domingues, P.H.; Nanduri, L.S.Y.; Seget, K.; Venkateswaran, S.V.; Agorku, D.; Viganó, C.; von Schubert, C.; Nigg, E.A.; Swanton, C.; Sotillo, R.; et al. Cellular Prion Protein PrPC and Ecto-5′-Nucleotidase Are Markers of the Cellular Stress Response to Aneuploidy. Cancer Res. 2017, 77, 2914–2926. [Google Scholar] [CrossRef] [Green Version]
- Qin, K.; Zhao, L.; Ash, R.D.; McDonough, W.F.; Zhao, R.Y. ATM-Mediated Transcriptional Elevation of Prion in Response to Copper-Induced Oxidative Stress. J. Biol. Chem. 2009, 284, 4582–4593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boddy, A.M.; Huang, W.; Aktipis, A. Life History Trade-Offs in Tumors. Curr. Pathobiol. Rep. 2018, 6, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef]
- Taniguchi, K.; Karin, M. NF-ΚB, Inflammation, Immunity and Cancer: Coming of Age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.-R.; Mu, T.-C.; Gao, Z.-X.; Wang, J.; Sy, M.-S.; Li, C.-Y. Prion Protein Is Required for Tumor Necrosis Factor α (TNFα)-Triggered Nuclear Factor ΚB (NF-ΚB) Signaling and Cytokine Production. J. Biol. Chem. 2017, 292, 18747–18759. [Google Scholar] [CrossRef] [Green Version]
- Assi, K.; Mills, J.; Owen, D.; Ong, C.; St Arnaud, R.; Dedhar, S.; Salh, B. Integrin-Linked Kinase Regulates Cell Proliferation and Tumour Growth in Murine Colitis-Associated Carcinogenesis. Gut 2008, 57, 931–940. [Google Scholar] [CrossRef] [PubMed]
- Pammer, J.; Cross, H.S.; Frobert, Y.; Tschachler, E.; Oberhuber, G. The Pattern of Prion-Related Protein Expression in the Gastrointestinal Tract. Virchows Arch. Int. J. Pathol. 2000, 436, 466–472. [Google Scholar] [CrossRef]
- Petit, C.S.V.; Barreau, F.; Besnier, L.; Gandille, P.; Riveau, B.; Chateau, D.; Roy, M.; Berrebi, D.; Svrcek, M.; Cardot, P.; et al. Requirement of Cellular Prion Protein for Intestinal Barrier Function and Mislocalization in Patients with Inflammatory Bowel Disease. Gastroenterology 2012, 143, 122–132.e15. [Google Scholar] [CrossRef]
- Cai, J.; Zhang, N.; Zheng, Y.; de Wilde, R.F.; Maitra, A.; Pan, D. The Hippo Signaling Pathway Restricts the Oncogenic Potential of an Intestinal Regeneration Program. Genes Dev. 2010, 24, 2383–2388. [Google Scholar] [CrossRef] [Green Version]
- Johnson, R.; Halder, G. The Two Faces of Hippo: Targeting the Hippo Pathway for Regenerative Medicine and Cancer Treatment. Nat. Rev. Drug Discov. 2014, 13, 63–79. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Secondary structure of human PrPC. The alpha-cleavage occurs at position 110/111 and generates the N1 and C1 fragments. The beta-cleavage occurs in the vicinity of octapeptide repeats, which bind copper ions, and generates the N2 and C2 fragments. PrPC can also be shed from the plasma membrane through the action of ADAM10. The alpha helices (H) and beta sheets (Β) are indicated (adapted from [7]).
Figure 1.
Secondary structure of human PrPC. The alpha-cleavage occurs at position 110/111 and generates the N1 and C1 fragments. The beta-cleavage occurs in the vicinity of octapeptide repeats, which bind copper ions, and generates the N2 and C2 fragments. PrPC can also be shed from the plasma membrane through the action of ADAM10. The alpha helices (H) and beta sheets (Β) are indicated (adapted from [7]).

Figure 2.
Summary of the data supporting a link between PrPC expression and resistance to cytotoxic agents.
Figure 2.
Summary of the data supporting a link between PrPC expression and resistance to cytotoxic agents.

Figure 3.
Summary of the interplay between PrPC and hypoxia and of the contribution of PrPC to angiogenesis. PrPC is induced under ischemic conditions and prevents the degradation of HIF1α by the proteasome. On the other hand, under hypoxia, HIF1α promotes the transcription of the PRNP gene, favours the sequestration of the GRP78 E3-ligase, leading to enhanced PrPC protein stability, and is associated with enhanced release of exosomal PrPC. PrPC may further sustain angiogenesis by controlling the levels of PDGFC.
Figure 3.
Summary of the interplay between PrPC and hypoxia and of the contribution of PrPC to angiogenesis. PrPC is induced under ischemic conditions and prevents the degradation of HIF1α by the proteasome. On the other hand, under hypoxia, HIF1α promotes the transcription of the PRNP gene, favours the sequestration of the GRP78 E3-ligase, leading to enhanced PrPC protein stability, and is associated with enhanced release of exosomal PrPC. PrPC may further sustain angiogenesis by controlling the levels of PDGFC.

Figure 4.
Elevated PrPC is associated with increased invasion and migration of cancer cells and metastasis. PrPC promotes an upregulation of EMT transcription factors (EMT TF), a switch from E-cadherin to N-cadherin expression as well as an induction of matrix metalloproteases, themselves fostering the remodelling of the extracellular matrix (ECM) (top panel). This action may be fostered by the interaction between PrPC and its ligand STI1. It is also supported by pro-PrPC, through its interaction with Filamin-A and the active cleaved fragment of Notch, NICD (Notch Intra Cellular Domain) or by exosomal PrPC (bottom panel).
Figure 4.
Elevated PrPC is associated with increased invasion and migration of cancer cells and metastasis. PrPC promotes an upregulation of EMT transcription factors (EMT TF), a switch from E-cadherin to N-cadherin expression as well as an induction of matrix metalloproteases, themselves fostering the remodelling of the extracellular matrix (ECM) (top panel). This action may be fostered by the interaction between PrPC and its ligand STI1. It is also supported by pro-PrPC, through its interaction with Filamin-A and the active cleaved fragment of Notch, NICD (Notch Intra Cellular Domain) or by exosomal PrPC (bottom panel).

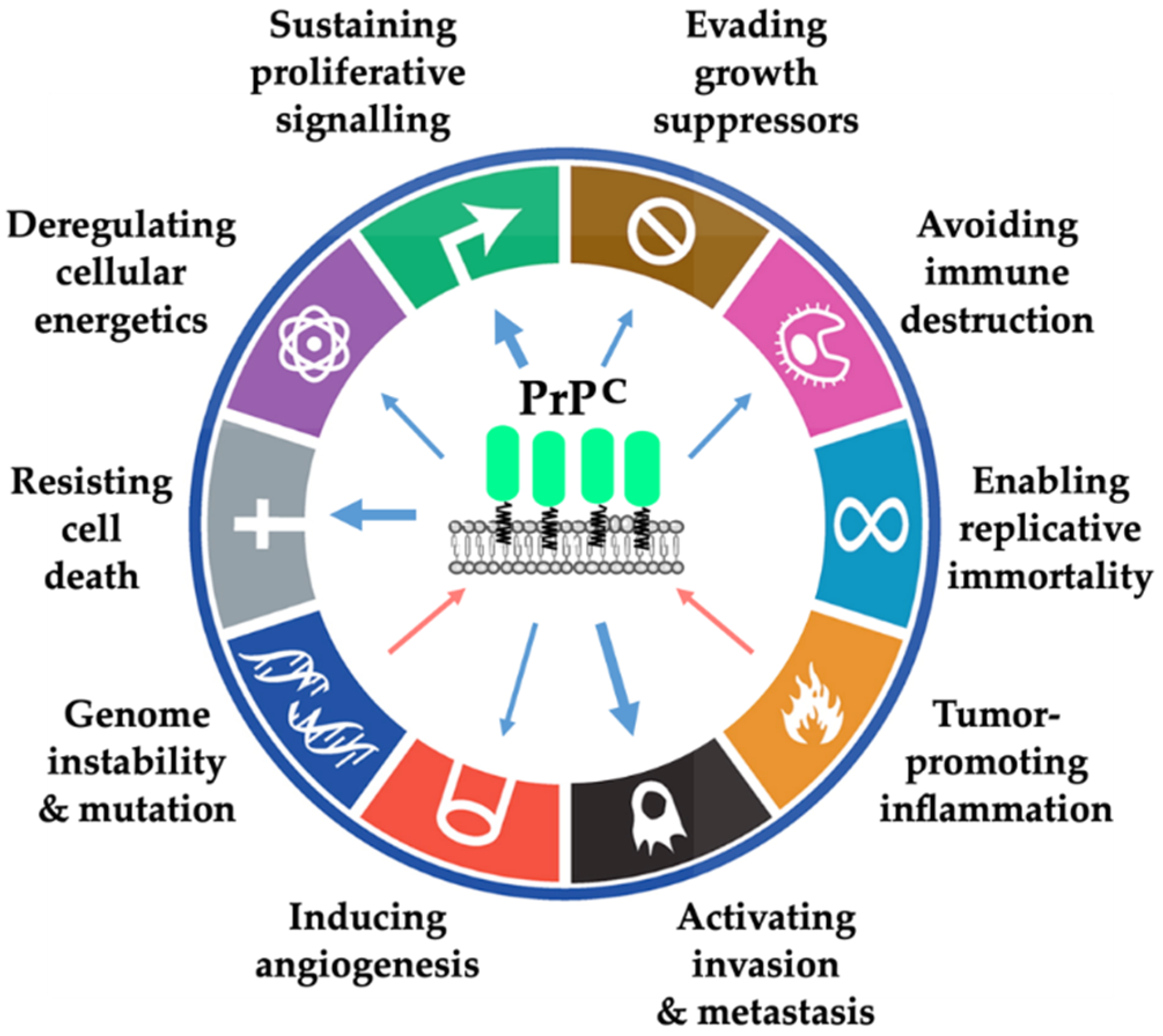
Figure 5.
Summary of the data linking PrPC with the different hallmarks of cancer. Blue arrows indicate a contribution of PrPC to the hallmarks and red arrows indicate a regulation of PrPC as a consequence of the hallmark. The thickness of the arrow is related to the amount of evidence supporting the link. The hallmarks of cancer have been adapted from Hanahan and Weinberg [14].
Figure 5.
Summary of the data linking PrPC with the different hallmarks of cancer. Blue arrows indicate a contribution of PrPC to the hallmarks and red arrows indicate a regulation of PrPC as a consequence of the hallmark. The thickness of the arrow is related to the amount of evidence supporting the link. The hallmarks of cancer have been adapted from Hanahan and Weinberg [14].


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Summary of the binding partners involved in the PrPC contribution to a given hallmark of cancer, in relation with the cancer type.
Table 1.
Summary of the binding partners involved in the PrPC contribution to a given hallmark of cancer, in relation with the cancer type.
| Hallmark | Partner | Cancer Type | Reference |
|---|---|---|---|
| Sustaining proliferative signalling | NOTCH1 | Pancreatic | [18] |
| Sustaining proliferative signalling | STI1 | Glioblastoma | [21] |
| Resisting cell death | 37LRP | Gastric | [62] |
| Activating invasion and metastasis | STI1 | Colorectal | [87] |
| Activating invasion and metastasis | Filamin-A | Melanoma | [88] |
Table 2.
Summary of the signalling pathways through which PrPC contributes to a given hallmark of cancer, in relation with the cancer type.
Table 2.
Summary of the signalling pathways through which PrPC contributes to a given hallmark of cancer, in relation with the cancer type.
| Hallmark | Partner | Cancer Type | Reference |
|---|---|---|---|
| Sustaining proliferative signalling | PI3K/AKT- CyclinD1 | Gastric | [15] |
| Sustaining proliferative signalling | NOTCH1 | Pancreatic | [18] |
| Sustaining proliferative signalling | ILK | Colorectal | [26] |
| Evading growth suppressors | NF2 | Schwannoma | [24] |
| Evading growth suppressors | TGFβ | Colorectal | [26] |
| Resisting cell death | MDR1 | Gastric | [54] |
| Resisting cell death | PI3K/AKT | Gastric | [59] |
| Resisting cell death | BCL2 | Gastric | [42,54,58] |
| Resisting cell death | BCL2 | Breast | [51] |
| Resisting cell death | BCL2 | Glioma | [63] |
| Resisting cell death | Survivin/cIAP-1/XIAP | Colorectal | [25] |
| Resisting cell death | Par4 | Glioblastoma | [23] |
| Resisting cell death | Soluble PrPC | Breast | [66] |
| Inducing angiogenesis | Hypoxia | Colorectal | [31] |
| Activating invasion and metastasis | MMPs | Breast | [52,82] |
| Activating invasion and metastasis | MMPs | Gastric | [83] |
| Activating invasion and metastasis | NOTCH1 | Pancreatic | [18] |
| Activating invasion and metastasis | JNK | Lung | [84] |
| Reprogramming of energy metabolism | GLUT1 | Colorectal | [29] |
| Evading immune surveillance | TGFβ | Colorectal | [26] |
| Evading immune surveillance | IDO | Colorectal | [26] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Mouillet-Richard, S.; Ghazi, A.; Laurent-Puig, P. The Cellular Prion Protein and the Hallmarks of Cancer. Cancers 2021, 13, 5032. https://doi.org/10.3390/cancers13195032
AMA Style
Mouillet-Richard S, Ghazi A, Laurent-Puig P. The Cellular Prion Protein and the Hallmarks of Cancer. Cancers. 2021; 13(19):5032. https://doi.org/10.3390/cancers13195032
Chicago/Turabian StyleMouillet-Richard, Sophie, Alexandre Ghazi, and Pierre Laurent-Puig. 2021. "The Cellular Prion Protein and the Hallmarks of Cancer" Cancers 13, no. 19: 5032. https://doi.org/10.3390/cancers13195032
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.