
Disruption of miRNA-mRNA Networks Defines Novel Molecular Signatures for Penile Carcinogenesis

 ,
,  , , and
, , and
Abstract
:Simple Summary
Abstract
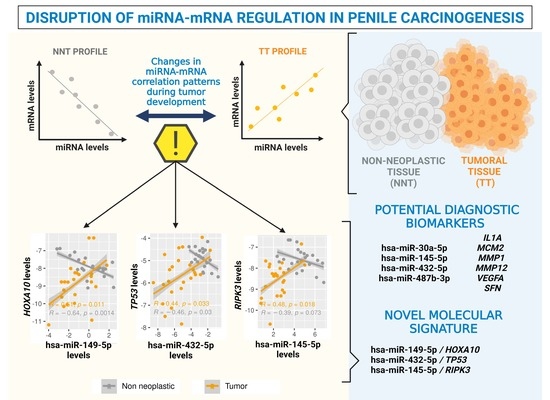
1. Introduction
2. Materials and Methods
2.1. Patients and Sample Collection
2.2. Total RNA Extraction
2.3. miRNA Expression Profiling by Microarray Analysis
2.4. Validation of DEmiRs by Quantitative Reverse Transcription Polymerase Chain Reaction (qRT-PCR)
2.5. Array Express Dataset for In Silico Analysis of miRNA Expression in PeC
2.6. Pathway Enrichment Analysis of the Predicted Targets of the DEmiRs and Establishment of miRNA-mRNA Interaction Networks
2.7. mRNA Expression Profiling by a High-Throughput Nanofluidic qRT-PCR Platform
2.8. GEO Dataset for In Silico Analysis of Deregulated Gene Expression in PeC
2.9. Gene Set Enrichment Analysis (GSEA) of Deregulated Genes in PeC
2.10. DNA Extraction and Human Papillomavirus (HPV) Detection
2.11. Statistical Analyses
3. Results
3.1. Patients, Treatment, and Follow Up
3.2. miRNAs Expression Profiles, Interaction Networks of miRNA and Predicted Target Genes and Pathway Enrichment Analysis
3.3. Deregulated mRNA Expression Profiling in PeC
3.4. Penile Carcinogenesis Alters Expression Profiles of miRNA-mRNA Pairs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Christodoulidou, M.; Sahdev, V.; Houssein, S.; Muneer, A. Epidemiology of penile cancer. Curr. Probl. Cancer 2015, 39, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Coelho, R.W.P.; Pinho, J.D.; Moreno, J.S.; Garbis, D.; do Nascimento, A.M.T.; Larges, J.S.; Calixto, J.R.R.; Ramalho, L.N.Z.; da Silva, A.A.M.; Nogueira, L.R.; et al. Penile cancer in Maranhao, Northeast Brazil: The highest incidence globally? BMC Urol. 2018, 18, 50. [Google Scholar] [CrossRef]
- Mulherkar, R.; Hasan, S.; Wegner, R.E.; Verma, V.; Glaser, S.M.; Kalash, R.; Beriwal, S.; Horne, Z.D. National patterns of care for early-stage penile cancers in the United States: How is radiation and brachytherapy utilized? Brachytherapy 2019, 18, 503–509. [Google Scholar] [CrossRef]
- Ornellas, A.A.; Kinchin, E.W.; Nobrega, B.L.; Wisnescky, A.; Koifman, N.; Quirino, R. Surgical treatment of invasive squamous cell carcinoma of the penis: Brazilian National Cancer Institute long-term experience. J. Surg. Oncol. 2008, 97, 487–495. [Google Scholar] [CrossRef]
- Gopman, J.M.; Djajadiningrat, R.S.; Baumgarten, A.S.; Espiritu, P.N.; Horenblas, S.; Zhu, Y.; Protzel, C.; Pow-Sang, J.M.; Kim, T.; Sexton, W.J.; et al. Predicting postoperative complications of inguinal lymph node dissection for penile cancer in an international multicentre cohort. BJU Int. 2015, 116, 196–201. [Google Scholar] [CrossRef]
- Thomas, A.; Necchi, A.; Muneer, A.; Tobias-Machado, M.; Tran, A.T.H.; Van Rompuy, A.S.; Spiess, P.E.; Albersen, M. Penile cancer. Nat. Rev. Dis. Primers 2021, 7, 11. [Google Scholar] [CrossRef] [PubMed]
- Emmanuel, A.; Nettleton, J.; Watkin, N.; Berney, D.M. The molecular pathogenesis of penile carcinoma-current developments and understanding. Virchows Arch. 2019, 475, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Kuasne, H.; Colus, I.M.; Busso, A.F.; Hernandez-Vargas, H.; Barros-Filho, M.C.; Marchi, F.A.; Scapulatempo-Neto, C.; Faria, E.F.; Lopes, A.; Guimaraes, G.C.; et al. Genome-wide methylation and transcriptome analysis in penile carcinoma: Uncovering new molecular markers. Clin. Epigenet. 2015, 7, 46. [Google Scholar] [CrossRef] [Green Version]
- Barzon, L.; Cappellesso, R.; Peta, E.; Militello, V.; Sinigaglia, A.; Fassan, M.; Simonato, F.; Guzzardo, V.; Ventura, L.; Blandamura, S.; et al. Profiling of expression of human papillomavirus-related cancer miRNAs in penile squamous cell carcinomas. Am. J. Pathol. 2014, 184, 3376–3383. [Google Scholar] [CrossRef]
- Munoz, J.J.; Drigo, S.A.; Barros-Filho, M.C.; Marchi, F.A.; Scapulatempo-Neto, C.; Pessoa, G.S.; Guimaraes, G.C.; Trindade Filho, J.C.; Lopes, A.; Arruda, M.A.; et al. Down-Regulation of SLC8A1 as a Putative Apoptosis Evasion Mechanism by Modulation of Calcium Levels in Penile Carcinoma. J. Urol. 2015, 194, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wei, P.; Shen, X.; Zhang, Y.; Xu, B.; Zhou, J.; Fan, S.; Hao, Z.; Shi, H.; Zhang, X.; et al. MicroRNA Expression Profile in Penile Cancer Revealed by Next-Generation Small RNA Sequencing. PLoS ONE 2015, 10, e0131336. [Google Scholar] [CrossRef]
- Hartz, J.M.; Engelmann, D.; Furst, K.; Marquardt, S.; Spitschak, A.; Goody, D.; Protzel, C.; Hakenberg, O.W.; Putzer, B.M. Integrated Loss of miR-1/miR-101/miR-204 Discriminates Metastatic from Nonmetastatic Penile Carcinomas and Can Predict Patient Outcome. J. Urol. 2016, 196, 570–578. [Google Scholar] [CrossRef] [PubMed]
- Kuasne, H.; Barros-Filho, M.C.; Busso-Lopes, A.; Marchi, F.A.; Pinheiro, M.; Munoz, J.J.; Scapulatempo-Neto, C.; Faria, E.F.; Guimaraes, G.C.; Lopes, A.; et al. Integrative miRNA and mRNA analysis in penile carcinomas reveals markers and pathways with potential clinical impact. Oncotarget 2017, 8, 15294–15306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peta, E.; Cappellesso, R.; Masi, G.; Sinigaglia, A.; Trevisan, M.; Grassi, A.; Di Camillo, B.; Vassarotto, E.; Fassina, A.; Palu, G.; et al. Down-regulation of microRNA-146a is associated with high-risk human papillomavirus infection and epidermal growth factor receptor overexpression in penile squamous cell carcinoma. Hum. Pathol. 2017, 61, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Marchi, F.A.; Martins, D.C.; Barros-Filho, M.C.; Kuasne, H.; Busso Lopes, A.F.; Brentani, H.; Trindade Filho, J.C.S.; Guimaraes, G.C.; Faria, E.F.; Scapulatempo-Neto, C.; et al. Multidimensional integrative analysis uncovers driver candidates and biomarkers in penile carcinoma. Sci. Rep. 2017, 7, 6707. [Google Scholar] [CrossRef]
- Pinho, J.D.; Silva, G.E.B.; Teixeira Junior, A.A.L.; Belfort, M.R.C.; Mendes, J.M.; Cunha, I.W.D.; Quintana, L.G.; Calixto, J.R.R.; Nogueira, L.R.; Coelho, R.W.P.; et al. MIR-107, MIR-223-3P and MIR-21-5P Reveals Potential Biomarkers in Penile Cancer. Asian Pac. J. Cancer Prev. 2020, 21, 391–397. [Google Scholar] [CrossRef]
- Ayoubian, H.; Heinzelmann, J.; Holters, S.; Khalmurzaev, O.; Pryalukhin, A.; Loertzer, P.; Heinzelbecker, J.; Lohse, S.; Geppert, C.; Loertzer, H.; et al. miRNA Expression Characterizes Histological Subtypes and Metastasis in Penile Squamous Cell Carcinoma. Cancers 2021, 13, 1480. [Google Scholar] [CrossRef]
- Silva, J.D.; Nogueira, L.; Coelho, R.; Deus, A.; Khayat, A.; Marchi, R.; Oliveira, E.; Santos, A.P.D.; Cavalli, L.; Pereira, S. HPV-associated penile cancer: Impact of copy number alterations in miRNA/mRNA interactions and potential druggable targets. Cancer Biomark. 2021, 1–14. [Google Scholar] [CrossRef]
- Pinho, J.D.; Silva, G.E.B.; Teixeira Júnior, A.A.L.; Belfort, M.R.D.C.; Mendes, J.M.M.; Calixto, J.D.R.R.; Nogueira, L.R.; Burbano, R.R.; Khayat, A.S. Downregulation of miR-145 is associated with perineural invasion in penile carcinoma. Transl. Androl. Urol. 2021, 10, 2019–2026. [Google Scholar] [CrossRef]
- Fabian, M.R.; Sonenberg, N. The mechanics of miRNA-mediated gene silencing: A look under the hood of miRISC. Nat. Struct. Mol. Biol. 2012, 19, 586–593. [Google Scholar] [CrossRef]
- Friedman, R.C.; Farh, K.K.H.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2008, 19, 92–105. [Google Scholar] [CrossRef] [Green Version]
- Brierley, J.D.; Gospodarowicz, M.K.; Wittekind, C. TNM Classification of Malignant Tumors, 8th ed.; Wiley-Blackwell: Hoboken, NJ, USA, 2016. [Google Scholar]
- Hakenberg, O.W.; Compérat, E.; Minhas, S.; Necchi, A.; Protzel, C.; Watkin, N. The European Association of Urology (EAU) Guidelines on Penile Cancer. Available online: http://uroweb.org/guideline/penile-cancer/#10 (accessed on 5 June 2018).
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Licursi, V.; Conte, F.; Fiscon, G.; Paci, P. MIENTURNET: An interactive web tool for microRNA-target enrichment and network-based analysis. BMC Bioinform. 2019, 20, 545. [Google Scholar] [CrossRef] [Green Version]
- Andersen, C.L.; Jensen, J.L.; Orntoft, T.F. Normalization of real-time quantitative reverse transcription-PCR data: A model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res. 2004, 64, 5245–5250. [Google Scholar] [CrossRef] [Green Version]
- Saiki, R.K.; Gelfand, D.H.; Stoffel, S.; Scharf, S.J.; Higuchi, R.; Horn, G.T.; Mullis, K.B.; Erlich, H.A. Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase. Science 1988, 239, 487–491. [Google Scholar] [CrossRef]
- Kleter, B.; van Doorn, L.J.; Schrauwen, L.; Molijn, A.; Sastrowijoto, S.; ter Schegget, J.; Lindeman, J.; ter Harmsel, B.; Burger, M.; Quint, W. Development and clinical evaluation of a highly sensitive PCR-reverse hybridization line probe assay for detection and identification of anogenital human papillomavirus. J. Clin. Microbiol. 1999, 37, 2508–2517. [Google Scholar] [CrossRef] [Green Version]
- de Sanjose, S.; Serrano, B.; Tous, S.; Alejo, M.; Lloveras, B.; Quiros, B.; Clavero, O.; Vidal, A.; Ferrandiz-Pulido, C.; Pavon, M.A.; et al. Burden of Human Papillomavirus (HPV)-Related Cancers Attributable to HPVs 6/11/16/18/31/33/45/52 and 58. JNCI Cancer Spectr. 2018, 2, pky045. [Google Scholar] [CrossRef] [Green Version]
- Chaux, A.; Munari, E.; Katz, B.; Sharma, R.; Lecksell, K.; Cubilla, A.L.; Burnett, A.L.; Netto, G.J. The epidermal growth factor receptor is frequently overexpressed in penile squamous cell carcinomas: A tissue microarray and digital image analysis study of 112 cases. Hum. Pathol. 2013, 44, 2690–2695. [Google Scholar] [CrossRef]
- Arya, M.; Thrasivoulou, C.; Henrique, R.; Millar, M.; Hamblin, R.; Davda, R.; Aare, K.; Masters, J.R.; Thomson, C.; Muneer, A.; et al. Targets of Wnt/ß-Catenin Transcription in Penile Carcinoma. PLoS ONE 2015, 10, e0124395. [Google Scholar] [CrossRef]
- Svoronos, A.A.; Engelman, D.M.; Slack, F.J. OncomiR or Tumor Suppressor? The Duplicity of MicroRNAs in Cancer. Cancer Res. 2016, 76, 3666–3670. [Google Scholar] [CrossRef] [Green Version]
- Saliminejad, K.; Khorram Khorshid, H.R.; Soleymani Fard, S.; Ghaffari, S.H. An overview of microRNAs: Biology, functions, therapeutics, and analysis methods. J. Cell. Physiol. 2019, 234, 5451–5465. [Google Scholar] [CrossRef]
- Gobin, E.; Bagwell, K.; Wagner, J.; Mysona, D.; Sandirasegarane, S.; Smith, N.; Bai, S.; Sharma, A.; Schleifer, R.; She, J.X. A pan-cancer perspective of matrix metalloproteases (MMP) gene expression profile and their diagnostic/prognostic potential. BMC Cancer 2019, 19, 581. [Google Scholar] [CrossRef] [Green Version]
- May, M.; Burger, M.; Otto, W.; Hakenberg, O.W.; Wieland, W.F.; May, D.; Hofstadter, F.; Gotz, S.; Niessl, N.; Fritsche, H.M.; et al. Ki-67, mini-chromosome maintenance 2 protein (MCM2) and geminin have no independent prognostic relevance for cancer-specific survival in surgically treated squamous cell carcinoma of the penis. BJU Int. 2013, 112, E383–E390. [Google Scholar] [CrossRef] [Green Version]
- Nayak, S.; Goel, M.M.; Chandra, S.; Bhatia, V.; Mehrotra, D.; Kumar, S.; Makker, A.; Rath, S.K.; Agarwal, S.P. VEGF-A immunohistochemical and mRNA expression in tissues and its serum levels in potentially malignant oral lesions and oral squamous cell carcinomas. Oral. Oncol. 2012, 48, 233–239. [Google Scholar] [CrossRef]
- Saleh, A.D.; Cheng, H.; Martin, S.E.; Si, H.; Ormanoglu, P.; Carlson, S.; Clavijo, P.E.; Yang, X.; Das, R.; Cornelius, S.; et al. Integrated Genomic and Functional microRNA Analysis Identifies miR-30-5p as a Tumor Suppressor and Potential Therapeutic Nanomedicine in Head and Neck Cancer. Clin. Cancer Res. 2019, 25, 2860–2873. [Google Scholar] [CrossRef] [Green Version]
- Song, Z.; Lin, Y.; Ye, X.; Feng, C.; Lu, Y.; Yang, G.; Dong, C. Expression of IL-1alpha and IL-6 is Associated with Progression and Prognosis of Human Cervical Cancer. Med. Sci. Monit. 2016, 22, 4475–4481. [Google Scholar] [CrossRef] [Green Version]
- Inoue, A.; Obayashi, K.; Sonoda, Y.; Nakamura, A.; Ueno, T.; Kuhara, S.; Tashiro, K. Regulation of matrix metalloproteinase-1 and alpha-smooth muscle actin expression by interleukin-1 alpha and tumour necrosis factor alpha in hepatic stellate cells. Cytotechnology 2017, 69, 461–468. [Google Scholar] [CrossRef]
- Tang, R.F.; Wang, S.X.; Zhang, F.R.; Peng, L.; Wang, S.X.; Xiao, Y.; Zhang, M. Interleukin-1alpha, 6 regulate the secretion of vascular endothelial growth factor A, C in pancreatic cancer. Hepatobiliary Pancreat. Dis. Int. 2005, 4, 460–463. [Google Scholar]
- Laurila, E.M.; Kallioniemi, A. The diverse role of miR-31 in regulating cancer associated phenotypes. Genes Chromosomes Cancer 2013, 52, 1103–1113. [Google Scholar] [CrossRef]
- Yuan, Y.; Wang, Z.; Chen, M.; Jing, Y.; Shu, W.; Xie, Z.; Li, Z.; Xu, J.; He, F.; Jiao, P.; et al. Macrophage-Derived Exosomal miR-31-5p Promotes Oral Squamous Cell Carcinoma Tumourigenesis Through the Large Tumor Suppressor 2-Mediated Hippo Signalling Pathway. J. Biomed. Nanotechnol. 2021, 17, 822–837. [Google Scholar] [CrossRef]
- Zhong, Z.; Dong, Z.; Yang, L.; Chen, X.; Gong, Z. MicroRNA-31-5p modulates cell cycle by targeting human mutL homolog 1 in human cancer cells. Tumour Biol. 2013, 34, 1959–1965. [Google Scholar] [CrossRef]
- Yuan, Y.; Shi, X.; Li, B.; Peng, M.; Zhu, T.; Lv, G.; Liu, L.; Jin, H.; Li, L.; Qin, D. Integrated analysis of key microRNAs /TFs /mRNAs/ in HPV-positive cervical cancer based on microRNA sequencing and bioinformatics analysis. Pathol. Res. Pract. 2020, 216, 152952. [Google Scholar] [CrossRef]
- Carrera, M.; Bitu, C.C.; de Oliveira, C.E.; Cervigne, N.K.; Graner, E.; Manninen, A.; Salo, T.; Coletta, R.D. HOXA10 controls proliferation, migration and invasion in oral squamous cell carcinoma. Int. J. Clin. Exp. Pathol. 2015, 8, 3613–3623. [Google Scholar] [PubMed]
- Wei, R.; Rodrìguez, R.A.; Mullor, M.D.M.R.; Tan, Z.; Gui, Y.; Hu, J.; Zhu, T.; Huang, X.; Zhu, Y.; Xu, J. Analyzing the prognostic value of DKK1 expression in human cancers based on bioinformatics. Ann. Transl. Med. 2020, 8, 552. [Google Scholar] [CrossRef] [PubMed]
- Vijayakumar, G.; Narwal, A.; Kamboj, M.; Sen, R. Association of SOX2, OCT4 and WNT5A Expression in Oral Epithelial Dysplasia and Oral Squamous Cell Carcinoma: An Immunohistochemical Study. Head Neck Pathol. 2020, 14, 749–757. [Google Scholar] [CrossRef]
- Torbrand, C.; Wigertz, A.; Drevin, L.; Folkvaljon, Y.; Lambe, M.; Hakansson, U.; Kirrander, P. Socioeconomic factors and penile cancer risk and mortality; a population-based study. BJU Int. 2017, 119, 254–260. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patients | Total Sample n (%) | Microarray Sample n (%) | Validation Sample n (%) |
|---|---|---|---|
| Number of patients | 24 | 11 | 13 |
| Age at surgery-Mean (SD) years old | 61.8 (16.1) | 61.6 (14.2) | 61.9 (18.2) |
| Follow up-Median (range) months | 39.8 (2–68) | 47.5 (8–62) | 34.9 (2–61) |
| Smoking history | 14 (58.3) | 8 (72.7) | 6 (46.1) |
| cT | |||
| cT1 | 3 (12.5) | 2 (18.2) | 1 (7.7) |
| cT2 | 13 (5.2) | 4 (36.4) | 9 (69.2) |
| cT3 | 8 (33.3) | 5 (45.4) | 3 (23.1) |
| cN | |||
| cN0 | 9 (37.5) | 5 (45.4) | 4 (30.8) |
| cN1 | 8 (33.3) | 3 (27.3) | 5 (38.4) |
| cN2 | 4 (16.7) | 3 (27.3) | 1 (7.7) |
| cN3 | 3 (12.5) | 0 (0) | 3 (23.1) |
| Penectomy | |||
| Partial | 17 (70.8) | 7 (63.6) | 10 (76.9) |
| Total | 7 (29.2) | 4 (36.4) | 3 (23.1) |
| Grade | |||
| I | 3 (12.5) | 2 (18.2) | 1 (7.7) |
| II | 13 (54.2) | 6 (54.5) | 7 (53.9) |
| III | 8 (33.3) | 3 (27.3) | 5 (38.4) |
| T Stage | |||
| pT1 | 4 (16.6) | 3 (27.2) | 1 (7.7) |
| pT2 | 13 (54.2) | 4 (36.4) | 9 (69.2) |
| pT3 | 7 (29.2) | 4 (36.4) | 3 (23.1) |
| Inguinal Lymphadenectomy | 16 (66.7) | 7 (63.6) | 9 (69.2) |
| Pelvic Lymphadenectomy | 4 (16.7) | 2 (18.1) | 2 (15.4) |
| Lymph node metastasis | 12 (50.0) | 6 (54.5) | 6 (46.2) |
| Presence of HPV infection | 8 (33.3) | 3 (27.3) | 5 (38.4) |
| Tumor Size-Mean (SD) cm | 4.75 (2.27) | 4.93 (2.50) | 4.59 (2.15) |
| Lymphovascular invasion | 6 (25.0) | 3 (27.3) | 3 (23.1) |
| Perineural invasion | 10 (41.7) | 4 (36.4) | 6 (46.2) |
| Group Risk (EAU) | |||
| Low | 1 (4.2) | 1 (9.1) | 0 (0) |
| Intermediate | 3 (12.5) | 2 (18. 2) | 1 (7.7) |
| High | 20 (83.3) | 8 (72.7) | 12 (92.3) |
| Regulation | DEmiR | Microarray | qRT-PCR | |||
|---|---|---|---|---|---|---|
| FC | FDR | FC | SD | FDR | ||
| Downregulated | miR-432-5p | 0.07 | 0.0048 * | 0.41 | 0.51 | 0.004 * |
| miR-487b-3p | 0.12 | 0.0079 * | 0.44 | 0.45 | 0.004 * | |
| miR-145-5p | 0.18 | 0.0088 * | 0.33 | 0.37 | 0.005 * | |
| miR-30a-5p | 0.21 | 0.0067 * | 0.57 | 0.32 | 0.002 * | |
| miR-149-5p | 0.29 | 0.0308 * | 0.79 | 1.04 | 0.097 | |
| Upregulated | miR-200a-5p | 2.93 | 0.0048 * | 3.48 | 1.28 | 0.004 * |
| miR-224-5p | 6.59 | 0.0083 * | 5.70 | 3.19 | 0.001 * | |
| miR-31-3p | 12.38 | 0.0002 * | 41.62 | 41.43 | 0.004 * | |
| miR-31-5p | 38.32 | 0.0048 * | 122.28 | 127.75 | 0.001 * | |
| Regulation in Our Study | miRNA | E-MTAB-3087 Dataset |
|---|---|---|
| log2 (Normalized Counts + 1) | ||
| Downregulated | miR-30a-5p * | −8.165 |
| miR-432-5p * | −7.947 | |
| miR-487b-3p * | −6.236 | |
| miR-145-5p * | −3.425 | |
| Upregulated | miR-200a-5p | −0.551 |
| miR-224-5p | −5.815 | |
| miR-31-3p | −3.342 | |
| miR-31-5p * | 2.914 |
| Reg | DEG | FC | FDR | DEG | FC | FDR | DEG | FC | FDR |
|---|---|---|---|---|---|---|---|---|---|
| Down | ABCB1 | 0.24 | <0.001 * | LATS2 | 0.58 | 0.004 * | RIPK3 | 0.53 | <0.001 * |
| ALDH1A1 | 0.09 | <0.001 * | MDM2 | 0.71 | 0.003 * | SAV1 | 0.45 | <0.001 * | |
| BCL2 | 0.31 | <0.001 * | MLH1 | 0.77 | 0.007 * | STAT3 | 0.66 | <0.001 * | |
| CCND1 | 0.61 | 0.007 * | NANOG | 0.33 | 0.005 * | STK4 | 0.65 | 0.002 * | |
| EGR1 | 0.19 | <0.001 * | NRP1 | 0.48 | <0.001 * | TCF7L2 | 0.36 | <0.001 * | |
| FGF2 | 0.18 | <0.001 * | PEBP1 | 0.53 | <0.001 * | TLR4 | 0.28 | <0.001 * | |
| FOS | 0.18 | <0.001 * | PITX2 | 0.30 | <0.001 * | TP53 | 0.57 | 0.003 * | |
| HOXA9 | 0.51 | 0.003 * | PPARGC1A | 0.11 | <0.001 * | TWIST1 | 0.18 | <0.001 * | |
| KAT6A | 0.47 | <0.001 * | PTEN | 0.55 | <0.001 * | ZEB1 | 0.23 | <0.001 * | |
| KLF4 | 0.22 | <0.001 * | RECK | 0.24 | <0.001 * | ||||
| KMT2A | 0.40 | <0.001 * | RHOA | 0.66 | <0.001 * | ||||
| Up | IL1A | 13.39 | <0.001 * | MMP1 | 28.00 | <0.001 * | SFN | 4.26 | 0.004 * |
| MCM2 | 2.14 | <0.001 * | MMP12 | 9.97 | <0.001 * | VEGFA | 2.06 | <0.001 * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Furuya, T.K.; Murta, C.B.; Murillo Carrasco, A.G.; Uno, M.; Sichero, L.; Villa, L.L.; Cardilli, L.; Coelho, R.F.; Guglielmetti, G.B.; Cordeiro, M.D.; et al. Disruption of miRNA-mRNA Networks Defines Novel Molecular Signatures for Penile Carcinogenesis. Cancers 2021, 13, 4745. https://doi.org/10.3390/cancers13194745
Furuya TK, Murta CB, Murillo Carrasco AG, Uno M, Sichero L, Villa LL, Cardilli L, Coelho RF, Guglielmetti GB, Cordeiro MD, et al. Disruption of miRNA-mRNA Networks Defines Novel Molecular Signatures for Penile Carcinogenesis. Cancers. 2021; 13(19):4745. https://doi.org/10.3390/cancers13194745
Chicago/Turabian StyleFuruya, Tatiane Katsue, Claudio Bovolenta Murta, Alexis Germán Murillo Carrasco, Miyuki Uno, Laura Sichero, Luisa Lina Villa, Leonardo Cardilli, Rafael Ferreira Coelho, Giuliano Betoni Guglielmetti, Mauricio Dener Cordeiro, and et al. 2021. "Disruption of miRNA-mRNA Networks Defines Novel Molecular Signatures for Penile Carcinogenesis" Cancers 13, no. 19: 4745. https://doi.org/10.3390/cancers13194745