Oxidative Stress and Apoptotic Responses Elicited by Nostoc-Synthesized Silver Nanoparticles against Different Cancer Cell Lines

Abstract
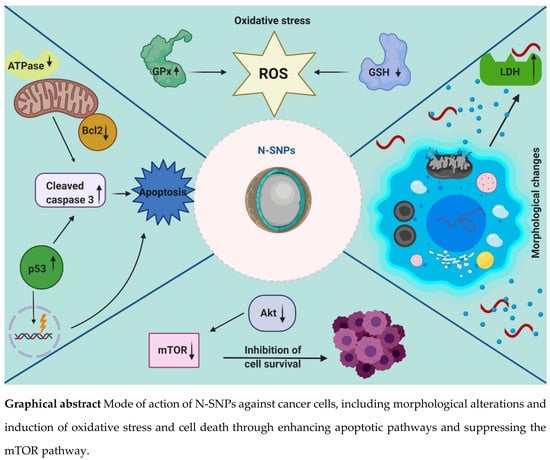
:
1. Introduction
2. Material and Methods
2.1. Materials
2.2. Methods
2.2.1. Preparation of N-SNP Suspension
2.2.2. Cell Culture
2.2.3. MTT Assay
2.2.4. Morphological Change Estimation
2.2.5. Transmission Electron Microscopy (TEM)
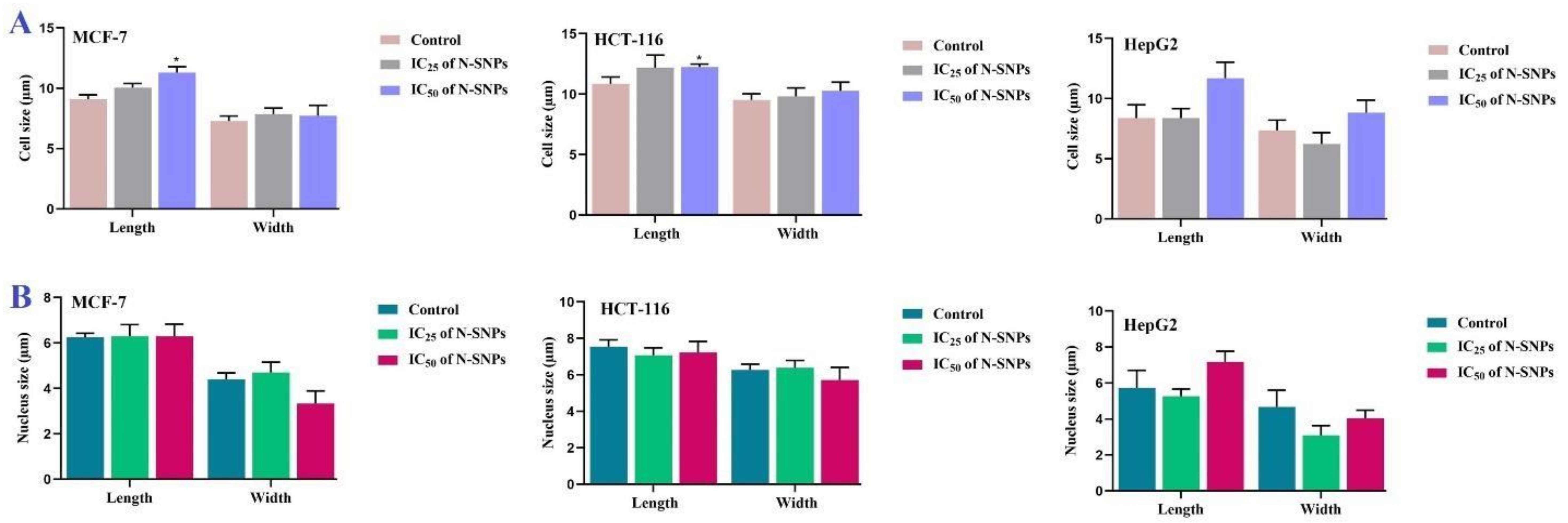
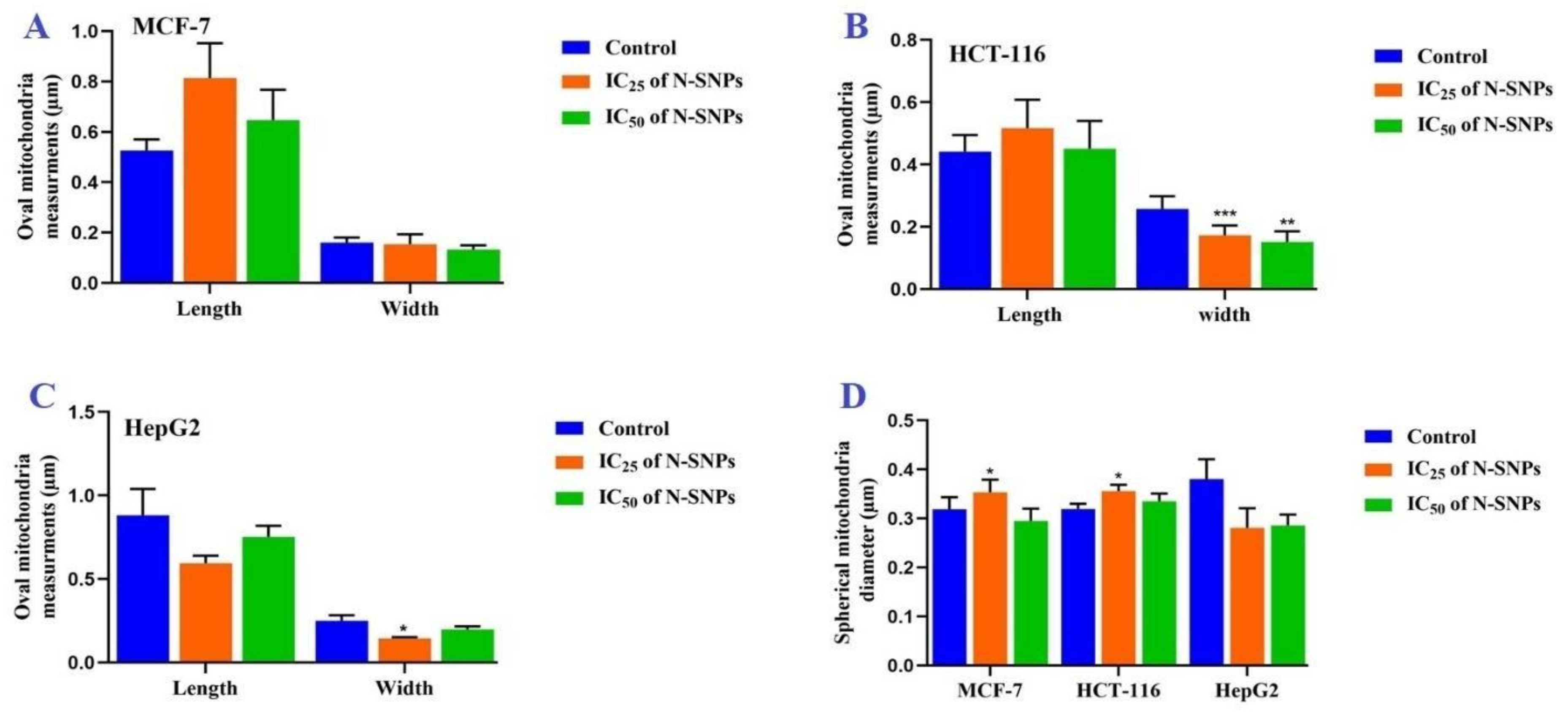
2.2.6. Morphometric Measurements
2.2.7. Membrane Integrity
2.2.8. Estimation of ATPase Activity
2.2.9. Evaluation of Antioxidative Marker Levels
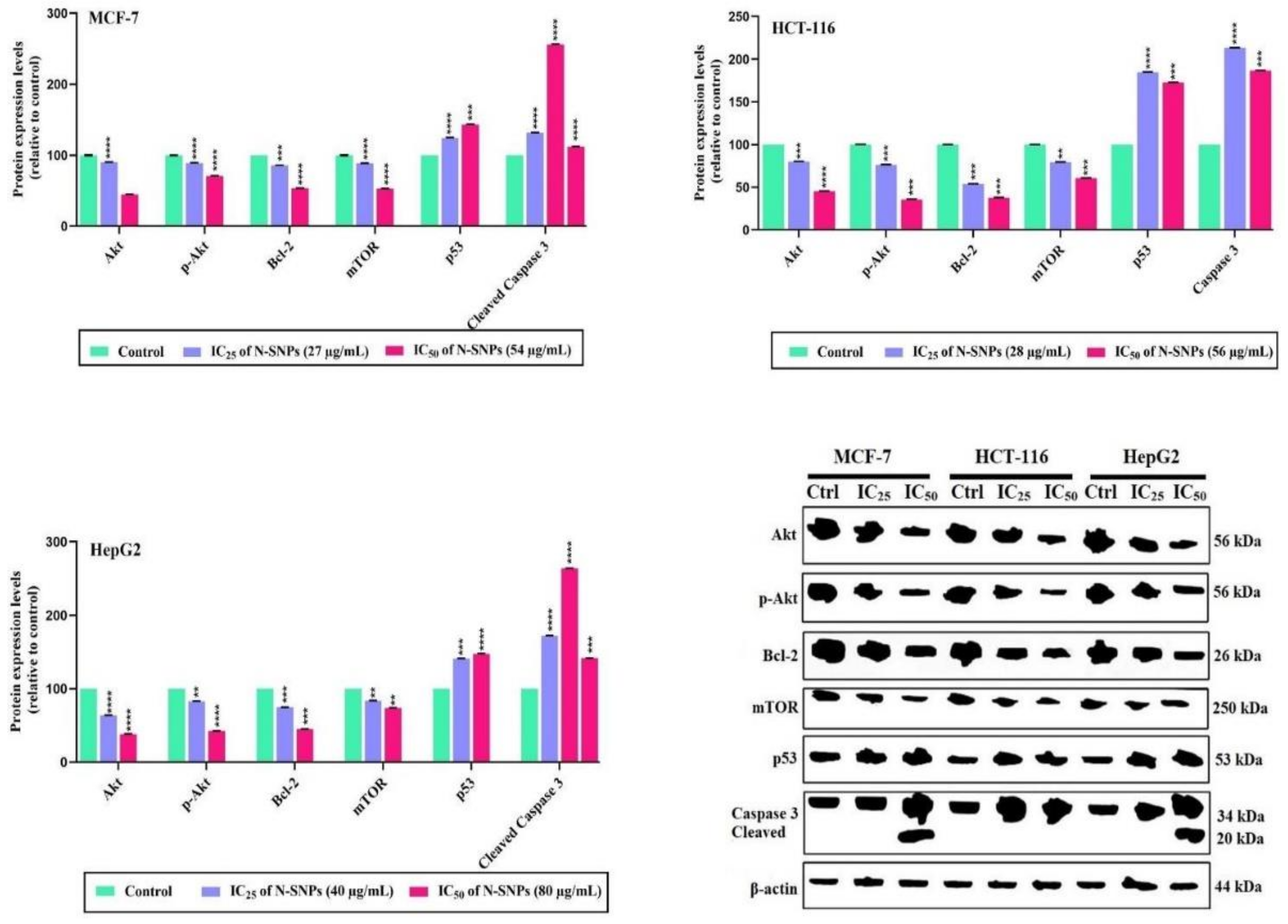
2.2.10. Western Blotting Analysis
2.2.11. Statistical Analysis
3. Results
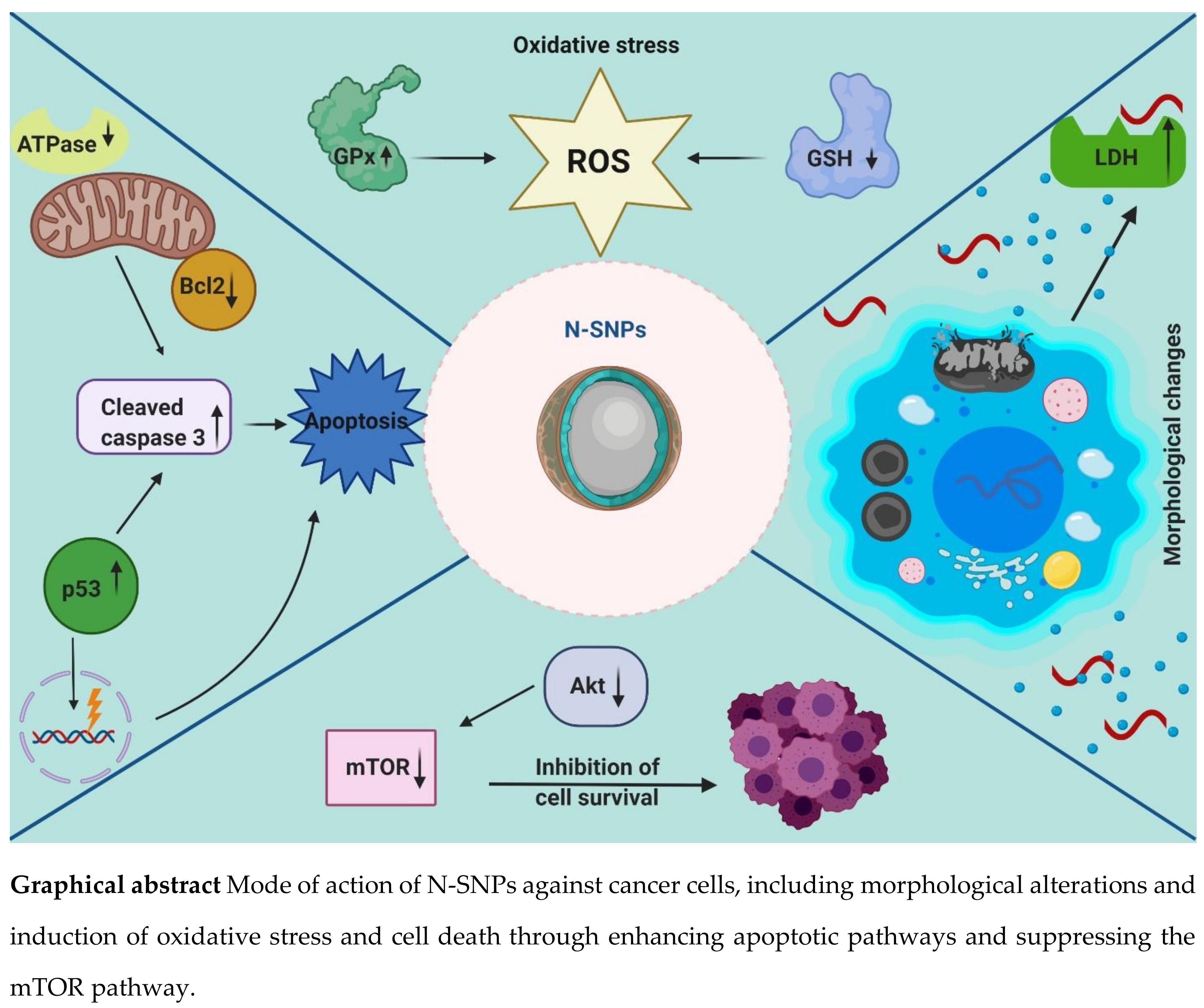
3.1. Cytotoxic Activity of N-SNPs Against MCF-7, HCT-116, and HepG2 Cells
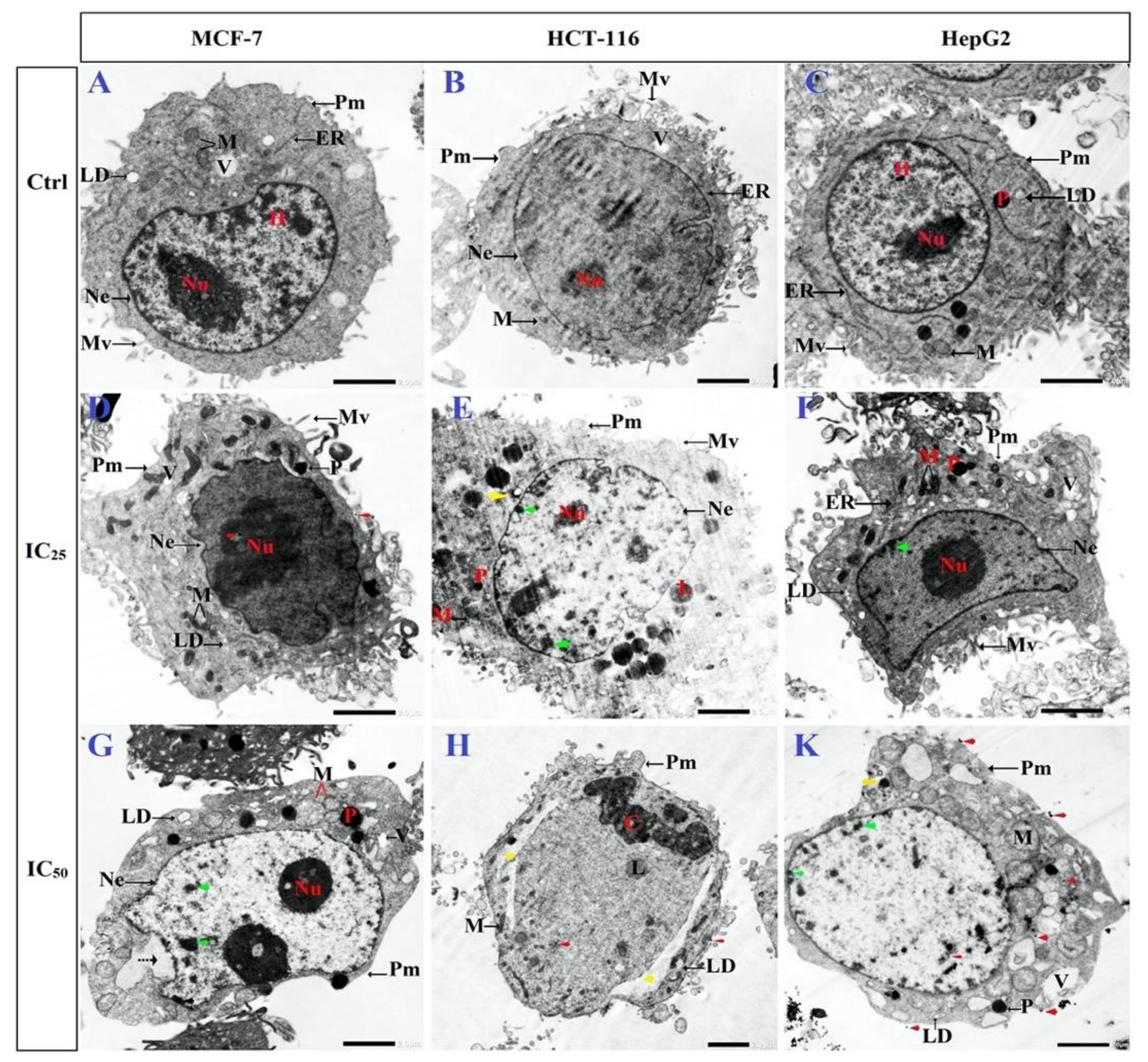
3.2. Morphological Changes of Cells Treated with N-SNPs
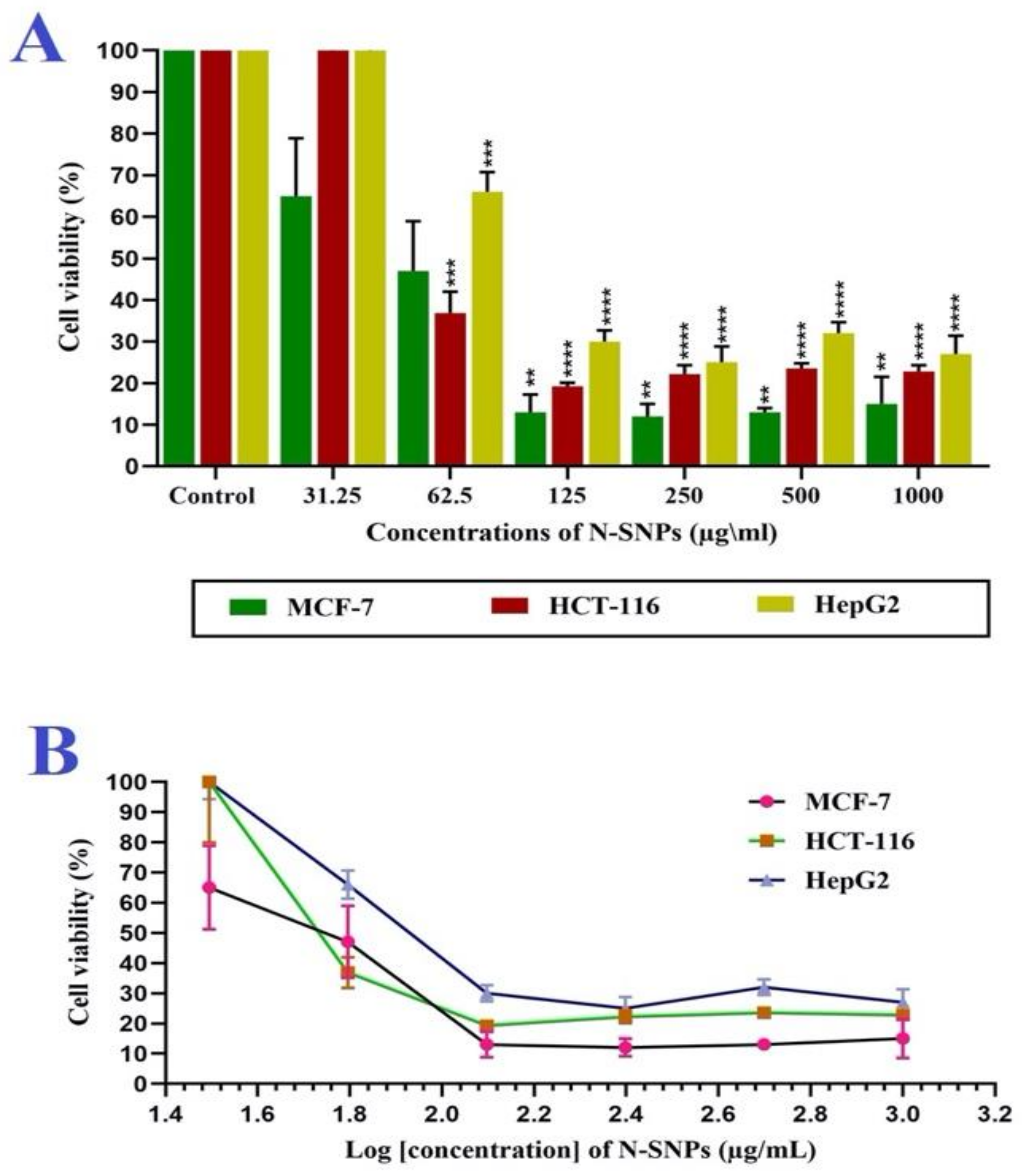
3.3. Membrane Integrity
3.4. ATPase Assay
3.5. Antioxidative Markers
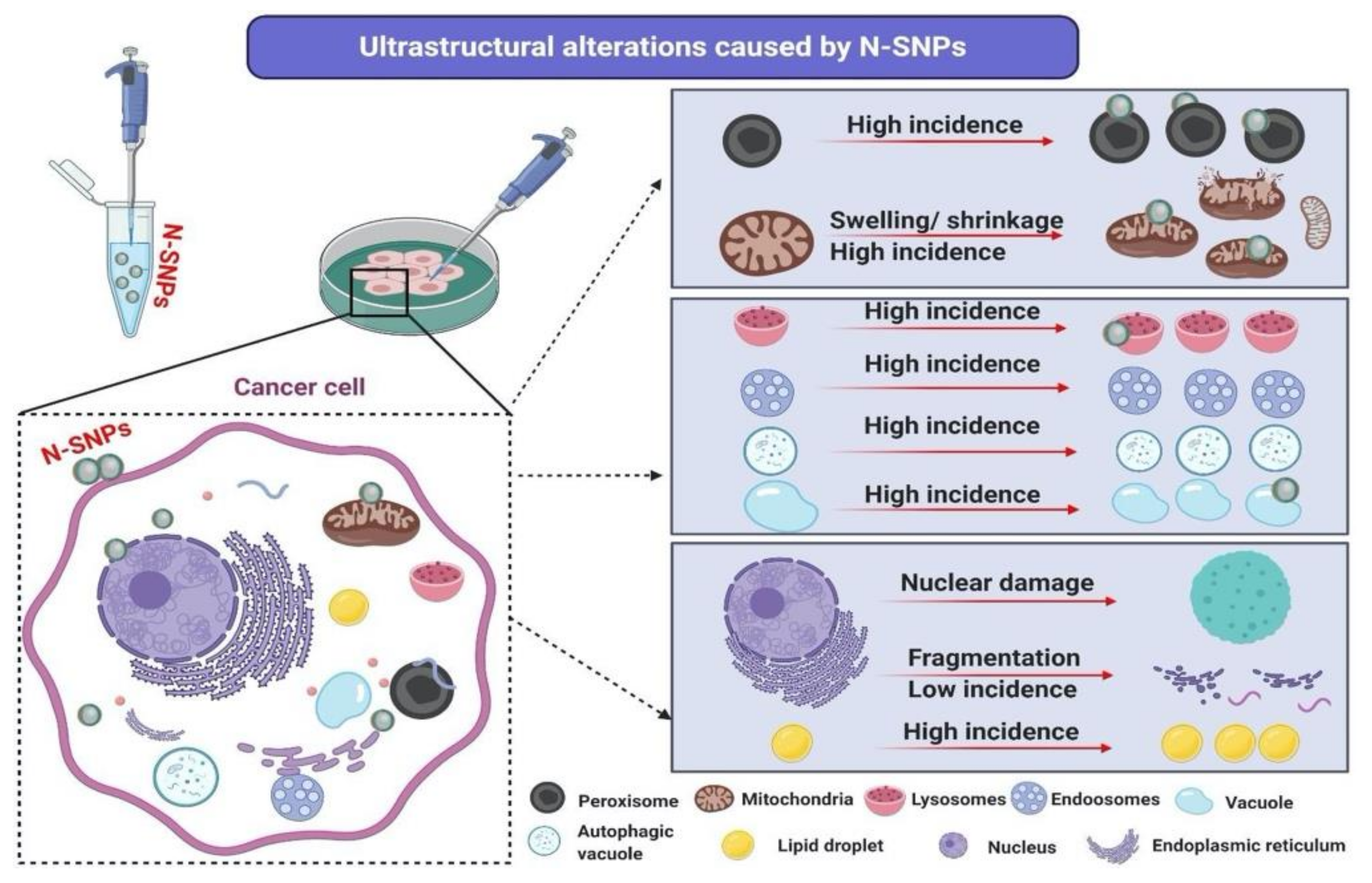
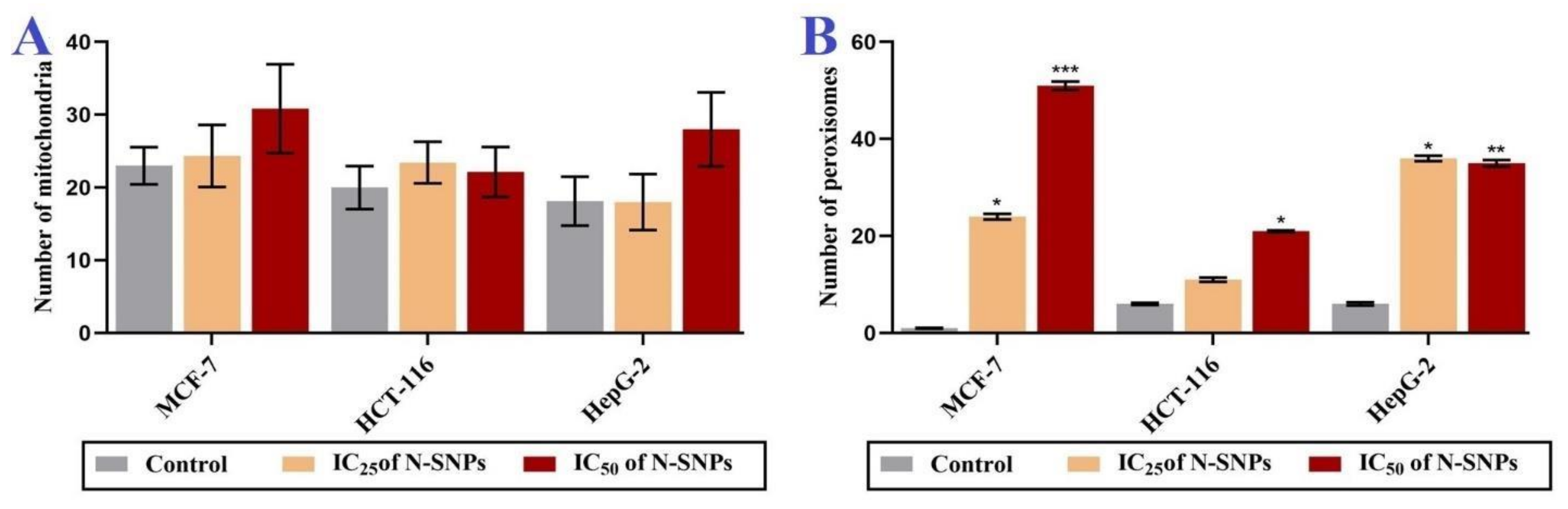
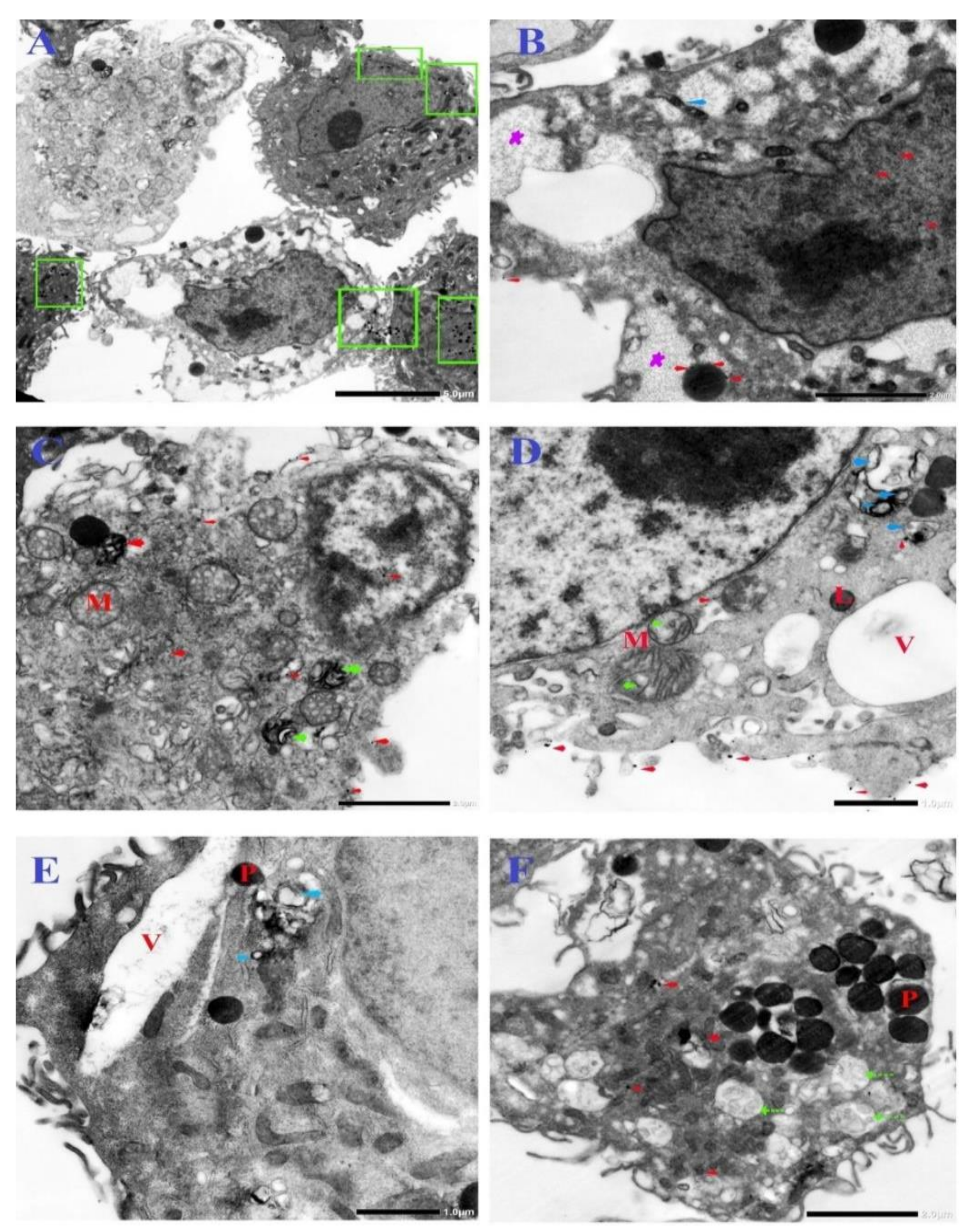
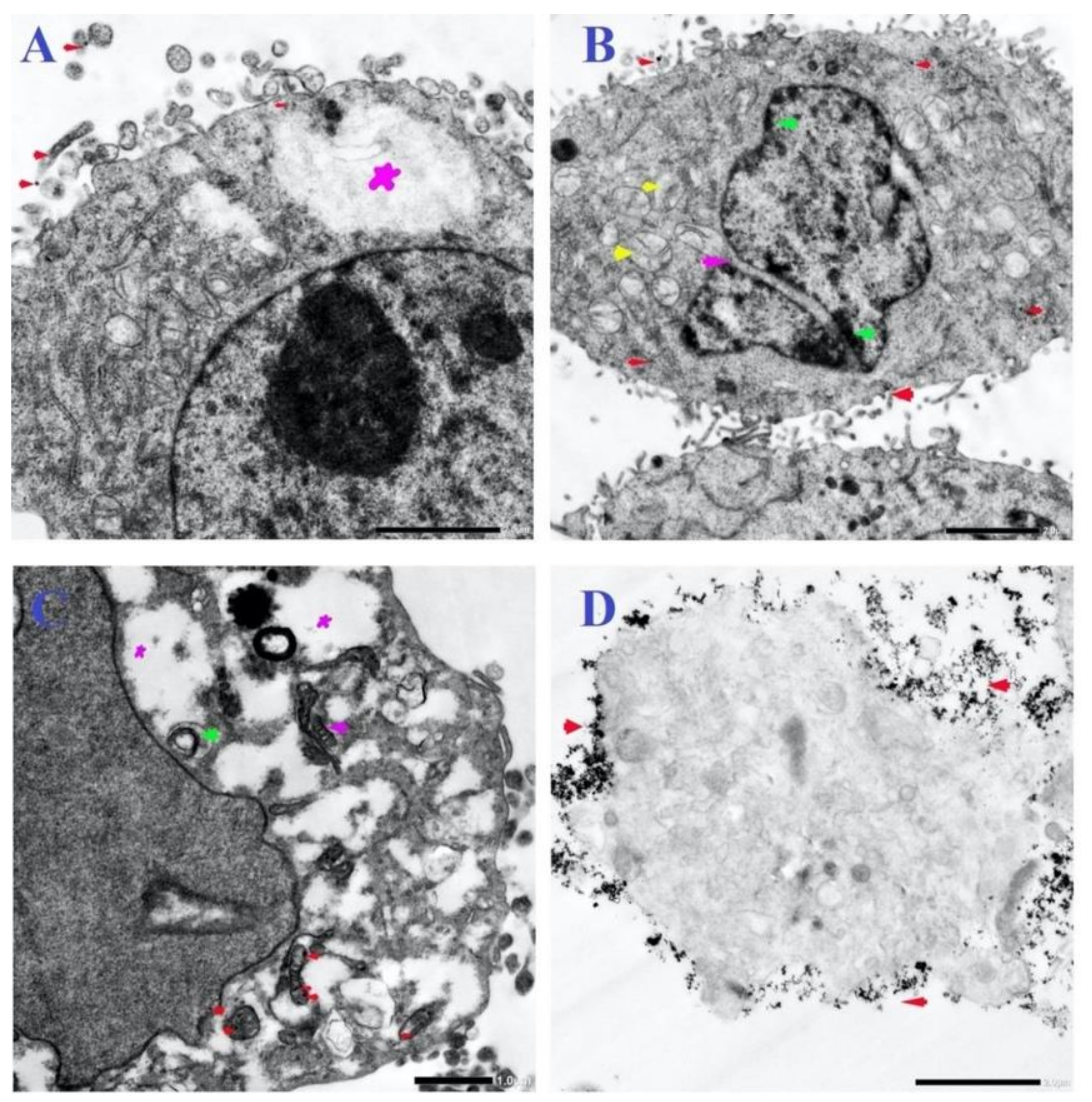
3.6. Ultrastructural Changes of Treated Cells with N-SNPs
3.7. Western Blotting Technique
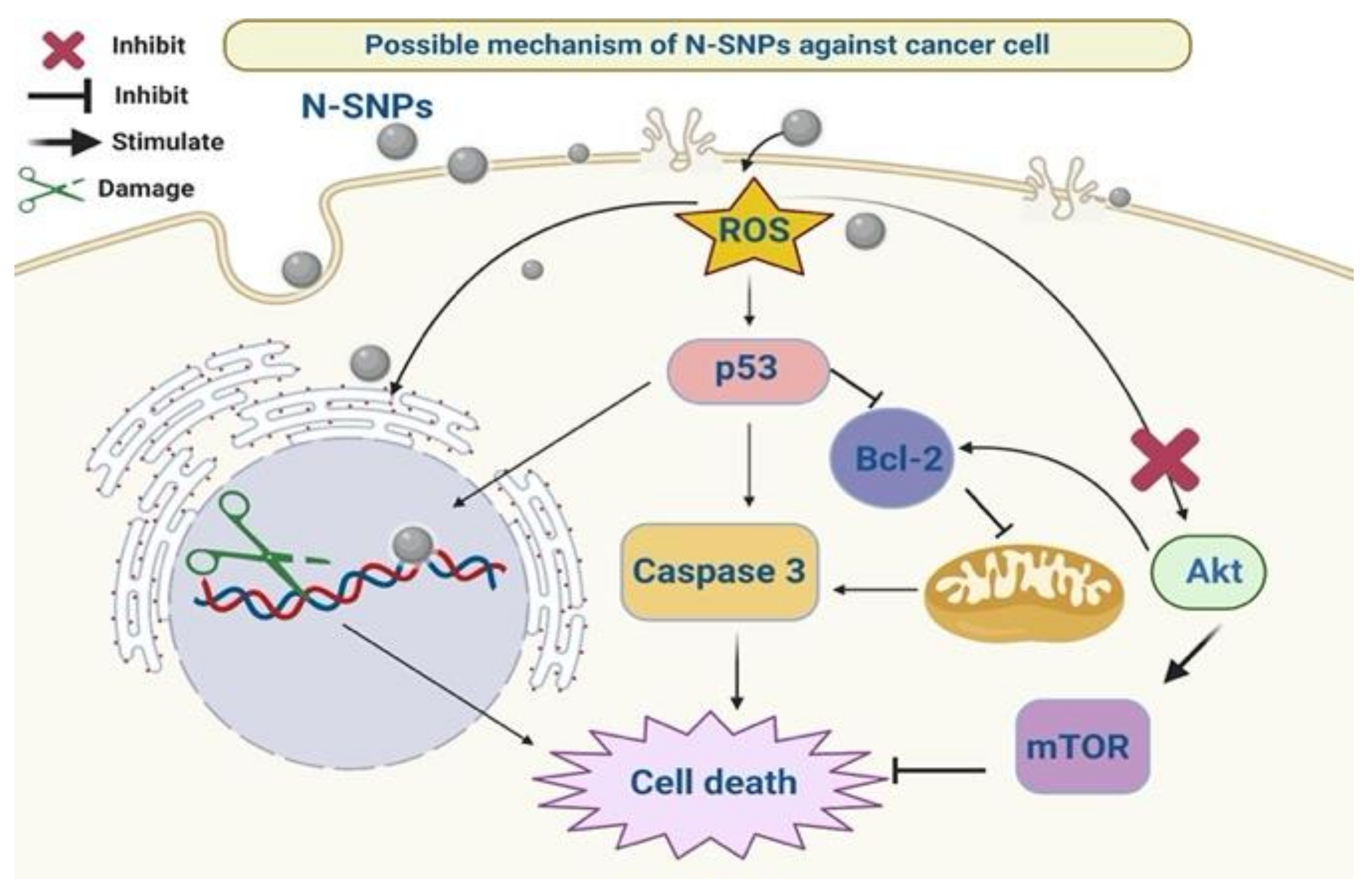
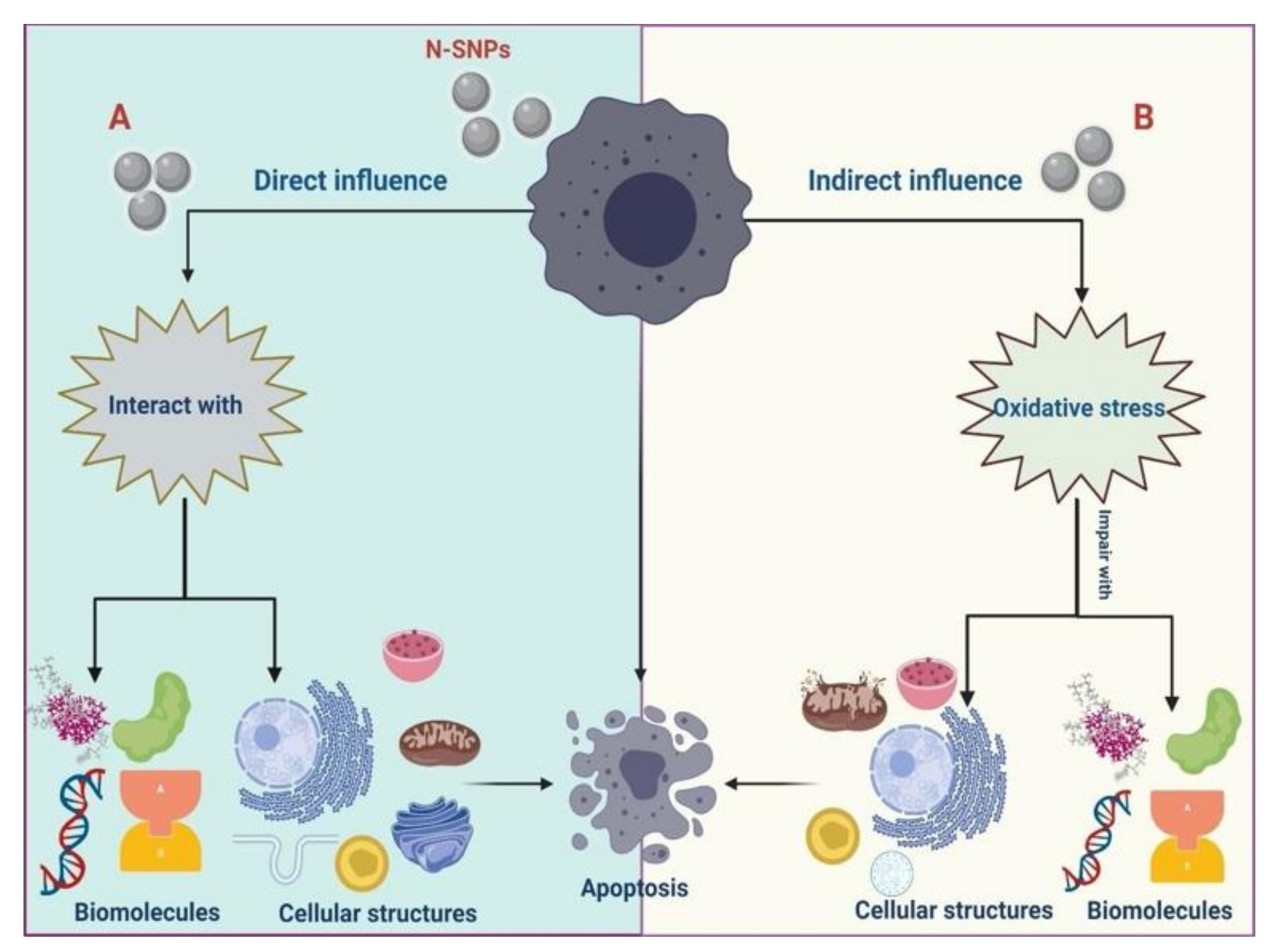
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| NPs | Nanoparticles |
| SNPs | Silver nanoparticles |
| nm | Nanometer |
| min | Minute |
| µL | Microliter |
| mg | Milligram |
| mL | Milliliter |
| µg | Microgram |
| mM | Millimolar |
| h | Hour |
| rpm | Revolutions per minute |
| kV | Kilovolt |
| kb | Kilobase |
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2018, 144, 1941–1953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yezhelyev, M.V.; Gao, X.; Xing, Y.; Al-Hajj, A.; Nie, S.; O’Regan, R.M. Emerging use of nanoparticles in diagnosis and treatment of breast cancer. Lancet Oncol. 2006, 7, 657–667. [Google Scholar] [CrossRef]
- Stephens, L.; Williams, R.L.; Hawkins, P. Phosphoinositide 3-kinases as drug targets in cancer. Curr. Opin. Pharmacol. 2005, 5, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Rajeswaran, A.; Trojan, A.; Burnand, B.; Giannelli, M. Efficacy and side effects of cisplatin- and carboplatin-based doublet chemotherapeutic regimens versus non-platinum-based doublet chemotherapeutic regimens as first line treatment of metastatic non-small cell lung carcinoma: A systematic review of randomized controlled trials. Lung Cancer 2008, 59, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Navya, P.N.; Kaphle, A.; Srinivas, S.P.; Bhargava, S.K.; Rotello, V.M.; Daima, H.K. Current trends and challenges in cancer management and therapy using designer nanomaterials. Nano Converg. 2019, 6, 23. [Google Scholar] [CrossRef] [Green Version]
- Goldberg, M.S. Improving cancer immunotherapy through nanotechnology. Nat. Rev. Cancer 2019, 19, 587–602. [Google Scholar] [CrossRef]
- Rosenblum, D.; Joshi, N.; Tao, W.; Karp, J.M.; Peer, D. Progress and challenges towards targeted delivery of cancer therapeutics. Nat. Commun. 2018, 9, 1410. [Google Scholar] [CrossRef] [Green Version]
- McNamara, K.; Tofail, S.A.M. Nanosystems: The use of nanoalloys, metallic, bimetallic, and magnetic nanoparticles in biomedical applications. Phys. Chem. Chem. Phys. 2015, 17, 27981–27995. [Google Scholar] [CrossRef]
- Sanpui, P.; Chattopadhyay, A.; Ghosh, S.S. Induction of Apoptosis in Cancer Cells at Low Silver Nanoparticle Concentrations using Chitosan Nanocarrier. ACS Appl. Mater. Interfaces 2011, 3, 218–228. [Google Scholar] [CrossRef]
- Lohcharoenkal, W.; Wang, L.; Chen, Y.C.; Rojanasakul, Y. Protein Nanoparticles as Drug Delivery Carriers for Cancer Therapy. BioMed Res. Int. 2014, 2014, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.J.; Nallathamby, P.D.; Browning, L.M.; Osgood, C.J.; Xu, X.-H.N. In Vivo Imaging of Transport and Biocompatibility of Single Silver Nanoparticles in Early Development of Zebrafish Embryos. ACS Nano 2007, 1, 133–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamida, R.S.; Ali, M.A.; Goda, D.A.; Khalil, M.; Al-Zaban, M.I. Novel Biogenic Silver Nanoparticle-Induced Reactive Oxygen Species Inhibit the Biofilm Formation and Virulence Activities of Methicillin-Resistant Staphylococcus aureus (MRSA) Strain. Front. Bioeng. Biotechnol. 2020, 8, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Draz, M.S.; Shafiee, H. Applications of gold nanoparticles in virus detection. Theranostics 2018, 8, 1985–2017. [Google Scholar] [CrossRef]
- Tiwari, S.B.; Amiji, S.B.T.A.M.M. A Review of Nanocarrier-Based CNS Delivery Systems. Curr. Drug Deliv. 2006, 3, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Sharifi, M.; Avadi, M.R.; Attar, F.; Dashtestani, F.; Ghorchian, H.; Rezayat, S.M.; Saboury, A.A.; Falahati, M. Cancer diagnosis using nanomaterials based electrochemical nanobiosensors. Biosens. Bioelectron. 2019, 126, 773–784. [Google Scholar] [CrossRef]
- Huang, T.; Nallathamby, P.D.; Xu, X.-H.N. Photostable Single-Molecule Nanoparticle Optical Biosensors for Real-Time Sensing of Single Cytokine Molecules and Their Binding Reactions. J. Am. Chem. Soc. 2008, 130, 17095–17105. [Google Scholar] [CrossRef] [Green Version]
- Thakkar, K.; Mhatre, S.S.; Parikh, R.Y. Biological synthesis of metallic nanoparticles. Nanomed. Nanotechnol. Boil. Med. 2010, 6, 257–262. [Google Scholar] [CrossRef]
- Gour, A.; Jain, N.K. Advances in green synthesis of nanoparticles. Artif. Cells, Nanomed. Biotechnol. 2019, 47, 844–851. [Google Scholar] [CrossRef] [Green Version]
- Dhand, C.; Dwivedi, N.; Ying, A.N.J.; Lakshminarayanan, R.; Ramakrishna, S.; Loh, X.J.; Verma, N.K.; Beuerman, R.W. Methods and strategies for the synthesis of diverse nanoparticles and their applications: A comprehensive overview. RSC Adv. 2015, 5, 105003–105037. [Google Scholar] [CrossRef]
- Shankar, S.; Rai, A.; Ahmad, A.; Sastry, M. Rapid synthesis of Au, Ag, and bimetallic Au core–Ag shell nanoparticles using Neem (Azadirachta indica) leaf broth. J. Colloid Interface Sci. 2004, 275, 496–502. [Google Scholar] [CrossRef]
- RajeshKumar, S.; Malarkodi, C.; Vanaja, M.; Annadurai, G. Anticancer and enhanced antimicrobial activity of biosynthesizd silver nanoparticles against clinical pathogens. J. Mol. Struct. 2016, 1116, 165–173. [Google Scholar] [CrossRef]
- Raman, J.; Reddy, G.R.; Lakshmanan, H.; Selvaraj, V.; Gajendran, B.; Nanjian, R.; Chinnasamy, A.; Sabaratnam, V. Mycosynthesis and characterization of silver nanoparticles from Pleurotus djamor var. roseus and their in vitro cytotoxicity effect on PC3 cells. Process. Biochem. 2015, 50, 140–147. [Google Scholar] [CrossRef]
- Mittal, A.K.; Chisti, Y.; Banerjee, U.C. Synthesis of metallic nanoparticles using plant extracts. Biotechnol. Adv. 2013, 31, 346–356. [Google Scholar] [CrossRef] [PubMed]
- Bin-Meferij, M.M.; Hamida, R.S. Biofabrication And Antitumor Activity Of Silver Nanoparticles Utilizing Novel Nostoc sp. Bahar M. Int. J. Nanomed. 2019, 14, 9019–9029. [Google Scholar] [CrossRef] [Green Version]
- Asmathunisha, N.; Kathiresan, K. A review on biosynthesis of nanoparticles by marine organisms. Colloids Surf. B Biointerfaces 2013, 103, 283–287. [Google Scholar] [CrossRef] [PubMed]
- R, D.; Revaprasadu, N. An in vitro and in vivo bio-interaction responses and biosafety evaluation of novel Au–ZnTe core–shell nanoparticles. Toxicol. Res. 2016, 5, 1078–1089. [Google Scholar] [CrossRef] [Green Version]
- Khorrami, S.; Zarrabi, A.; Khaleghi, M.; Danaei, M.; Mozafari, M.R. Selective cytotoxicity of green synthesized silver nanoparticles against the MCF-7 tumor cell line and their enhanced antioxidant and antimicrobial properties. Int. J. Nanomed. 2018, 13, 8013–8024. [Google Scholar] [CrossRef] [Green Version]
- El-Naggar, N.E.-A.; Hussein, M.H.; El-Sawah, A.A. Phycobiliprotein-mediated synthesis of biogenic silver nanoparticles, characterization, in vitro and in vivo assessment of anticancer activities. Sci. Rep. 2018, 8, 8925. [Google Scholar] [CrossRef]
- Hamida, R.S.; Ali, M.A.; Goda, D.A.; Khalil, M.I.; Redhwan, A. Cytotoxic effect of green silver nanoparticles against ampicillin-resistant Klebsiella pneumoniae. RSC Adv. 2020, 10, 21136–21146. [Google Scholar] [CrossRef]
- Mori, Y.; Ono, T.; Miyahira, Y.; Nguyen, V.Q.; Matsui, T.; Ishihara, M. Antiviral activity of silver nanoparticle/chitosan composites against H1N1 influenza A virus. Nanoscale Res. Lett. 2013, 8, 93. [Google Scholar] [CrossRef] [Green Version]
- Mazurak, V.C.; Burrell, R.E.; Tredget, E.E.; Clandinin, M.T.; Field, C.J. The effect of treating infected skin grafts with Acticoat™ on immune cells. Burns 2007, 33, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Hamida, R.S.; Abdelmeguid, N.E.; Ali, M.A.; Bin-Meferij, M.M.; Kahlil, M. Synthesis of Silver Nanoparticles Using a Novel Cyanobacteria Desertifilum sp. extract: Their Antibacterial and Cytotoxicity Effects. Int. J. Nanomed. 2020, 15, 49–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurunathan, S.; Qasim, M.; Park, C.H.; Yoo, H.; Kim, J.-H.; Hong, K. Cytotoxic Potential and Molecular Pathway Analysis of Silver Nanoparticles in Human Colon Cancer Cells HCT116. Int. J. Mol. Sci. 2018, 19, 2269. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.-G.; Han, Y.-H.; Kankala, R.K.; Wang, S.-B.; Chen, A. Subcellular Performance of Nanoparticles in Cancer Therapy. Int. J. Nanomed. 2020, 15, 675–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnamoorthy, K.; Moon, J.Y.; Hyun, H.B.; Cho, S.K.; Kim, S.-J. Mechanistic investigation on the toxicity of MgO nanoparticles toward cancer cells. J. Mater. Chem. 2012, 22, 24610. [Google Scholar] [CrossRef]
- Satapathy, S.R.; Mohapatra, P.; Preet, R.; Das, D.; Sarkar, B.; Choudhuri, T.; Wyatt, M.D.; Kundu, C.N. Silver-based nanoparticles induce apoptosis in human colon cancer cells mediated through p53. Nanomedicine 2013, 8, 1307–1322. [Google Scholar] [CrossRef] [PubMed]
- Gopinath, P.; Gogoi, S.K.; Chattopadhyay, A.; Ghosh, S.S. Implications of silver nanoparticle induced cell apoptosis forin vitrogene therapy. Nanotechnology 2008, 19, 075104. [Google Scholar] [CrossRef] [PubMed]
- Thompson, C. Apoptosis in the pathogenesis and treatment of disease. Science 1995, 267, 1456–1462. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Buttacavoli, M.; Albanese, N.N.; Di Cara, G.; Alduina, R.; Faleri, C.; Gallo, M.; Pizzolanti, G.; Gallo, G.; Feo, S.; Baldi, F.; et al. Anticancer activity of biogenerated silver nanoparticles: An integrated proteomic investigation. Oncotarget 2017, 9, 9685–9705. [Google Scholar] [CrossRef] [Green Version]
- Abrahamse, H.; Harith, M.A.; El-Hussein, A.; Tynga, I. Photodynamic ability of silver nanoparticles in inducing cytotoxic effects in breast and lung cancer cell lines. Int. J. Nanomed. 2014, 9, 3771. [Google Scholar] [CrossRef] [Green Version]
- Sabaratnam, V.; Gurunathan, S.; Raman, J.; Malek, S.N.A.; A John, P.; Vikineswary, S. Green synthesis of silver nanoparticles using Ganoderma neo-japonicum Imazeki: A potential cytotoxic agent against breast cancer cells. Int. J. Nanomed. 2013, 8, 4399–4413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrés, M.; Fierro, J. Antimicrobial Mechanism of Action of Transferrins: Selective Inhibition of H+-ATPase. Antimicrob. Agents Chemother. 2010, 54, 4335–4342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siddiqi, N.J.; Abdelhalim, M.A.K.; El-Ansary, A.; Alhomida, A.S.; Ong, W.Y. Identification of potential biomarkers of gold nanoparticle toxicity in rat brains. J. Neuroinflamm. 2012, 9, 123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beutler, E.; Duron, O.; Kelly, B.M. Improved method for the determination of blood glutathione. J. Lab. Clin. Med. 1963, 61, 882–888. [Google Scholar] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Reddy, N.J.; Vali, D.N.; Rani, M.; Rani, S.S. Evaluation of antioxidant, antibacterial and cytotoxic effects of green synthesized silver nanoparticles by Piper longum fruit. Mater. Sci. Eng. C 2014, 34, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Al Sufyani, N.M.; Hussien, N.A.; Hawsawi, Y. Characterization and Anticancer Potential of Silver Nanoparticles Biosynthesized from Olea chrysophylla and Lavandula dentata Leaf Extracts on HCT116 Colon Cancer Cells. J. Nanomater. 2019, 2019, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Firdhouse, J.; Lalitha, P. Apoptotic efficacy of biogenic silver nanoparticles on human breast cancer MCF-7 cell lines. Prog. Biomater. 2015, 4, 113–121. [Google Scholar] [CrossRef] [Green Version]
- Burd, J.; Usategui-Gomez, M. A colorimetric assay for serum lactate dehydrogenase. Clin. Chim. Acta 1973, 46, 223–227. [Google Scholar] [CrossRef]
- Chan, F.K.; Moriwaki, K.; De Rosa, M.J. Detection of Necrosis by Release of Lactate Dehydrogenase Activity; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 2013; Volume 979, pp. 65–70. [Google Scholar]
- Gurunathan, S.; Qasim, M.; Park, C.H.; Yoo, H.; Choi, D.Y.; Song, H.; Seo, H.G.; Kim, J.-H.; Hong, K. Cytotoxicity and Transcriptomic Analysis of Silver Nanoparticles in Mouse Embryonic Fibroblast Cells. Int. J. Mol. Sci. 2018, 19, 3618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, Y.-G.; Zhang, S.; Hwang, J.-Y.; Kong, I.-K. Silver Nanoparticles Potentiates Cytotoxicity and Apoptotic Potential of Camptothecin in Human Cervical Cancer Cells. Oxidative Med. Cell. Longev. 2018, 2018, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Bu, W.; Ding, H.; Li, Q.; Wang, D.; Bi, H.; Guo, D. Cytotoxic effect of zinc oxide nanoparticles on murine photoreceptor cells via potassium channel block and Na+/K+-ATP ase inhibition. Cell Prolif. 2017, 50, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- I Kandil, E.; El-Sonbaty, S.M.; Moawed, F.S.; Khedr, O.M. Anticancer redox activity of gallium nanoparticles accompanied with low dose of gamma radiation in female mice. Tumor Boil. 2018, 40, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Hayes, J.D.; McLellan, L.I. Glutathione and glutathione-dependent enzymes represent a co-ordinately regulated defence against oxidative stress. Free. Radic. Res. 1999, 31, 273–300. [Google Scholar] [CrossRef]
- Mccord, J.M.; Fridovich, I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J. Boil. Chem. 1969, 244, 6049–6055. [Google Scholar]
- Petkovic, J.; Žegura, B.; Filipič, M. Influence of TiO2nanoparticles on cellular antioxidant defense and its involvement in genotoxicity in HepG2 cells. In Proceedings of the Journal of Physics: Conference Series, Grenoble, France, July 2011; IOP Publisher: Bristol, UK, 2011; pp. 12037–12045. [Google Scholar]
- Al-Sheddi, E.S.; Farshori, N.N.; Al-Oqail, M.M.; Al-Massarani, S.M.; Saquib, Q.; Wahab, R.; Musarrat, J.; Al-Khedhairy, A.; Siddiqui, M. Anticancer Potential of Green Synthesized Silver Nanoparticles Using Extract of Nepeta deflersiana against Human Cervical Cancer Cells (HeLA). Bioinorg. Chem. Appl. 2018, 2018, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Kodiha, M.; Wang, Y.M.; Hutter, E.; Maysinger, D.; Stochaj, U. Off to the Organelles - Killing Cancer Cells with Targeted Gold Nanoparticles. Theranostics 2015, 5, 357–370. [Google Scholar] [CrossRef]
- Manke, A.; Wang, L.; Rojanasakul, Y. Mechanisms of Nanoparticle-Induced Oxidative Stress and Toxicity. BioMed Res. Int. 2013, 2013, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Guo, M.; Lin, Z.; Zhao, M.; Xia, Y.; Wang, C.; Xu, T.; Zhu, B. Multifunctional selenium nanoparticles with Galangin-induced HepG2 cell apoptosis through p38 and AKT signalling pathway. R. Soc. Open Sci. 2018, 5, 180509. [Google Scholar] [CrossRef] [Green Version]
- Vieira, L.F.D.A.; Lins, M.P.; Viana, I.M.M.N.; Dos Santos, J.E.; Smaniotto, S.; Reis, M.D.D.S. Metallic nanoparticles reduce the migration of human fibroblasts in vitro. Nanoscale Res. Lett. 2017, 12, 200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sathishkumar, P.; Vennila, K.; Jayakumar, R.; Yusoff, A.R.M.; Hadibarata, T.; Palvannan, T. Phyto-synthesis of silver nanoparticles using Alternanthera tenella leaf extract: An effective inhibitor for the migration of human breast adenocarcinoma (MCF-7) cells. Bioprocess Biosyst. Eng. 2016, 39, 651–659. [Google Scholar] [CrossRef]
- Barkhade, T.; Mahapatra, S.K.; Banerjee, I. Study of mitochondrial swelling, membrane fluidity and ROS production induced by nano-TiO2 and prevented by Fe incorporation. Toxicol. Res. 2019, 8, 711–722. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Piett, C.; Farkas, S.; Qazzaz, M.; Syed, N. Silver nanoparticles (AgNPs) cause degeneration of cytoskeleton and disrupt synaptic machinery of cultured cortical neurons. Mol. Brain 2013, 6, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baranowski, Z.; Hrebenda, B.; Cieslawska, M.; Beylina, S. Division of Phusarum mitochondria during starvation. Cell Biol. Int. Rep. 1991, 15, 197–204. [Google Scholar] [CrossRef]
- Kepsutlu, B.; Wycisk, V.; Achazi, K.; Kapishnikov, S.; Pérez-Berná, A.J.; Guttmann, P.; Cossmer, A.; Pereiro, E.; Ewers, H.; Ballauff, M.; et al. Cells Undergo Major Changes in the Quantity of Cytoplasmic Organelles after Uptake of Gold Nanoparticles with Biologically Relevant Surface Coatings. ACS Nano 2020, 14, 2248–2264. [Google Scholar] [CrossRef]
- Ostaszewska, T.; Śliwiński, J.; Kamaszewski, M.; Sysa, P.; Chojnacki, M. Cytotoxicity of silver and copper nanoparticles on rainbow trout (Oncorhynchus mykiss) hepatocytes. Environ. Sci. Pollut. Res. 2017, 25, 908–915. [Google Scholar] [CrossRef] [Green Version]
- Herrero, E.; Ros, J.; Belli, G.; Cabiscol, E. Redox control and oxidative stress in yeast cells. Biochim. Biophys. Acta (BBA) Gen. Subj. 2008, 1780, 1217–1235. [Google Scholar] [CrossRef]
- Khatchadourian, A.; Maysinger, D. Lipid Droplets: Their Role in Nanoparticle-Induced Oxidative Stress. Mol. Pharm. 2009, 6, 1125–1137. [Google Scholar] [CrossRef] [Green Version]
- Erlandson, R.A. Ultrastructural Pathology of the Cell and Matrix. Am. J. Surg. Pathol. 1997, 21, 1391. [Google Scholar] [CrossRef]
- Zoncu, R.; Efeyan, A.; Sabatini, D.M. mTOR: From growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Boil. 2010, 12, 21–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lunova, M.; Smolková, B.; Lynnyk, A.; Uzhytchak, M.; Jirsa, M.; Kubinová, Š.; Dejneka, A.; Lunov, O. Targeting the mTOR Signaling Pathway Utilizing Nanoparticles: A Critical Overview. Cancers 2019, 11, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuang, H.; Yang, P.; Yang, L.; Aguilar, Z.P.; Xu, H. Size dependent effect of ZnO nanoparticles on endoplasmic reticulum stress signaling pathway in murine liver. J. Hazard. Mater. 2016, 317, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Borkowska, M.; Siek, M.; Kolygina, D.V.; Sobolev, Y.I.; Lach, S.; Kumar, S.; Cho, Y.-K.; Kandere-Grzybowska, K.; Grzybowski, B.A. Targeted crystallization of mixed-charge nanoparticles in lysosomes induces selective death of cancer cells. Nat. Nanotechnol. 2020, 15, 1–11. [Google Scholar] [CrossRef]
- Kim, S.; Ryu, D.-Y. Silver nanoparticle-induced oxidative stress, genotoxicity and apoptosis in cultured cells and animal tissues. J. Appl. Toxicol. 2012, 33, 78–89. [Google Scholar] [CrossRef]
- Song, K.; Xu, P.; Meng, Y.; Geng, F.; Li, J.; Li, Z.; Xing, J.; Chen, J.; Kong, B.-H. Smart gold nanoparticles enhance killing effect on cancer cells. Int. J. Oncol. 2012, 42, 597–608. [Google Scholar] [CrossRef] [Green Version]
- Hafner, A.; Bulyk, M.L.; Jambhekar, A.; Lahav, G. The multiple mechanisms that regulate p53 activity and cell fate. Nat. Rev. Mol. Cell Boil. 2019, 20, 199–210. [Google Scholar] [CrossRef]
- Nakajima, Y.-I.; Kuranaga, E. Caspase-dependent non-apoptotic processes in development. Cell Death Differ. 2017, 24, 1422–1430. [Google Scholar] [CrossRef]
- Hird, A.W.; Tron, A.E. Recent advances in the development of Mcl-1 inhibitors for cancer therapy. Pharmacol. Ther. 2019, 198, 59–67. [Google Scholar] [CrossRef]
- Kalkavan, H.; Green, D.R. MOMP, cell suicide as a BCL-2 family business. Cell Death Differ. 2017, 25, 46–55. [Google Scholar] [CrossRef]
- Wu, Y.; Ma, J.; Sun, Y.; Tang, M.; Kong, L. Effect and mechanism of PI3K/AKT/mTOR signaling pathway in the apoptosis of GC-1 cells induced by nickel nanoparticles. Chemosphere 2020, 126913. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Han, X.; Han, X. The Cardioprotection of the Insulin-Mediated PI3K/Akt/mTOR Signaling Pathway. Am. J. Cardiovasc. Drugs 2014, 14, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Jian, J.; Li, S.; Zhang, Y.; Liu, L. TPX2 silencing exerts anti-tumor effects on hepatocellular carcinoma by regulating the PI3K/AKT signaling pathway. Int. J. Mol. Med. 2019, 44, 2113–2122. [Google Scholar] [CrossRef] [PubMed]
- Kovács, D.; Igaz, N.; Keskeny, C.; Bélteky, P.; Tóth, T.; Gáspár, R.; Madarász, D.; Rázga, Z.; Kónya, Z.; Boros, I.M.; et al. Silver nanoparticles defeat p53-positive and p53-negative osteosarcoma cells by triggering mitochondrial stress and apoptosis. Sci. Rep. 2016, 6, 27902. [Google Scholar] [CrossRef] [Green Version]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Type | Product Code | Company |
|---|---|---|---|
| Akt | Rabbit monoclonal | EPR16798 | Abcam |
| p-Akt | Rabbit polyclonal | ab8805 | Abcam |
| Bcl-2 | Rabbit polyclonal | ab59348 | Abcam |
| mTOR | Rabbit monoclonal | ab32028 | Abcam |
| p53 | Rabbit polyclonal | ab131442 | Abcam |
| Caspase 3 | Rabbit polyclonal | ab13847 | Abcam |
| β-actin | Rabbit polyclonal | ab228001 | Abcam |
| Cell | IC25 | IC50 |
|---|---|---|
| MCF-7 | 27 ± 0.02 | 54 ± 0.08 |
| HCT-116 | 28 ± 0.04 | 56 ± 0.03 |
| HepG2 | 40 ± 0.08 | 80 ± 0.02 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hamida, R.S.; Albasher, G.; Bin-Meferij, M.M. Oxidative Stress and Apoptotic Responses Elicited by Nostoc-Synthesized Silver Nanoparticles against Different Cancer Cell Lines. Cancers 2020, 12, 2099. https://doi.org/10.3390/cancers12082099
Hamida RS, Albasher G, Bin-Meferij MM. Oxidative Stress and Apoptotic Responses Elicited by Nostoc-Synthesized Silver Nanoparticles against Different Cancer Cell Lines. Cancers. 2020; 12(8):2099. https://doi.org/10.3390/cancers12082099
Chicago/Turabian StyleHamida, Reham Samir, Gadah Albasher, and Mashael Mohammed Bin-Meferij. 2020. "Oxidative Stress and Apoptotic Responses Elicited by Nostoc-Synthesized Silver Nanoparticles against Different Cancer Cell Lines" Cancers 12, no. 8: 2099. https://doi.org/10.3390/cancers12082099