The Bradykinin-BDKRB1 Axis Regulates Aquaporin 4 Gene Expression and Consequential Migration and Invasion of Malignant Glioblastoma Cells via a Ca2+-MEK1-ERK1/2-NF-κB Mechanism

 ,
,
Abstract
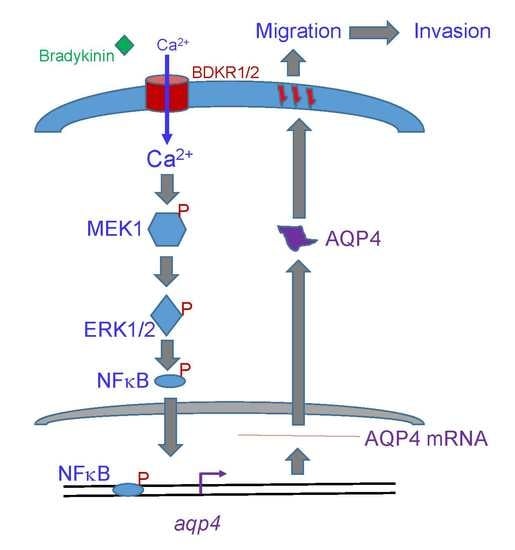
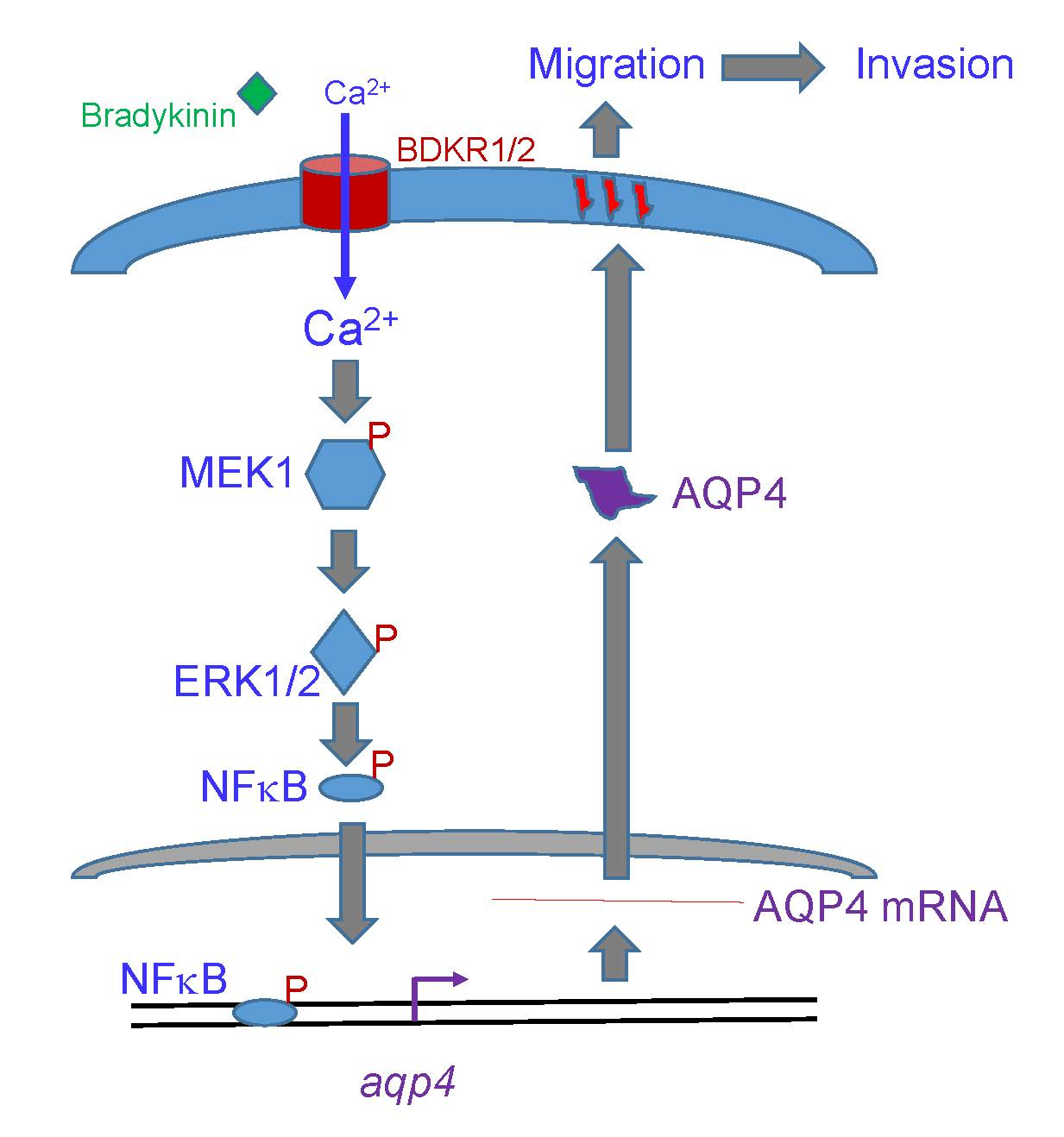
:
1. Introduction
2. Results
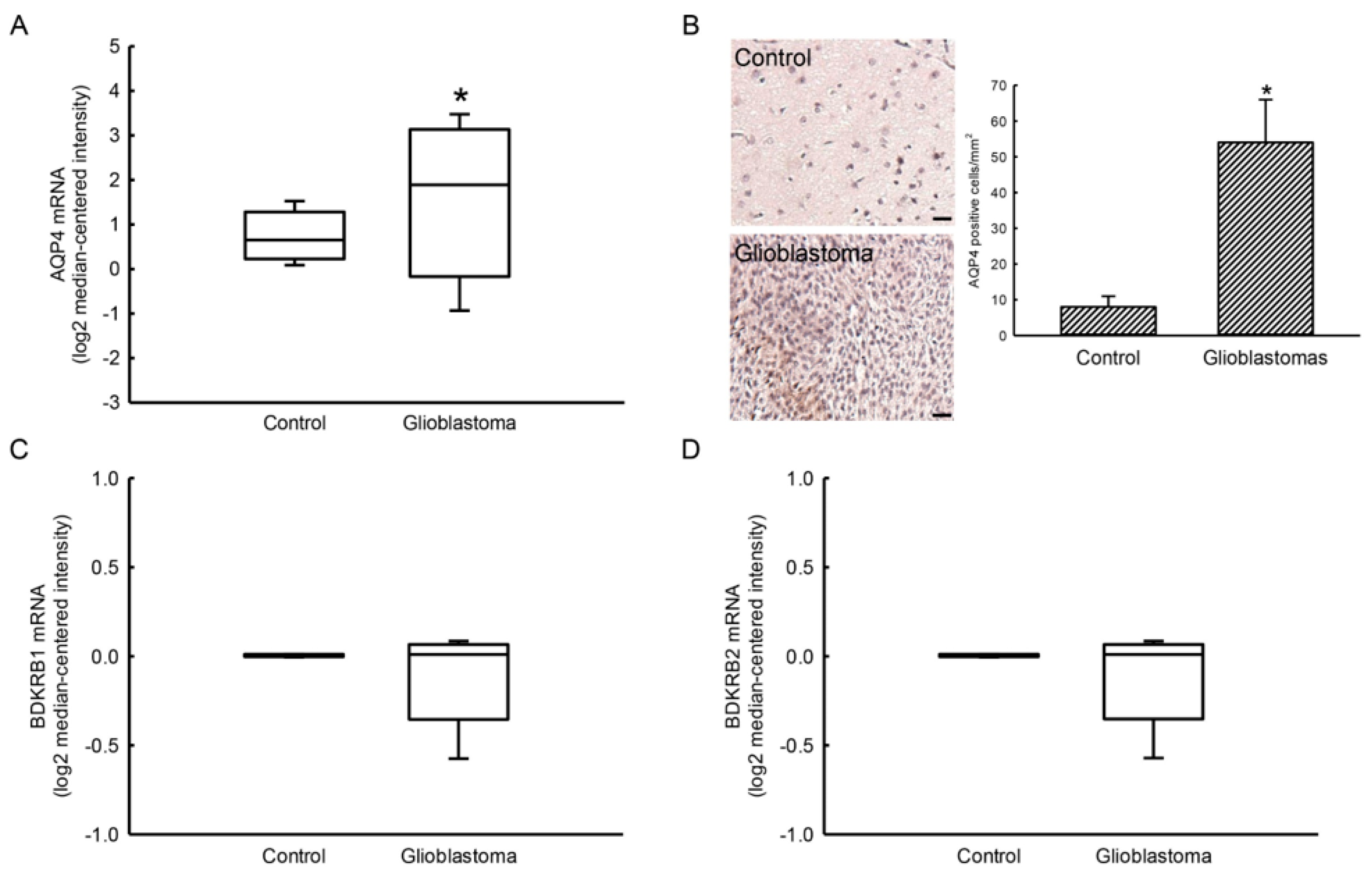
2.1. Expressions of AQP4 and BDKRB1/2 mRNAs in Human Glioblastomas
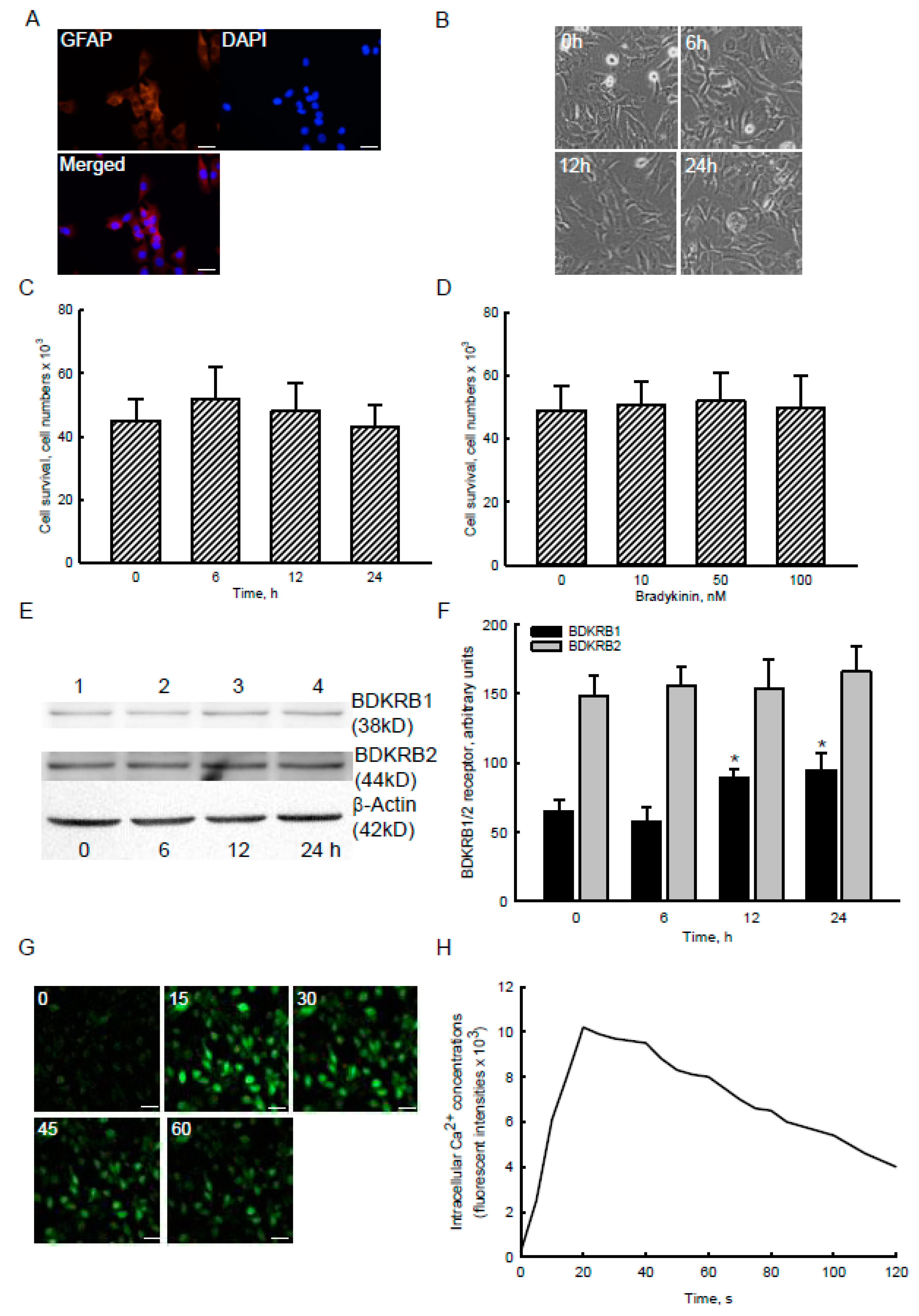
2.2. Bradykinin Specifically Increased Levels of BDKRB1 and Stimulated Ca2+ Influx without Affecting Cell Survival in Human Malignant Glioblastoma Cells
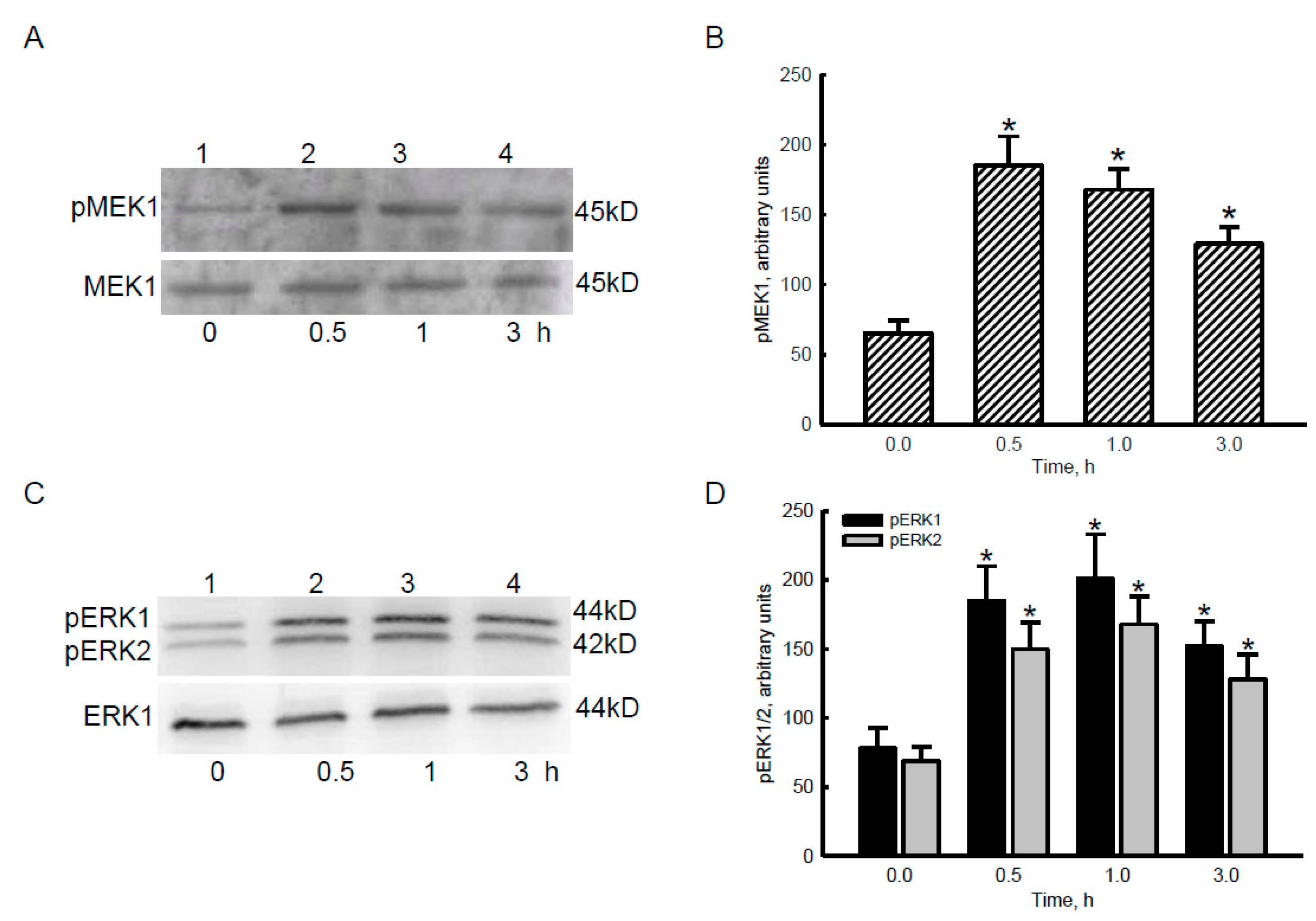
2.3. Bradykinin Successively Activated MEK1 and ERK1/2 in Human Malignant Glioblastoma Cells
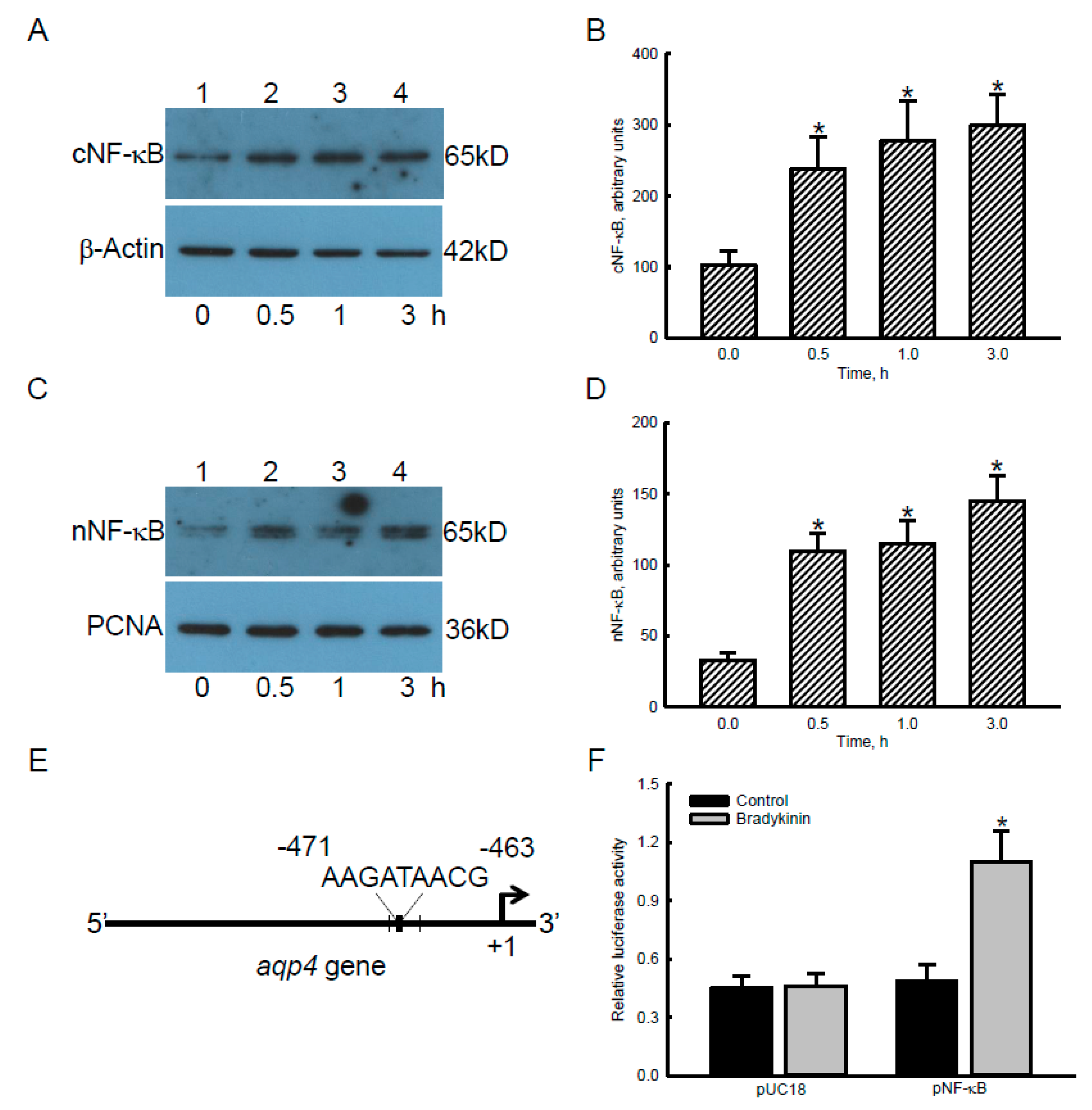
2.4. Bradykinin Accordingly Triggered Translocation and Transactivation Activity of NF-κB in Human Malignant Glioblastoma Cells
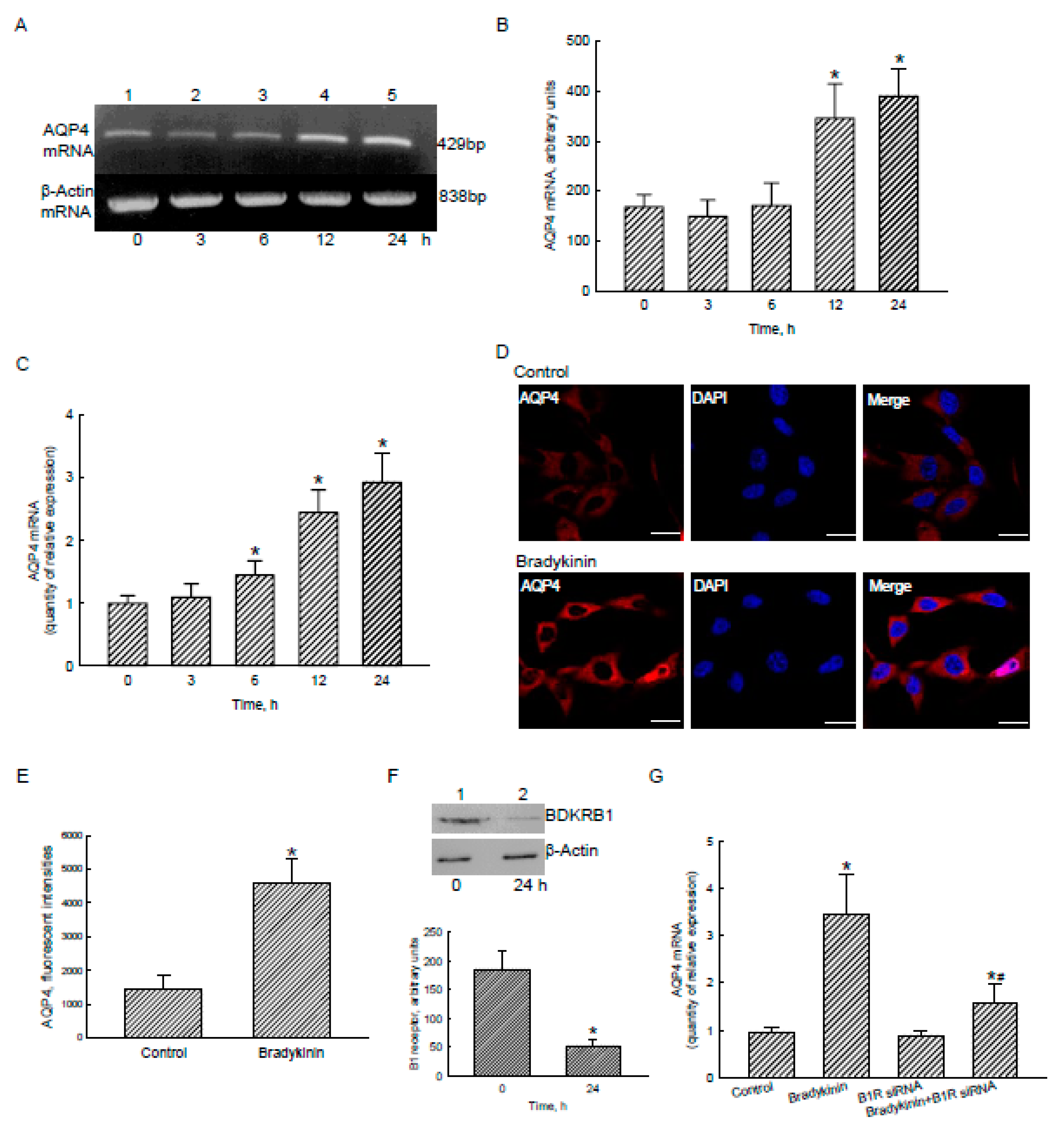
2.5. Bradykinin Induced AQP4 mRNA and Protein Expression via Activation of BDKRB1
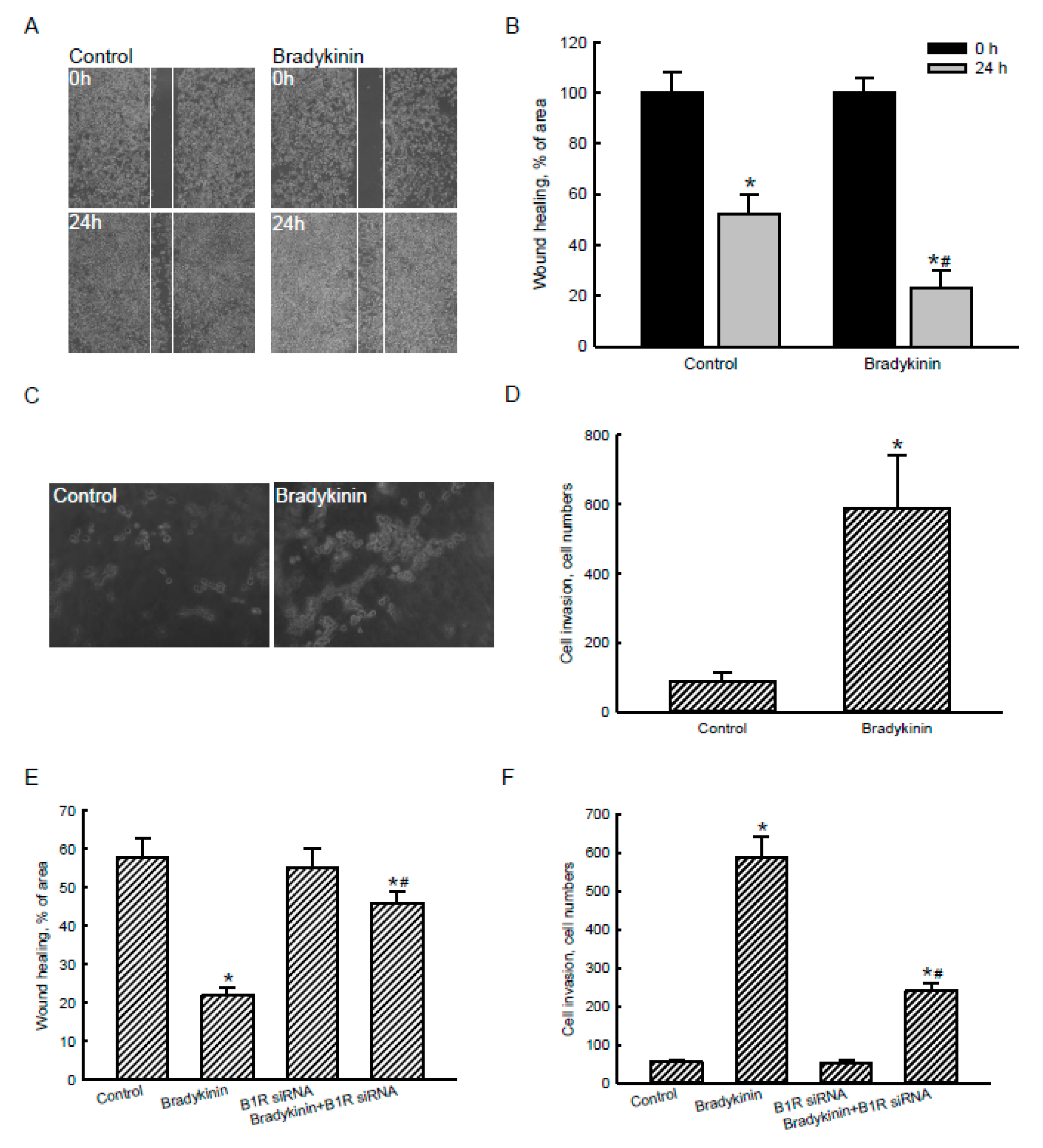
2.6. Bradykinin Stimulated Migration and Invasion of Human Malignant Glioblastoma Cells via Activation of BDKRB1
2.7. The Bradykinin-Induced Ca2+ Influx, AQP4 mRNA Expression, Wound Healing, and Cell Migration and Invasion Were Further Confirmed in Murine GL261 Glioblastoma Cells
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Data Downloading and Preprocessing
4.3. Immunohistochemical (IHC) Analysis of AQP4
4.4. Culture of Malignant Glioblastoma Cells and Drug Treatments
4.5. Cell Morphology and Cell Survival
4.6. Immunoblot Analyses
4.7. Extraction and Immunodetection of Nuclear Proteins
4.8. Measurement of Ca2+ Influx
4.9. Bioinformatic Approach
4.10. NF-κB Reporter Assay
4.11. Reverse-Transcription (RT) and Quantitative Polymerase Chain Reaction (qPCR) Analyses
4.12. Distribution of AQP4 in Human Glioblastoma Cells
4.13. Knockdown of BDKRB1
4.14. Wound-Healing Assay
4.15. Matrigel-Based Invasion Assay
4.16. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Louis, D.N.; Perry, A.; Reifenberger, G.; Von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization classification of tumors of the central nervous system: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [Green Version]
- Sturm, D.; Bender, S.; Jones, D.T.; Lichter, P.; Grill, J.; Becher, O.; Hawkins, C.; Majewski, J.; Jones, C.; Costello, J.F.; et al. Paediatric and adult glioblastoma: Multiform (epi)genomic culprits emerge. Nat. Rev. Cancer 2014, 14, 92–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stupp, R.; Mason, W.P.; Van Den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. New Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Hottinger, A.F.; Pacheco, P.; Stupp, R. Tumor treating fields: A novel treatment modality and its use in brain tumors. Neuro Oncol. 2016, 18, 1338–1349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Zhang, M.; Gan, H.; Wang, H.; Lee, J.H.; Fang, D.; Kitange, G.J.; He, L.; Hu, Z.; Parney, I.F.; et al. A novel enhancer regulates MGMT expression and promotes temozolomide resistance in glioblastoma. Nat. Commun. 2018, 9, 2949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Q.; Mittal, S.; Berens, M.E. Targeting adaptive glioblastoma: An overview of proliferation and invasion. Neuro Oncol. 2014, 16, 1575–1584. [Google Scholar] [CrossRef] [Green Version]
- Roth, P.; Weller, M. Challenges to targeting epidermal growth factor receptor in glioblastoma: Escape mechanisms and combinatorial treatment strategies. Neuro Oncol. 2014, 16 (Suppl. 8), viii14–viii19. [Google Scholar] [CrossRef]
- Costa-Neto, C.M.; Dillenburg-Pilla, P.; Heinrich, T.A.; Parreiras-e-Silva, L.T.; Pereira, M.G.; Reis, R.I.; Souza, P.P. Participation of kallikrein-kinin system in different pathologies. Int. Immunopharmacol. 2008, 8, 135–142. [Google Scholar] [CrossRef]
- Ekdahl, K.N.; Teramura, Y.; Hamad, O.A.; Asif, S.; Duehrkop, C.; Fromell, K.; Gustafson, E.; Hong, J.; Kozarcanin, H.; Magnusson, P.U.; et al. Dangerous liaisons: Complement, coagulation, and kallikrein/kinin cross-talk act as a linchpin in the events leading to thromboinflammation. Immunol. Rev. 2016, 274, 245–269. [Google Scholar] [CrossRef]
- Guevara-Lora, I. Kinin-mediated inflammation in neurodegenerative disorders. Neurochem. Int. 2012, 61, 72–78. [Google Scholar] [CrossRef]
- Petho, G.; Reeh, P.W. Sensory and signaling mechanisms of bradykinin, eicosanoids, platelet-activating factor, and nitric oxide in peripheral nociceptors. Physiol. Rev. 2012, 92, 1699–1775. [Google Scholar] [CrossRef] [PubMed]
- Ufnal, M.; Skrzypecki, J. Blood borne hormones in a cross-talk between peripheral and brain mechanisms regulating blood pressure, the role of circumventricular organs. Neuropeptides 2014, 48, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Ratajczak, M.Z.; Zuba-Surma, E.; Kucia, M.; Reca, R.; Wojakowski, W.; Ratajczak, J. The pleiotropic effects of the SDF-1-CXCR4 axis in organogenesis, regeneration and tumorigenesis. Leukemia 2006, 20, 1915–1924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montana, V.; Sontheimer, H. Bradykinin promotes the chemotactic invasion of primary brain tumors. J. Neurosci. 2011, 31, 4858–4867. [Google Scholar] [CrossRef] [Green Version]
- Nicoletti, N.F.; Sénécal, J.; Da Silva, V.D.; Roxo, M.R.; Ferreira, N.P.; De Morais, R.L.T.; Pesquero, J.B.; Campos, M.M.; Couture, R.; Morrone, F.B. Primary role for kinin b1 and b2 receptors in glioma proliferation. Mol. Neurobiol. 2017, 54, 7869–7882. [Google Scholar] [CrossRef]
- Oliveira, M.N.; Pillat, M.M.; Motaln, H.; Ulrich, H.; Lah, T.T. Kinin-B1 receptor stimulation promotes invasion and is involved in cell-cell interaction of co-cultured glioblastoma and mesenchymal stem cells. Sci. Rep. 2018, 8, 1299. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.S.; Hsu, J.W.; Lin, H.Y.; Lai, S.W.; Huang, B.R.; Tsai, C.F.; Lu, D.Y. Bradykinin B1 receptor contributes to interleukin-8 production and glioblastoma migration through interaction of STAT3 and SP-1. Neuropharmacology 2019, 144, 143–154. [Google Scholar] [CrossRef]
- Prados, M.D.; Schold, S.C., Jr.; Fine, H.A.; Jaeckle, K.; Hochberg, F.; Mechtler, L.; Fetell, M.R.; Phuphanich, S.; Feun, L.; Janus, T.J.; et al. A randomized, double-blind, placebo-controlled, phase 2 study of RMP-7 in combination with carboplatin administered intravenously for the treatment of recurrent malignant glioma. Neuro Oncol. 2003, 5, 96–103. [Google Scholar] [CrossRef]
- Van Tellingen, O.; Yetkin-Arik, B.; De Gooijer, M.C.; Wesseling, P.; Wurdinger, T.; De Vries, H.E. Overcoming the blood-brain tumor barrier for effective glioblastoma treatment. Drug Resist. Updates 2015, 19, 1–12. [Google Scholar] [CrossRef]
- Jung, J.S.; Bhat, R.V.; Preston, G.M.; Guggino, W.B.; Baraban, J.M.; Agre, P. Molecular characterization of an aquaporin cDNA from brain: Candidate osmoreceptor and regulator of water balance. Proc. Natl. Acad. Sci. USA 1994, 91, 13052–13056. [Google Scholar] [CrossRef] [Green Version]
- Nico, B.; Mangieri, D.; Tamma, R.; Longo, V.; Annese, T.; Crivellato, E.; Pollo, B.; Maderna, E.; Ribatti, D.; Salmaggi, A. Aquaporin-4 contributes to the resolution of peritumoural brain oedema in human glioblastoma multiforme after combined chemotherapy and radiotherapy. Eur. J. Cancer 2009, 45, 3315–3325. [Google Scholar] [CrossRef] [PubMed]
- Ding, T.; Zhou, Y.; Sun, K.; Jiang, W.; Li, W.; Liu, X.; Tian, C.; Li, Z.; Ying, G.; Fu, L.; et al. Knockdown a water channel protein, aquaporin-4, induced glioblastoma cell apoptosis. PLoS ONE 2013, 8, e66751. [Google Scholar] [CrossRef] [PubMed]
- Watkins, S.; Robel, S.; Kimbrough, I.F.; Robert, S.M.; Ellis-Davies, G.; Sontheimer, H. Disruption of astrocyte-vascular coupling and the blood-brain barrier by invading glioma cells. Nat. Commun. 2014, 5, 4196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warth, A.; Kröger, S.; Wolburg, H. Redistribution of aquaporin-4 in human glioblastoma correlates with loss of agrinimmunoreactivity from brain capillary basal laminae. Acta Neuropathol. 2004, 107, 311–318. [Google Scholar] [CrossRef]
- Tome-Garcia, J.; Erfani, P.; Nudelman, G.; Tsankov, A.M.; Katsyv, I.; Tejero, R.; Zhang, B.; Walsh, M.; Friedel, R.H.; Zaslavsky, E.; et al. Analysis of chromatin accessibility uncovers TEAD1 as a regulator of migration in human glioblastoma. Nat. Commun. 2018, 9, 4020. [Google Scholar] [CrossRef] [Green Version]
- Monti, J.; Gross, V.; Luft, F.C.; Franca Milia, A.; Schulz, H.; Dietz, R.; Sharma, A.M.; Hübner, N. Expression analysis using oligonucleotide microarrays in mice lacking bradykinin type 2 receptors. Hypertension 2001, 38, E1–E3. [Google Scholar] [CrossRef] [Green Version]
- Hus-Citharel, A.; Bouby, N.; Iturrioz, X.; Llorens-Cortes, C. Multiple cross talk between angiotensin II, bradykinin, and insulin signaling in the cortical thick ascending limb of rat kidney. Endocrinology 2010, 151, 3181–3194. [Google Scholar] [CrossRef] [Green Version]
- Wiegert, J.S.; Bading, H. Activity-dependent calcium signaling and ERK-MAP kinases in neurons: A link to structural plasticity of the nucleus and gene transcription regulation. Cell Calcium 2011, 49, 296–305. [Google Scholar] [CrossRef]
- Zanotto-Filho, A.; Gonçalves, R.M.; Klafke, K.; De Souza, P.O.; Dillenburg, F.C.; Carro, L.; Gelain, D.P.; Moreira, J.C. Inflammatory landscape of human brain tumors reveals an NFκB dependent cytokine pathway associated with mesenchymal glioblastoma. Cancer Lett. 2017, 390, 176–187. [Google Scholar] [CrossRef]
- Chen, B.; Liu, J.; Ho, T.T.; Ding, X.; Mo, Y.Y. ERK-mediated NF-κB activation through ASIC1 in response to acidosis. Oncogenesis 2016, 5, e279. [Google Scholar] [CrossRef]
- Soubannier, V.; Stifani, S. NF-κB signalling in glioblastoma. Biomedicines 2017, 5, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terzuoli, E.; Meini, S.; Cucchi, P.; Catalani, C.; Cialdai, C.; Maggi, C.A.; Giachetti, A.; Ziche, M.; Donnini, S. Antagonism of bradykinin b2 receptor prevents inflammatory responses in human endothelial cells by quenching the nf-κb pathway activation. PLoS ONE 2014, 9, e84358. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Lin, W.; Zheng, M.; Liu, Q.; He, B.; Luo, C.; Lu, X.; Pei, Z.; Su, H.; Yao, X. Alterations in AQP4 expression and polarization in the course of motor neuron degeneration in SOD1G93A mice. Mol. Med. Rep. 2017, 16, 1739–1746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCoy, E.; Sontheimer, H. MAPK induces AQP1 expression in astrocytes following injury. Glia 2010, 58, 209–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.; Ye, Y.; Zhang, X.; Song, J. Bradykinin stimulates IL-6 production and cell invasion in colorectal cancer cells. Oncol. Rep. 2014, 32, 1709–1714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Zhou, Y.; Wei, R.; Jiang, G.; Li, F.; Chen, X.; Wang, X.; Ma, D.; Xi, L. Bradykinin promotes proliferation, migration, and invasion of cervical cancer cells through STAT3 signaling pathways. Oncol. Rep. 2019, 42, 2521–2527. [Google Scholar] [CrossRef]
- Chen, Y.; Yu, Y.; Sun, S.; Wang, Z.; Liu, P.; Jiang, J. Bradykinin promotes migration and invasion of hepatocellular carcinoma cells through TRPM7 and MMP2. Exp. Cell Res. 2016, 349, 68–76. [Google Scholar] [CrossRef]
- Da Costa, P.L.; Sirois, P.; Tannock, I.F.; Chammas, R. The role of kinin receptors in cancer and therapeutic opportunities. Cancer Lett. 2014, 345, 27–38. [Google Scholar] [CrossRef] [Green Version]
- Chio, C.C.; Wei, L.; Chen, T.G.; Tsai, S.H.; Shieh, J.P.; Yeh, P.S.; Chen, R.M. Neuron-derived orphan receptor 1 transduces survival signals in neuronal cells in response to hypoxia-induced apoptotic insults. J. Neurosurg. 2016, 124, 1654–1664. [Google Scholar] [CrossRef] [Green Version]
- Yeh, P.S.; Wang, W.; Chang, Y.A.; Lin, C.J.; Wang, J.J.; Chen, R.M. Honokiol induces autophagy of neuroblastoma cells through activating the PI3K/Akt/mTOR and endoplasmic reticular stress/ERK1/2 signaling pathways and suppressing cell migration. Cancer Lett. 2016, 370, 66–77. [Google Scholar] [CrossRef]
- Lin, M.C.; Lee, Y.W.; Tseng, Y.Y.; Lin, Y.W.; Chen, J.T.; Liu, S.H.; Chen, R.M. Honokiol induces apoptosis and successive apoptotic insults to neuroblastomas via a p53-dependent mechanism. Am. J. Chin. Med. 2019, 47, 859–912. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.J.; Wang, W.; Lin, Y.L.; Liu, S.H.; Chen, R.M. Oxidative stress-induced apoptotic insults to rat osteoblasts are attenuated by nitric oxide pretreatment via GATA-5-involved regulation of Bcl-XL gene expression and protein translocation. Arch. Toxicol. 2016, 90, 905–916. [Google Scholar] [CrossRef] [PubMed]
- Liao, M.H.; Lin, P.I.; Ho, W.P.; Chan, W.P.; Chen, T.L.; Chen, R.M. Participation of GATA-3 in regulation of bone healing through transcriptionally upregulating bcl-xL gene expression. Exp. Mol. Med. 2017, 49, e398. [Google Scholar] [CrossRef] [PubMed]
- Chio, C.C.; Tai, Y.T.; Mohanraj, M.; Liu, S.H.; Yang, S.T.; Chen, R.M. Honokiol improves temozolomide-induced apoptotic insults to malignant glioma cells via an intrinsic mitochondria-dependent pathway. Phytomedicine 2018, 49, 41–51. [Google Scholar] [CrossRef]
- Chen, R.M.; Lin, Y.L.; Chou, C.W. GATA-3 transduces survival signals in osteoblasts through upregulation of bcl-xL gene expression. J. Bone Miner. Res. 2010, 25, 2193–2204. [Google Scholar] [CrossRef]
- Rivero-GutiérrezA, B.; Anzola, A.; Martínez-Augustin, O.; De Medina, F.S. Stain-free detection as loading control alternative to Ponceau and housekeeping protein immunodetection in Western blotting. Anal. Biochem. 2014, 467, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.M.; Chen, T.L.; Lin, Y.L.; Chen, T.G.; Tai, Y.T. Ketamine reduces nitric oxide biosynthesis in human umbilical vein endothelial cells through downregulating endothelial nitric oxide synthase expression and intracellular calcium levels. Crit. Care Med. 2005, 33, 1044–1049. [Google Scholar] [CrossRef]
- Farré, D.; Roset, R.; Huerta, M.; Adsuara, J.E.; Roselló, L.; Albà, M.M.; Messeguer, X. Identification of patterns in biological sequences at the ALGGEN server: PROMO and MALGEN. Nucleic Acids Res. 2003, 31, 3651–3653. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.C.; Lin, K.H.; Tai, Y.T.; Chen, J.T.; Chen, R.M. Lipoteichoic acid-induced TNF-α and IL-6 gene expressions and oxidative stress production in macrophages are suppressed by ketamine through downregulating toll-like receptor 2-mediated activation of ERK1/2 and NFκB. Shock 2010, 33, 485–492. [Google Scholar] [CrossRef]
- Chio, C.C.; Lin, J.W.; Cheng, H.A.; Chiu, W.T.; Wang, Y.H.; Wang, J.J.; Hsing, C.H.; Chen, R.M. MicroRNA-210 targets antiapoptotic Bcl-2 expression and mediates hypoxia-induced apoptosis of neuroblastoma cells. Arch. Toxicol. 2013, 87, 458–468. [Google Scholar] [CrossRef]
- Ho, M.H.; Yao, C.J.; Liao, M.H.; Lin, P.I.; Liu, S.H.; Chen, R.M. Chitosan nanofiber scaffold improves bone healing via stimulating trabecular bone production due to upregulation of the Runx2/osteocalcin/alkaline phosphatase signaling pathway. Int. J. Nanomed. 2015, 10, 5941–5954. [Google Scholar] [CrossRef] [Green Version]
- Gresz, V.; Kwon, T.H.; Hurley, P.T.; Varga, G.; Zelles, T.; Nielsen, S.; Case, R.M.; Steward, M.C. Identification and localization of aquaporin water channels in human salivary glands. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 281, G247–G254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oshio, K.; Binder, D.K.; Yang, B.; Schecter, S.; Verkman, A.S.; Manley, G.T. Expression of aquaporin water channels in mouse spinal cord. Neuroscience 2004, 127, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.W.; Chen, J.T.; Hong, C.Y.; Lin, Y.L.; Wang, K.T.; Yao, C.J.; Lai, G.M.; Chen, R.M. Honokiol traverses the blood-brain barrier and induces apoptosis of neuroblastoma cells via an intrinsic bax-mitochondrion-cytochrome c-caspase protease pathway. Neuro Oncol. 2012, 14, 302–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albini, A.; Iwamoto, Y.; Kleinman, H.K.; Martin, G.R.; Aaronson, S.A.; Kozlowski, J.M.; McEwan, R.N. A rapid in vitro assay for quantitating the invasive potential of tumor cells. Cancer Res. 1987, 47, 3239–3245. [Google Scholar] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Activities | Control | Bradykinin |
|---|---|---|
| Cell survival, cell numbers × 103 | 46 ± 9 | 43 ± 10 |
| [Ca2+]i, fluorescent intensities × 103 | 0.8 ± 0.2 | 16.2 ± 3.5 * |
| AQP4 mRNA, quantity of relative expression | 1 ± 0.2 | 3.2 ± 0.7 * |
| Wound healing, % of area | 57 ± 11 | 23 ± 5 * |
| Cell invasion, cell numbers | 49 ± 9 | 578 ± 99 * |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, D.-P.; Lee, Y.-W.; Chen, J.-T.; Lin, Y.-W.; Chen, R.-M. The Bradykinin-BDKRB1 Axis Regulates Aquaporin 4 Gene Expression and Consequential Migration and Invasion of Malignant Glioblastoma Cells via a Ca2+-MEK1-ERK1/2-NF-κB Mechanism. Cancers 2020, 12, 667. https://doi.org/10.3390/cancers12030667
Sun D-P, Lee Y-W, Chen J-T, Lin Y-W, Chen R-M. The Bradykinin-BDKRB1 Axis Regulates Aquaporin 4 Gene Expression and Consequential Migration and Invasion of Malignant Glioblastoma Cells via a Ca2+-MEK1-ERK1/2-NF-κB Mechanism. Cancers. 2020; 12(3):667. https://doi.org/10.3390/cancers12030667
Chicago/Turabian StyleSun, Ding-Ping, Yuan-Wen Lee, Jui-Tai Chen, Yung-Wei Lin, and Ruei-Ming Chen. 2020. "The Bradykinin-BDKRB1 Axis Regulates Aquaporin 4 Gene Expression and Consequential Migration and Invasion of Malignant Glioblastoma Cells via a Ca2+-MEK1-ERK1/2-NF-κB Mechanism" Cancers 12, no. 3: 667. https://doi.org/10.3390/cancers12030667