Biphasic Mathematical Model of Cell–Drug Interaction That Separates Target-Specific and Off-Target Inhibition and Suggests Potent Targeted Drug Combinations for Multi-Driver Colorectal Cancer Cells



Abstract
:1. Introduction
2. Results
2.1. Viability of HT-29 Cells Is Sensitive to Four Protein Kinase Inhibitors
2.2. IC50 Values Do Not Accurately Reflect How HT-29 Cells Respond to Targeted Therapy
2.3. Biological Meaning of Biphasic Inhibition
2.4. Src, IR/IGF-1R/AKT Signaling and the MAP Kinase Pathway Are All Partially Responsible for Driving HT-29 Cell Proliferation and Viability
2.5. HT-29 Cell Viability Can Be Effectively and Synergistically Blocked by Combinations of Drugs, Each Blocking an Independent Driver
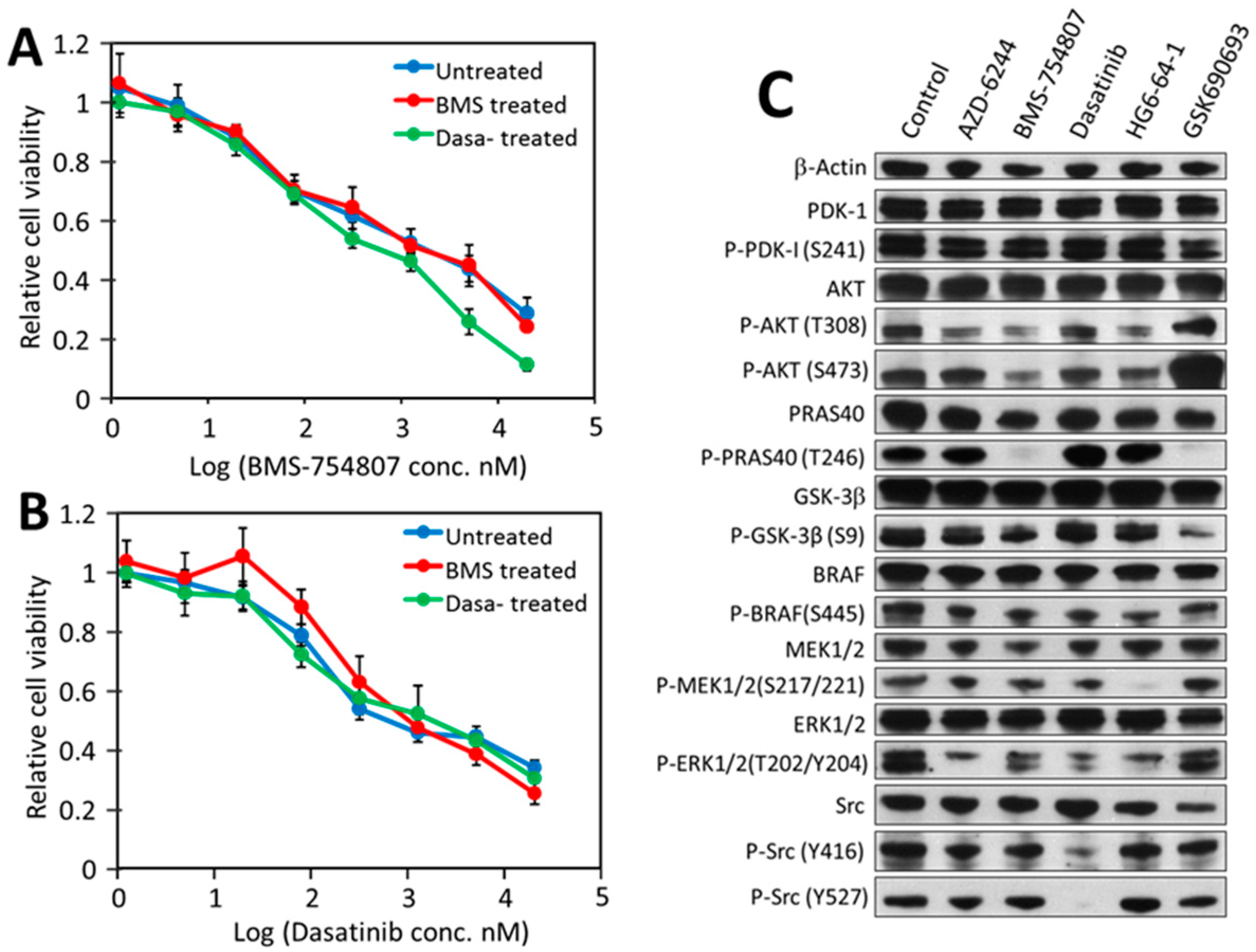
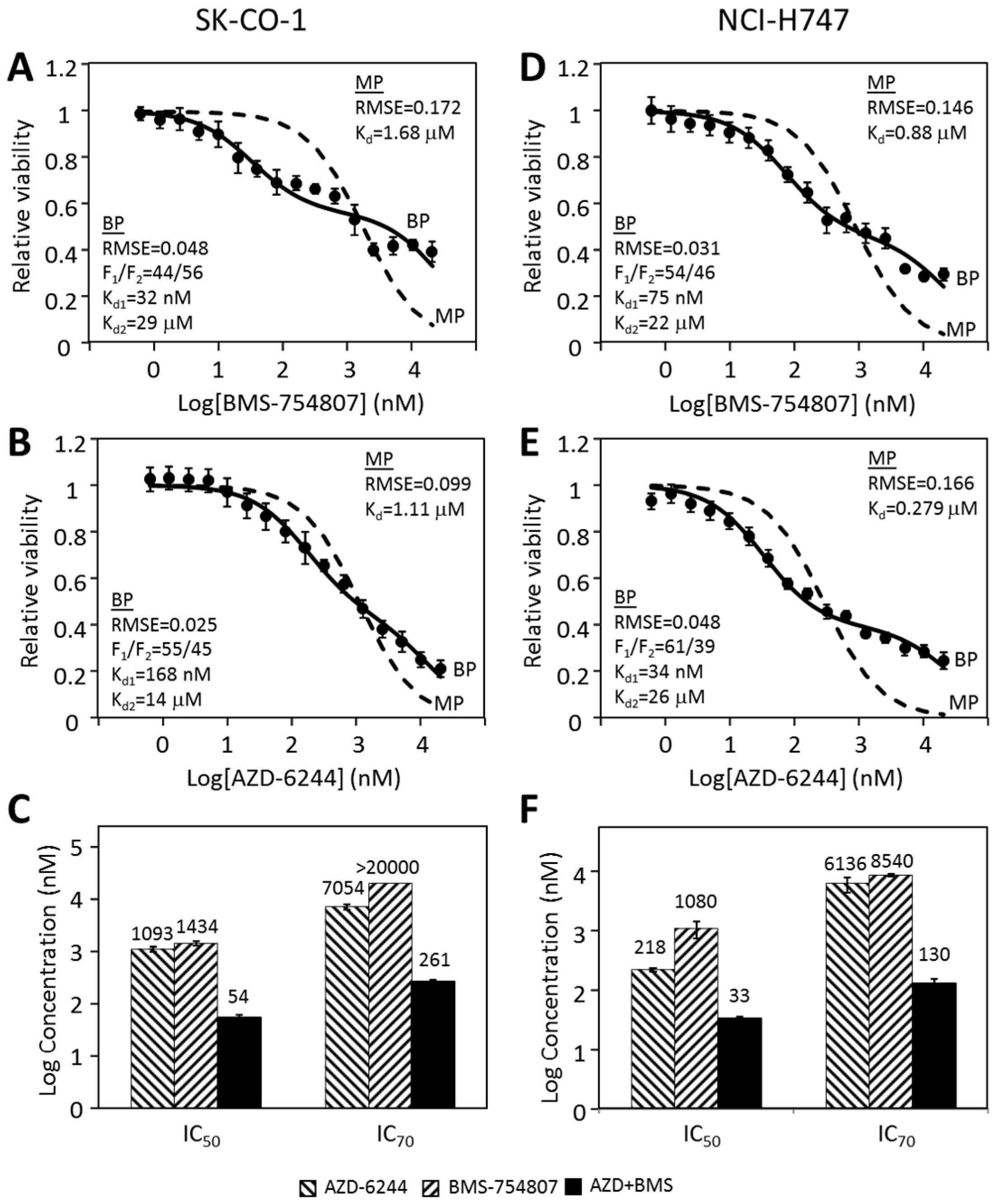
2.6. Other CRC Cancer Cell Lines Follow a Similar Pattern in Their Responses to Individual Kinase Inhibitors and Inhibitor Combinations
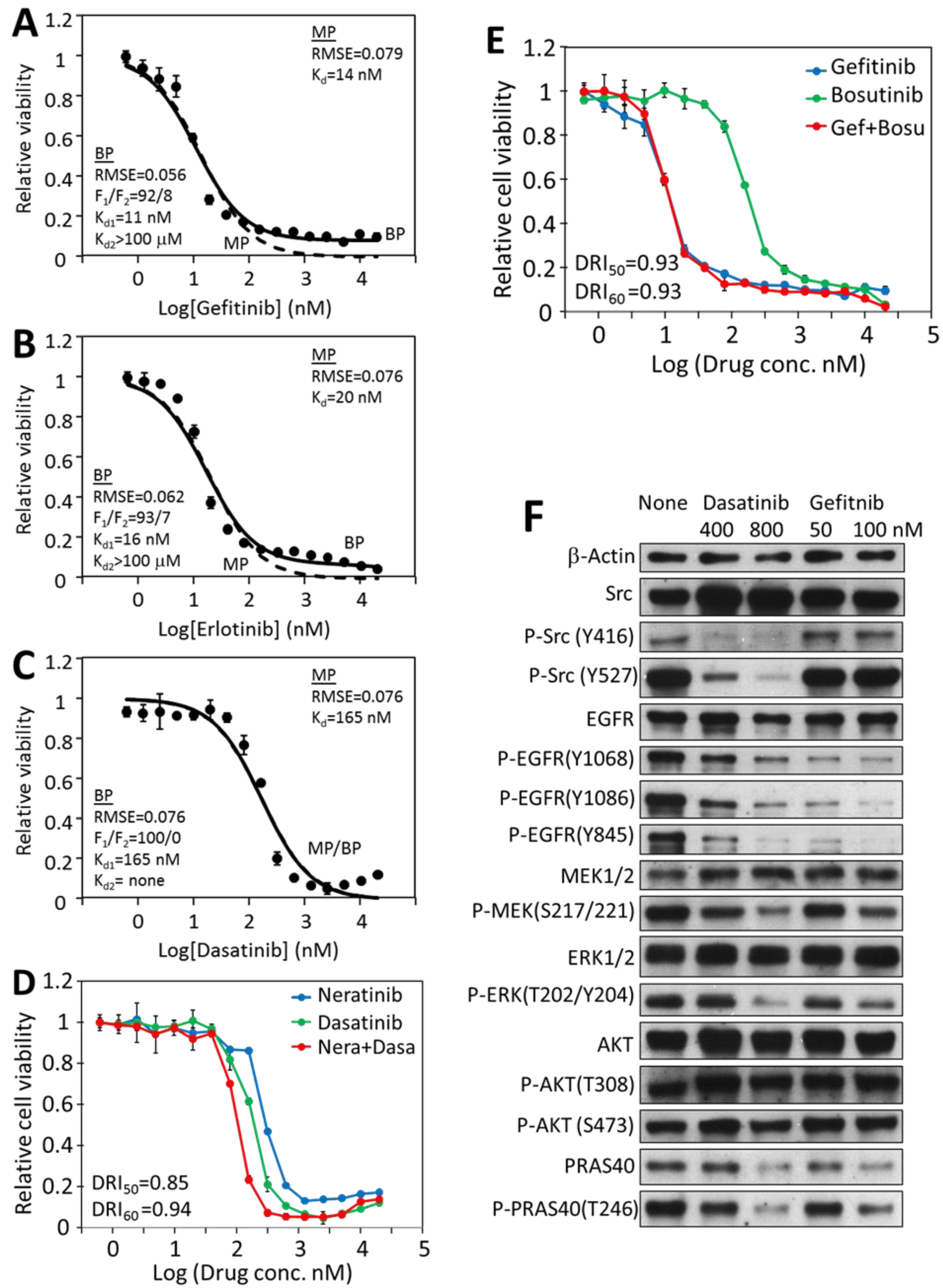
2.7. Mono-Driver Cancer Cell Responses to Targeted Therapy Is Monophasic
2.8. Inhibition of Cancer Cell Viability by Dasatinib Is Mostly Biphasic
3. Discussion
3.1. Multi-Driver Cancers Are a Major Challenge to Targeted Therapy
3.2. Multi-Driver Proliferation Challenges Traditional Pharmacological Metrics for Analyzing Cancer Cell–Drug Response
3.3. Biphasic Analysis Provides a Basis for Formulating Combination Targeted Therapy for Multi-Driver Cancers
3.4. Biphasic Analysis Provides the Tool to Quantitatively Assess the Target-Specific and Off-Target Effects of Targeted Therapeutics
4. Materials and Methods
4.1. Cell Lines, Media and Drugs
4.2. Cell Culture and Viability Assays
4.3. Drug Synergy Analysis and Combination Index Calculation
4.4. Curve Fitting by Single Target Equation and Biphasic Equation
4.5. Western Blot Analysis of Drug Effects on Cell Signaling
4.6. Monophasic and Biphasic Analyses of Genomics of Drug Sensitivity in Cancer Cell Responses to Dasatinib
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BP | biphasic |
| CML | chronic myeloid leukemia |
| CRC | colorectal cancer |
| DRI | dose reduction index |
| IGF-1R | insulin-like growth factor 1 receptor |
| IR | insulin receptor |
| MAP kinase | mitogen-activated protein kinase |
| MP | monophasic |
| PTK | protein tyrosine kinase |
| RMSE | root mean square error |
References
- Druker, B.J. Translation of the Philadelphia chromosome into therapy for CML. Blood 2008, 112, 4808–4817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Figueroa-Magalhães, M.C.; Jelovac, D.; Connolly, R.; Wolff, A.C. Treatment of HER2-positive breast cancer. Breast 2014, 23, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Da Cunha Santos, G.; Shepherd, F.A.; Tsao, M.S. EGFR mutations and lung cancer. Annu. Rev. Pathol. 2011, 6, 49–69. [Google Scholar] [CrossRef] [Green Version]
- Martincorena, I.; Raine, K.M.; Gerstung, M.; Dawson, K.J.; Haase, K.; Van Loo, P.; Davies, H.; Stratton, M.R.; Campbell, P.J. Universal Patterns of Selection in Cancer and Somatic Tissues. Cell 2017, 171, 1029–1041. [Google Scholar] [CrossRef]
- Tomasetti, C.; Marchionni, L.; Nowak, M.A.; Parmigiani, G.; Vogelstein, B. Only three driver gene mutations are required for the development of lung and colorectal cancers. Proc. Natl. Acad. Sci. USA 2015, 112, 118–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef]
- Tolcher, A.W.; Peng, W.; Calvo, E. Rational Approaches for Combination Therapy Strategies Targeting the MAP Kinase Pathway in Solid Tumors. Mol. Cancer Ther. 2018, 17, 3–16. [Google Scholar] [CrossRef] [Green Version]
- Brenner, H.; Kloor, M.; Pox, C.P. Colorectal cancer. Lancet 2014, 383, 1490–1502. [Google Scholar] [CrossRef]
- Loree, J.M.; Kopetz, S. Recent developments in the treatment of metastatic colorectal cancer. Ther. Adv. Med. Oncol. 2017, 9, 551–564. [Google Scholar] [CrossRef]
- Seeber, A.; Gastl, G. Targeted Therapy of Colorectal Cancer. Oncol. Res. Treat. 2016, 39, 796–802. [Google Scholar] [CrossRef]
- Bozic, I.; Antal, T.; Ohtsuki, H.; Carter, H.; Kim, D.; Chen, S.; Karchin, R.; Kinzler, K.W.; Vogelstein, B.; Nowak, M.A. Accumulation of driver and passenger mutations during tumor progression. Proc. Natl. Acad. Sci. USA 2010, 107, 18545–18550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Network, C.G.A. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karoulia, Z.; Gavathiotis, E.; Poulikakos, P.I. New perspectives for targeting RAF kinase in human cancer. Nat. Rev. Cancer 2017, 17, 676–691. [Google Scholar] [CrossRef] [PubMed]
- Frame, M.C. Src in cancer: Deregulation and consequences for cell behaviour. Biochim. Biophys. Acta 2002, 1602, 114–130. [Google Scholar] [CrossRef]
- Johnson, F.M.; Gallick, G.E. SRC family nonreceptor tyrosine kinases as molecular targets for cancer therapy. Anticancer Agents Med. Chem. 2007, 7, 651–659. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Yu, D. Targeting Src family kinases in anti-cancer therapies: Turning promise into triumph. Trends Pharmacol. Sci. 2012, 33, 122–128. [Google Scholar] [CrossRef] [Green Version]
- Puls, L.N.; Eadens, M.; Messersmith, W. Current status of SRC inhibitors in solid tumor malignancies. Oncologist 2011, 16, 566–578. [Google Scholar] [CrossRef] [Green Version]
- O’Neil, J.; Benita, Y.; Feldman, I.; Chenard, M.; Roberts, B.; Liu, Y.; Li, J.; Kral, A.; Lejnine, S.; Loboda, A.; et al. An Unbiased Oncology Compound Screen to Identify Novel Combination Strategies. Mol. Cancer Ther. 2016, 15, 1155–1162. [Google Scholar] [CrossRef] [Green Version]
- Wali, V.B.; Langdon, C.G.; Held, M.A.; Platt, J.T.; Patwardhan, G.A.; Safonov, A.; Aktas, B.; Pusztai, L.; Stern, D.F.; Hatzis, C. Systematic Drug Screening Identifies Tractable Targeted Combination Therapies in Triple-Negative Breast Cancer. Cancer Res. 2017, 77, 566–578. [Google Scholar] [CrossRef] [Green Version]
- Menden, M.P.; Wang, D.; Mason, M.J.; Szalai, B.; Bulusu, K.C.; Guan, Y.; Yu, T.; Kang, J.; Jeon, M.; Wolfinger, R.; et al. Community assessment to advance computational prediction of cancer drug combinations in a pharmacogenomic screen. Nat. Commun. 2019, 10, 2674. [Google Scholar] [CrossRef] [Green Version]
- Holbeck, S.L.; Camalier, R.; Crowell, J.A.; Govindharajulu, J.P.; Hollingshead, M.; Anderson, L.W.; Polley, E.; Rubinstein, L.; Srivastava, A.; Wilsker, D.; et al. The National Cancer Institute ALMANAC: A Comprehensive Screening Resource for the Detection of Anticancer Drug Pairs with Enhanced Therapeutic Activity. Cancer Res. 2017, 77, 3564–3576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitzgerald, J.B.; Schoeberl, B.; Nielsen, U.B.; Sorger, P.K. Systems biology and combination therapy in the quest for clinical efficacy. Nat. Chem. Biol. 2006, 2, 458–466. [Google Scholar] [CrossRef] [PubMed]
- Palmer, A.C.; Sorger, P.K. Combination Cancer Therapy Can Confer Benefit via Patient-to-Patient Variability without Drug Additivity or Synergy. Cell 2017, 171, 1678–1691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fallahi-Sichani, M.; Honarnejad, S.; Heiser, L.M.; Gray, J.W.; Sorger, P.K. Metrics other than potency reveal systematic variation in responses to cancer drugs. Nat. Chem. Biol. 2013, 9, 708–714. [Google Scholar] [CrossRef] [Green Version]
- Shoemaker, R.H. The NCI60 human tumour cell line anticancer drug screen. Nat. Rev. Cancer 2006, 6, 813–823. [Google Scholar] [CrossRef]
- Forbes, S.A.; Beare, D.; Boutselakis, H.; Bamford, S.; Bindal, N.; Tate, J.; Cole, C.G.; Ward, S.; Dawson, E.; Ponting, L.; et al. COSMIC: Somatic cancer genetics at high-resolution. Nucleic Acids Res. 2017, 45, D777–D783. [Google Scholar] [CrossRef]
- Windham, T.C.; Parikh, N.U.; Siwak, D.R.; Summy, J.M.; McConkey, D.J.; Kraker, A.J.; Gallick, G.E. Src activation regulates anoikis in human colon tumor cell lines. Oncogene 2002, 21, 7797–7807. [Google Scholar] [CrossRef] [Green Version]
- Herr, R.; Köhler, M.; Andrlová, H.; Weinberg, F.; Möller, Y.; Halbach, S.; Lutz, L.; Mastroianni, J.; Klose, M.; Bittermann, N.; et al. B-Raf inhibitors induce epithelial differentiation in BRAF-mutant colorectal cancer cells. Cancer Res. 2015, 75, 216–229. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Song, Y.; Cui, L.; Wen, Z.; Lu, X. Inositol hexaphosphate suppresses growth and induces apoptosis in HT-29 colorectal cancer cells in culture: PI3K/Akt pathway as a potential target. Int. J. Clin. Exp. Pathol. 2015, 8, 1402–1410. [Google Scholar]
- Lee, H.S.; Cho, H.J.; Kwon, G.T.; Park, J.H. Kaempferol Downregulates Insulin-like Growth Factor-I Receptor and ErbB3 Signaling in HT-29 Human Colon Cancer Cells. J. Cancer Prev. 2014, 19, 161–169. [Google Scholar] [CrossRef]
- Shen, J.; Li, L.; Yang, T.; Cheng, N.; Sun, G. Drug Sensitivity Screening and Targeted Pathway Analysis Reveal a Multi-Driver Proliferative Mechanism and Suggest a Strategy of Combination Targeted Therapy for Colorectal Cancer Cells. Molecules 2019, 24, 623. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Cui, Y.; Shen, J.; Dobson, H.; Sun, G. Evidence for activated Lck protein tyrosine kinase as the driver of proliferation in acute myeloid leukemia cell, CTV-1. Leuk. Res. 2019, 78, 12–20. [Google Scholar] [CrossRef] [Green Version]
- Gray, N.S. Type II Raf Kinase Inhibitors. International Patent Number WO2011090738A2, 28 July 2011. [Google Scholar]
- Bernabé, R.; Patrao, A.; Carter, L.; Blackhall, F.; Dean, E. Selumetinib in the treatment of non-small-cell lung cancer. Future Oncol. 2016, 12, 2545–2560. [Google Scholar] [CrossRef]
- Araujo, J.; Logothetis, C. Dasatinib: A potent SRC inhibitor in clinical development for the treatment of solid tumors. Cancer Treat. Rev. 2010, 36, 492–500. [Google Scholar] [CrossRef] [Green Version]
- Carboni, J.M.; Wittman, M.; Yang, Z.; Lee, F.; Greer, A.; Hurlburt, W.; Hillerman, S.; Cao, C.; Cantor, G.H.; Dell-John, J.; et al. BMS-754807, a small molecule inhibitor of insulin-like growth factor-1R/IR. Mol. Cancer Ther. 2009, 8, 3341–3349. [Google Scholar] [CrossRef] [Green Version]
- Rhodes, N.; Heerding, D.A.; Duckett, D.R.; Eberwein, D.J.; Knick, V.B.; Lansing, T.J.; McConnell, R.T.; Gilmer, T.M.; Zhang, S.Y.; Robell, K.; et al. Characterization of an Akt kinase inhibitor with potent pharmacodynamic and antitumor activity. Cancer Res. 2008, 68, 2366–2374. [Google Scholar] [CrossRef] [Green Version]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Lin, X.; Nam, N.H.; Parang, K.; Sun, G. Determination of the substrate-docking site of protein tyrosine kinase C-terminal Src kinase. Proc. Natl. Acad. Sci. USA 2003, 100, 14707–14712. [Google Scholar] [CrossRef] [Green Version]
- Tiwari, R.K.; Brown, A.; Sadeghiani, N.; Shirazi, A.N.; Bolton, J.; Tse, A.; Verkhivker, G.; Parang, K.; Sun, G. Design, Synthesis, and Evaluation of Dasatinib-Amino Acid and Dasatinib-Fatty Acid Conjugates as Protein Tyrosine Kinase Inhibitors. ChemMedChem 2017, 12, 86–99. [Google Scholar] [CrossRef]
- Levy, D.S.; Kahana, J.A.; Kumar, R. AKT inhibitor, GSK690693, induces growth inhibition and apoptosis in acute lymphoblastic leukemia cell lines. Blood 2009, 113, 1723–1729. [Google Scholar] [CrossRef]
- Altomare, D.A.; Zhang, L.; Deng, J.; Di Cristofano, A.; Klein-Szanto, A.J.; Kumar, R.; Testa, J.R. GSK690693 delays tumor onset and progression in genetically defined mouse models expressing activated Akt. Clin Cancer Res. 2010, 16, 486–496. [Google Scholar] [CrossRef] [Green Version]
- Chan, T.O.; Tsichlis, P.N. PDK2: A complex tail in one Akt. Sci. Signal. 2001, 2001, pe1. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [Green Version]
- Chou, T.C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [Green Version]
- Guagnano, V.; Furet, P.; Spanka, C.; Bordas, V.; Le Douget, M.; Stamm, C.; Brueggen, J.; Jensen, M.R.; Schnell, C.; Schmid, H.; et al. Discovery of 3-(2,6-dichloro-3,5-dimethoxy-phenyl)-1-{6-[4-(4-ethyl-piperazin-1-yl)-phenylamino]-pyrimidin-4-yl}-1-methyl-urea (NVP-BGJ398), a potent and selective inhibitor of the fibroblast growth factor receptor family of receptor tyrosine kinase. J. Med. Chem. 2011, 54, 7066–7083. [Google Scholar] [CrossRef]
- Nishii, H.; Chiba, T.; Morikami, K.; Fukami, T.A.; Sakamoto, H.; Ko, K.; Koyano, H. Discovery of 6-benzyloxyquinolines as c-Met selective kinase inhibitors. Bioorg. Med. Chem. Lett. 2010, 20, 1405–1409. [Google Scholar] [CrossRef]
- Tsao, M.S.; Sakurada, A.; Cutz, J.C.; Zhu, C.Q.; Kamel-Reid, S.; Squire, J.; Lorimer, I.; Zhang, T.; Liu, N.; Daneshmand, M.; et al. Erlotinib in lung cancer—Molecular and clinical predictors of outcome. N. Engl. J. Med. 2005, 353, 133–144. [Google Scholar] [CrossRef]
- Amann, J.; Kalyankrishna, S.; Massion, P.P.; Ohm, J.E.; Girard, L.; Shigematsu, H.; Peyton, M.; Juroske, D.; Huang, Y.; Stuart Salmon, J.; et al. Aberrant epidermal growth factor receptor signaling and enhanced sensitivity to EGFR inhibitors in lung cancer. Cancer Res. 2005, 65, 226–235. [Google Scholar]
- Baro, M.; de Llobet, L.I.; Figueras, A.; Skvortsova, I.; Mesia, R.; Balart, J. Dasatinib worsens the effect of cetuximab in combination with fractionated radiotherapy in FaDu- and A431-derived xenografted tumours. Br. J. Cancer 2014, 111, 1310–1318. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Kalyankrishna, S.; Wislez, M.; Thilaganathan, N.; Saigal, B.; Wei, W.; Ma, L.; Wistuba, I.I.; Johnson, F.M.; Kurie, J.M. SRC-family kinases are activated in non-small cell lung cancer and promote the survival of epidermal growth factor receptor-dependent cell lines. Am. J. Pathol. 2007, 170, 366–376. [Google Scholar] [CrossRef] [Green Version]
- Neuber, S.; Jäger, S.; Meyer, M.; Wischmann, V.; Koch, P.J.; Moll, R.; Schmidt, A. c-Src mediated tyrosine phosphorylation of plakophilin 3 as a new mechanism to control desmosome composition in cells exposed to oxidative stress. Cell Tissue Res. 2015, 359, 799–816. [Google Scholar] [CrossRef] [PubMed]
- Di Veroli, G.Y.; Fornari, C.; Goldlust, I.; Mills, G.; Koh, S.B.; Bramhall, J.L.; Richards, F.M.; Jodrell, D.I. An automated fitting procedure and software for dose-response curves with multiphasic features. Sci. Rep. 2015, 5, 14701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iorio, F.; Knijnenburg, T.A.; Vis, D.J.; Bignell, G.R.; Menden, M.P.; Schubert, M.; Aben, N.; Gonçalves, E.; Barthorpe, S.; Lightfoot, H.; et al. A Landscape of Pharmacogenomic Interactions in Cancer. Cell 2016, 166, 740–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellis, L.M.; Hicklin, D.J. Resistance to Targeted Therapies: Refining Anticancer Therapy in the Era of Molecular Oncology. Clin. Cancer Res. 2009, 15, 7471–7478. [Google Scholar] [CrossRef] [Green Version]
- Corso, S.; Giordano, S. Targeted therapies in cancer and mechanisms of resistance. J. Mol. Med. 2014, 92, 677–679. [Google Scholar] [CrossRef] [Green Version]
- Neel, D.S.; Bivona, T.G. Resistance is futile: Overcoming resistance to targeted therapies in lung adenocarcinoma. NPJ Precis. Oncol. 2017, 1, 1–6. [Google Scholar] [CrossRef]
- Gorre, M.E.; Mohammed, M.; Ellwood, K.; Hsu, N.; Paquette, R.; Rao, P.N.; Sawyers, C.L. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science 2001, 293, 876–880. [Google Scholar] [CrossRef] [Green Version]
- Prahallad, A.; Sun, C.; Huang, S.; Di Nicolantonio, F.; Salazar, R.; Zecchin, D.; Beijersbergen, R.L.; Bardelli, A.; Bernards, R. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 2012, 483, 100–103. [Google Scholar] [CrossRef] [Green Version]
- Sun, G. Biphasic Analysis of Drug Response to Targeted Therapy by Triple Negative Breast Cancer Cells; University of Rhode Island: Kingston, RI, USA, 2020. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Target | IC50 (µM) |
|---|---|---|
| HG6-64-1 | BRAF | 0.243 ± 0.02 |
| Dasatinib | Src, Abl, PDGFR | 0.403 ± 0.06 |
| BMS-754807 | IR, IGF-1R, Met | 0.990 ± 0.09 |
| AZD-6244 | Mek | 1.334 ± 0.18 |
| Crizotinib | Met | 8.326 ± 0.99 |
| Sunitinib | PDGFRs | 11.17 ± 0.61 |
| BX-912 | PDK1 | 12.82 ± 2.12 |
| BGJ398 | FGFRs | 16.49 ± 1.71 |
| Gefitinib | EGFR | >20 |
| Pazopanib | PDGFR, VEGFR | >20 |
| AZD-6482 | PI 3-K | >20 |
| Masitinib | PDGFR, DDR1, Abl | >20 |
| Erlotinib | EGFR | >20 |
| GSK-690693 | Akt | >20 |
| Imatinib | Abl | >20 |
| Lapatinib | EGFR, ErbB2 | >20 |
| Linsitinib | IR, IGF-1R | >20 |
| Neratinib | EGFRs | >20 |
| Drug | Target | IC50 (nM) |
|---|---|---|
| Afatinib | EGFR, ErbB2/4 | 7.8 ± 0.2 |
| Gefitinib | EGFR | 12.1 ± 0.4 |
| Erlotinib | EGFR | 15.4 ± 0.5 |
| Dasatinib | Src, Abl, PDGFRs, etc | 140 ± 9 |
| Bosutinib | Src, Abl, | 273 ± 10 |
| Neratinib | EGFR, ErbB2/3/4 | 512 ± 12 |
| Lapatinib | ErbB2/3 | 585 ± 31 |
| Sorafenib | DDR1/2, FLT3 | 3286 ± 44 |
| Linifanib | PDGFRs | 6871 ± 333 |
| BMS-754807 | IR, IGF-1R, Met | 10,571 ± 797 |
| Linsitinib | IR, IGF-1R | 18,358 ± 1069 |
| BGJ398 | FGFRs | >20,000 |
| Nilotinib | Abl, DDR1 | >20,000 |
| Sunitinib | PDGFRs | >20,000 |
| Crizotinib | Met | >20,000 |
| HG6-64-1 | BRAF | >20,000 |
| AZD-6244 | Mek | >20,000 |
| BX-912 | PDK1 | >20,000 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shen, J.; Li, L.; Yang, T.; Cohen, P.S.; Sun, G. Biphasic Mathematical Model of Cell–Drug Interaction That Separates Target-Specific and Off-Target Inhibition and Suggests Potent Targeted Drug Combinations for Multi-Driver Colorectal Cancer Cells. Cancers 2020, 12, 436. https://doi.org/10.3390/cancers12020436
Shen J, Li L, Yang T, Cohen PS, Sun G. Biphasic Mathematical Model of Cell–Drug Interaction That Separates Target-Specific and Off-Target Inhibition and Suggests Potent Targeted Drug Combinations for Multi-Driver Colorectal Cancer Cells. Cancers. 2020; 12(2):436. https://doi.org/10.3390/cancers12020436
Chicago/Turabian StyleShen, Jinyan, Li Li, Tao Yang, Paul S. Cohen, and Gongqin Sun. 2020. "Biphasic Mathematical Model of Cell–Drug Interaction That Separates Target-Specific and Off-Target Inhibition and Suggests Potent Targeted Drug Combinations for Multi-Driver Colorectal Cancer Cells" Cancers 12, no. 2: 436. https://doi.org/10.3390/cancers12020436