GLUL Ablation Can Confer Drug Resistance to Cancer Cells via a Malate-Aspartate Shuttle-Mediated Mechanism

 , , ,
, , ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
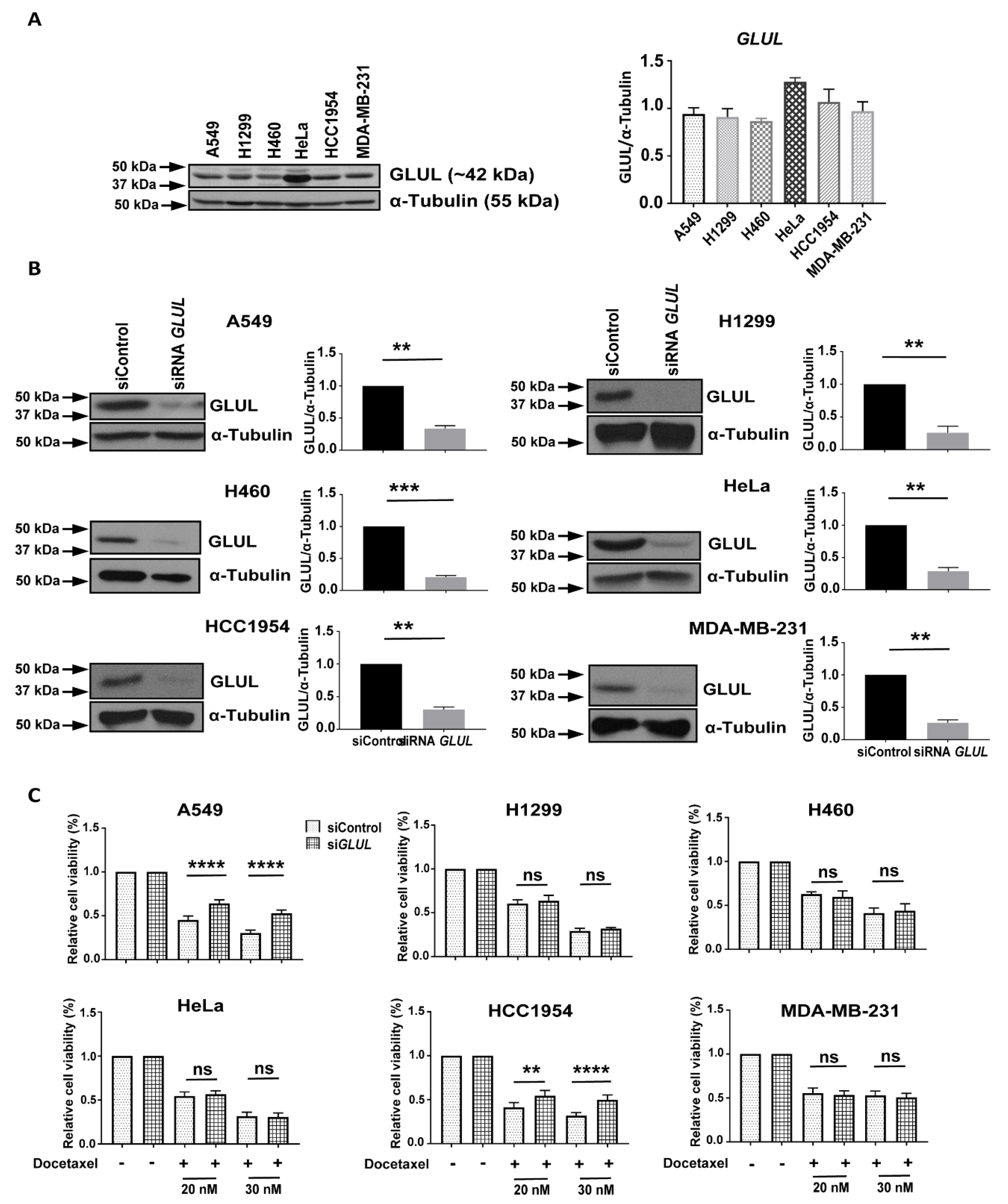
2.1. Transient GLUL Knockdown Induces Drug Resistance
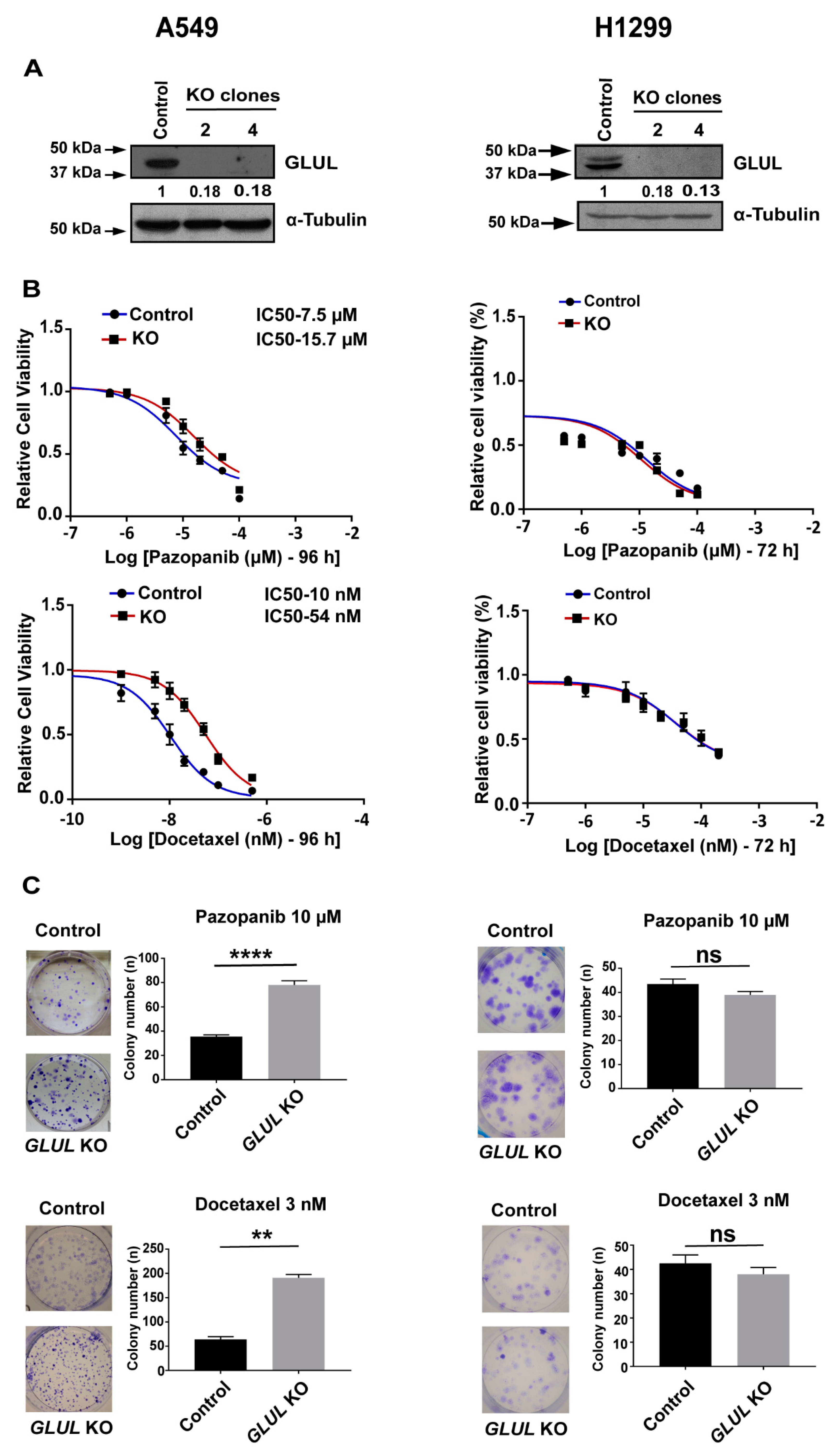
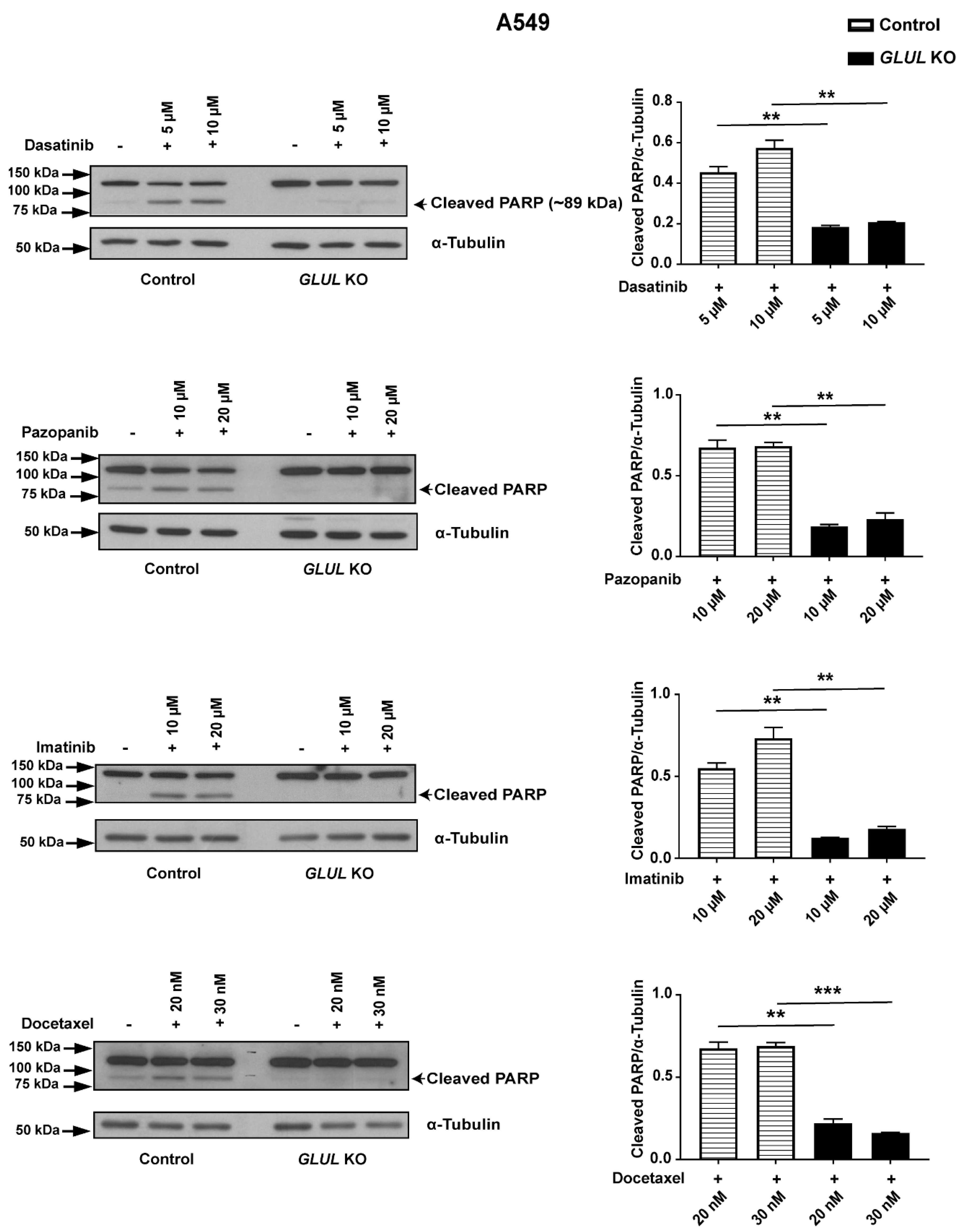
2.2. GLUL Knockout Cells Display Drug Resistance
2.3. GLUL KO-Associated Drug Resistance Correlates with Apoptosis
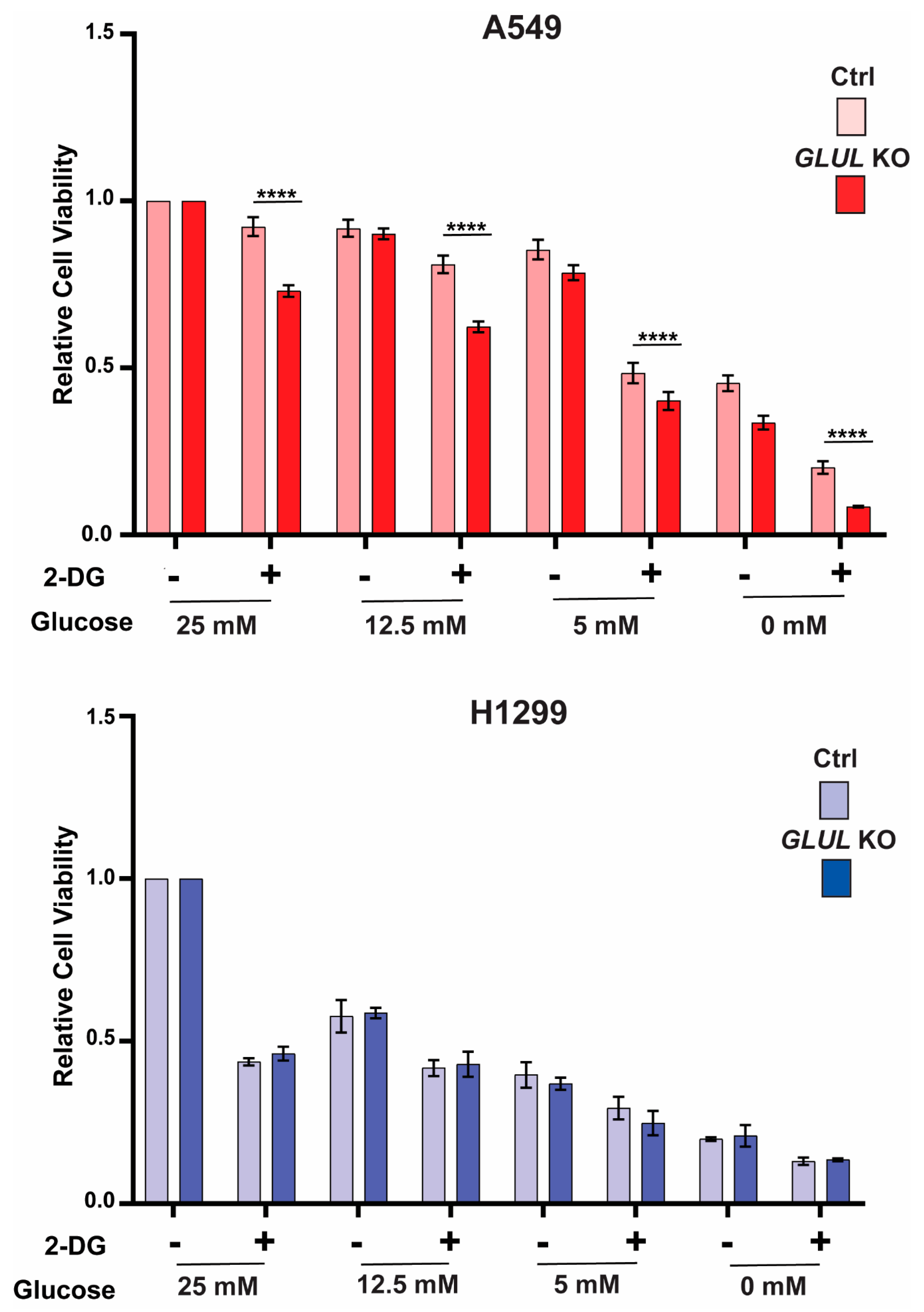
2.4. GLUL Knockout Cells Display Glucose Dependence
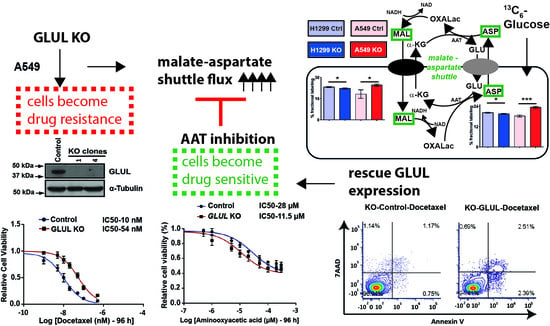
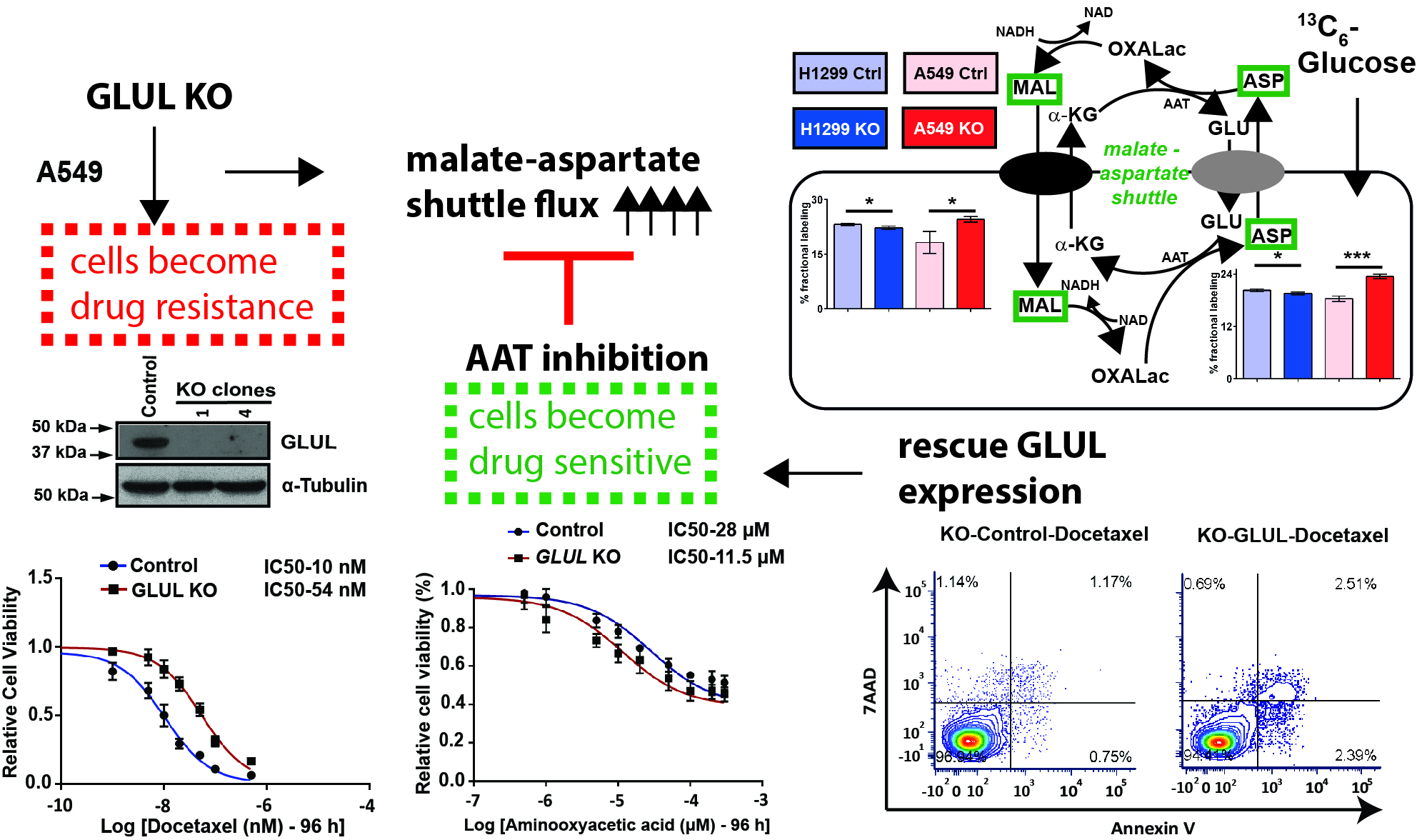
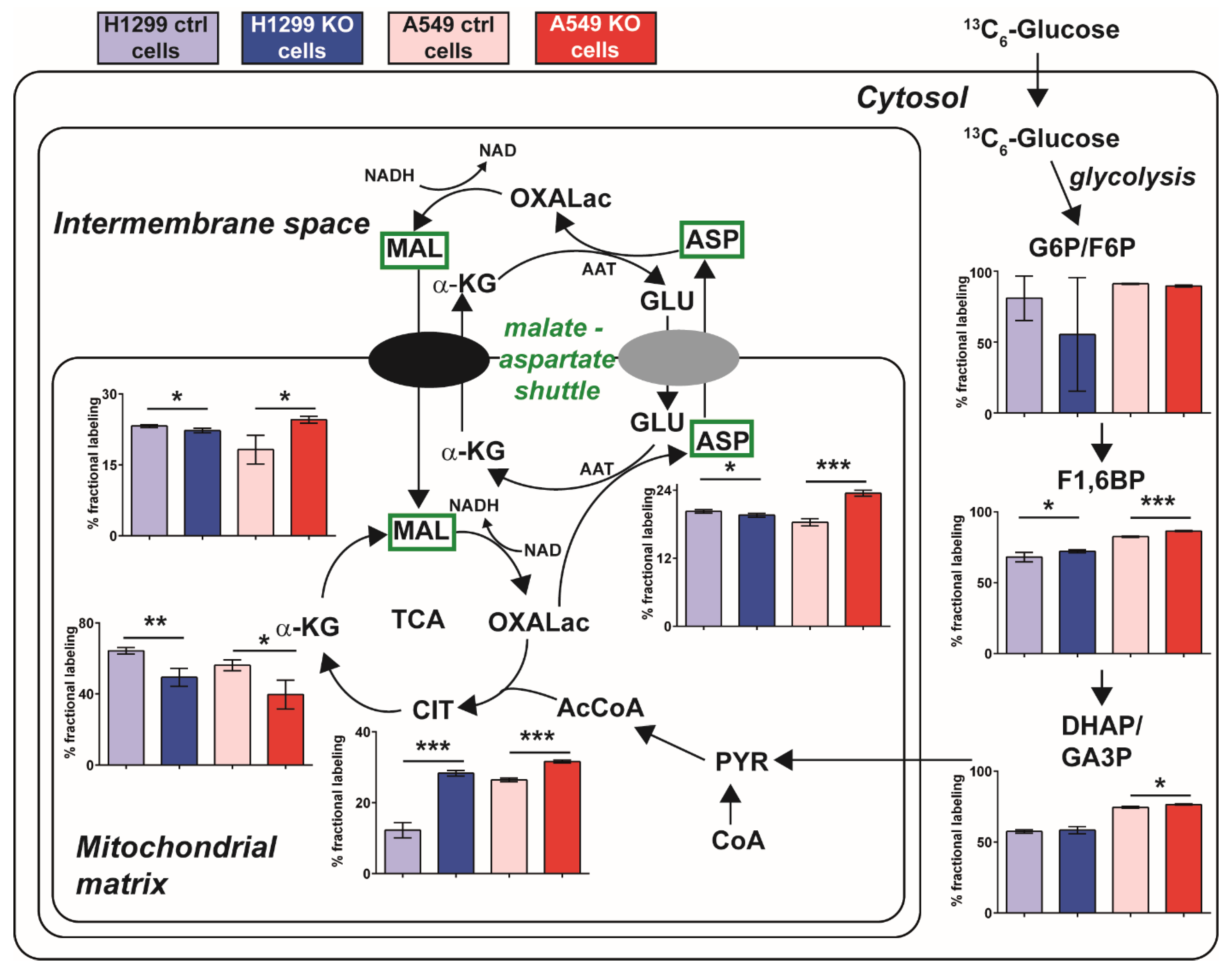
2.5. A Metabolic Phenotype Involving the Malate-Aspartate Shuttle Supports Drug Resistance Associated with GLUL KO
2.6. Inhibition of the Malate-Aspartate Shuttle Reveals Metabolic Vulnerability Associated with Resistance
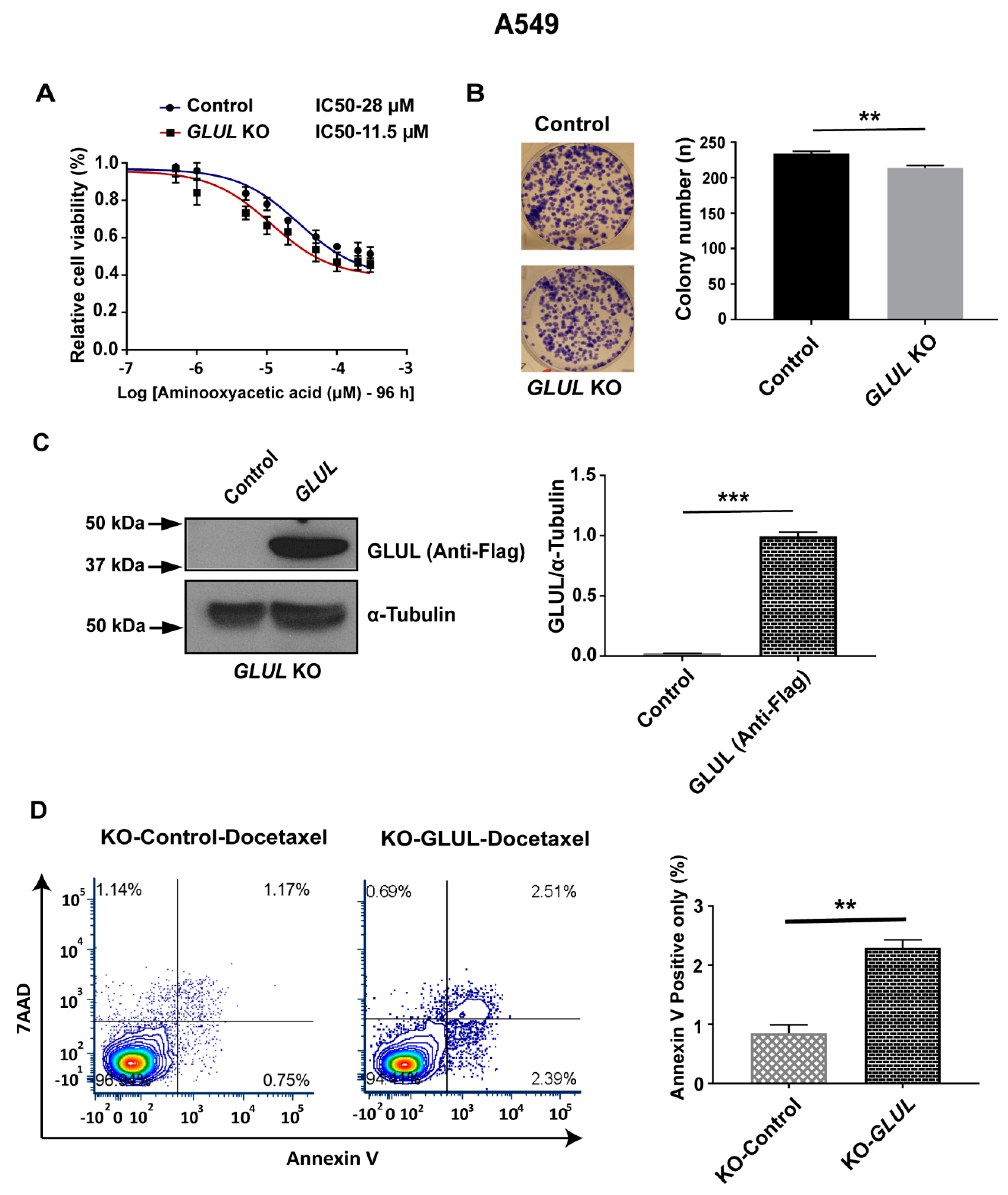
2.7. Rescuing GLUL Expression Sensitizes A549 KO Cells to Drugs
3. Discussion
4. Material and Methods
4.1. Cell Culture and Reagents
4.2. GLUL siRNA Transfection
4.3. Establishment of Inducible GLUL KD Cell Lines by shRNA Expression
4.4. Establishment of Stable GLUL KO Cell Lines by CRISPR/Cas9
4.5. GLUL Re-Expression in A549 GLUL KO Cells
4.6. MTS Assay and Western Blot Analysis
4.7. Clonogenic Assay
4.8. Flow Cytometry (FACS)-Based Apoptosis Analysis
4.9. Metabolic Profiling and Stable Isotope Labeling and Tracing
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Stronach, E.A.; Alfraidi, A.; Rama, N.; Datler, C.; Studd, J.B.; Agarwal, R.; Guney, T.G.; Gourley, C.; Hennessy, B.T.; Mills, G.B.; et al. HDAC4-regulated STAT1 activation mediates platinum resistance in ovarian cancer. Cancer Res. 2011, 71, 4412–4422. [Google Scholar] [CrossRef] [Green Version]
- Patel, C.; Stenke, L.; Varma, S.; Lindberg, M.L.; Bjorkholm, M.; Sjoberg, J.; Viktorsson, K.; Lewensohn, R.; Landgren, O.; Gottesman, M.M.; et al. Multidrug resistance in relapsed acute myeloid leukemia: EVIDENCE of biological heterogeneity. Cancer 2013, 119, 3076–3083. [Google Scholar] [CrossRef] [Green Version]
- Haffner, M.C.; Mosbruger, T.; Esopi, D.M.; Fedor, H.; Heaphy, C.M.; Walker, D.A.; Adejola, N.; Gurel, M.; Hicks, J.; Meeker, A.K.; et al. Tracking the clonal origin of lethal prostate cancer. J. Clin. Investig. 2013, 123, 4918–4922. [Google Scholar] [CrossRef] [Green Version]
- Leary, M.; Heerboth, S.; Lapinska, K.; Sarkar, S. Sensitization of Drug Resistant Cancer Cells: A Matter of Combination Therapy. Cancers 2018, 10, 483. [Google Scholar] [CrossRef] [Green Version]
- Gottesman, M.M. Mechanisms of cancer drug resistance. Annu. Rev. Med. 2002, 53, 615–627. [Google Scholar] [CrossRef] [Green Version]
- Ueda, K.; Cardarelli, C.; Gottesman, M.M.; Pastan, I. Expression of a full-length cDNA for the human “MDR1” gene confers resistance to colchicine, doxorubicin, and vinblastine. Proc. Natl. Acad. Sci. USA 1987, 84, 3004–3008. [Google Scholar] [CrossRef] [Green Version]
- Higgins, C.F. ABC transporters: From microorganisms to man. Annu. Rev. Cell Biol. 1992, 8, 67–113. [Google Scholar] [CrossRef]
- Chan, H.S.; Grogan, T.M.; Haddad, G.; DeBoer, G.; Ling, V. P-glycoprotein expression: Critical determinant in the response to osteosarcoma chemotherapy. J. Natl. Cancer Inst. 1997, 89, 1706–1715. [Google Scholar] [CrossRef] [Green Version]
- Ota, E.; Abe, Y.; Oshika, Y.; Ozeki, Y.; Iwasaki, M.; Inoue, H.; Yamazaki, H.; Ueyama, Y.; Takagi, K.; Ogata, T.; et al. Expression of the multidrug resistance-associated protein (MRP) gene in non-small-cell lung cancer. Br. J. Cancer 1995, 72, 550–554. [Google Scholar] [CrossRef] [Green Version]
- Bordow, S.B.; Haber, M.; Madafiglio, J.; Cheung, B.; Marshall, G.M.; Norris, M.D. Expression of the multidrug resistance-associated protein (MRP) gene correlates with amplification and overexpression of the N-myc oncogene in childhood neuroblastoma. Cancer Res. 1994, 54, 5036–5040. [Google Scholar]
- Callaghan, R.; Luk, F.; Bebawy, M. Inhibition of the multidrug resistance P-glycoprotein: Time for a change of strategy? Drug Metab. Dispos. Biol. Fate Chem. 2014, 42, 623–631. [Google Scholar] [CrossRef] [Green Version]
- Zaal, E.A.; Berkers, C.R. The Influence of Metabolism on Drug Response in Cancer. Front. Oncol. 2018, 8, 500. [Google Scholar] [CrossRef]
- Iwamoto, H.; Abe, M.; Yang, Y.; Cui, D.; Seki, T.; Nakamura, M.; Hosaka, K.; Lim, S.; Wu, J.; He, X.; et al. Cancer Lipid Metabolism Confers Antiangiogenic Drug Resistance. Cell Metab. 2018, 28, 104–117.e105. [Google Scholar] [CrossRef] [Green Version]
- Staubert, C.; Bhuiyan, H.; Lindahl, A.; Broom, O.J.; Zhu, Y.; Islam, S.; Linnarsson, S.; Lehtio, J.; Nordstrom, A. Rewired metabolism in drug-resistant leukemia cells: A metabolic switch hallmarked by reduced dependence on exogenous glutamine. J. Biol. Chem. 2015, 290, 8348–8359. [Google Scholar] [CrossRef] [Green Version]
- Staubert, C.; Krakowsky, R.; Bhuiyan, H.; Witek, B.; Lindahl, A.; Broom, O.; Nordstrom, A. Increased lanosterol turnover: A metabolic burden for daunorubicin-resistant leukemia cells. Med. Oncol. 2016, 33, 6. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- DeBerardinis, R.J.; Lum, J.J.; Hatzivassiliou, G.; Thompson, C.B. The biology of cancer: Metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008, 7, 11–20. [Google Scholar] [CrossRef] [Green Version]
- Wise, D.R.; Thompson, C.B. Glutamine addiction: A new therapeutic target in cancer. Trends Biochem. Sci. 2010, 35, 427–433. [Google Scholar] [CrossRef] [Green Version]
- Daye, D.; Wellen, K.E. Metabolic reprogramming in cancer: Unraveling the role of glutamine in tumorigenesis. Semin. Cell Dev. Biol. 2012, 23, 362–369. [Google Scholar] [CrossRef]
- Eelen, G.; Dubois, C.; Cantelmo, A.R.; Goveia, J.; Bruning, U.; DeRan, M.; Jarugumilli, G.; van Rijssel, J.; Saladino, G.; Comitani, F.; et al. Role of glutamine synthetase in angiogenesis beyond glutamine synthesis. Nature 2018, 561, 63–69. [Google Scholar] [CrossRef]
- Long, J.; Lang, Z.W.; Wang, H.G.; Wang, T.L.; Wang, B.E.; Liu, S.Q. Glutamine synthetase as an early marker for hepatocellular carcinoma based on proteomic analysis of resected small hepatocellular carcinomas. Hepatobil. Pancreat. Dis. Int. HBPD Int. 2010, 9, 296–305. [Google Scholar] [PubMed]
- Wang, Y.; Fan, S.; Lu, J.; Zhang, Z.; Wu, D.; Wu, Z.; Zheng, Y. GLUL Promotes Cell Proliferation in Breast Cancer. J. Cell. Biochem. 2017, 118, 2018–2025. [Google Scholar] [CrossRef] [PubMed]
- Rosati, A.; Poliani, P.L.; Todeschini, A.; Cominelli, M.; Medicina, D.; Cenzato, M.; Simoncini, E.L.; Magrini, S.M.; Buglione, M.; Grisanti, S.; et al. Glutamine synthetase expression as a valuable marker of epilepsy and longer survival in newly diagnosed glioblastoma multiforme. Neuro-oncology 2013, 15, 618–625. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.; Wang, Y.; Zhang, Z.; Lu, J.; Wu, Z.; Shan, Q.; Sun, C.; Wu, D.; Li, M.; Sheng, N.; et al. High expression of glutamate-ammonia ligase is associated with unfavorable prognosis in patients with ovarian cancer. J. Cell. Biochem. 2018, 119, 6008–6015. [Google Scholar] [CrossRef]
- Easlon, E.; Tsang, F.; Skinner, C.; Wang, C.; Lin, S.J. The malate-aspartate NADH shuttle components are novel metabolic longevity regulators required for calorie restriction-mediated life span extension in yeast. Genes Dev. 2008, 22, 931–944. [Google Scholar] [CrossRef] [Green Version]
- Greenhouse, W.V.; Lehninger, A.L. Occurrence of the malate-aspartate shuttle in various tumor types. Cancer Res. 1976, 36, 1392–1396. [Google Scholar]
- Kaufmann, S.H.; Desnoyers, S.; Ottaviano, Y.; Davidson, N.E.; Poirier, G.G. Specific proteolytic cleavage of poly(ADP-ribose) polymerase: An early marker of chemotherapy-induced apoptosis. Cancer Res. 1993, 53, 3976–3985. [Google Scholar]
- D’Amours, D.; Sallmann, F.R.; Dixit, V.M.; Poirier, G.G. Gain-of-function of poly(ADP-ribose) polymerase-1 upon cleavage by apoptotic proteases: Implications for apoptosis. J. Cell Sci. 2001, 114, 3771–3778. [Google Scholar]
- Wick, A.N.; Drury, D.R.; Nakada, H.I.; Wolfe, J.B. Localization of the primary metabolic block produced by 2-deoxyglucose. J. Biol. Chem. 1957, 224, 963–969. [Google Scholar]
- Vila, J.; Thomasset, N.; Navarro, C.; Dore, J.F. In vitro and in vivo anti-tumor activity of L-glutamic acid gamma-monohydroxamate against L1210 leukemia and B16 melanoma. Int. J. Cancer 1990, 45, 737–743. [Google Scholar] [CrossRef]
- Thomasset, N.; Goetsch, L.; Hamedi-Sangsari, F.; Tournaire, R.; Malley, S.; Navarro, C.; Dore, J.F.; Vila, J. Inhibition of malate-aspartate shuttle by the antitumor drug L-glutamic acid gamma-monohydroxamate in L1210 leukemia cells. Int. J. Cancer 1992, 51, 329–332. [Google Scholar] [CrossRef] [PubMed]
- Thornburg, J.M.; Nelson, K.K.; Clem, B.F.; Lane, A.N.; Arumugam, S.; Simmons, A.; Eaton, J.W.; Telang, S.; Chesney, J. Targeting aspartate aminotransferase in breast cancer. Breast Cancer Res. BCR 2008, 10, R84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muthu, M.; Nordstrom, A. Current Status and Future Prospects of Clinically Exploiting Cancer-specific Metabolism-Why Is Tumor Metabolism Not More Extensively Translated into Clinical Targets and Biomarkers? Int. J. Mol. Sci. 2019, 20, 1835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kauppinen, R.A.; Sihra, T.S.; Nicholls, D.G. Aminooxyacetic acid inhibits the malate-aspartate shuttle in isolated nerve terminals and prevents the mitochondria from utilizing glycolytic substrates. Biochim. Biophys. Acta 1987, 930, 173–178. [Google Scholar] [CrossRef]
- Cluntun, A.A.; Lukey, M.J.; Cerione, R.A.; Locasale, J.W. Glutamine Metabolism in Cancer: Understanding the Heterogeneity. Trends Cancer 2017, 3, 169–180. [Google Scholar] [CrossRef] [Green Version]
- Yuneva, M.O.; Fan, T.W.; Allen, T.D.; Higashi, R.M.; Ferraris, D.V.; Tsukamoto, T.; Mates, J.M.; Alonso, F.J.; Wang, C.; Seo, Y.; et al. The metabolic profile of tumors depends on both the responsible genetic lesion and tissue type. Cell Metab. 2012, 15, 157–170. [Google Scholar] [CrossRef] [Green Version]
- Davidson, S.M.; Papagiannakopoulos, T.; Olenchock, B.A.; Heyman, J.E.; Keibler, M.A.; Luengo, A.; Bauer, M.R.; Jha, A.K.; O’Brien, J.P.; Pierce, K.A.; et al. Environment Impacts the Metabolic Dependencies of Ras-Driven Non-Small Cell Lung Cancer. Cell Metab. 2016, 23, 517–528. [Google Scholar] [CrossRef]
- Lanczky, A.; Nagy, A.; Bottai, G.; Munkacsy, G.; Szabo, A.; Santarpia, L.; Gyorffy, B. miRpower: A web-tool to validate survival-associated miRNAs utilizing expression data from 2178 breast cancer patients. Breast Cancer Res. Treat. 2016, 160, 439–446. [Google Scholar] [CrossRef]
- Jurikova, M.; Danihel, L.; Polak, S.; Varga, I. Ki67, PCNA, and MCM proteins: Markers of proliferation in the diagnosis of breast cancer. Acta Histochem. 2016, 118, 544–552. [Google Scholar] [CrossRef]
- Safer, B.; Smith, C.M.; Williamson, J.R. Control of the transport of reducing equivalents across the mitochondrial membrane in perfused rat heart. J. Mol. Cell. Cardiol. 1971, 2, 111–124. [Google Scholar] [CrossRef]
- Safer, B. The Metabolic Significance of the Malate-Aspartate Cycle in Heart. Circ. Res. 1975, 37, 527–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohrig, F.; Schulze, A. The multifaceted roles of fatty acid synthesis in cancer. Nat. Rev. Cancer 2016, 16, 732–749. [Google Scholar] [CrossRef] [PubMed]
- Pick, A.M.; Nystrom, K.K. Pazopanib for the treatment of metastatic renal cell carcinoma. Clin. Ther. 2012, 34, 511–520. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Peng, W.; Wu, T.; Deng, P.; Zhao, Y.L. Increased glutamine anabolism sensitizes non-small cell lung cancer to gefitinib treatment. Cell Death Discov. 2018, 4, 24. [Google Scholar] [CrossRef]
- Schoffski, P. Pazopanib in the treatment of soft tissue sarcoma. Expert Rev. Anticancer Ther. 2012, 12, 711–723. [Google Scholar] [CrossRef]
- Yvon, A.M.; Wadsworth, P.; Jordan, M.A. Taxol suppresses dynamics of individual microtubules in living human tumor cells. Mol. Biol. Cell 1999, 10, 947–959. [Google Scholar] [CrossRef] [Green Version]
- Pruefer, F.G.; Lizarraga, F.; Maldonado, V.; Melendez-Zajgla, J. Participation of Omi Htra2 serine-protease activity in the apoptosis induced by cisplatin on SW480 colon cancer cells. J. Chemother. (Florence Italy) 2008, 20, 348–354. [Google Scholar] [CrossRef]
- Sanjana, N.E.; Shalem, O.; Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 2014, 11, 783–784. [Google Scholar] [CrossRef] [Green Version]
- Shalem, O.; Sanjana, N.E.; Hartenian, E.; Shi, X.; Scott, D.A.; Mikkelson, T.; Heckl, D.; Ebert, B.L.; Root, D.E.; Doench, J.G.; et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 2014, 343, 84–87. [Google Scholar] [CrossRef] [Green Version]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muthu, M.; Kumar, R.; Syed Khaja, A.S.; Gilthorpe, J.D.; Persson, J.L.; Nordström, A. GLUL Ablation Can Confer Drug Resistance to Cancer Cells via a Malate-Aspartate Shuttle-Mediated Mechanism. Cancers 2019, 11, 1945. https://doi.org/10.3390/cancers11121945
Muthu M, Kumar R, Syed Khaja AS, Gilthorpe JD, Persson JL, Nordström A. GLUL Ablation Can Confer Drug Resistance to Cancer Cells via a Malate-Aspartate Shuttle-Mediated Mechanism. Cancers. 2019; 11(12):1945. https://doi.org/10.3390/cancers11121945
Chicago/Turabian StyleMuthu, Magesh, Ranjeet Kumar, Azharuddin Sajid Syed Khaja, Jonathan D. Gilthorpe, Jenny L. Persson, and Anders Nordström. 2019. "GLUL Ablation Can Confer Drug Resistance to Cancer Cells via a Malate-Aspartate Shuttle-Mediated Mechanism" Cancers 11, no. 12: 1945. https://doi.org/10.3390/cancers11121945