Liver Cancer Cell Lines Treated with Doxorubicin under Normoxia and Hypoxia: Cell Viability and Oncologic Protein Profile

Abstract
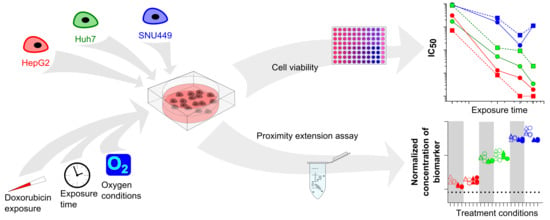
:
1. Introduction
2. Results
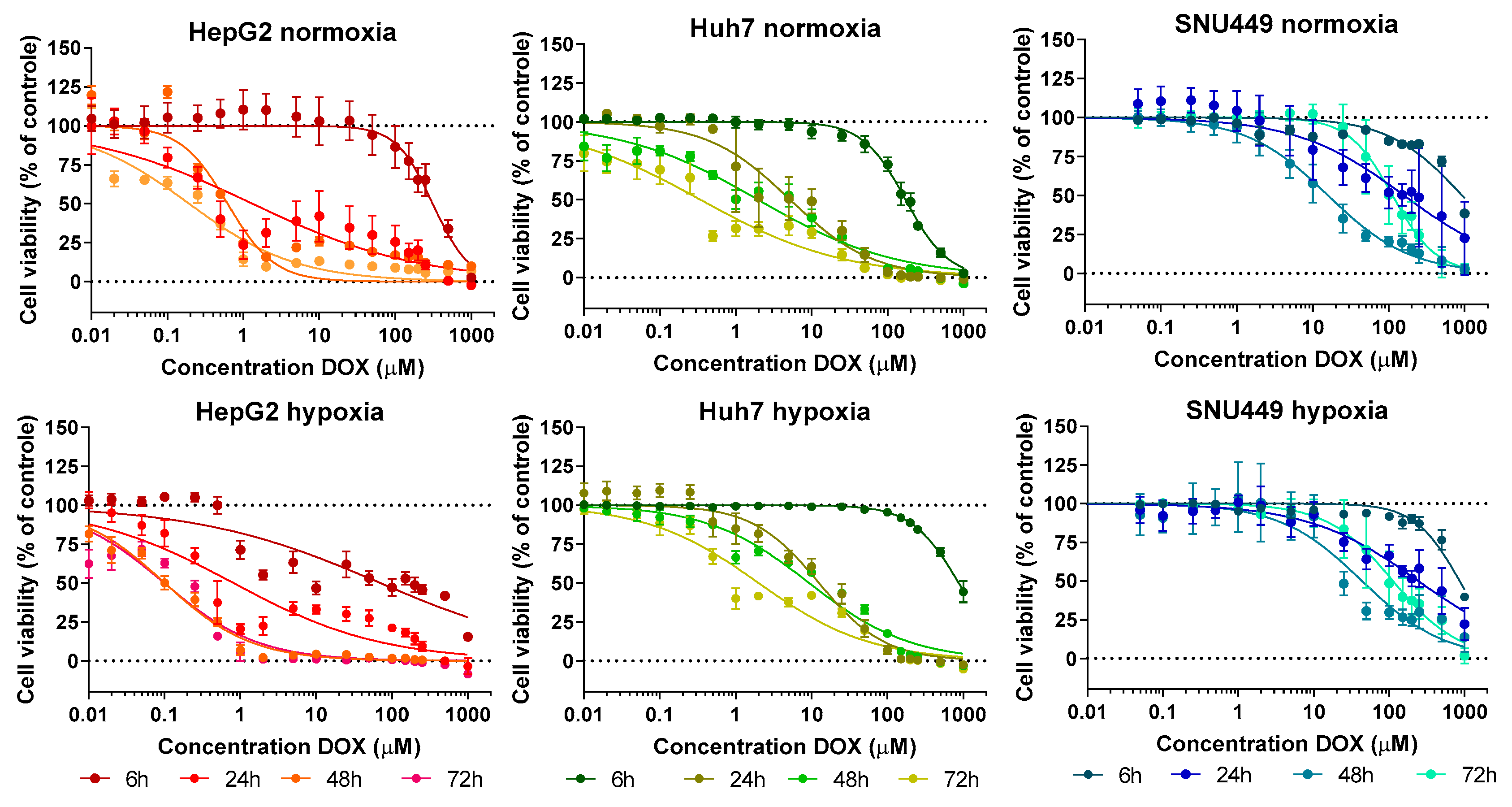
2.1. Cell Viability—Treatment with DOX under Hypoxic or Normoxic Conditions
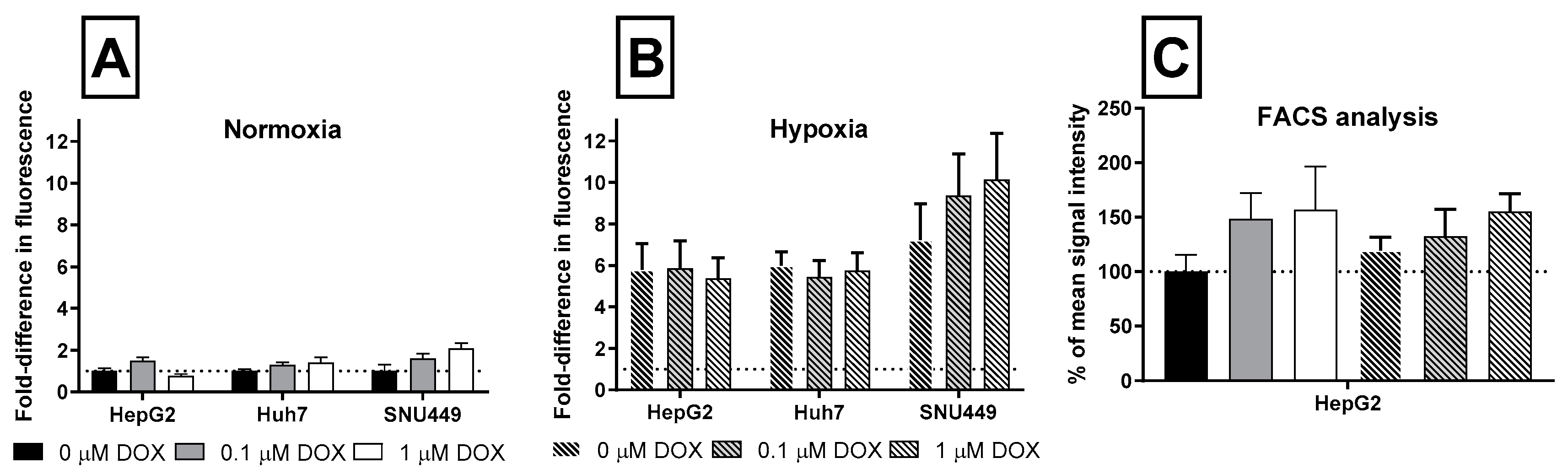
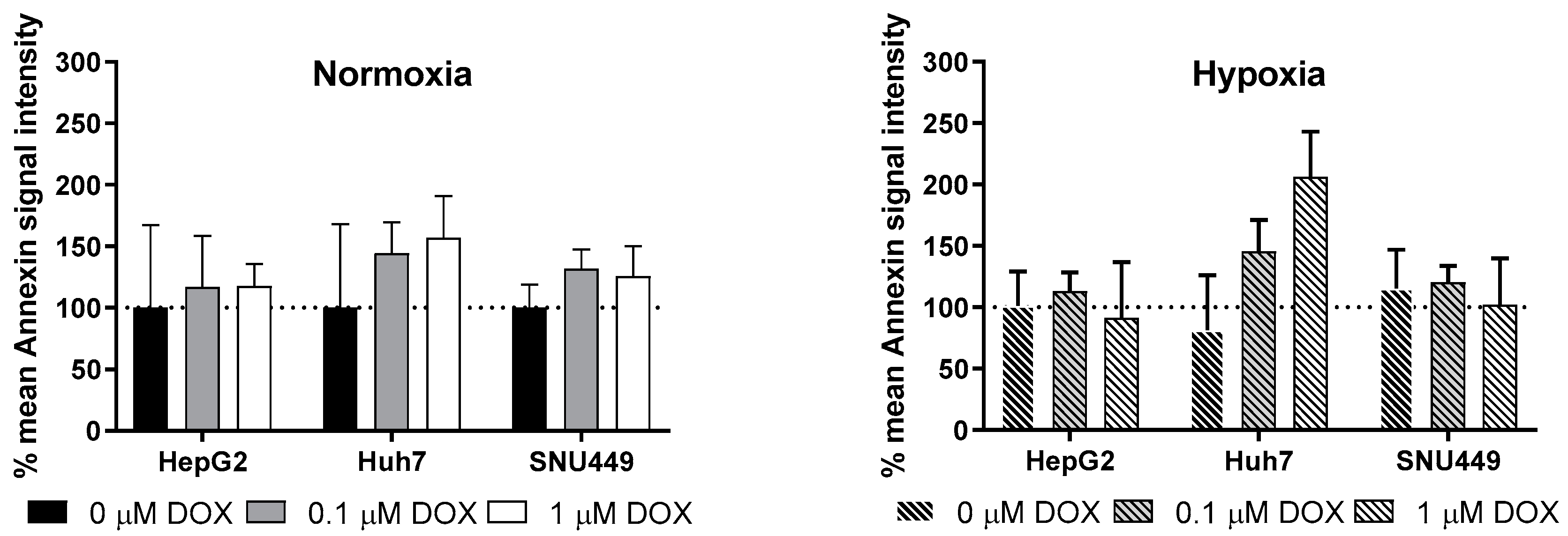
2.2. Oxidative Stress and Apoptosis
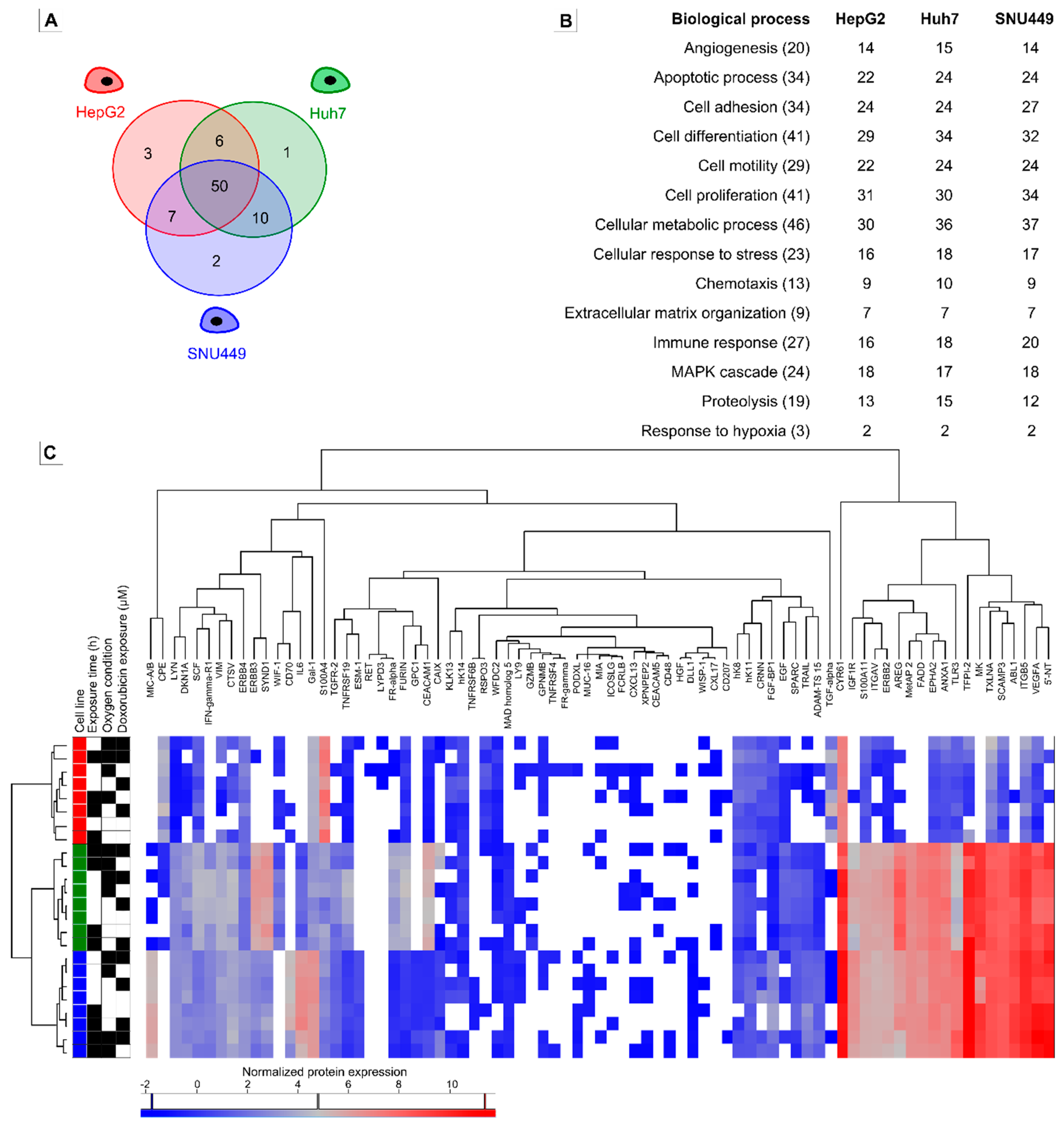
2.3. Oncologic Protein Profile
3. Discussion
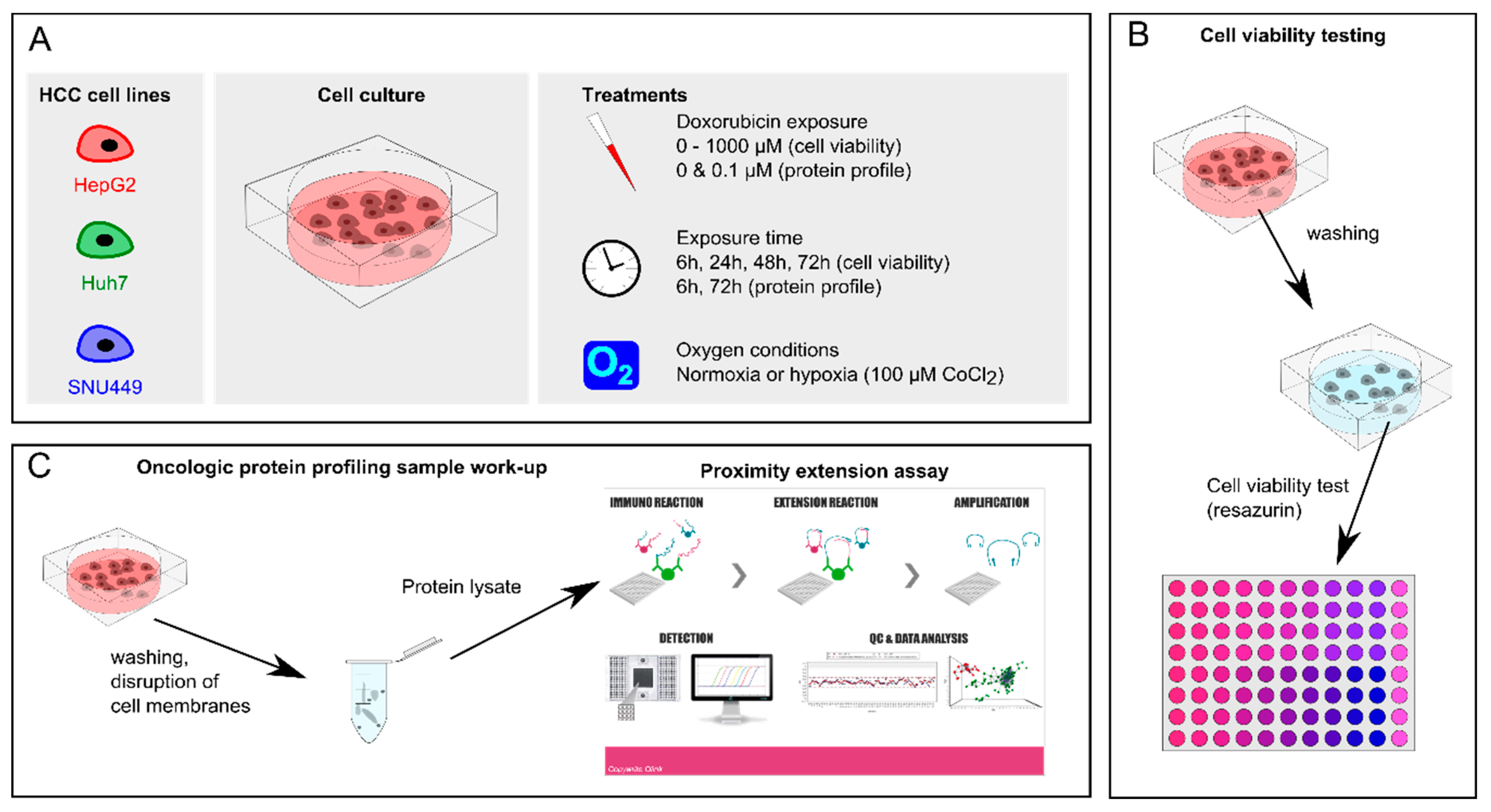
4. Materials and Methods
4.1. Chemicals
4.2. Cell Culture
4.3. Cell Viability Assay
4.4. HIF1α and PDK1 in Cell ELISA Assay
4.5. Oxidative Stress Measurement
4.6. Annexin V Flow Cytometry
4.7. Oncologic Protein Profile
4.8. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Pecorelli, A.; Lenzi, B.; Gramenzi, A.; Garuti, F.; Farinati, F.; Giannini, E.G.; Ciccarese, F.; Piscaglia, F.; Rapaccini, G.L.; Di Marco, M.; et al. Curative therapies are superior to standard of care (transarterial chemoembolization) for intermediate stage hepatocellular carcinoma. Liver Int. 2017, 37, 423–433. [Google Scholar] [CrossRef]
- Galle, P.R.; Tovoli, F.; Foerster, F.; Wörns, M.A.; Cucchetti, A.; Bolondi, L. The treatment of intermediate stage tumours beyond TACE: From surgery to systemic therapy. J. Hepatol. 2017, 67, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Gaba, R.C.; Lewandowski, R.J.; Hickey, R.; Baerlocher, M.O.; Cohen, E.I.; Dariushnia, S.R.; Janne d’Othee, B.; Padia, S.A.; Salem, R.; Wang, D.S.; et al. Transcatheter Therapy for Hepatic Malignancy: Standardization of Terminology and Reporting Criteria. J. Vasc. Interv. Radiol. 2016, 27, 457–473. [Google Scholar] [CrossRef] [Green Version]
- European Association for the Study of the Liver. EASL Clinical Practice Guidelines: Management of hepatocellular carcinoma. J. Hepatol. 2018. [Google Scholar] [CrossRef]
- Hepatocellular Carcinoma: Diagnosis and Treatment, 3rd ed.; Carr, B.I. (Ed.) Springer International Publishing: Cham, Switzerland, 2016. [Google Scholar] [CrossRef]
- El Fouly, A.; Ertle, J.; El Dorry, A.; Shaker, M.K.; Dechene, A.; Abdella, H.; Mueller, S.; Barakat, E.; Lauenstein, T.; Bockisch, A.; et al. In intermediate stage hepatocellular carcinoma: Radioembolization with yttrium 90 or chemoembolization? Liver Int. 2015, 35, 627–635. [Google Scholar] [CrossRef] [PubMed]
- Lencioni, R.; de Baere, T.; Soulen, M.C.; Rilling, W.S.; Geschwind, J.F. Lipiodol transarterial chemoembolization for hepatocellular carcinoma: A systematic review of efficacy and safety data. Hepatology 2016, 64, 106–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danesi, R.; Fogli, S.; Gennari, A.; Conte, P.; Del Tacca, M. Pharmacokinetic-pharmacodynamic relationships of the anthracycline anticancer drugs. Clin. Pharmacokinet. 2002, 41, 431–444. [Google Scholar] [CrossRef] [PubMed]
- Minotti, G.; Menna, P.; Salvatorelli, E.; Cairo, G.; Gianni, L. Anthracyclines: Molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol. Rev. 2004, 56, 185–229. [Google Scholar] [CrossRef]
- Tacar, O.; Sriamornsak, P.; Dass, C.R. Doxorubicin: An update on anticancer molecular action, toxicity and novel drug delivery systems. J. Pharm. Pharmacol. 2013, 65, 157–170. [Google Scholar] [CrossRef]
- Asghar, U.; Meyer, T. Are there opportunities for chemotherapy in the treatment of hepatocellular cancer? J. Hepatol. 2012, 56, 686–695. [Google Scholar] [CrossRef] [Green Version]
- Sieghart, W.; Hucke, F.; Peck-Radosavljevic, M. Transarterial chemoembolization: Modalities, indication, and patient selection. J. Hepatol. 2015, 62, 1187–1195. [Google Scholar] [CrossRef] [Green Version]
- Corbet, C.; Feron, O. Tumour acidosis: From the passenger to the driver’s seat. Nat. Rev. Cancer 2017, 17, 577–593. [Google Scholar] [CrossRef]
- Copple, B.L. Hypoxia stimulates hepatocyte epithelial to mesenchymal transition by hypoxia-inducible factor and transforming growth factor-beta-dependent mechanisms. Liver Int. 2010, 30, 669–682. [Google Scholar] [CrossRef]
- Vaupel, P.; Mayer, A. Tumor Hypoxia: Causative Mechanisms, Microregional Heterogeneities, and the Role of Tissue-Based Hypoxia Markers; Springer: Cham, Switzerland, 2016; pp. 77–86. [Google Scholar]
- Vaupel, P.; Multhoff, G. Accomplices of the Hypoxic Tumor Microenvironment Compromising Antitumor Immunity: Adenosine, Lactate, Acidosis, Vascular Endothelial Growth Factor, Potassium Ions, and Phosphatidylserine. Front. Immunol. 2017, 8, 1887. [Google Scholar] [CrossRef] [Green Version]
- Wilson, G.K.; Tennant, D.A.; McKeating, J.A. Hypoxia inducible factors in liver disease and hepatocellular carcinoma: Current understanding and future directions. J. Hepatol. 2014, 61, 1397–1406. [Google Scholar] [CrossRef] [Green Version]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- Zhang, Y.F.; Wei, W.; Wang, J.H.; Xu, L.; Jian, P.E.; Xiao, C.Z.; Zhong, X.P.; Shi, M.; Guo, R.P. Transarterial chemoembolization combined with sorafenib for the treatment of hepatocellular carcinoma with hepatic vein tumor thrombus. Oncotargets Ther. 2016, 9, 4239–4246. [Google Scholar] [CrossRef]
- Choi, G.H.; Shim, J.H.; Kim, M.J.; Ryu, M.H.; Ryoo, B.Y.; Kang, Y.K.; Shin, Y.M.; Kim, K.M.; Lim, Y.S.; Lee, H.C. Sorafenib alone versus sorafenib combined with transarterial chemoembolization for advanced-stage hepatocellular carcinoma: Results of propensity score analyses. Radiology 2013, 269, 603–611. [Google Scholar] [CrossRef]
- Meyer, T.; Fox, R.; Ma, Y.T.; Ross, P.J.; James, M.W.; Sturgess, R.; Stubbs, C.; Stocken, D.D.; Wall, L.; Watkinson, A.; et al. Sorafenib in combination with transarterial chemoembolisation in patients with unresectable hepatocellular carcinoma (TACE 2): A randomised placebo-controlled, double-blind, phase 3 trial. Lancet Gastroenterol. Hepatol. 2017, 2, 565–575. [Google Scholar] [CrossRef]
- Chau, D.K.; Chen, G.G.; Zhang, H.; Leung, B.C.; Chun, S.; Lai, P.B. Differential functions of C- and N-terminal hepatitis B x protein in liver cells treated with doxorubicin in normoxic or hypoxic condition. PLoS ONE 2012, 7, e50118. [Google Scholar] [CrossRef]
- Chen, Y.L.; Yang, T.Y.; Chen, K.C.; Wu, C.L.; Hsu, S.L.; Hsueh, C.M. Hypoxia can impair doxorubicin resistance of non-small cell lung cancer cells by inhibiting MRP1 and P-gp expression and boosting the chemosensitizing effects of MRP1 and P-gp blockers. Cell. Oncol. 2016, 39, 411–433. [Google Scholar] [CrossRef]
- Wang, C.; Tang, Z.; Zhao, Y.; Yao, R.; Li, L.; Sun, W. Three-dimensional in vitro cancer models: A short review. Biofabrication 2014, 6, 022001. [Google Scholar] [CrossRef]
- Ao, L.; Guo, Y.; Song, X.K.; Guan, Q.Z.; Zheng, W.C.; Zhang, J.H.; Huang, H.Y.; Zou, Y.; Guo, Z.; Wang, X.L. Evaluating hepatocellular carcinoma cell lines for tumour samples using within-sample relative expression orderings of genes. Liver Int. 2017, 37, 1688–1696. [Google Scholar] [CrossRef]
- Pang, R.T.K.; Poon, T.C.W.; Wong, N.; Lai, P.B.S.; Wong, N.L.Y.; Chan, C.M.L.; Yu, J.W.S.; Chan, A.T.C.; Sung, J.J.Y. Comparison of protein expression patterns between hepatocellular carcinoma cell lines and a hepatoblastoma cell line. Clin. Proteom. 2004, 1, 313–331. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.; Sirota, M.; Fan-Minogue, H.; Hadley, D.; Butte, A.J. Relating hepatocellular carcinoma tumor samples and cell lines using gene expression data in translational research. BMC Med. Genom. 2015, 8 (Suppl. 2), S5. [Google Scholar] [CrossRef]
- American Type Culture Collection. Available online: www.lgcstandards-atcc.org (accessed on 5 November 2018).
- Olink Proteomics: Oncology Panel. Available online: https://www.olink.com/ (accessed on 5 November 2018).
- Zhou, Q.Y.; Tu, C.Y.; Shao, C.X.; Wang, W.K.; Zhu, J.D.; Cai, Y.; Mao, J.Y.; Chen, W. GC7 blocks epithelial-mesenchymal transition and reverses hypoxia-induced chemotherapy resistance in hepatocellular carcinoma cells. Am. J. Transl. Res. 2017, 9, 2608–2617. [Google Scholar]
- Zhang, Y.; Lu, Y.; Zhang, C.; Huang, D.; Wu, W.; Zhang, Y.; Shen, J.; Cai, Y.; Chen, W.; Yao, W. FSCN1 increases doxorubicin resistance in hepatocellular carcinoma through promotion of epithelial-mesenchymal transition. Int. J. Oncol. 2018. [Google Scholar] [CrossRef]
- Chang, A.Y.; Wang, M. In-vitro growth inhibition of chemotherapy and molecular targeted agents in hepatocellular carcinoma. Anti-Cancer Drugs 2013, 24, 251–259. [Google Scholar] [CrossRef]
- Barraud, L.; Merle, P.; Soma, E.; Lefrancois, L.; Guerret, S.; Chevallier, M.; Dubernet, C.; Couvreur, P.; Trepo, C.; Vitvitski, L. Increase of doxorubicin sensitivity by doxorubicin-loading into nanoparticles for hepatocellular carcinoma cells in vitro and in vivo. J. Hepatol. 2005, 42, 736–743. [Google Scholar] [CrossRef]
- Hu, Q.D.; Chen, W.; Yan, T.L.; Ma, T.; Chen, C.L.; Liang, C.; Zhang, Q.; Xia, X.F.; Liu, H.; Zhi, X.; et al. NSC 74859 enhances doxorubicin cytotoxicity via inhibition of epithelial-mesenchymal transition in hepatocellular carcinoma cells. Cancer Lett. 2012, 325, 207–213. [Google Scholar] [CrossRef]
- Lee, T.K.; Lau, T.C.; Ng, I.O. Doxorubicin-induced apoptosis and chemosensitivity in hepatoma cell lines. Cancer Chemother. Pharmacol. 2002, 49, 78–86. [Google Scholar] [CrossRef]
- Qi, W.W.; Yu, H.Y.; Guo, H.; Lou, J.; Wang, Z.M.; Liu, P.; Sapin-Minet, A.; Maincent, P.; Hong, X.C.; Hu, X.M.; et al. Doxorubicin-loaded glycyrrhetinic acid modified recombinant human serum albumin nanoparticles for targeting liver tumor chemotherapy. Mol. Pharm. 2015, 12, 675–683. [Google Scholar] [CrossRef]
- Bains, O.S.; Szeitz, A.; Lubieniecka, J.M.; Cragg, G.E.; Grigliatti, T.A.; Riggs, K.W.; Reid, R.E. A correlation between cytotoxicity and reductase-mediated metabolism in cell lines treated with doxorubicin and daunorubicin. J. Pharmacol. Exp. Ther. 2013, 347, 375–387. [Google Scholar] [CrossRef]
- Levasseur, L.M.; Slocum, H.K.; Rustum, Y.M.; Greco, W.R. Modeling of the time-dependency of in vitro drug cytotoxicity and resistance. Cancer Res. 1998, 58, 5749–5761. [Google Scholar]
- Cox, J.; Weinman, S. Mechanisms of doxorubicin resistance in hepatocellular carcinoma. Hepatic Oncol. 2016, 3, 57–59. [Google Scholar] [CrossRef]
- Buschauer, S.; Koch, A.; Wiggermann, P.; Muller, M.; Hellerbrand, C. Hepatocellular carcinoma cells surviving doxorubicin treatment exhibit increased migratory potential and resistance to doxorubicin re-treatment in vitro. Oncol. Lett. 2018, 15, 4635–4640. [Google Scholar] [CrossRef]
- Delgado, J.L.; Hsieh, C.M.; Chan, N.L.; Hiasa, H. Topoisomerases as anticancer targets. Biochem. J. 2018, 475, 373–398. [Google Scholar] [CrossRef]
- The UniProt Consortium. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar] [CrossRef]
- Baral, E.; Auer, G. In vitro effect of doxorubicin on non-proliferating and proliferating epithelial cells. Int. J. Radiat. Oncol. Biol. Phys. 1990, 19, 963–965. [Google Scholar] [CrossRef]
- Campiglio, M.; Somenzi, G.; Olgiati, C.; Beretta, G.; Balsari, A.; Zaffaroni, N.; Valagussa, P.; Menard, S. Role of proliferation in HER2 status predicted response to doxorubicin. Int. J. Cancer 2003, 105, 568–573. [Google Scholar] [CrossRef]
- Eom, Y.W.; Kim, M.A.; Park, S.S.; Goo, M.J.; Kwon, H.J.; Sohn, S.; Kim, W.H.; Yoon, G.; Choi, K.S. Two distinct modes of cell death induced by doxorubicin: Apoptosis and cell death through mitotic catastrophe accompanied by senescence-like phenotype. Oncogene 2005, 24, 4765–4777. [Google Scholar] [CrossRef]
- Wei, L.; Surma, M.; Gough, G.; Shi, S.; Lambert-Cheatham, N.; Chang, J.; Shi, J. Dissecting the Mechanisms of Doxorubicin and Oxidative Stress-Induced Cytotoxicity: The Involvement of Actin Cytoskeleton and ROCK1. PLoS ONE 2015, 10, e0131763. [Google Scholar] [CrossRef]
- Czeczuga-Semeniuk, E.; Wolczynski, S.; Dabrowska, M.; Dzieciol, J.; Anchim, T. The effect of doxorubicin and retinoids on proliferation, necrosis and apoptosis in MCF-7 breast cancer cells. Folia Histochem. Cytobiol. 2004, 42, 221–227. [Google Scholar]
- Zhou, W.; Dosey, T.L.; Biechele, T.; Moon, R.T.; Horwitz, M.S.; Ruohola-Baker, H. Assessment of hypoxia inducible factor levels in cancer cell lines upon hypoxic induction using a novel reporter construct. PLoS ONE 2011, 6, e27460. [Google Scholar] [CrossRef]
- Blancher, C.; Moore, J.W.; Talks, K.L.; Houlbrook, S.; Harris, A.L. Relationship of hypoxia-inducible factor (HIF)-1alpha and HIF-2alpha expression to vascular endothelial growth factor induction and hypoxia survival in human breast cancer cell lines. Cancer Res. 2000, 60, 7106–7113. [Google Scholar]
- Pastorekova, S.; Ratcliffe, P.J.; Pastorek, J. Molecular mechanisms of carbonic anhydrase IX-mediated pH regulation under hypoxia. BJU Int. 2008, 101 (Suppl. 4), 8–15. [Google Scholar] [CrossRef]
- Koch, S.; Mayer, F.; Honecker, F.; Schittenhelm, M.; Bokemeyer, C. Efficacy of cytotoxic agents used in the treatment of testicular germ cell tumours under normoxic and hypoxic conditions in vitro. Br. J. Cancer 2003, 89, 2133. [Google Scholar] [CrossRef]
- Li, J.-q.; Wu, X.; Gan, L.; Yang, X.-l.; Miao, Z.-h. Hypoxia induces universal but differential drug resistance and impairs anticancer mechanisms of 5-fluorouracil in hepatoma cells. Acta Pharmacol. Sin. 2017, 38, 1642. [Google Scholar] [CrossRef]
- Available online: https://www.nature.com/articles/aps201779#supplementary-information (accessed on 19 June 2019).
- Bowyer, C.; Lewis, A.L.; Lloyd, A.W.; Phillips, G.J.; Macfarlane, W.M. Hypoxia as a target for drug combination therapy of liver cancer. Anti-Cancer Drugs 2017, 28, 771–780. [Google Scholar] [CrossRef] [Green Version]
- Frederiksen, L.J.; Siemens, D.R.; Heaton, J.P.; Maxwell, L.R.; Adams, M.A.; Graham, C.H. Hypoxia induced resistance to doxorubicin in prostate cancer cells is inhibited by low concentrations of glyceryl trinitrate. J. Urol. 2003, 170, 1003–1007. [Google Scholar] [CrossRef]
- Strese, S.; Fryknäs, M.; Larsson, R.; Gullbo, J. Effects of hypoxia on human cancer cell line chemosensitivity. BMC Cancer 2013, 13, 331. [Google Scholar] [CrossRef]
- Mai, Y.; Yu, J.J.; Bartholdy, B.; Xu-Monette, Z.Y.; Knapp, E.E.; Yuan, F.; Chen, H.; Ding, B.B.; Yao, Z.; Das, B.; et al. An oxidative stress-based mechanism of doxorubicin cytotoxicity suggests new therapeutic strategies in ABC-DLBCL. Blood 2016, 128, 2797–2807. [Google Scholar] [CrossRef] [Green Version]
- Kotake-Nara, E.; Saida, K. Characterization of CoCl2-induced reactive oxygen species (ROS): Inductions of neurite outgrowth and endothelin-2/vasoactive intestinal contractor in PC12 cells by CoCl2 are ROS dependent, but those by MnCl2 are not. Neurosci. Lett. 2007, 422, 223–227. [Google Scholar] [CrossRef]
- He, Y.; Gan, X.; Zhang, L.; Liu, B.; Zhu, Z.; Li, T.; Zhu, J.; Chen, J.; Yu, H. CoCl2 induces apoptosis via a ROS-dependent pathway and Drp1-mediated mitochondria fission in periodontal ligament stem cells. Am. J. Physiol. Cell Physiol. 2018, 315, C389–C397. [Google Scholar] [CrossRef]
- Storz, P.; Doppler, H.; Ferran, C.; Grey, S.T.; Toker, A. Functional dichotomy of A20 in apoptotic and necrotic cell death. Biochem. J. 2005, 387, 47–55. [Google Scholar] [CrossRef] [Green Version]
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Eble, J.M.; Moon, E.; Yuan, H.; Weitzel, D.H.; Landon, C.D.; Nien, C.Y.; Hanna, G.; Rich, J.N.; Provenzale, J.M.; et al. Tumor cells upregulate normoxic HIF-1alpha in response to doxorubicin. Cancer Res. 2013, 73, 6230–6242. [Google Scholar] [CrossRef]
- Kelly, T.J.; Souza, A.L.; Clish, C.B.; Puigserver, P. A hypoxia-induced positive feedback loop promotes hypoxia-inducible factor 1alpha stability through miR-210 suppression of glycerol-3-phosphate dehydrogenase 1-like. Mol. Cell. Biol. 2011, 31, 2696–2706. [Google Scholar] [CrossRef]
- Blokhina, O.; Virolainen, E.; Fagerstedt, K.V. Antioxidants, oxidative damage and oxygen deprivation stress: A review. Ann. Bot. 2003, 91, 179–194. [Google Scholar] [CrossRef]
- Costantini, S.; Di Bernardo, G.; Cammarota, M.; Castello, G.; Colonna, G. Gene expression signature of human HepG2 cell line. Gene 2013, 518, 335–345. [Google Scholar] [CrossRef]
- Lopez-Terrada, D.; Cheung, S.W.; Finegold, M.J.; Knowles, B.B. Hep G2 is a hepatoblastoma-derived cell line. Hum. Pathol. 2009, 40, 1512–1515. [Google Scholar] [CrossRef]
- Capes-Davis, A.; Theodosopoulos, G.; Atkin, I.; Drexler, H.G.; Kohara, A.; MacLeod, R.A.; Masters, J.R.; Nakamura, Y.; Reid, Y.A.; Reddel, R.R.; et al. Check your cultures! A list of cross-contaminated or misidentified cell lines. Int. J. Cancer 2010, 127, 1–8. [Google Scholar] [CrossRef]
- Shiraga, K.; Sakaguchi, K.; Senoh, T.; Ohta, T.; Ogawa, S.; Sawayama, T.; Mouri, H.; Fujiwara, A.; Tsuji, T. Modulation of doxorubicin sensitivity by cyclosporine A in hepatocellular carcinoma cells and their doxorubicin-resistant sublines. J. Gastroenterol. Hepatol. 2001, 16, 460–466. [Google Scholar] [CrossRef]
- Chang, J.E.; Shim, W.S.; Yang, S.G.; Kwak, E.Y.; Chong, S.; Kim, D.D.; Chung, S.J.; Shim, C.K. Liver cancer targeting of Doxorubicin with reduced distribution to the heart using hematoporphyrin-modified albumin nanoparticles in rats. Pharm. Res. 2012, 29, 795–805. [Google Scholar] [CrossRef]
- Lou, B.; Fan, J.; Wang, K.; Chen, W.; Zhou, X.; Zhang, J.; Lin, S.; Lv, F.; Chen, Y. N1-guanyl-1,7-diaminoheptane (GC7) enhances the therapeutic efficacy of doxorubicin by inhibiting activation of eukaryotic translation initiation factor 5A2 (eIF5A2) and preventing the epithelial-mesenchymal transition in hepatocellular carcinoma cells. Exp. Cell Res. 2013, 319, 2708–2717. [Google Scholar] [CrossRef]
- Wu, D.; Yotnda, P. Induction and testing of hypoxia in cell culture. JoVE 2011. [Google Scholar] [CrossRef]
- Rampersad, S.N. Multiple applications of Alamar Blue as an indicator of metabolic function and cellular health in cell viability bioassays. Sensors 2012, 12, 12347–12360. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time | HepG2 | Huh7 | SNU449 |
|---|---|---|---|
| 6 h | 1.01 ± 0.045 | 1.01 ± 0.0055 * | 0.99 ± 0.022 |
| 24 h | 1.03 ± 0.14 | 1.04 ± 0.064 | 0.95 ± 0.077 * |
| 48 h | 0.90 ± 0.072 * | 1.06 ± 0.0091 * | 0.76 ± 0.082 * |
| 72 h | 0.81 ± 0.1 * | 0.99 ± 0.009 * | 1.02 ± 0.044 |
| Time | HepG2 | Huh7 | SNU449 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Normoxia | Hypoxia | Rhyp/norm | Normoxia | Hypoxia | Rhyp/norm | Normoxia | Hypoxia | Rhyp/norm | |
| 6 h | 310 ± 12 | 70 ± 11 * | 0.2 | 170 ± 3.3 a | 870 ± 15 *,a | 5.1 | 920 ± 72 a,b | 860 ± 32 a | 0.9 |
| 24 h | 1.3 ± 0.18c | 0.81 ± 0.087 | 0.6 | 5.2 ± 0.49 c | 12 ± 0.78 c | 2.4 | 160 ± 17 a,b,c | 240 ± 16 *,a,b,c | 1.5 |
| 48 h | 0.62 ± 0.06 c | 0.1 ± 0.0046 | 0.2 | 2 ± 0.18 a,c | 9.2 ± 0.58 c | 4.7 | 16 ± 0.73 c,d | 44 ± 3.7 c,d | 2.8 |
| 72 h | 0.19 ± 0.017 c | 0.099 ± 0.0089 | 0.5 | 0.34 ± 0.038 c | 2 ± 0.17 c | 5.8 | 110 ± 3.9 a,b,c,e | 110 ± 5.5 a,b,c,d,e | 1.0 |
| Protein | Biological Process † | HepG2 vs. Huh7 | HepG2 vs. SNU449 | Huh7 vs. SNU449 |
|---|---|---|---|---|
| 5′-nucleotidase (5′-NT; P21589) | Cell adhesion; Cellular metabolic process | ---- * | ---- * | -- * |
| A disintegrin and metalloproteinase with thrombospondin motifs 15 (ADAM-TS 15; Q8TE58) | - * | -- * | -- * | |
| Alpha-taxilin (TXLNA; P40222) | Cell proliferation; Cellular response to stress | --- * | --- * | - |
| Amphiregulin (AREG; P15514) | Cell differentiation; Cell proliferation; Cellular metabolic process | ---- * | --- * | ++ * |
| Annexin A1 (ANXA1; P04083) | Apoptotic process; Cell adhesion; Cell differentiation; Cell motility; Cell proliferation; Cellular metabolic process; Cellular response to stress; Chemotaxis; Immune response | --- * | --- * | + |
| Carbonic anhydrase 9 (CAIX; Q16790) | Cellular metabolic process; Cellular response to stress; Response to hypoxia | -- | -- * | + |
| Carboxypeptidase E (CPE; P16870) | Cellular metabolic process; Proteolysis | +++ * | NA | NA |
| Carcinoembryonic antigen-related cell adhesion molecule 1 (CEACAM1; P13688) | Angiogenesis; Cell adhesion; Cell differentiation; Cell motility; Cell proliferation; Cellular metabolic process; Immune response; MAPK cascade | --- * | - * | +++ * |
| Carcinoembryonic antigen-related cell adhesion molecule 5 (CEACAM5; P06731) | Apoptotic process; Cell adhesion; Cell differentiation | NA | NA | NA |
| Cathepsin L2 (CTSV; O60911) | Cell differentiation; Cell proliferation; Cellular metabolic process; Cellular response to stress; Extracellular matrix organization; Proteolysis | -- * | -- * | + |
| CD160 antigen (CD160; O95971) | Cell proliferation; Immune response; | ND | ND | ND |
| CD27 antigen (CD27; P26842) | Apoptotic process; Cell differentiation; Cell proliferation; Cellular metabolic process; Cellular response to stress; Immune response; MAPK cascade | ND | ND | ND |
| CD48 antigen (CD48; P09326) | Cell motility | + | - | - |
| CD70 antigen (CD70; P32970) | Apoptotic process; Cell proliferation; Immune response | NA | --- * | NA |
| Cornulin (CRNN; Q9UBG3) | Cell adhesion | + | - | - |
| C-type lectin domain family 4 member K (CD207; Q9UJ71) | NA | - | NA | |
| C-X-C motif chemokine 13 (CXCL13; O43927) | Angiogenesis; Cell adhesion; Cell motility; Cell proliferation; Chemotaxis; Immune response | - | - | + |
| Cyclin-dependent kinase inhibitor 1 (DKN1A; P38936) | Apoptotic process; Cell differentiation; Cell proliferation; Cellular metabolic process; Cellular response to stress | -- * | -- * | + |
| Delta-like protein 1 (DLL1; O00548) | Angiogenesis; Apoptotic process; Cell adhesion; Cell differentiation; Cell proliferation; Cellular metabolic process; Immune response | NA | NA | - |
| Disintegrin and metalloproteinase domain-containing protein 8 (ADAM 8; P78325) | Angiogenesis; Apoptotic process; Cell adhesion; Cell differentiation; Cell motility; Cellular metabolic process; Cellular response to stress; Chemotaxis; Extracellular matrix organization; Immune response; MAPK cascade; Proteolysis; Response to hypoxia | ND | ND | ND |
| Endothelial cell-specific molecule 1 (ESM-1; Q9NQ30) | Angiogenesis; Cell proliferation | NA | -- | NA |
| Ephrin type-A receptor 2 (EPHA2; P29317) | Angiogenesis; Apoptotic process; Cell adhesion; Cell differentiation; Cell motility; Cell proliferation; Cellular metabolic process; Cellular response to stress; Chemotaxis; MAPK cascade | --- * | --- * | + * |
| FAS-associated death domain protein (FADD; Q13158) | Apoptotic process; Cell adhesion; Cell differentiation; Cell motility; Cell proliferation; Cellular metabolic process; Immune response; Proteolysis | NA | NA | + * |
| Fc receptor-like B (FCRLB; Q6BAA4) | Immune response | + | NA | NA |
| Fibroblast growth factor-binding protein 1 (FGF-BP1; Q14512) | Cell proliferation; Cellular response to stress; | ++ * | -- * | -- * |
| Folate receptor alpha (FR-alpha; P15328) | Cell differentiation; Cell motility; Cellular metabolic process; Cellular response to stress | --- * | -- * | ++ * |
| Folate receptor gamma (FR-gamma; P41439) | NA | NA | NA | |
| Furin (FURIN; P09958) | Cell motility; Cell proliferation; Cellular metabolic process; Extracellular matrix organization; Proteolysis | --- * | + | +++ * |
| Galectin-1 (Gal-1; P09382) | Apoptotic process; Cell adhesion; Cell differentiation; Immune response | ++ * | -- * | -- * |
| Glypican-1 (GPC1; P35052) | Cell differentiation; Cellular metabolic process; Chemotaxis | NA | NA | NA |
| Granzyme B (GZMB; P10144) | Apoptotic process; Immune response; Proteolysis | - | NA | NA |
| Granzyme H (GZMH; P20718) | Apoptotic process; Immune response; Proteolysis | ND | ND | ND |
| Hepatocyte growth factor (HGF; P14210) | Angiogenesis; Apoptotic process; Cell differentiation; Cell motility; Cell proliferation; Cellular metabolic process; Cellular response to stress; Chemotaxis; MAPK cascade; Proteolysis | - | NA | NA |
| ICOS ligand (ICOSLG; O75144) | Cell adhesion; Cell proliferation; Immune response | NA | - | NA |
| Insulin-like growth factor 1 receptor (IGF1R; P08069) | Apoptotic process; Cell motility; Cell proliferation; Cellular metabolic process; Cellular response to stress; Immune response; MAPK cascade | --- | --- | -- * |
| Integrin alpha-V (ITGAV; P06756) | Angiogenesis; Apoptotic process; Cell adhesion; Cell differentiation; Cell motility; Cell proliferation; Cellular metabolic process; Chemotaxis; Extracellular matrix organization; MAPK cascade | --- * | --- * | + * |
| Integrin beta-5 (ITGB5; P18084) | Cell adhesion; Cell differentiation; Extracellular matrix organization | --- * | --- * | - * |
| Interferon gamma receptor 1 (IFN-gamma-R1; P15260) | Immune response | NA | NA | ++ * |
| Interleukin-6 (IL6; P05231) | Angiogenesis; Apoptotic process; Cell adhesion; Cell differentiation; Cell motility; Cell proliferation; Cellular metabolic process; Cellular response to stress; Chemotaxis; Immune response; MAPK cascade; Proteolysis | NA | NA | --- * |
| Kallikrein-11 (hK11; Q9UBX7) | ++ * | + | -- * | |
| Kallikrein-13 (KLK13; Q9UKR3) | Proteolysis | + | + | - |
| Kallikrein-14 (hK14; Q9P0G3) | Proteolysis | + | + * | + |
| Kallikrein-8 (hK8; O60259) | Cell differentiation; Cell proliferation; Cellular response to stress | ++ * | + * | - |
| Kit ligand (SCF; P21583) | Apoptotic process; Cell adhesion; Cell differentiation; Cell motility; Cell proliferation; Cellular metabolic process; MAPK cascade | -- * | -- * | ++ * |
| Ly6/PLAUR domain-containing protein 3 (LYPD3; O95274) | Cell adhesion | NA | NA | NA |
| Melanoma-derived growth regulatory protein (MIA; Q16674) | Cell proliferation | + | - | - |
| Mesothelin (MSLN; Q13421) | Cell adhesion | ND | ND | ND |
| Methionine aminopeptidase 2 (MetAP 2; P50579) | Cellular metabolic process; Proteolysis | NA | NA | + |
| MHC class I polypeptide-related sequence A and B (MIC-A/B; Q29983,Q29980) | Cell adhesion; Cellular response to stress; Immune response | NA | NA | --- * |
| Midkine (MK; P21741) | Apoptotic process; Cell differentiation; Cell motility; Cellular metabolic process | NA | NA | ++ * |
| Mothers against decapentaplegic homolog 5 (MAD homolog 5; Q99717) | Cell differentiation; Cellular metabolic process | NA | NA | + |
| Mucin-16 (MUC-16; Q8WXI7) | Cell adhesion; Cellular metabolic process; | NA | NA | + |
| Nectin-4 (PVRL4; Q96NY8) | Cell adhesion | ND | ND | ND |
| Pancreatic prohormone (PPY; P01298) | Cellular response to stress | ND | ND | ND |
| Podocalyxin (PODXL; O00592) | Cell adhesion; Cell differentiation; Cell motility | NA | + | NA |
| Pro-epidermal growth factor (EGF; P01133) | Angiogenesis; Cell motility; Cell proliferation; Cellular metabolic process; MAPK cascade; Proteolysis | -- * | - | ++ * |
| Protein CYR61 (CYR61; O00622) | Angiogenesis; Apoptotic process; Cell adhesion; Cell differentiation; Cell motility; Cell proliferation; Cellular metabolic process; Chemotaxis; Extracellular matrix organization; MAPK cascade; Proteolysis | -- * | -- * | -- * |
| Protein S100-A11 (S100A11; P31949) | Cell proliferation; Cellular metabolic process | --- * | --- * | + |
| Protein S100-A4 (S100A4; P26447) | Cell differentiation | ++ * | +++ * | + |
| Proto-oncogene tyrosine-protein kinase receptor Ret (RET; P07949) | Apoptotic process; Cell adhesion; Cell differentiation; Cell motility; Cellular metabolic process; MAPK cascade; Proteolysis | NA | NA | NA |
| Protransforming growth factor alpha (TGF-alpha; P01135) | Cell proliferation; Cellular metabolic process; MAPK cascade | +++ * | +++ * | + |
| Receptor tyrosine-protein kinase erbB-2 (ERBB2; P04626) | Angiogenesis | --- * | --- * | + * |
| Receptor tyrosine-protein kinase erbB-3 (ERBB3; P21860) | Apoptotic process; Cell adhesion; Cell differentiation; Cell motility; Cell proliferation; Cellular metabolic process; MAPK cascade | --- | -- | ++ * |
| Receptor tyrosine-protein kinase erbB-4 (ERBB4; Q15303) | Apoptotic process; Cell differentiation; Cell motility; Cell proliferation; Cellular metabolic process; MAPK cascade | + | -- * | -- * |
| R-spondin-3 (RSPO3; Q9BXY4) | Angiogenesis | ++ * | ++ | - |
| Secretory carrier-associated membrane protein 3 (Secretory carrier membrane protein 3) (SCAMP3; O14828) | --- * | --- * | - | |
| Seizure 6-like protein (SEZ6L; Q9BYH1) | ND | ND | ND | |
| SPARC (SPARC; P09486) | Angiogenesis; Cell motility; Cell proliferation; Extracellular matrix organization; | -- * | -- * | - |
| Syndecan-1 (SYND1; P18827) | Cell differentiation; Cell motility; Cellular metabolic process | NA | NA | ++ * |
| T-cell leukemia/lymphoma protein 1A (TCL1A; P56279) | ND | ND | ND | |
| TGF-beta receptor type-2 (TGFR-2; P37173) | Angiogenesis; Apoptotic process; Cell adhesion; Cell differentiation; Cell motility; Cell proliferation; Cellular metabolic process | -- * | -- * | + * |
| Tissue factor pathway inhibitor 2 (TFPI-2; P48307) | ---- * | ---- * | -- * | |
| T-lymphocyte surface antigen Ly-9 (LY9; Q9HBG7) | Cell adhesion; Cell differentiation; Immune response | + | - | - |
| Toll-like receptor 3 (TLR3; O15455) | Apoptotic process; Cell differentiation; Cellular metabolic process; Cellular response to stress; Immune response; MAPK cascade | -- * | --- * | -- * |
| Transmembrane glycoprotein NMB (GPNMB; Q14956) | Cell adhesion; Cell differentiation; Immune response | + | + | - |
| Tumor necrosis factor ligand superfamily member 10 (TRAIL; P50591) | Apoptotic process; Cellular metabolic process; Immune response; Proteolysis | -- * | -- * | -- * |
| Tumor necrosis factor ligand superfamily member 13 (TNFSF13; O75888) | Cell proliferation; Cellular metabolic process | ND | ND | ND |
| Tumor necrosis factor ligand superfamily member 6 (FASLG; P48023) | Angiogenesis; Apoptotic process; Cell differentiation; Cell proliferation; Cellular metabolic process; Immune response; Proteolysis | ND | ND | ND |
| Tumor necrosis factor receptor superfamily member 19 (TNFRSF19; Q9NS68) | Apoptotic process; Cellular metabolic process; Cellular response to stress; MAPK cascade | -- * | - * | ++ * |
| Tumor necrosis factor receptor superfamily member 4 (TNFRSF4; P43489) | Apoptotic process; Cell adhesion; Cell proliferation; Cellular metabolic process; Immune response; MAPK cascade | NA | + | NA |
| Tumor necrosis factor receptor superfamily member 6B (TNFRSF6B; O95407) | Apoptotic process; Cell proliferation; Cellular metabolic process; Immune response; MAPK cascade | NA | NA | NA |
| Tyrosine-protein kinase ABL1 (ABL1; P00519) | Apoptotic process; Cell adhesion; Cell differentiation; Cell motility; Cell proliferation; Cellular metabolic process; Cellular response to stress; Extracellular matrix organization; Immune response; MAPK cascade | ---- | ---- | - |
| Tyrosine-protein kinase Lyn (LYN; P07948) | Apoptotic process; Cell adhesion; Cell differentiation; Cell motility; Cell proliferation; Cellular metabolic process; Cellular response to stress; Chemotaxis; Immune response; MAPK cascade | -- * | -- * | - |
| Vascular endothelial growth factor A (VEGFA; P15692) | Angiogenesis; Apoptotic process; Cell adhesion; Cell differentiation; Cell motility; Cell proliferation; Cellular metabolic process; Cellular response to stress; Chemotaxis; MAPK cascade; Proteolysis; Response to hypoxia | --- * | ---- * | -- * |
| Vascular endothelial growth factor receptor 2 (VEGFR-2; P35968) | Angiogenesis; Apoptotic process; Cell adhesion; Cell differentiation; Cell motility; Cell proliferation; Cellular metabolic process; Cellular response to stress; Chemotaxis; Extracellular matrix organization; MAPK cascade | ND | ND | ND |
| Vascular endothelial growth factor receptor 3 (VEGFR-3; P35916) | Angiogenesis; Apoptotic process; Cell motility; Cell proliferation; Cellular metabolic process; Cellular response to stress; MAPK cascade | ND | ND | ND |
| VEGF coregulated chemokine 1 (CXL17; Q6UXB2) | Angiogenesis; Cell differentiation; Chemotaxis | + | NA | NA |
| Vimentin (VIM; P08670) | Cell differentiation | -- * | -- * | + * |
| WAP four-disulfide core domain protein 2 (WFDC2; Q14508) | Proteolysis | + | + | - |
| Wnt inhibitory factor 1 (WIF-1; Q9Y5W5) | Cell differentiation | -- * | NA | NA |
| WNT1-inducible-signaling pathway protein 1 (WISP-1; O95388) | Cell adhesion | NA | + | NA |
| Xaa-Pro aminopeptidase 2 (XPNPEP2; O43895) | + | - | - |
| Cell Lines | Experimental Conditions | |||||
| Incubation | 24 h | 24 h | 36 h | 48 h | 72 h | 48 h |
| Reference | [33] | [67] | [35] | [34] | [68] | [69] |
| Cell line | IC50 (µM ± St. dev) | |||||
| HepaRG | 1.73 ± 0.38 | - | - | - | - | |
| HepG2 | 2.33 ± 0.05 | - | 3.85 ± 0.59 | 1.91 (1.52–2.4) | 0.029 ± 0.002 | 0.288 (0.25–0.32) |
| HepG2.2.15 | 3.15 ± 0.92 | - | - | - | - | |
| HLE | - | - | - | - | 0.67 ± 0.06 | |
| HLF | - | - | - | - | 0.76 ± 0.04 | |
| HT-17 | - | - | - | - | 6.0 ± 1.9 | |
| Huh-7 | 2.55 ± 0.10 | - | 0.47 ± 0.9 | 3.38 (2.57–4.46) | 0.37 ± 0.01 | 0.36 (0.29–0.42) |
| Li-7 | - | - | - | - | 0.46 ± 0.01 | |
| PLC/DOR | - | 48.63 ± 17.04 | - | - | - | |
| PLC/PRF/5 | - | 0.93 ± 0.29 | - | - | - | |
| PLC/PRF/6 | - | - | - | - | 1.2 ± 0.04 | |
| sk-Hep1 | - | - | - | - | 0.031 ± 0.002 | |
| SNU449 | - | - | - | 24.86 (17.97–34.40) | - | 1.30 (0.84–0.90) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dubbelboer, I.R.; Pavlovic, N.; Heindryckx, F.; Sjögren, E.; Lennernäs, H. Liver Cancer Cell Lines Treated with Doxorubicin under Normoxia and Hypoxia: Cell Viability and Oncologic Protein Profile. Cancers 2019, 11, 1024. https://doi.org/10.3390/cancers11071024
Dubbelboer IR, Pavlovic N, Heindryckx F, Sjögren E, Lennernäs H. Liver Cancer Cell Lines Treated with Doxorubicin under Normoxia and Hypoxia: Cell Viability and Oncologic Protein Profile. Cancers. 2019; 11(7):1024. https://doi.org/10.3390/cancers11071024
Chicago/Turabian StyleDubbelboer, Ilse R., Natasa Pavlovic, Femke Heindryckx, Erik Sjögren, and Hans Lennernäs. 2019. "Liver Cancer Cell Lines Treated with Doxorubicin under Normoxia and Hypoxia: Cell Viability and Oncologic Protein Profile" Cancers 11, no. 7: 1024. https://doi.org/10.3390/cancers11071024