
Antitumor Effects of Ursolic Acid through Mediating the Inhibition of STAT3/PD-L1 Signaling in Non-Small Cell Lung Cancer Cells


Abstract
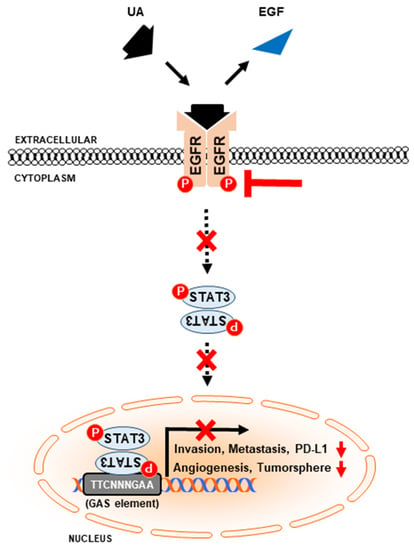
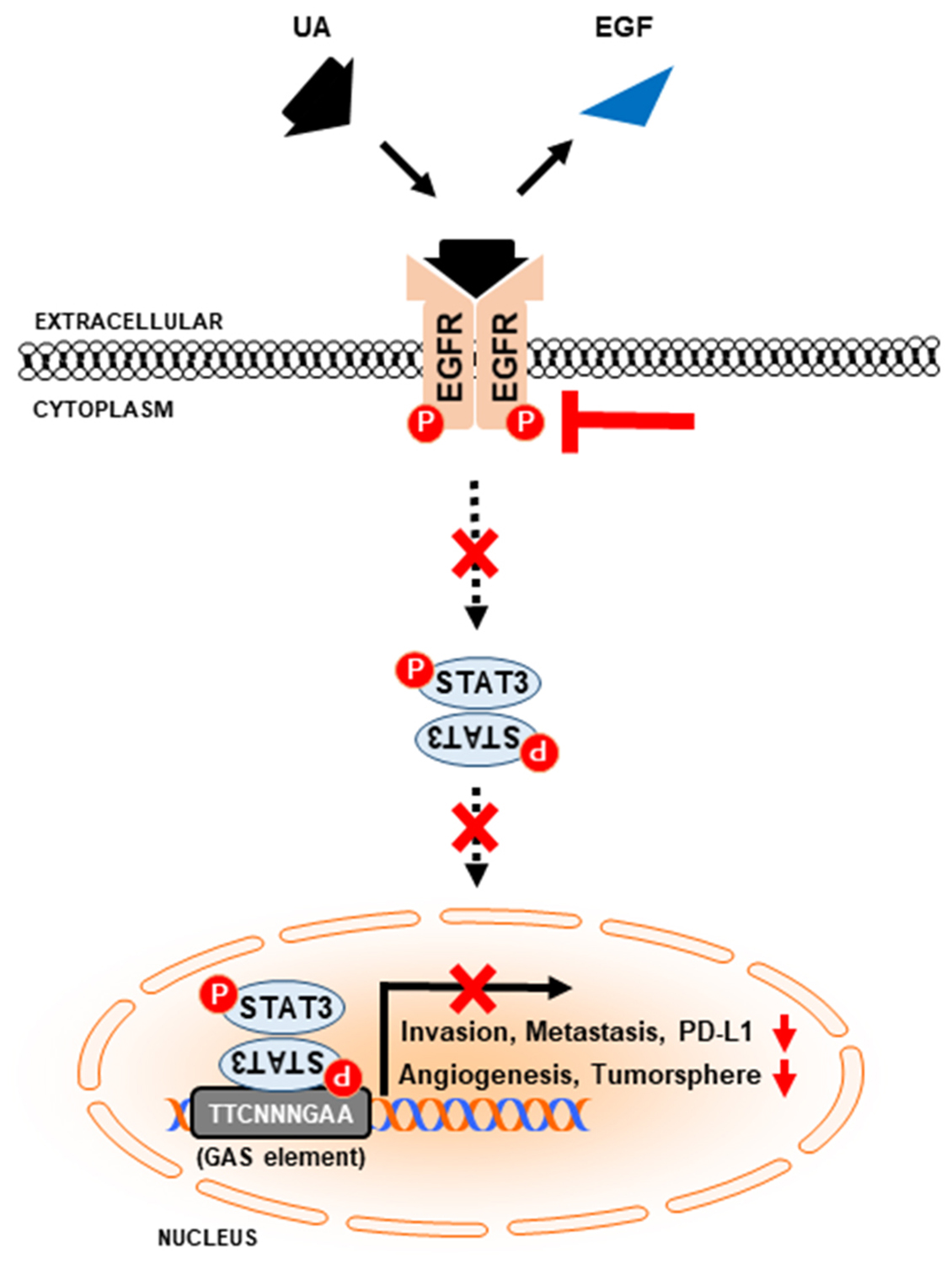
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Antibodies and Cell Culture Reagents
2.2. Cell Culture and Treatment
2.3. Cell Proliferation Inhibition
2.4. DAPI Staining and Morphological Analysis
2.5. Western Blotting
2.6. Reverse Transcriptase–Quantitative Polymerase Chain Reaction (RT-qPCR)
2.7. Cell Cycle Analysis
2.8. Apoptosis Analysis
2.9. In Vitro Angiogenesis Assay
2.10. Matrigel Invasion Assay
2.11. Wound Healing Assay
2.12. Tumorsphere Formation Assay
2.13. siRNA Transfection
2.14. Molecular Docking
2.15. Chromatin Immunoprecipitation (ChIP) Assay
2.16. Statistical Analyses
3. Results
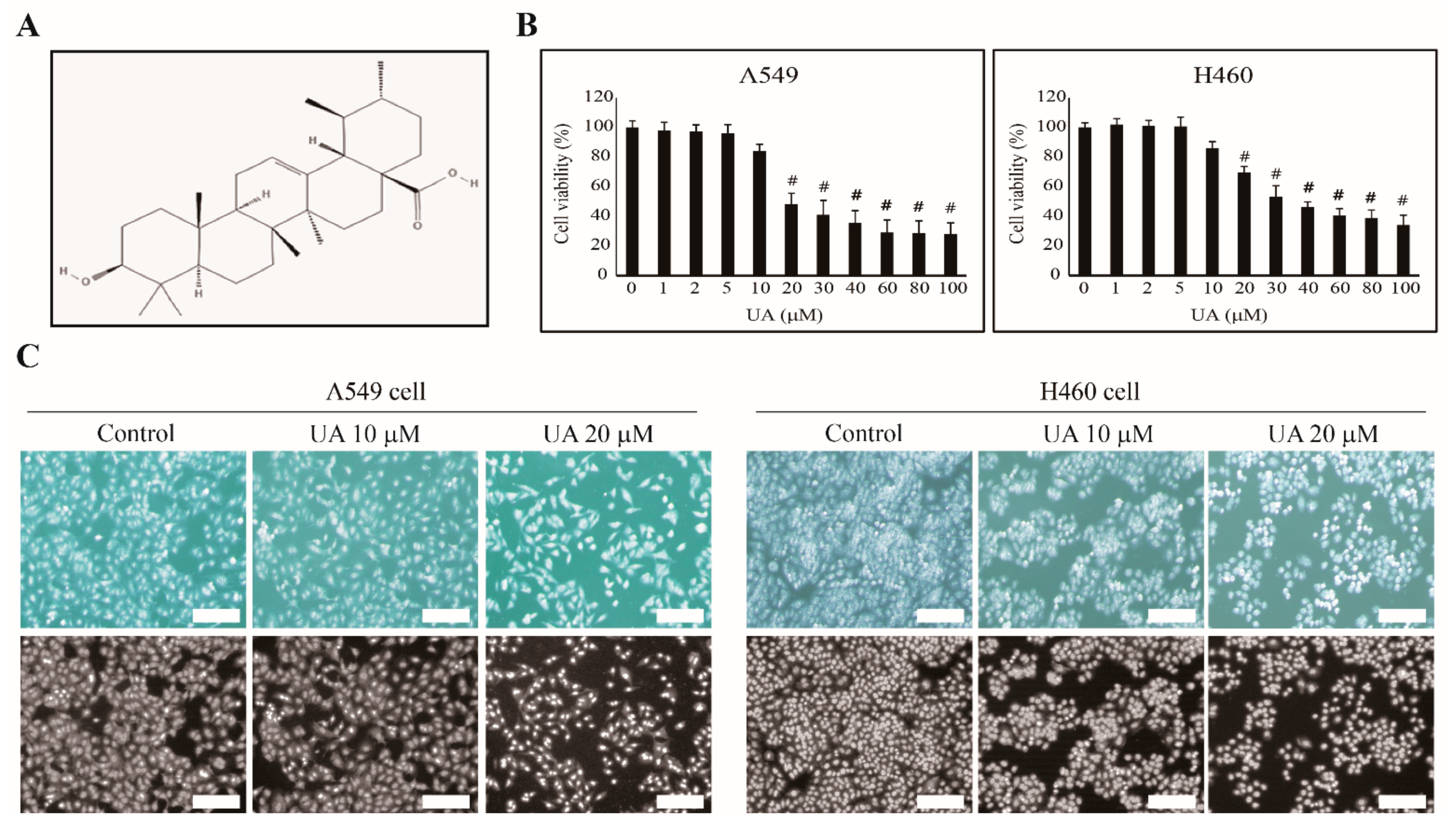
3.1. UA Inhibits NSCLC Cell Proliferation in a Concentration-Dependent Manner
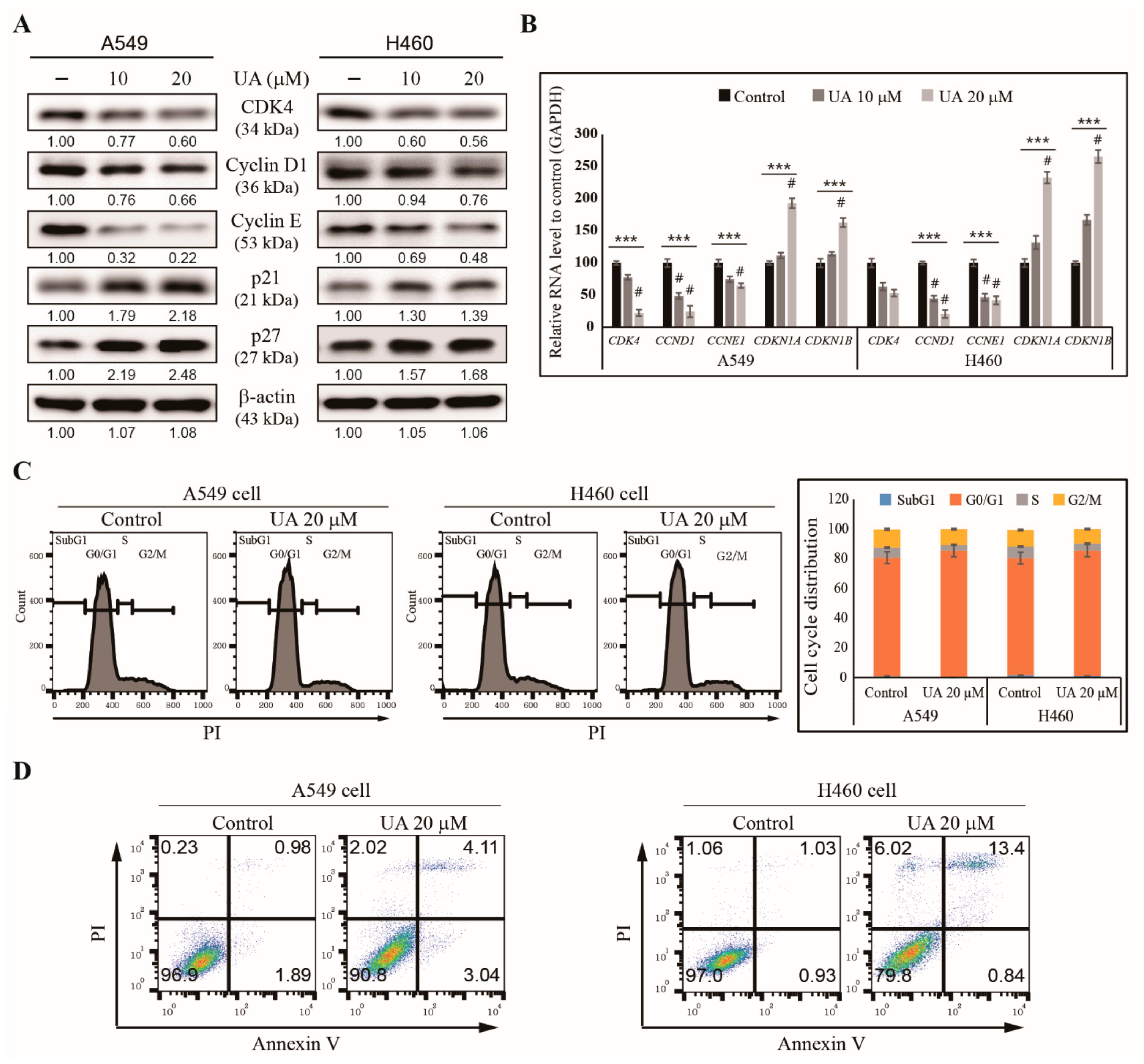
3.2. UA Induces Cell Cycle Arrest and Apoptosis in NSCLC Cells
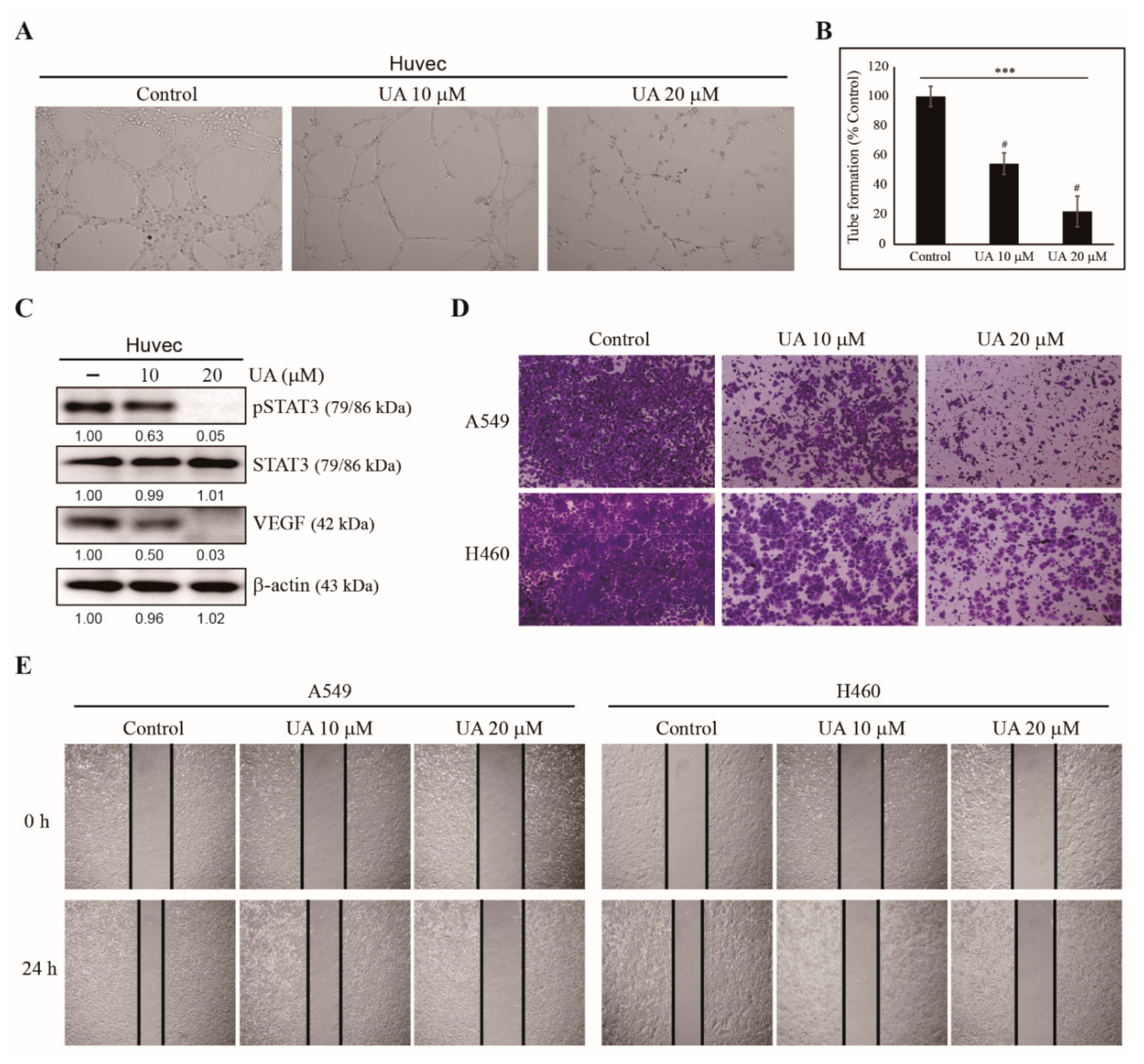
3.3. UA Inhibits Angiogenesis, Invasion, and Migration of NSCLC Cells
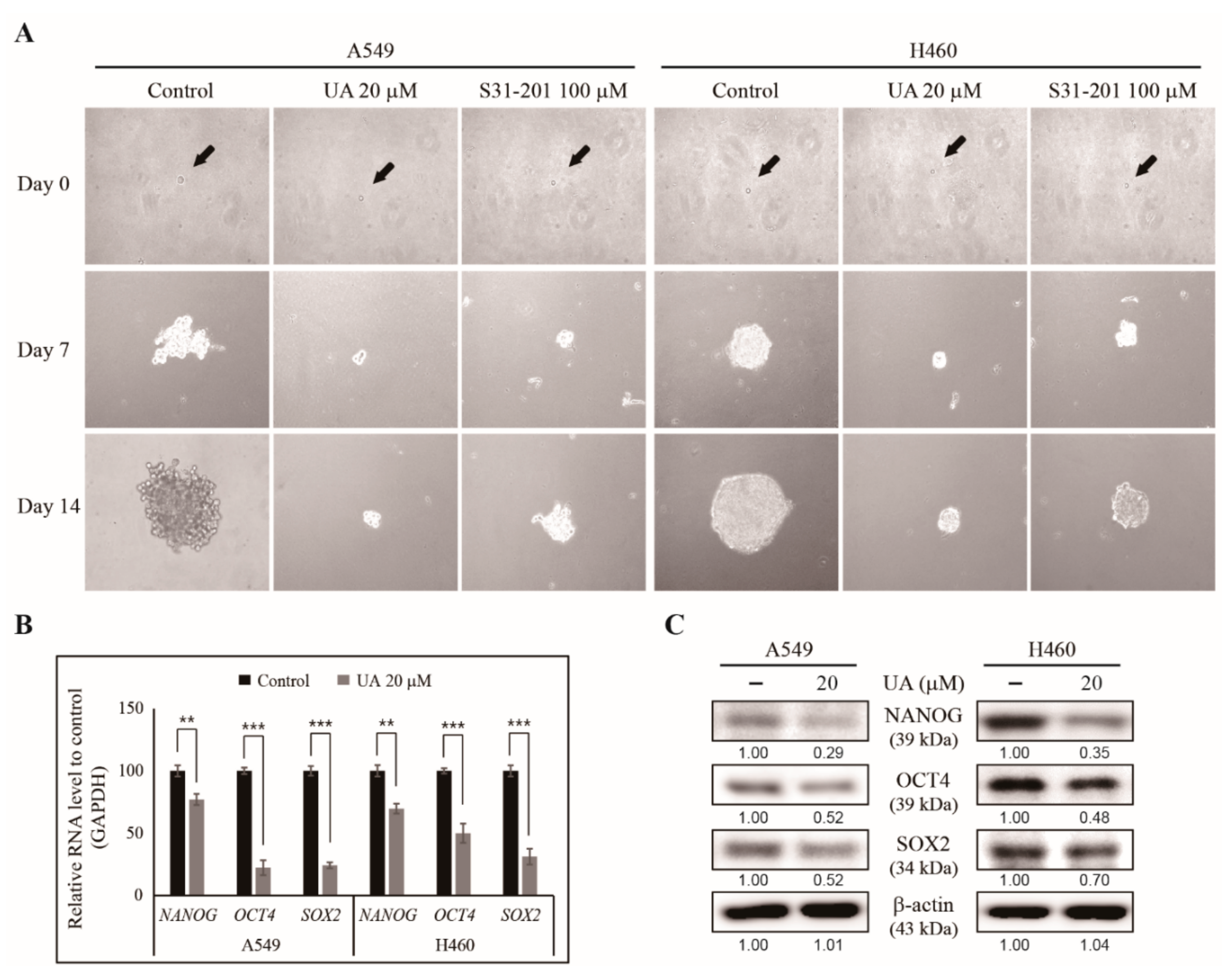
3.4. UA Suppresses Cancer Stemness by Inhibiting Tumorsphere Formation in NSCLC Cells
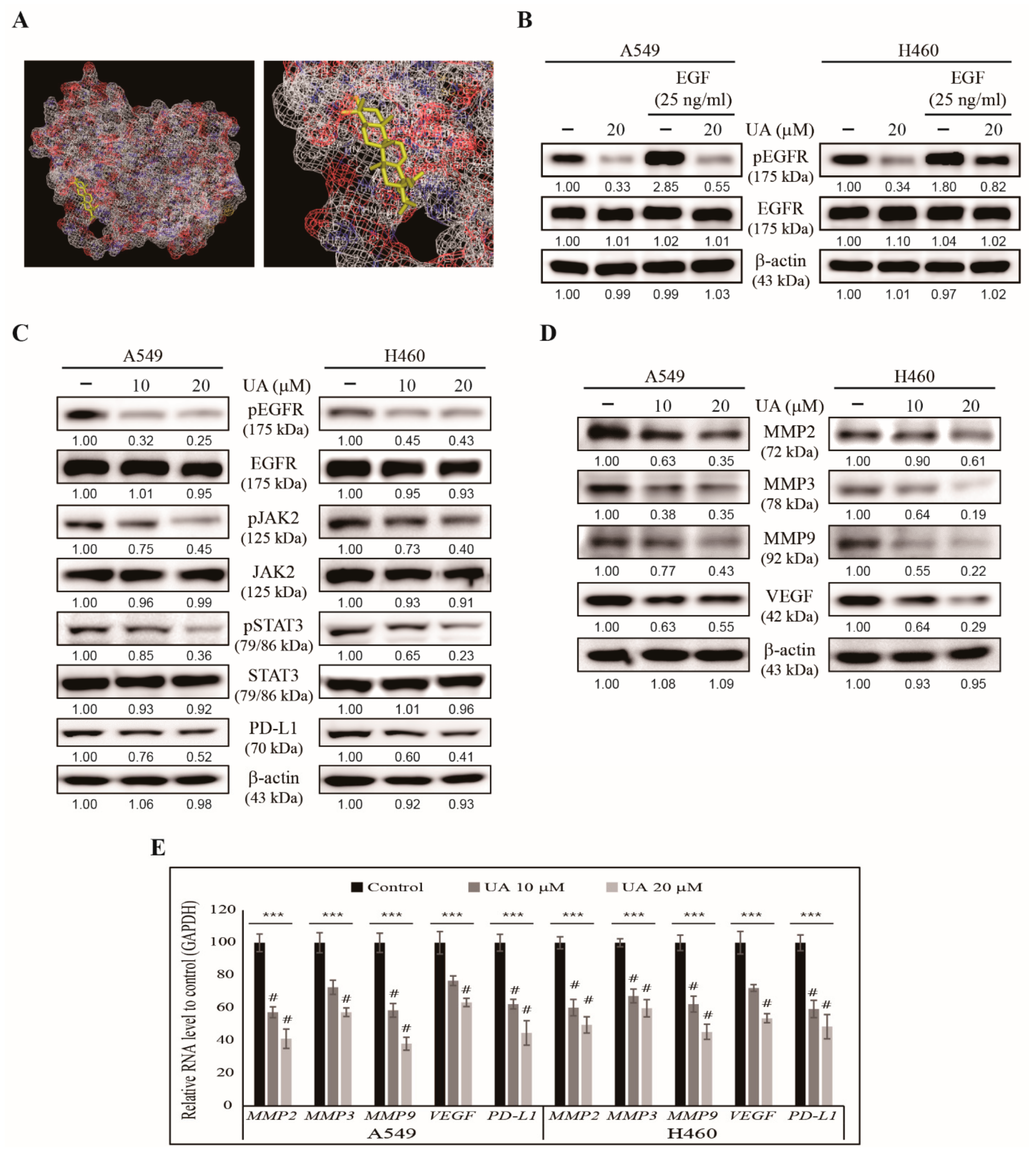
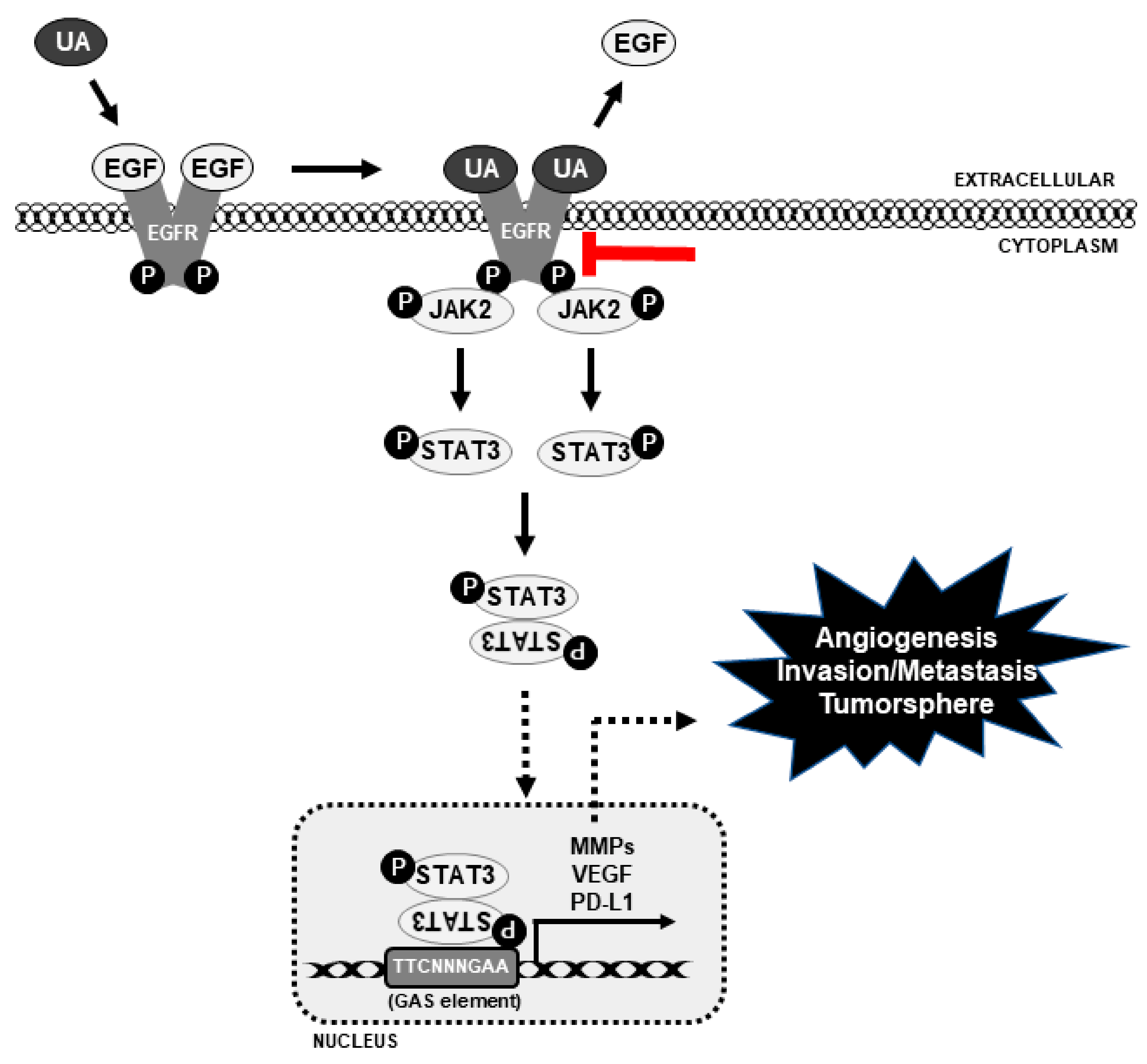
3.5. UA Inhibits PD-L1 Expression Through the EGFR/JAK2/STAT3 Pathway in NSCLC Cells
3.6. UA Downregulates the Binding of STAT3 to MMP2 and PD-L1 Promoters
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Molina, J.R.; Yang, P.; Cassivi, S.D.; Schild, S.E.; Adjei, A.A. Non-small cell lung cancer: Epidemiology, risk factors, treatment, and survivorship. Mayo Clin. Proc. 2008, 83, 584–594. [Google Scholar] [CrossRef]
- Mendelsohn, J.; Baselga, J. Status of epidermal growth factor receptor antagonists in the biology and treatment of cancer. J. Clin. Oncol. 2003, 21, 2787–2799. [Google Scholar] [CrossRef] [PubMed]
- Thatcher, N.; Chang, A.; Parikh, P.; Rodrigues Pereira, J.; Ciuleanu, T.; von Pawel, J.; Thongprasert, S.; Tan, E.H.; Pemberton, K.; Archer, V.; et al. Gefitinib plus best supportive care in previously treated patients with refractory advanced non-small-cell lung cancer: Results from a randomised, placebo-controlled, multicentre study (Iressa Survival Evaluation in Lung Cancer). Lancet 2005, 366, 1527–1537. [Google Scholar] [CrossRef]
- Chatterjee, S.; Heukamp, L.C.; Siobal, M.; Schottle, J.; Wieczorek, C.; Peifer, M.; Frasca, D.; Koker, M.; Konig, K.; Meder, L.; et al. Tumor VEGF:VEGFR2 autocrine feed-forward loop triggers angiogenesis in lung cancer. J. Clin. Investig. 2013, 123, 1732–1740. [Google Scholar] [CrossRef] [Green Version]
- Deng, C.; Zhang, P.; Harper, J.W.; Elledge, S.J.; Leder, P. Mice lacking p21CIP1/WAF1 undergo normal development, but are defective in G1 checkpoint control. Cell 1995, 82, 675–684. [Google Scholar] [CrossRef] [Green Version]
- Abbas, T.; Dutta, A. p21 in cancer: Intricate networks and multiple activities. Nat. Rev. Cancer 2009, 9, 400–414. [Google Scholar] [CrossRef]
- Musgrove, E.A.; Hunter, L.J.; Lee, C.S.; Swarbrick, A.; Hui, R.; Sutherland, R.L. Cyclin D1 overexpression induces progestin resistance in T-47D breast cancer cells despite p27(Kip1) association with cyclin E-Cdk2. J. Biol. Chem. 2001, 276, 47675–47683. [Google Scholar] [CrossRef] [Green Version]
- Pfeffer, C.M.; Singh, A.T.K. Apoptosis: A target for anticancer therapy. Int. J. Mol. Sci. 2018, 19, 448. [Google Scholar] [CrossRef] [Green Version]
- Sp, N.; Kang, D.Y.; Kim, D.H.; Yoo, J.S.; Jo, E.S.; Rugamba, A.; Jang, K.J.; Yang, Y.M. Tannic acid inhibits non-small cell lung cancer (NSCLC) stemness by inducing G0/G1 cell cycle arrest and intrinsic apoptosis. Anticancer Res. 2020, 40, 3209–3220. [Google Scholar] [CrossRef]
- Sp, N.; Kang, D.Y.; Kim, D.H.; Park, J.H.; Lee, H.G.; Kim, H.J.; Darvin, P.; Park, Y.M.; Yang, Y.M. Nobiletin inhibits CD36-dependent tumor angiogenesis, migration, invasion, and sphere formation through the Cd36/Stat3/Nf-Kb signaling axis. Nutrients 2018, 10, 772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sp, N.; Kang, D.Y.; Joung, Y.H.; Park, J.H.; Kim, W.S.; Lee, H.K.; Song, K.D.; Park, Y.M.; Yang, Y.M. Nobiletin inhibits angiogenesis by regulating Src/FAK/STAT3-mediated signaling through PXN in ER(+) breast cancer cells. Int. J. Mol. Sci. 2017, 18, 935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Folkman, J. Tumor angiogenesis: Therapeutic implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar] [CrossRef]
- Parangi, S.; O’Reilly, M.; Christofori, G.; Holmgren, L.; Grosfeld, J.; Folkman, J.; Hanahan, D. Antiangiogenic therapy of transgenic mice impairs de novo tumor growth. Proc. Natl. Acad. Sci. USA 1996, 93, 2002–2007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishida, N.; Yano, H.; Nishida, T.; Kamura, T.; Kojiro, M. Angiogenesis in cancer. Vasc. Health Risk Manag. 2006, 2, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Nelson, A.R.; Fingleton, B.; Rothenberg, M.L.; Matrisian, L.M. Matrix metalloproteinases: Biologic activity and clinical implications. J. Clin. Oncol. 2000, 18, 1135–1149. [Google Scholar] [CrossRef]
- P, N.S.; Darvin, P.; Yoo, Y.B.; Joung, Y.H.; Kang, D.Y.; Kim, D.N.; Hwang, T.S.; Kim, S.Y.; Kim, W.S.; Lee, H.K.; et al. The combination of methylsulfonylmethane and tamoxifen inhibits the Jak2/STAT5b pathway and synergistically inhibits tumor growth and metastasis in ER-positive breast cancer xenografts. BMC Cancer 2015, 15, 474. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Shi, P.; Zhao, G.; Xu, J.; Peng, W.; Zhang, J.; Zhang, G.; Wang, X.; Dong, Z.; Chen, F.; et al. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct. Target. Ther. 2020, 5, 8. [Google Scholar] [CrossRef] [Green Version]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.L.; Sun, Y.F.; Wang, B.L.; Shen, M.N.; Zhou, Y.; Chen, J.W.; Hu, B.; Gong, Z.J.; Zhang, X.; Cao, Y.; et al. Sphere-forming culture enriches liver cancer stem cells and reveals Stearoyl-CoA desaturase 1 as a potential therapeutic target. BMC Cancer 2019, 19, 760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larzabal, L.; El-Nikhely, N.; Redrado, M.; Seeger, W.; Savai, R.; Calvo, A. Differential effects of drugs targeting cancer stem cell (CSC) and non-CSC populations on lung primary tumors and metastasis. PLoS ONE 2013, 8, e79798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez-Casal, R.; Bhattacharya, C.; Ganesh, N.; Bailey, L.; Basse, P.; Gibson, M.; Epperly, M.; Levina, V. Non-small cell lung cancer cells survived ionizing radiation treatment display cancer stem cell and epithelial-mesenchymal transition phenotypes. Mol. Cancer 2013, 12, 94. [Google Scholar] [CrossRef] [Green Version]
- Yakisich, J.S.; Azad, N.; Venkatadri, R.; Kulkarni, Y.; Wright, C.; Kaushik, V.; Iyer, A.K. Formation of tumorspheres with increased stemness without external mitogens in a lung cancer model. Stem Cells Int. 2016, 2016, 5603135. [Google Scholar] [CrossRef] [Green Version]
- Marotta, L.L.; Almendro, V.; Marusyk, A.; Shipitsin, M.; Schemme, J.; Walker, S.R.; Bloushtain-Qimron, N.; Kim, J.J.; Choudhury, S.A.; Maruyama, R.; et al. The JAK2/STAT3 signaling pathway is required for growth of CD44(+)CD24(-) stem cell-like breast cancer cells in human tumors. J. Clin. Investig. 2011, 121, 2723–2735. [Google Scholar] [CrossRef] [PubMed]
- Jin, W. Role of JAK/STAT3 signaling in the regulation of metastasis, the transition of cancer stem cells, and chemoresistance of cancer by epithelial-mesenchymal transition. Cells 2020, 9, 217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeda, S.; Okamoto, T.; Okano, S.; Umemoto, Y.; Tagawa, T.; Morodomi, Y.; Kohno, M.; Shimamatsu, S.; Kitahara, H.; Suzuki, Y.; et al. PD-L1 is upregulated by simultaneous amplification of the PD-L1 and JAK2 genes in non-small cell lung cancer. J. Thorac. Oncol. 2016, 11, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Zeng, Y.; Du, W.; Zhu, J.; Shen, D.; Liu, Z.; Huang, J.A. The EGFR pathway is involved in the regulation of PD-L1 expression via the IL-6/JAK/STAT3 signaling pathway in EGFR-mutated non-small cell lung cancer. Int. J. Oncol. 2016, 49, 1360–1368. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Diez, I.; Hernandez-Ruiz, E.; Andrades, E.; Gimeno, J.; Ferrandiz-Pulido, C.; Yebenes, M.; Garcia-Patos, V.; Pujol, R.M.; Hernandez-Munoz, I.; Toll, A. PD-L1 expression is increased in metastasizing squamous cell carcinomas and their metastases. Am. J. Dermatopathol. 2018, 40, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.B.; Yao, H.; Li, C.S.; Liang, L.X.; Zhang, Y.; Chen, Y.X.; Fang, J.Y.; Xu, J. Rise of PD-L1 expression during metastasis of colorectal cancer: Implications for immunotherapy. J. Dig. Dis. 2017, 18, 574–581. [Google Scholar] [CrossRef]
- Wei, X.L.; Luo, X.; Sheng, H.; Wang, Y.; Chen, D.L.; Li, J.N.; Wang, F.H.; Xu, R.H. PD-L1 expression in liver metastasis: Its clinical significance and discordance with primary tumor in colorectal cancer. J. Transl. Med. 2020, 18, 475. [Google Scholar] [CrossRef]
- Feng, X.M.; Su, X.L. Anticancer effect of ursolic acid via mitochondria-dependent pathways. Oncol. Lett. 2019, 17, 4761–4767. [Google Scholar] [CrossRef]
- Kashyap, D.; Tuli, H.S.; Sharma, A.K. Ursolic acid (UA): A metabolite with promising therapeutic potential. Life Sci. 2016, 146, 201–213. [Google Scholar] [CrossRef]
- Wei, Z.Y.; Chi, K.Q.; Wang, K.S.; Wu, J.; Liu, L.P.; Piao, H.R. Design, synthesis, evaluation, and molecular docking of ursolic acid derivatives containing a nitrogen heterocycle as anti-inflammatory agents. Bioorganic Med. Chem. Lett. 2018, 28, 1797–1803. [Google Scholar] [CrossRef] [PubMed]
- Yin, R.; Li, T.; Tian, J.X.; Xi, P.; Liu, R.H. Ursolic acid, a potential anticancer compound for breast cancer therapy. Crit. Rev. Food Sci. Nutr. 2018, 58, 568–574. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, F.; Yang, L.; Mei, Y.; Long, H.; Zhang, X.; Zhang, J.; Qimuge, S.; Su, X. Ursolic acid inhibits proliferation and induces apoptosis of cancer cells in vitro and in vivo. J. Biomed. Biotechnol. 2011, 2011, 419343. [Google Scholar] [CrossRef] [PubMed]
- Leng, S.; Hao, Y.; Du, D.; Xie, S.; Hong, L.; Gu, H.; Zhu, X.; Zhang, J.; Fan, D.; Kung, H.F. Ursolic acid promotes cancer cell death by inducing Atg5-dependent autophagy. Int. J. Cancer 2013, 133, 2781–2790. [Google Scholar] [CrossRef]
- Ruan, J.S.; Zhou, H.; Yang, L.; Wang, L.; Jiang, Z.S.; Sun, H.; Wang, S.M. Ursolic acid attenuates TGF-beta1-induced epithelial-mesenchymal transition in NSCLC by targeting integrin alphaVbeta5/MMPs signaling. Oncol. Res. 2019, 27, 593–600. [Google Scholar] [CrossRef]
- Wang, M.; Yu, H.; Wu, R.; Chen, Z.Y.; Hu, Q.; Zhang, Y.F.; Gao, S.H.; Zhou, G.B. Autophagy inhibition enhances the inhibitory effects of ursolic acid on lung cancer cells. Int. J. Mol. Med. 2020, 46, 1816–1826. [Google Scholar] [CrossRef] [PubMed]
- Sp, N.; Kang, D.Y.; Jo, E.S.; Rugamba, A.; Kim, W.S.; Park, Y.M.; Hwang, D.Y.; Yoo, J.S.; Liu, Q.; Jang, K.J.; et al. Tannic acid promotes TRAIL-induced extrinsic apoptosis by regulating mitochondrial ROS in human embryonic carcinoma cells. Cells 2020, 9, 282. [Google Scholar] [CrossRef] [Green Version]
- Nippin, S.P.; Kang, D.Y.; Kim, B.J.; Joung, Y.H.; Darvin, P.; Byun, H.J.; Kim, J.G.; Park, J.U.; Yang, Y.M. Methylsulfonylmethane induces G1 arrest and mitochondrial apoptosis in YD-38 gingival cancer cells. Anticancer Res. 2017, 37, 1637–1646. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Zhang, T.; Du, J. Ursolic acid sensitizes cisplatin-resistant HepG2/DDP cells to cisplatin via inhibiting Nrf2/ARE pathway. Drug Des. Dev. Ther. 2016, 10, 3471–3481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, M.Y.; Leung, H.W.; Yang, W.H.; Chen, W.H.; Lee, H.Z. Up-regulation of matrix metalloproteinase family gene involvement in ursolic acid-induced human lung non-small carcinoma cell apoptosis. Anticancer Res. 2007, 27, 145–153. [Google Scholar] [PubMed]
- Liu, T.; Ma, H.; Shi, W.; Duan, J.; Wang, Y.; Zhang, C.; Li, C.; Lin, J.; Li, S.; Lv, J.; et al. Inhibition of STAT3 signaling pathway by ursolic acid suppresses growth of hepatocellular carcinoma. Int. J. Oncol. 2017, 51, 555–562. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Meng, X.; Dong, Y. Ursolic acid nanoparticles inhibit cervical cancer growth in vitro and in vivo via apoptosis induction. Int. J. Oncol. 2017, 50, 1330–1340. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Wang, W.; Qian, L.; Zhang, Q.; Lai, D.; Qi, C. Ursolic acid inhibits the proliferation of human ovarian cancer stem-like cells through epithelial-mesenchymal transition. Oncol. Rep. 2015, 34, 2375–2384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, H.; Pi, J.; Yang, F.; Jiang, J.; Wang, X.; Bai, H.; Shao, M.; Huang, L.; Zhu, H.; Yang, P.; et al. Folate-chitosan nanoparticles loaded with ursolic acid confer anti-breast cancer activities in vitro and in vivo. Sci. Rep. 2016, 6, 30782. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.Y.; Sp, N.; Jo, E.S.; Rugamba, A.; Hong, D.Y.; Lee, H.G.; Yoo, J.S.; Liu, Q.; Jang, K.J.; Yang, Y.M. The inhibitory mechanisms of tumor PD-L1 expression by natural bioactive gallic acid in non-small-cell lung cancer (NSCLC) cells. Cancers 2020, 12, 727. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.H.; Sp, N.; Kang, D.Y.; Jo, E.S.; Rugamba, A.; Jang, K.J.; Yang, Y.M. Effect of methylsulfonylmethane on proliferation and apoptosis of A549 lung cancer cells through G2/M cell-cycle arrest and intrinsic cell death pathway. Anticancer Res. 2020, 40, 1905–1913. [Google Scholar] [CrossRef]
- Krock, B.L.; Skuli, N.; Simon, M.C. Hypoxia-induced angiogenesis: Good and evil. Genes Cancer 2011, 2, 1117–1133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Zijl, F.; Krupitza, G.; Mikulits, W. Initial steps of metastasis: Cell invasion and endothelial transmigration. Mutat. Res. 2011, 728, 23–34. [Google Scholar] [CrossRef]
- Niu, G.; Wright, K.L.; Huang, M.; Song, L.; Haura, E.; Turkson, J.; Zhang, S.; Wang, T.; Sinibaldi, D.; Coppola, D.; et al. Constitutive Stat3 activity up-regulates VEGF expression and tumor angiogenesis. Oncogene 2002, 21, 2000–2008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phi, L.T.H.; Sari, I.N.; Yang, Y.G.; Lee, S.H.; Jun, N.; Kim, K.S.; Lee, Y.K.; Kwon, H.Y. Cancer stem cells (CSCs) in drug resistance and their therapeutic implications in cancer treatment. Stem Cells Int. 2018, 2018, 5416923. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.H.; Yu, C.C.; Wang, B.Y.; Chang, W.W. Tumorsphere as an effective in vitro platform for screening anti-cancer stem cell drugs. Oncotarget 2016, 7, 1215–1226. [Google Scholar] [CrossRef] [Green Version]
- Amini, S.; Fathi, F.; Mobalegi, J.; Sofimajidpour, H.; Ghadimi, T. The expressions of stem cell markers: Oct4, Nanog, Sox2, nucleostemin, Bmi, Zfx, Tcl1, Tbx3, Dppa4, and Esrrb in bladder, colon, and prostate cancer, and certain cancer cell lines. Anat. Cell Biol. 2014, 47, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bethune, G.; Bethune, D.; Ridgway, N.; Xu, Z. Epidermal growth factor receptor (EGFR) in lung cancer: An overview and update. J. Thorac. Dis. 2010, 2, 48–51. [Google Scholar] [PubMed]
- Colomiere, M.; Ward, A.C.; Riley, C.; Trenerry, M.K.; Cameron-Smith, D.; Findlay, J.; Ackland, L.; Ahmed, N. Cross talk of signals between EGFR and IL-6R through JAK2/STAT3 mediate epithelial-mesenchymal transition in ovarian carcinomas. Br. J. Cancer 2009, 100, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.H.; Murphy, D.A.; Lassoued, W.; Thurston, G.; Feldman, M.D.; Lee, W.M. Activated STAT3 is a mediator and biomarker of VEGF endothelial activation. Cancer Biol. Ther. 2008, 7, 1994–2003. [Google Scholar] [CrossRef] [Green Version]
- Byun, H.J.; Darvin, P.; Kang, D.Y.; Sp, N.; Joung, Y.H.; Park, J.H.; Kim, S.J.; Yang, Y.M. Silibinin downregulates MMP2 expression via Jak2/STAT3 pathway and inhibits the migration and invasive potential in MDA-MB-231 cells. Oncol. Rep. 2017, 37, 3270–3278. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, D.Y.; Sp, N.; Lee, J.-M.; Jang, K.-J. Antitumor Effects of Ursolic Acid through Mediating the Inhibition of STAT3/PD-L1 Signaling in Non-Small Cell Lung Cancer Cells. Biomedicines 2021, 9, 297. https://doi.org/10.3390/biomedicines9030297
Kang DY, Sp N, Lee J-M, Jang K-J. Antitumor Effects of Ursolic Acid through Mediating the Inhibition of STAT3/PD-L1 Signaling in Non-Small Cell Lung Cancer Cells. Biomedicines. 2021; 9(3):297. https://doi.org/10.3390/biomedicines9030297
Chicago/Turabian StyleKang, Dong Young, Nipin Sp, Jin-Moo Lee, and Kyoung-Jin Jang. 2021. "Antitumor Effects of Ursolic Acid through Mediating the Inhibition of STAT3/PD-L1 Signaling in Non-Small Cell Lung Cancer Cells" Biomedicines 9, no. 3: 297. https://doi.org/10.3390/biomedicines9030297