Metabolic Profiling of Female Tg2576 Mouse Brains Provides Novel Evidence Supporting Intranasal Low-Dose Pioglitazone for Long-Term Treatment at an Early Stage of Alzheimer’s Disease


Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Preparation of PIO-Loaded PLGA-TPGS Nanoparticles
2.3. Inhalation Exposure to Aerosol Generated by Nebulization of PIO-Loaded PLGA-TPGS Nanoparticles
2.3.1. Animals
2.3.2. Aerosol Generation System for Mouse Inhalation
2.3.3. In-Vivo Comparative Study of Drug Distribution Profiles
2.4. Cell Culture
2.5. Analysis of In-Vitro Mitochondrial Respiration
2.6. Effect of Long-Term Inhalation Exposure to PIO-Loaded PLGA-TPGS Nanoparticles
2.6.1. Animal Husbandry and Long-Term Treatment
2.6.2. Preparation of PIO-Containing PB Pellets
2.6.3. Acclimation to PB Pellets and Inhalation Exposure
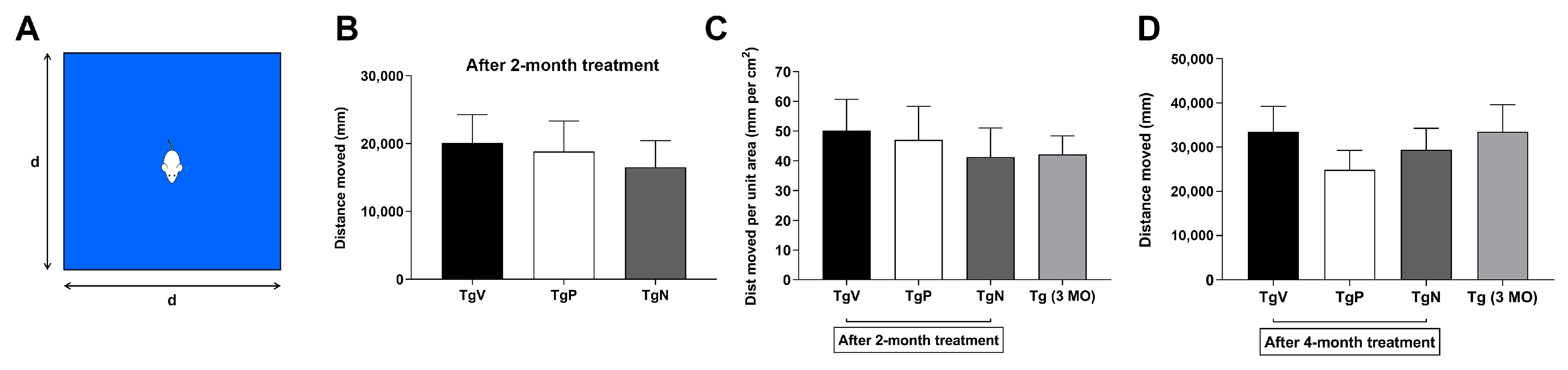
2.6.4. Spontaneous Activity in Open Field Test after 2-Month Treatment
2.6.5. Open Field Test at the End of the 4-Month Treatment
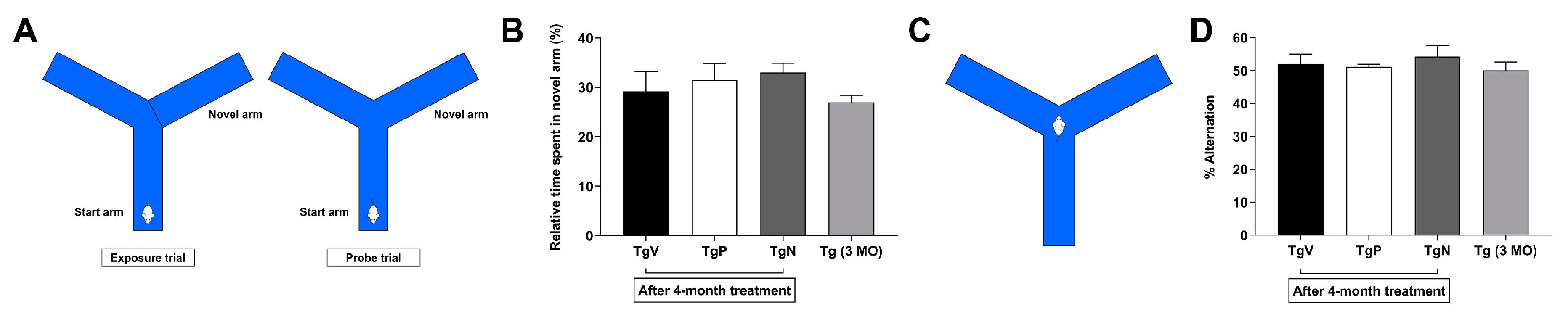
2.6.6. Forced Alternation in Y-Maze
2.6.7. Spontaneous Alternation in Y-Maze
2.6.8. Statistical Analyses for Behavioral Study
2.7. 1H-NMR-Based NMR Metabolic Profiling
2.7.1. Sample Collection
2.7.2. Extraction and Sample Preparation for NMR
2.7.3. Data Acquisition and Spectral Processing
2.7.4. Multivariate Data Analysis of Metabolic Data Matrix
3. Results
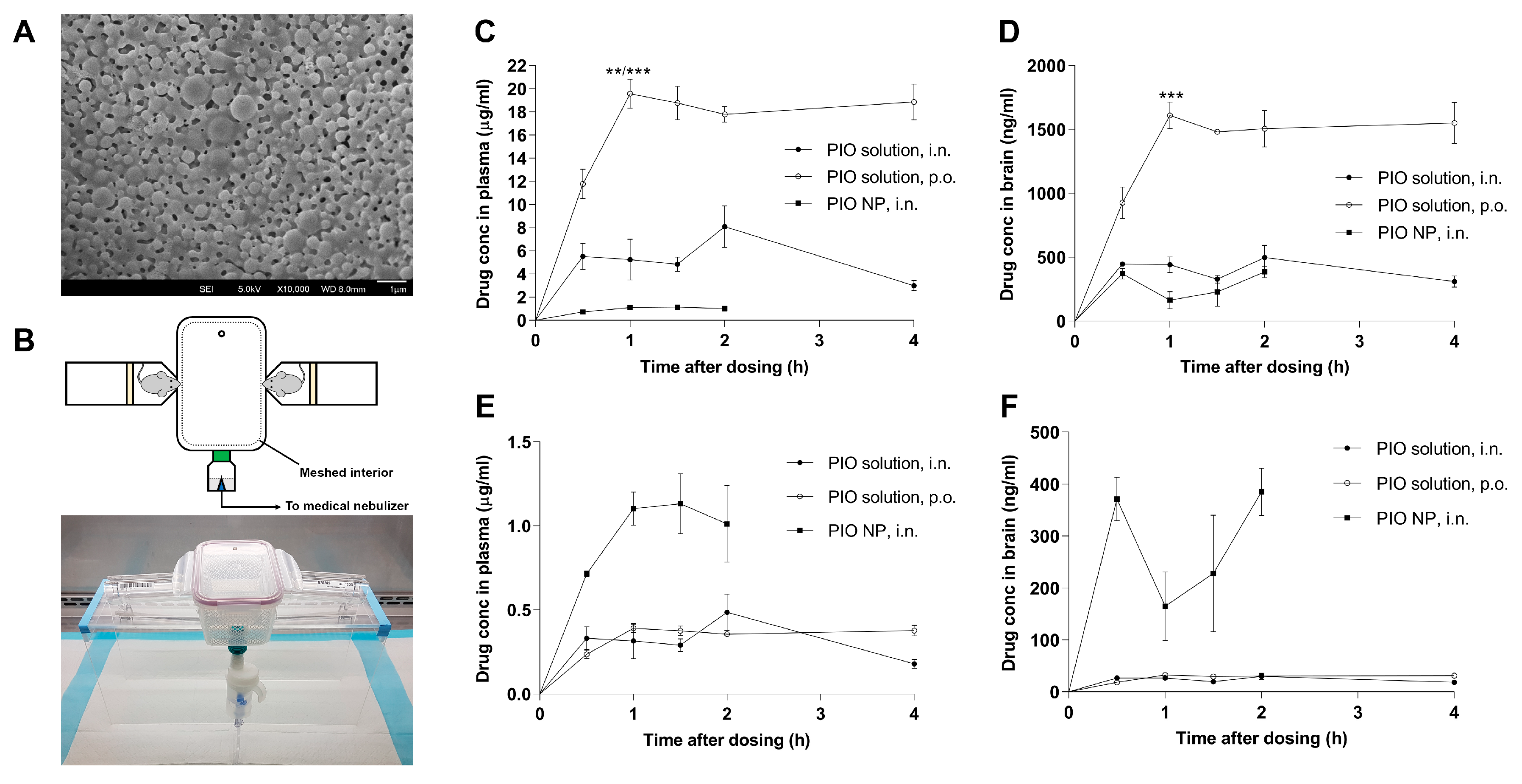
3.1. PIO-Loaded PLGA-TPGS Nanoparticles have Spherical Shape and are Stable When Resuspended
3.2. Inhalation Exposure to PIO-Loaded PLGA-TPGS Nanoparticles Revealed Comparatively Lower Systemic Exposure than Simple Nasal Instillation or Oral Administration of the PIO Solution
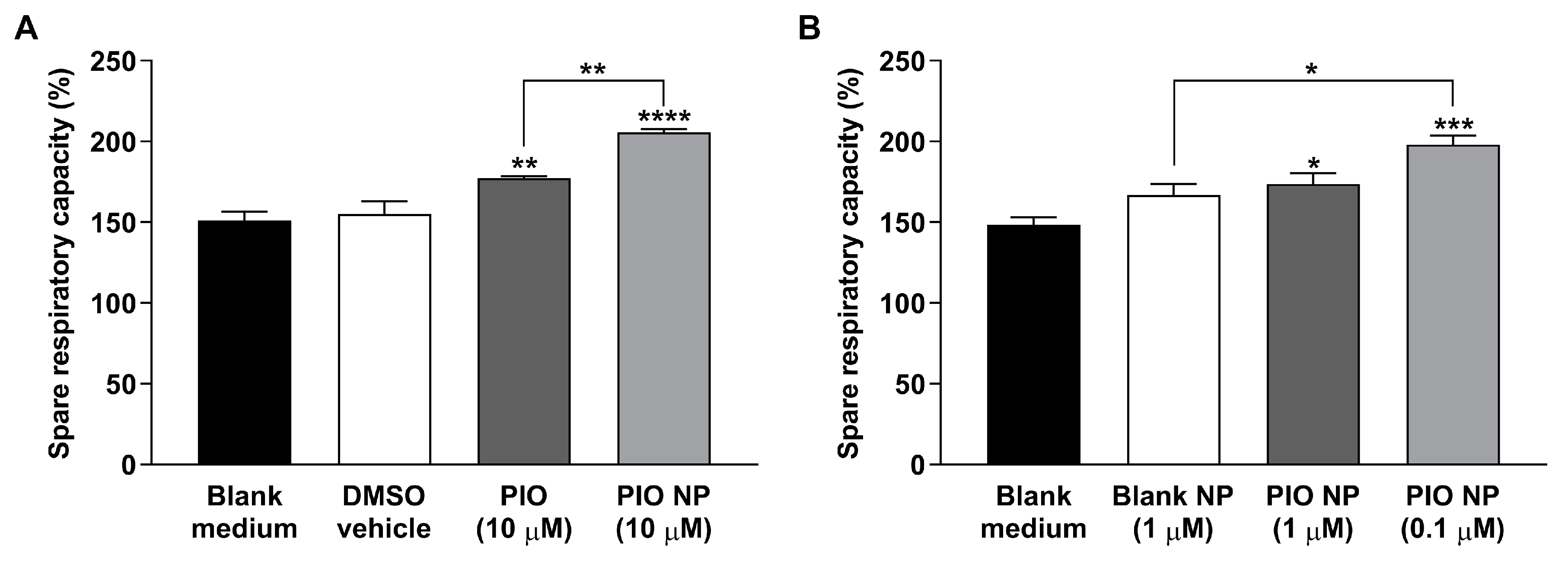
3.3. CHO-APP695 Cells Treated with PIO-Loaded PLGA-TPGS Nanoparticles Show Augmented Mitochondrial Spare Respiratory Capacity
3.4. Long-Term Oral PIO and PIO NP Inhalation Treatments Attenuate Hyperactivity in AD Mice
3.5. Long-Term Oral PIO and PIO NP Inhalation Treatments have No Obvious Effect on Spontaneous Alternation Behavior in AD Mice
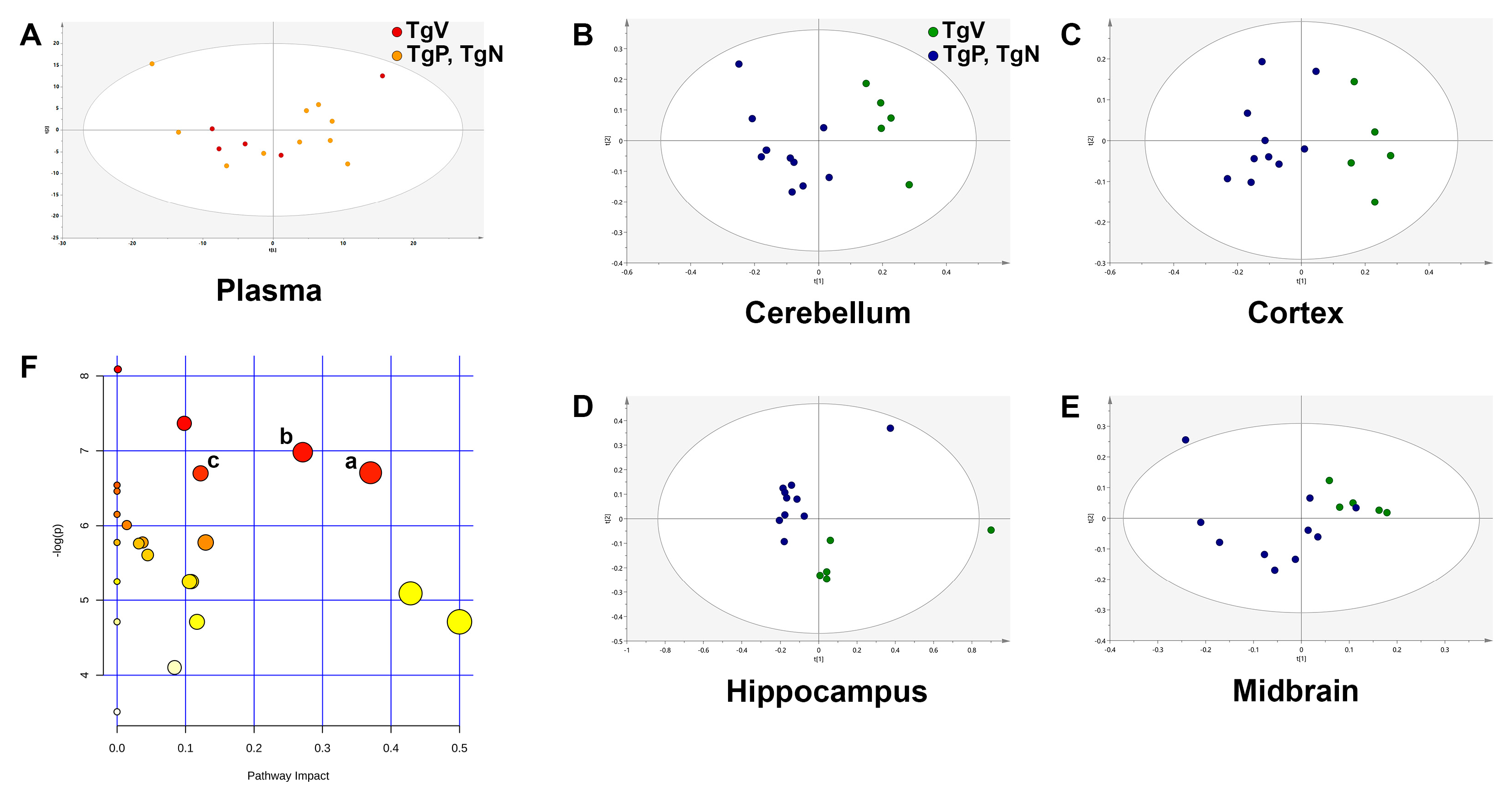
3.6. Metabolic Profiling Revealed Differences in Amino Acid Metabolism in Cortex Region after Long-Term Oral PIO and PIO NP Inhalation Treatments
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Masters, C.L.; Bateman, R.; Blennow, K.; Rowe, C.C.; Sperling, R.A.; Cummings, J.L. Alzheimer’s disease. Nat. Rev. Dis. Primer 2015, 1, 15056. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.-P.; Xie, Y.; Meng, X.-Y.; Kang, J.-S. History and progress of hypotheses and clinical trials for Alzheimer’s disease. Signal Transduct. Target. Ther. 2019, 4, 29. [Google Scholar] [CrossRef] [PubMed]
- Makin, S. The amyloid hypothesis on trial. Nature 2018, 559, S4–S7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuehn, B.M. In Alzheimer Research, Glucose Metabolism Moves to Center Stage. JAMA 2020, 323, 297–299. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhong, C. Decoding Alzheimer’s disease from perturbed cerebral glucose metabolism: implications for diagnostic and therapeutic strategies. Prog. Neurobiol. 2013, 108, 21–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kellar, D.; Craft, S. Brain insulin resistance in Alzheimer’s disease and related disorders: mechanisms and therapeutic approaches. Lancet Neurol. 2020, 19, 758–766. [Google Scholar] [CrossRef]
- Cummings, J.; Lee, G.; Ritter, A.; Sabbagh, M.; Zhong, K. Alzheimer’s disease drug development pipeline: 2020. Alzheimers Dement. N. Y. N 2020, 6, e12050. [Google Scholar] [CrossRef]
- Lebovitz, H.E. Thiazolidinediones: the Forgotten Diabetes Medications. Curr. Diab. Rep. 2019, 19, 151. [Google Scholar] [CrossRef] [Green Version]
- Nicolakakis, N.; Aboulkassim, T.; Ongali, B.; Lecrux, C.; Fernandes, P.; Rosa-Neto, P.; Tong, X.-K.; Hamel, E. Complete rescue of cerebrovascular function in aged Alzheimer’s disease transgenic mice by antioxidants and pioglitazone, a peroxisome proliferator-activated receptor gamma agonist. J. Neurosci. Off. J. Soc. Neurosci. 2008, 28, 9287–9296. [Google Scholar] [CrossRef]
- Papadopoulos, P.; Rosa-Neto, P.; Rochford, J.; Hamel, E. Pioglitazone improves reversal learning and exerts mixed cerebrovascular effects in a mouse model of Alzheimer’s disease with combined amyloid-β and cerebrovascular pathology. PLoS ONE 2013, 8, e68612. [Google Scholar] [CrossRef]
- Prakash, A.; Kumar, A. Role of nuclear receptor on regulation of BDNF and neuroinflammation in hippocampus of β-amyloid animal model of Alzheimer’s disease. Neurotox. Res. 2014, 25, 335–347. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.L.; Wong, L.R.; Pee, H.N.; Yang, S.; Ho, P.C.-L. Reverting Metabolic Dysfunction in Cortex and Cerebellum of APP/PS1 Mice, a Model for Alzheimer’s Disease by Pioglitazone, a Peroxisome Proliferator-Activated Receptor Gamma (PPARγ) Agonist. Mol. Neurobiol. 2019, 56, 7267–7283. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Hanyu, H.; Hirao, K.; Kanetaka, H.; Sakurai, H.; Iwamoto, T. Efficacy of PPAR-γ agonist pioglitazone in mild Alzheimer disease. Neurobiol. Aging 2011, 32, 1626–1633. [Google Scholar] [CrossRef] [PubMed]
- Geldmacher, D.S.; Fritsch, T.; McClendon, M.J.; Landreth, G. A randomized pilot clinical trial of the safety of pioglitazone in treatment of patients with Alzheimer disease. Arch. Neurol. 2011, 68, 45–50. [Google Scholar] [CrossRef]
- Schneider, L.S.; Mangialasche, F.; Andreasen, N.; Feldman, H.; Giacobini, E.; Jones, R.; Mantua, V.; Mecocci, P.; Pani, L.; Winblad, B.; et al. Clinical trials and late-stage drug development for Alzheimer’s disease: An appraisal from 1984 to 2014. J. Intern. Med. 2014, 275, 251–283. [Google Scholar] [CrossRef]
- Takeda and Zinfandel Pharmaceuticals Discontinue TOMMORROW Trial Following Planned Futility Analysis. Available online: https://www.takeda.com/newsroom/newsreleases/2018/takeda-tommorrow-trial/ (accessed on 2 June 2018).
- Maeshiba, Y.; Kiyota, Y.; Yamashita, K.; Yoshimura, Y.; Motohashi, M.; Tanayama, S. Disposition of the new antidiabetic agent pioglitazone in rats, dogs, and monkeys. Arzneimittelforschung 1997, 47, 29–35. [Google Scholar]
- Chang, K.L.; Pee, H.N.; Yang, S.; Ho, P.C. Influence of drug transporters and stereoselectivity on the brain penetration of pioglitazone as a potential medicine against Alzheimer’s disease. Sci. Rep. 2015, 5, 9000. [Google Scholar] [CrossRef] [Green Version]
- Tuccori, M.; Filion, K.B.; Yin, H.; Yu, O.H.; Platt, R.W.; Azoulay, L. Pioglitazone use and risk of bladder cancer: population based cohort study. BMJ 2016, 352, i1541. [Google Scholar] [CrossRef] [Green Version]
- Tang, H.; Shi, W.; Fu, S.; Wang, T.; Zhai, S.; Song, Y.; Han, J. Pioglitazone and bladder cancer risk: a systematic review and meta-analysis. Cancer Med. 2018, 7, 1070–1080. [Google Scholar] [CrossRef]
- Agrawal, M.; Saraf, S.; Saraf, S.; Antimisiaris, S.G.; Chougule, M.B.; Shoyele, S.A.; Alexander, A. Nose-to-brain drug delivery: An update on clinical challenges and progress towards approval of anti-Alzheimer drugs. J. Control. Release Off. J. Control. Release Soc. 2018, 281, 139–177. [Google Scholar] [CrossRef]
- Feng, Y.; He, H.; Li, F.; Lu, Y.; Qi, J.; Wu, W. An update on the role of nanovehicles in nose-to-brain drug delivery. Drug Discov. Today 2018. [Google Scholar] [CrossRef] [PubMed]
- McCall, R.L.; Sirianni, R.W. PLGA nanoparticles formed by single- or double-emulsion with vitamin E-TPGS. J. Vis. Exp. JoVE 2013, 51015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroeder, W.G.; Mitrescu, L.M.; Hart, M.L.; Unnithan, R.; Gilchrist, J.M.; Smith, E.E.; Shanley, C.; Benedict, K.M.; Taraba, L.; Volckens, J.; et al. Flexible low-cost system for small animal aerosol inhalation exposure to drugs, proteins, inflammatory agents, and infectious agents. BioTechniques 2009, 46, Piii–Pviii. [Google Scholar] [CrossRef] [PubMed]
- Hanson, L.R.; Fine, J.M.; Svitak, A.L.; Faltesek, K.A. Intranasal administration of CNS therapeutics to awake mice. J. Vis. Exp. JoVE 2013. [Google Scholar] [CrossRef] [Green Version]
- Wong, L.R.; Ho, P.C. Role of serum albumin as a nanoparticulate carrier for nose-to-brain delivery of R-flurbiprofen: implications for the treatment of Alzheimer’s disease. J. Pharm. Pharmacol. 2018, 70, 59–69. [Google Scholar] [CrossRef]
- Searcy, J.L.; Phelps, J.T.; Pancani, T.; Kadish, I.; Popovic, J.; Anderson, K.L.; Beckett, T.L.; Murphy, M.P.; Chen, K.-C.; Blalock, E.M.; et al. Long-term pioglitazone treatment improves learning and attenuates pathological markers in a mouse model of Alzheimer’s disease. J. Alzheimers Dis. JAD 2012, 30, 943–961. [Google Scholar] [CrossRef] [Green Version]
- Garamszegi, L.Z. A simple statistical guide for the analysis of behaviour when data are constrained due to practical or ethical reasons. Anim. Behav. 2016, 120, 223–234. [Google Scholar] [CrossRef]
- Wheelock, Å.M.; Wheelock, C.E. Trials and tribulations of ’omics data analysis: assessing quality of SIMCA-based multivariate models using examples from pulmonary medicine. Mol. Biosyst. 2013, 9, 2589–2596. [Google Scholar] [CrossRef] [Green Version]
- Govindaraju, V.; Young, K.; Maudsley, A.A. Proton NMR chemical shifts and coupling constants for brain metabolites. NMR Biomed. 2000, 13, 129–153. [Google Scholar] [CrossRef]
- Graham, S.F.; Holscher, C.; McClean, P.; Elliott, C.T.; Green, B.D. 1H NMR metabolomics investigation of an Alzheimer’s disease (AD) mouse model pinpoints important biochemical disturbances in brain and plasma. Metabolomics 2013, 9, 974–983. [Google Scholar] [CrossRef]
- Lalande, J.; Halley, H.; Balayssac, S.; Gilard, V.; Déjean, S.; Martino, R.; Francés, B.; Lassalle, J.-M.; Malet-Martino, M. 1H NMR metabolomic signatures in five brain regions of the AβPPswe Tg2576 mouse model of Alzheimer’s disease at four ages. J. Alzheimers Dis. JAD 2014, 39, 121–143. [Google Scholar] [CrossRef] [PubMed]
- Chong, J.; Soufan, O.; Li, C.; Caraus, I.; Li, S.; Bourque, G.; Wishart, D.S.; Xia, J. MetaboAnalyst 4.0: towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 2018, 46, W486–W494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durani, L.W.; Hamezah, H.S.; Ibrahim, N.F.; Yanagisawa, D.; Nasaruddin, M.L.; Mori, M.; Azizan, K.A.; Damanhuri, H.A.; Makpol, S.; Wan Ngah, W.Z.; et al. Tocotrienol-Rich Fraction of Palm Oil Improves Behavioral Impairments and Regulates Metabolic Pathways in AβPP/PS1 Mice. J. Alzheimers Dis. JAD 2018, 64, 249–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danhier, F.; Ansorena, E.; Silva, J.M.; Coco, R.; Le Breton, A.; Préat, V. PLGA-based nanoparticles: An overview of biomedical applications. J. Control. Release Off. J. Control. Release Soc. 2012, 161, 505–522. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.-S.; Zhao, L.; Zhang, Z.; Bhakta, G.; Yin Win, K.; Dong, Y.; Chien, S. Chemotherapeutic engineering: Vitamin E TPGS-emulsified nanoparticles of biodegradable polymers realized sustainable paclitaxel chemotherapy for 168h in vivo. Chem. Eng. Sci. 2007, 62, 6641–6648. [Google Scholar] [CrossRef]
- Reichel, A. Addressing central nervous system (CNS) penetration in drug discovery: basics and implications of the evolving new concept. Chem. Biodivers. 2009, 6, 2030–2049. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.L.; Pee, H.N.; Tan, W.P.; Dawe, G.S.; Holmes, E.; Nicholson, J.K.; Chan, E.C.Y.; Ho, P.C. Metabolic profiling of CHO-AβPP695 cells revealed mitochondrial dysfunction prior to amyloid-β pathology and potential therapeutic effects of both PPARγ and PPARα Agonisms for Alzheimer’s disease. J. Alzheimers Dis. JAD 2015, 44, 215–231. [Google Scholar] [CrossRef]
- González-Domínguez, R.; García-Barrera, T.; Vitorica, J.; Gómez-Ariza, J.L. Region-specific metabolic alterations in the brain of the APP/PS1 transgenic mice of Alzheimer’s disease. Biochim. Biophys. Acta 2014, 1842, 2395–2402. [Google Scholar] [CrossRef] [Green Version]
- Mir, M.; Ahmed, N.; Rehman, A.U. Recent applications of PLGA based nanostructures in drug delivery. Colloids Surf. B Biointerfaces 2017, 159, 217–231. [Google Scholar] [CrossRef]
- Jain, R.A. The manufacturing techniques of various drug loaded biodegradable poly(lactide-co-glycolide) (PLGA) devices. Biomaterials 2000, 21, 2475–2490. [Google Scholar] [CrossRef]
- Schwendeman, S.P.; Shah, R.B.; Bailey, B.A.; Schwendeman, A.S. Injectable controlled release depots for large molecules. J. Control. Release Off. J. Control. Release Soc. 2014, 190, 240–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halliday, M.; Radford, H.; Zents, K.A.M.; Molloy, C.; Moreno, J.A.; Verity, N.C.; Smith, E.; Ortori, C.A.; Barrett, D.A.; Bushell, M.; et al. Repurposed drugs targeting eIF2α-P-mediated translational repression prevent neurodegeneration in mice. Brain J. Neurol. 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krako, N.; Magnifico, M.C.; Arese, M.; Meli, G.; Forte, E.; Lecci, A.; Manca, A.; Giuffrè, A.; Mastronicola, D.; Sarti, P.; et al. Characterization of mitochondrial dysfunction in the 7PA2 cell model of Alzheimer’s disease. J. Alzheimers Dis. JAD 2013, 37, 747–758. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, D.G. Spare respiratory capacity, oxidative stress and excitotoxicity. Biochem. Soc. Trans. 2009, 37, 1385–1388. [Google Scholar] [CrossRef] [PubMed]
- Bell, S.M.; De Marco, M.; Barnes, K.; Shaw, P.J.; Ferraiuolo, L.; Blackburn, D.J.; Mortiboys, H.; Venneri, A. Deficits in Mitochondrial Spare Respiratory Capacity Contribute to the Neuropsychological Changes of Alzheimer’s Disease. J. Pers. Med. 2020, 10, 32. [Google Scholar] [CrossRef]
- Win, K.Y.; Feng, S.-S. Effects of particle size and surface coating on cellular uptake of polymeric nanoparticles for oral delivery of anticancer drugs. Biomaterials 2005, 26, 2713–2722. [Google Scholar] [CrossRef]
- Ghosh, S.; Patel, N.; Rahn, D.; McAllister, J.; Sadeghi, S.; Horwitz, G.; Berry, D.; Wang, K.X.; Swerdlow, R.H. The thiazolidinedione pioglitazone alters mitochondrial function in human neuron-like cells. Mol. Pharmacol. 2007, 71, 1695–1702. [Google Scholar] [CrossRef] [Green Version]
- Pagel-Langenickel, I.; Bao, J.; Joseph, J.J.; Schwartz, D.R.; Mantell, B.S.; Xu, X.; Raghavachari, N.; Sack, M.N. PGC-1alpha integrates insulin signaling, mitochondrial regulation, and bioenergetic function in skeletal muscle. J. Biol. Chem. 2008, 283, 22464–22472. [Google Scholar] [CrossRef] [Green Version]
- Mollo, N.; Nitti, M.; Zerillo, L.; Faicchia, D.; Micillo, T.; Accarino, R.; Secondo, A.; Petrozziello, T.; Calì, G.; Cicatiello, R.; et al. Pioglitazone Improves Mitochondrial Organization and Bioenergetics in Down Syndrome Cells. Front. Genet. 2019, 10, 606. [Google Scholar] [CrossRef] [Green Version]
- Yonutas, H.M.; Hubbard, W.B.; Pandya, J.D.; Vekaria, H.J.; Geldenhuys, W.J.; Sullivan, P.G. Bioenergetic restoration and neuroprotection after therapeutic targeting of mitoNEET: New mechanism of pioglitazone following traumatic brain injury. Exp. Neurol. 2020, 327, 113243. [Google Scholar] [CrossRef]
- Geldenhuys, W.J.; Leeper, T.C.; Carroll, R.T. mitoNEET as a novel drug target for mitochondrial dysfunction. Drug Discov. Today 2014, 19, 1601–1606. [Google Scholar] [CrossRef] [PubMed]
- Kerr, J.S.; Adriaanse, B.A.; Greig, N.H.; Mattson, M.P.; Cader, M.Z.; Bohr, V.A.; Fang, E.F. Mitophagy and Alzheimer’s Disease: Cellular and Molecular Mechanisms. Trends Neurosci. 2017, 40, 151–166. [Google Scholar] [CrossRef] [Green Version]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Eckland, D.A.; Danhof, M. Clinical pharmacokinetics of pioglitazone. Exp. Clin. Endocrinol. Diabetes 2000, 108, 234–242. [Google Scholar] [CrossRef]
- Miglio, G.; Rosa, A.C.; Rattazzi, L.; Collino, M.; Lombardi, G.; Fantozzi, R. PPARgamma stimulation promotes mitochondrial biogenesis and prevents glucose deprivation-induced neuronal cell loss. Neurochem. Int. 2009, 55, 496–504. [Google Scholar] [CrossRef] [PubMed]
- Miglio, G.; Rattazzi, L.; Rosa, A.C.; Fantozzi, R. PPARgamma stimulation promotes neurite outgrowth in SH-SY5Y human neuroblastoma cells. Neurosci. Lett. 2009, 454, 134–138. [Google Scholar] [CrossRef] [PubMed]
- Crenshaw, D.G.; Asin, K.; Gottschalk, W.K.; Liang, Z.; Zhang, N.; Roses, A.D. Effects of low doses of pioglitazone on resting-state functional connectivity in conscious rat brain. PLoS ONE 2015, 10, e0117973. [Google Scholar] [CrossRef] [Green Version]
- Umegaki, H. Therapeutic Potential of Antidiabetic Medications in the Treatment of Cognitive Dysfunction and Dementia. Drugs Aging 2016, 33, 399–409. [Google Scholar] [CrossRef]
- Hsiao, K.; Chapman, P.; Nilsen, S.; Eckman, C.; Harigaya, Y.; Younkin, S.; Yang, F.; Cole, G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 1996, 274, 99–102. [Google Scholar] [CrossRef]
- Rodriguez-Rivera, J.; Denner, L.; Dineley, K.T. Rosiglitazone reversal of Tg2576 cognitive deficits is independent of peripheral gluco-regulatory status. Behav. Brain Res. 2011, 216, 255–261. [Google Scholar] [CrossRef] [Green Version]
- Kawarabayashi, T.; Younkin, L.H.; Saido, T.C.; Shoji, M.; Ashe, K.H.; Younkin, S.G. Age-dependent changes in brain, CSF, and plasma amyloid (beta) protein in the Tg2576 transgenic mouse model of Alzheimer’s disease. J. Neurosci. Off. J. Soc. Neurosci. 2001, 21, 372–381. [Google Scholar] [CrossRef] [Green Version]
- Gillardon, F.; Rist, W.; Kussmaul, L.; Vogel, J.; Berg, M.; Danzer, K.; Kraut, N.; Hengerer, B. Proteomic and functional alterations in brain mitochondria from Tg2576 mice occur before amyloid plaque deposition. Proteomics 2007, 7, 605–616. [Google Scholar] [CrossRef] [PubMed]
- Varghese, M.; Zhao, W.; Wang, J.; Cheng, A.; Qian, X.; Chaudhry, A.; Ho, L.; Pasinetti, G.M. Mitochondrial bioenergetics is defective in presymptomatic Tg2576 AD mice. Transl. Neurosci. 2011, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zahs, K.R.; Ashe, K.H. “Too much good news”-are Alzheimer mouse models trying to tell us how to prevent, not cure, Alzheimer’s disease? Trends Neurosci. 2010, 33, 381–389. [Google Scholar] [CrossRef]
- King, D.L.; Arendash, G.W. Behavioral characterization of the Tg2576 transgenic model of Alzheimer’s disease through 19 months. Physiol. Behav. 2002, 75, 627–642. [Google Scholar] [CrossRef]
- Mega, M.S.; Cummings, J.L.; Fiorello, T.; Gornbein, J. The spectrum of behavioral changes in Alzheimer’s disease. Neurology 1996, 46, 130–135. [Google Scholar] [CrossRef]
- Shineman, D.W.; Basi, G.S.; Bizon, J.L.; Colton, C.A.; Greenberg, B.D.; Hollister, B.A.; Lincecum, J.; Leblanc, G.G.; Lee, L.B.H.; Luo, F.; et al. Accelerating drug discovery for Alzheimer’s disease: Best practices for preclinical animal studies. Alzheimers Res. Ther. 2011, 3, 28. [Google Scholar] [CrossRef]
- Button, K.S.; Ioannidis, J.P.A.; Mokrysz, C.; Nosek, B.A.; Flint, J.; Robinson, E.S.J.; Munafò, M.R. Power failure: why small sample size undermines the reliability of neuroscience. Nat. Rev. Neurosci. 2013, 14, 365–376. [Google Scholar] [CrossRef] [Green Version]
- Lalonde, R. The neurobiological basis of spontaneous alternation. Neurosci. Biobehav. Rev. 2002, 26, 91–104. [Google Scholar] [CrossRef]
- Phelan, M.M.; Caamaño-Gutiérrez, E.; Gant, M.S.; Grosman, R.X.; Madine, J. Using an NMR metabolomics approach to investigate the pathogenicity of amyloid-beta and alpha-synuclein. Metab. Off. J. Metab. Soc. 2017, 13, 151. [Google Scholar] [CrossRef] [Green Version]
- Kori, M.; Aydın, B.; Unal, S.; Arga, K.Y.; Kazan, D. Metabolic Biomarkers and Neurodegeneration: A Pathway Enrichment Analysis of Alzheimer’s Disease, Parkinson’s Disease, and Amyotrophic Lateral Sclerosis. Omics J. Integr. Biol. 2016, 20, 645–661. [Google Scholar] [CrossRef] [PubMed]
- Dedeoglu, A.; Choi, J.-K.; Cormier, K.; Kowall, N.W.; Jenkins, B.G. Magnetic resonance spectroscopic analysis of Alzheimer’s disease mouse brain that express mutant human APP shows altered neurochemical profile. Brain Res. 2004, 1012, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Barker, P.B. Various MRS application tools for Alzheimer disease and mild cognitive impairment. AJNR Am. J. Neuroradiol. 2014, 35, S4–S11. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, Z.; Ji, B.; Liu, L.; Wu, S.; Liu, X.; Wang, S.; Wang, L. Metabolite Profile of Alzheimer’s Disease in the Frontal Cortex as Analyzed by HRMAS 1H NMR. Front. Aging Neurosci. 2018, 10, 424. [Google Scholar] [CrossRef] [PubMed]
- Griffin, J.W.D.; Bradshaw, P.C. Amino Acid Catabolism in Alzheimer’s Disease Brain: Friend or Foe? Oxid. Med. Cell. Longev. 2017, 2017, 5472792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhinderwala, F.; Wase, N.; DiRusso, C.; Powers, R. Combining Mass Spectrometry and NMR Improves Metabolite Detection and Annotation. J. Proteome Res. 2018, 17, 4017–4022. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment Group | Plasma | Brain | |||||
|---|---|---|---|---|---|---|---|
| Cmax (µg/mL) | Tmax (h) | AUC0 → 4 h (μg·h/mL) a | Cmax (ng/mL) | Tmax (h) | AUC0 → 4 h (μg·h/mL) a | ||
| PIO, i.n. | 8.09 ± 1.80 0.48 ± 0.11 b | 2 | 20.06 1.21 b | 498 ± 96.5 30 ± 5.8 b | 2 | 1.52 0.09 b | 7.58% |
| PIO, p.o. | 19.55 ± 1.26 0.39 ± 0.03 b | 1 | 66.11 1.32 b | 1609 ± 104.9 32 ± 2.1 b | 1 | 5.44 0.11 b | 8.23% |
| PIO NP, i.n. | 1.13 ± 0.18 | 1.5 | 1.73 | 385 ± 45.5 | 2 | 0.48 | 27.75% |
| Brain Region | No. of Components | R2(cum) | Q2(cum) | p[CV-ANOVA] |
|---|---|---|---|---|
| Cerebellum | 1 predictive + 1 orthogonal | 0.945 | 0.887 | 0.00010 |
| Cortex | 1 predictive + 0 orthogonal | 0.861 | 0.806 | 0.00005 |
| Hippocampus | 1 predictive + 1 orthogonal | 0.894 | 0.818 | 0.00101 |
| Midbrain | 1 predictive + 4 orthogonal | 0.997 | 0.842 | 0.05442 |
| Brain Region | Metabolite | δ (ppm) | Signal a | FC | Adjusted p Value b |
|---|---|---|---|---|---|
| Cerebellum | Lactate | 1.31 | d, CH3 | 0.96 | NS |
| NAA | 2.01 | s, CH3 (acetyl moiety) | 0.95 | NS | |
| Glutamate | 2.12 | m, CH2 | 0.98 | NS | |
| GABA | 2.28 | t, CH2 | 1.09 | NS | |
| Creatine | 3.03 | s, N(CH3) | 0.98 | NS | |
| GPC | 3.21 | s, N(CH3)3 (choline moiety) | 0.82 | NS | |
| Taurine | 3.25 | t, CH2 | 0.98 | NS | |
| mIns | 3.61 | t, CH | 0.96 | NS | |
| Cortex | Valine | 1.03 | d, CH3 | 1.76 | 0.000791 |
| Alanine | 1.47 | d, CH3 | 1.54 | 0.001229 | |
| NAA | 2.01 | s, CH3 (acetyl moiety) | 1.46 | 0.004118 | |
| Glutamate | 2.12 | m, CH2 | 1.32 | NS | |
| GABA | 2.28 | t, CH2 | 1.67 | 0.00077 | |
| Creatine | 3.03 | s, N(CH3) | 1.42 | 0.004823 | |
| Choline | 3.18 | s, N(CH3)3 | 1.72 | 0.010769 | |
| GPC | 3.21 | s, N(CH3)3 (choline moiety) | 1.28 | NS | |
| Taurine | 3.25 | t, CH2 | 1.42 | 0.032589 | |
| Glycine | 3.55 | s, CH2 | 1.32 | NS | |
| mIns | 3.61 | t, CH | 1.39 | 0.007666 | |
| PE | 3.82 | m, CH2 | 1.27 | 0.004332 | |
| Lactate | 4.09 | q, CH | 1.26 | NS | |
| Ascorbate | 4.49 | d, CH | 1.60 | 0.001417 | |
| Adenosine | 8.23 | s, CH | 2.00 | <0.000001 | |
| Hippocampus | NAA | 2.01 | s, CH3 (acetyl moiety) | 1.02 | NS |
| Glutamate | 2.12 | m, CH2 | 1.03 | NS | |
| GABA | 2.28 | t, CH2 | 0.92 | NS | |
| Aspartate | 2.80 | dd, CH2 | 0.93 | NS | |
| Creatine | 3.03 | s, N(CH3) | 0.94 | NS | |
| GPC | 3.21 | s, N(CH3)3 (choline moiety) | 0.86 | NS | |
| Taurine | 3.42 | t, CH2 | 0.99 | NS | |
| mIns | 3.52 | dd, CH | 0.95 | NS | |
| Lactate | 4.09 | q, CH | 1.03 | NS | |
| Ascorbate | 4.49 | d, CH | 1.64 | 0.002485 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wong, L.R.; Wong, P.; Ho, P.C.-L. Metabolic Profiling of Female Tg2576 Mouse Brains Provides Novel Evidence Supporting Intranasal Low-Dose Pioglitazone for Long-Term Treatment at an Early Stage of Alzheimer’s Disease. Biomedicines 2020, 8, 589. https://doi.org/10.3390/biomedicines8120589
Wong LR, Wong P, Ho PC-L. Metabolic Profiling of Female Tg2576 Mouse Brains Provides Novel Evidence Supporting Intranasal Low-Dose Pioglitazone for Long-Term Treatment at an Early Stage of Alzheimer’s Disease. Biomedicines. 2020; 8(12):589. https://doi.org/10.3390/biomedicines8120589
Chicago/Turabian StyleWong, Ling Rong, Peiyan Wong, and Paul Chi-Lui Ho. 2020. "Metabolic Profiling of Female Tg2576 Mouse Brains Provides Novel Evidence Supporting Intranasal Low-Dose Pioglitazone for Long-Term Treatment at an Early Stage of Alzheimer’s Disease" Biomedicines 8, no. 12: 589. https://doi.org/10.3390/biomedicines8120589