
Ribose and Non-Ribose A2A Adenosine Receptor Agonists: Do They Share the Same Receptor Recognition Mechanism?





Abstract
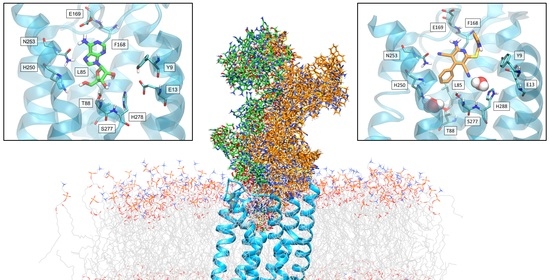
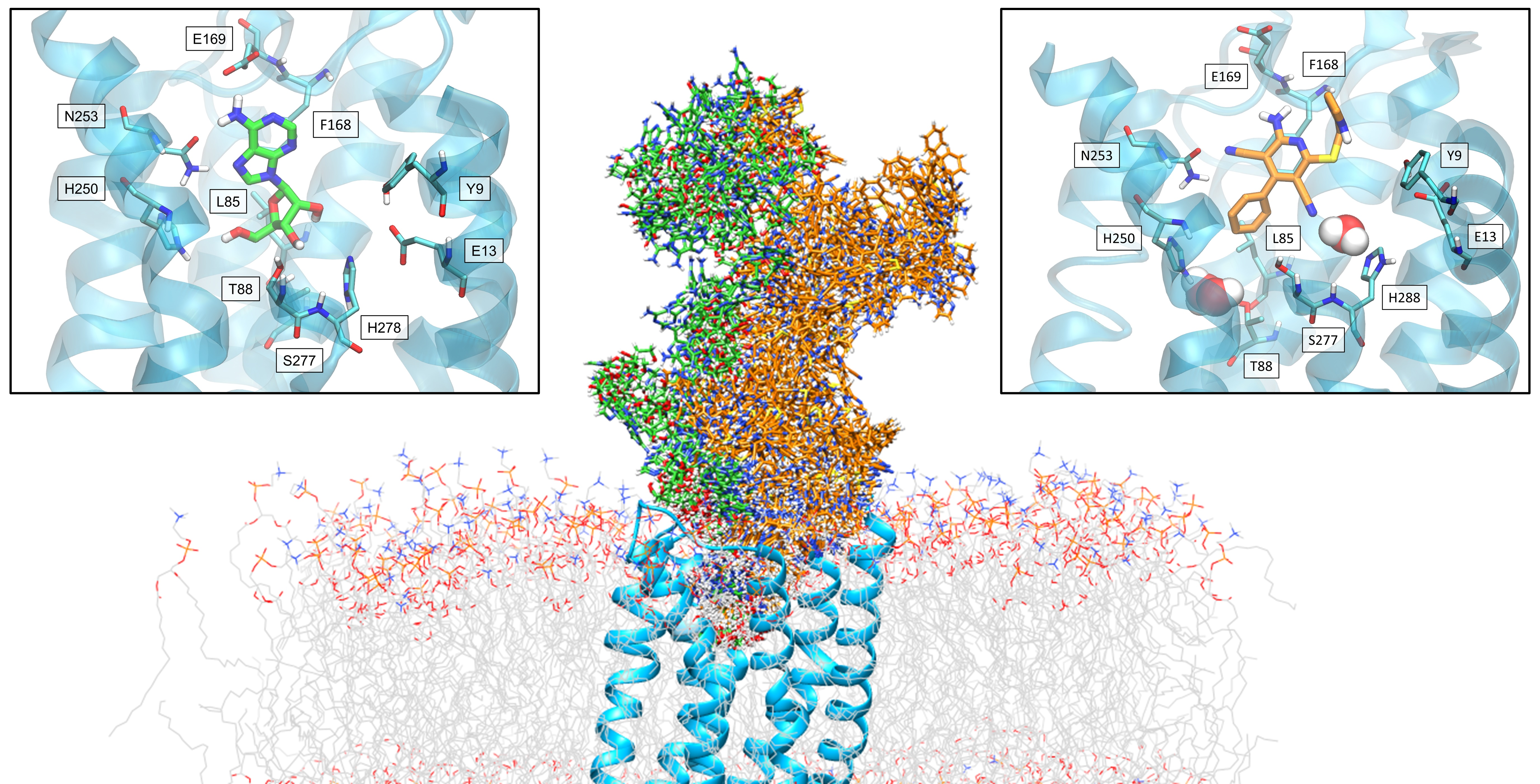
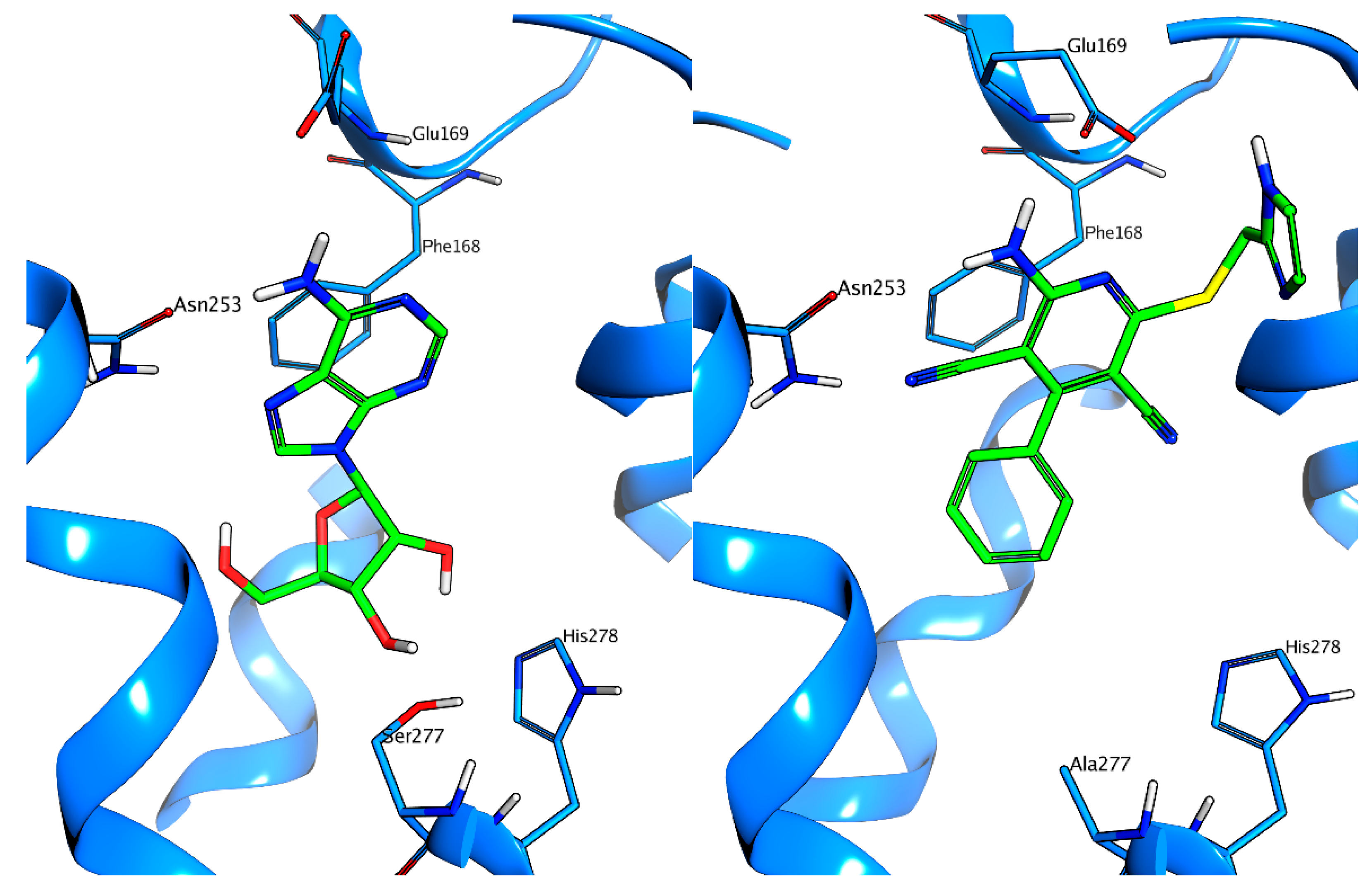
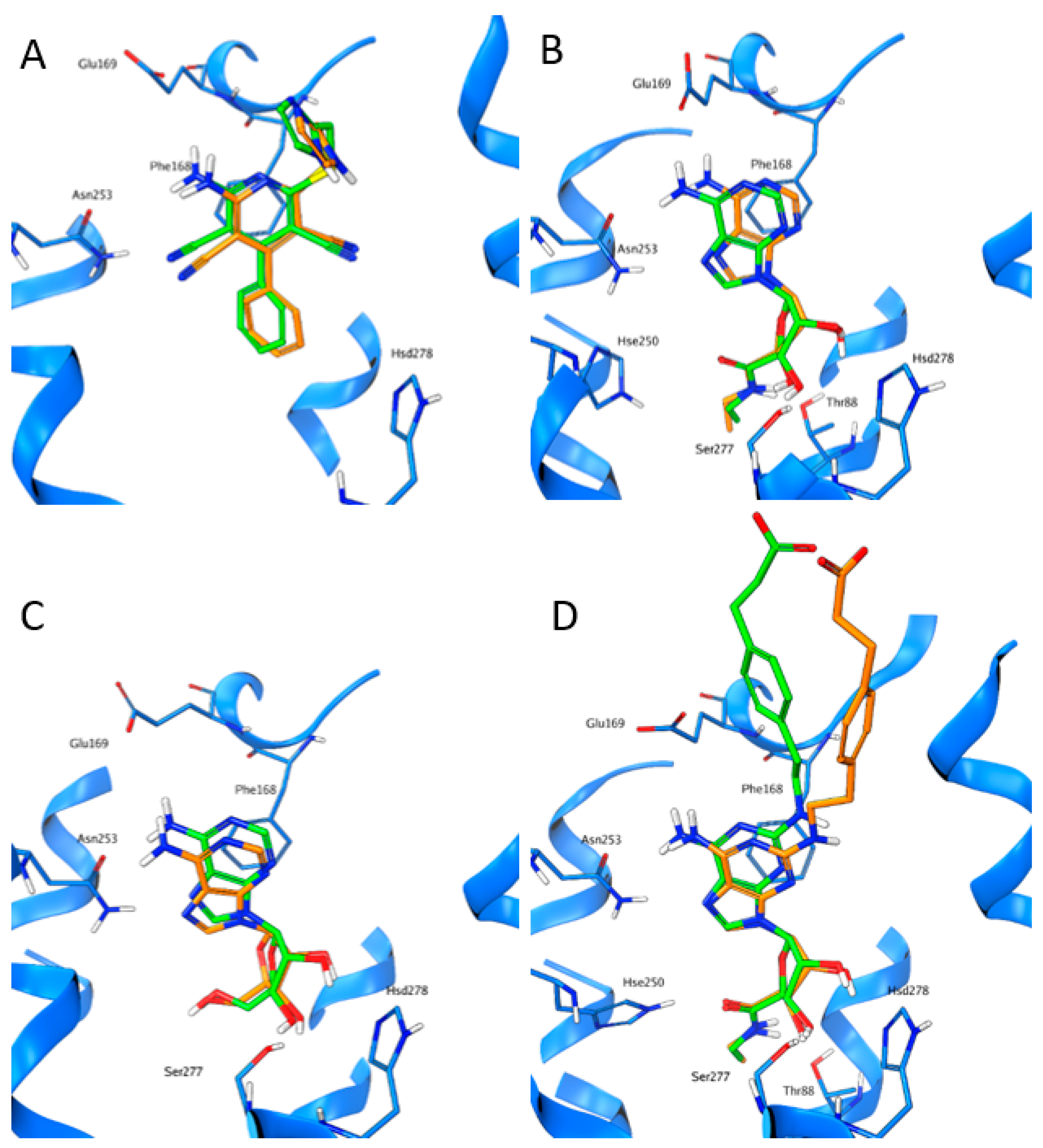
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. System Setup
2.2. Molecular Dynamics
2.3. Equilibration Phase
2.4. Supervised Molecular Dynamics (SuMD) Simulations
2.5. Trajectory Analysis
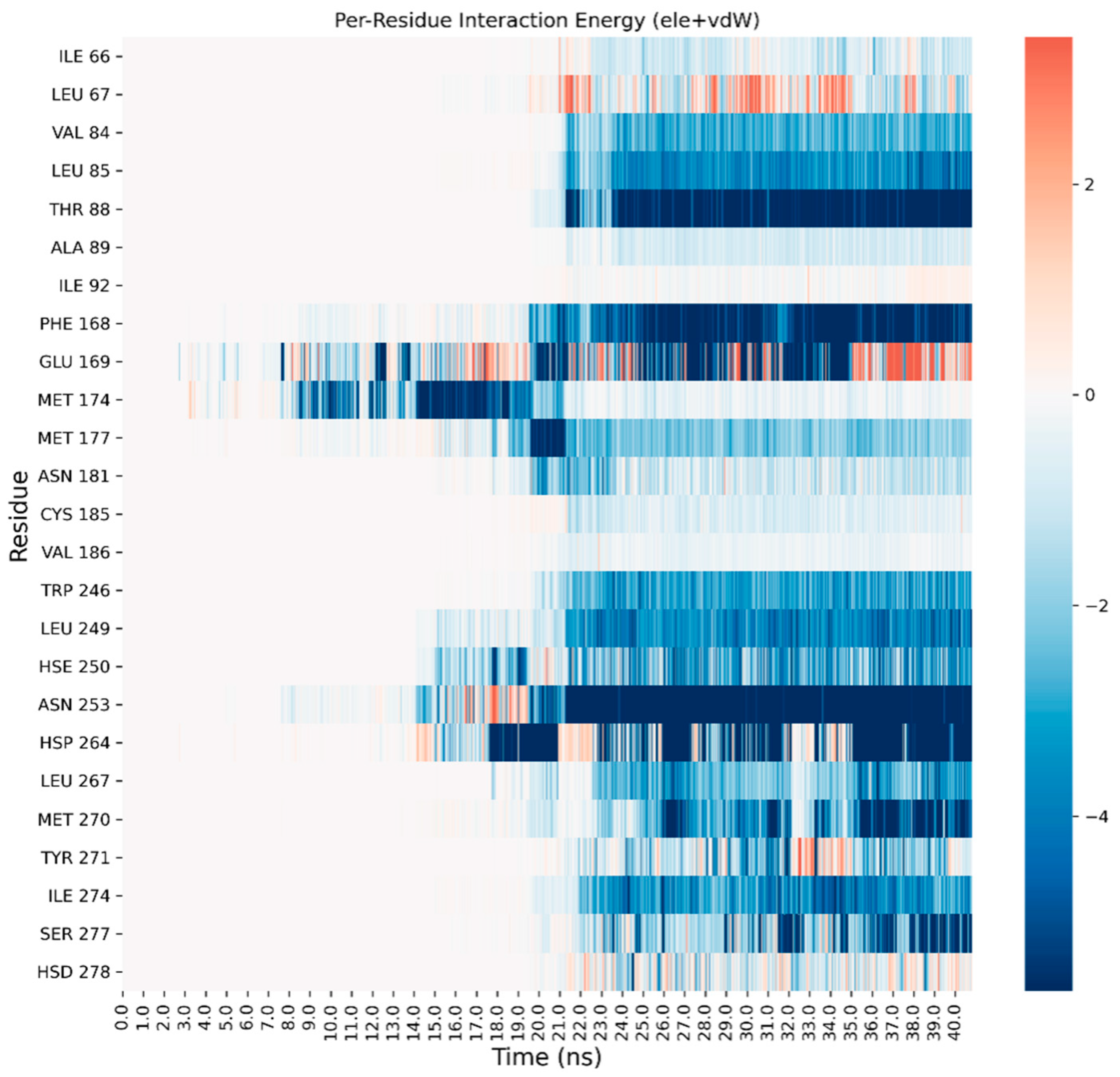
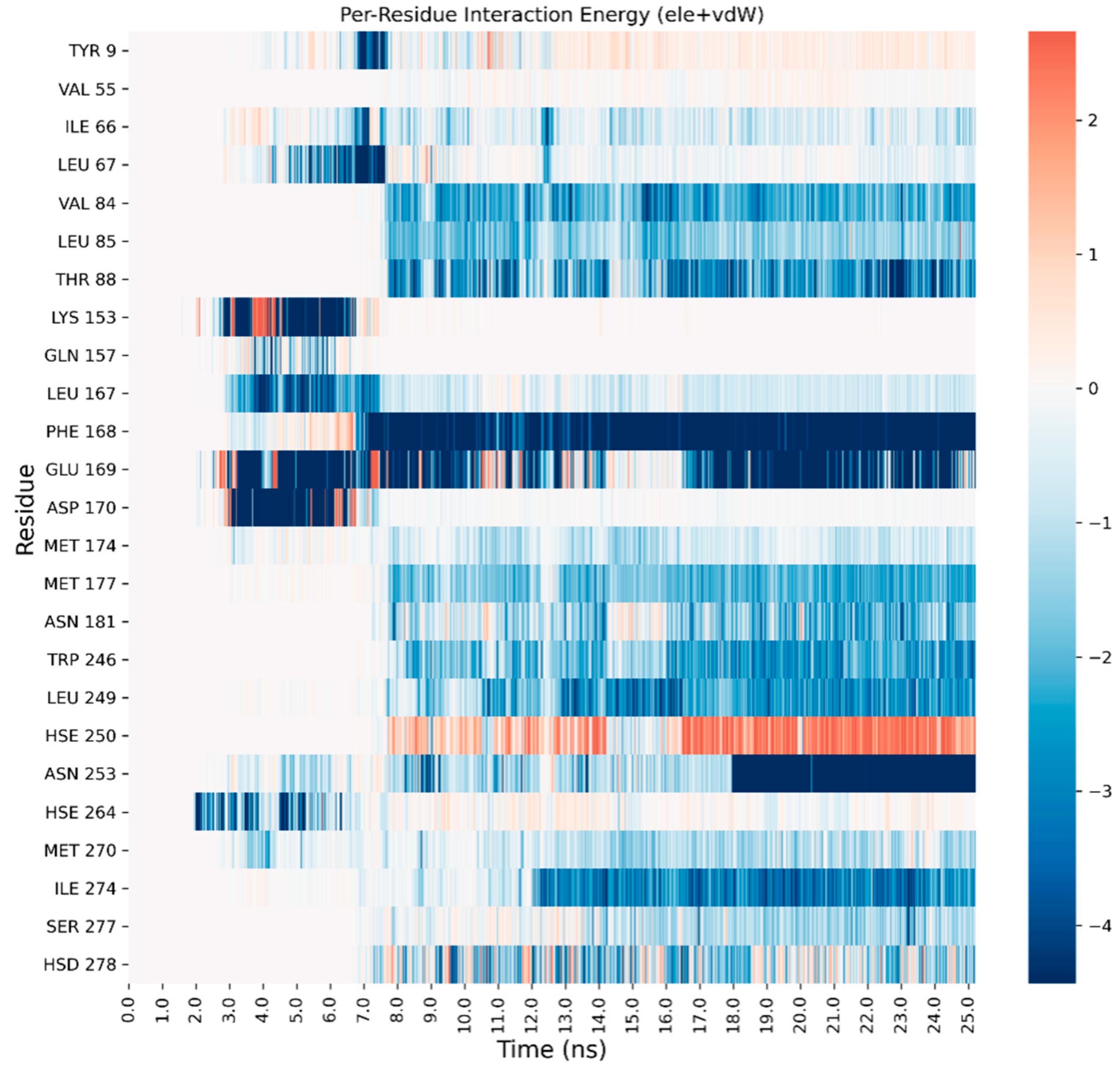
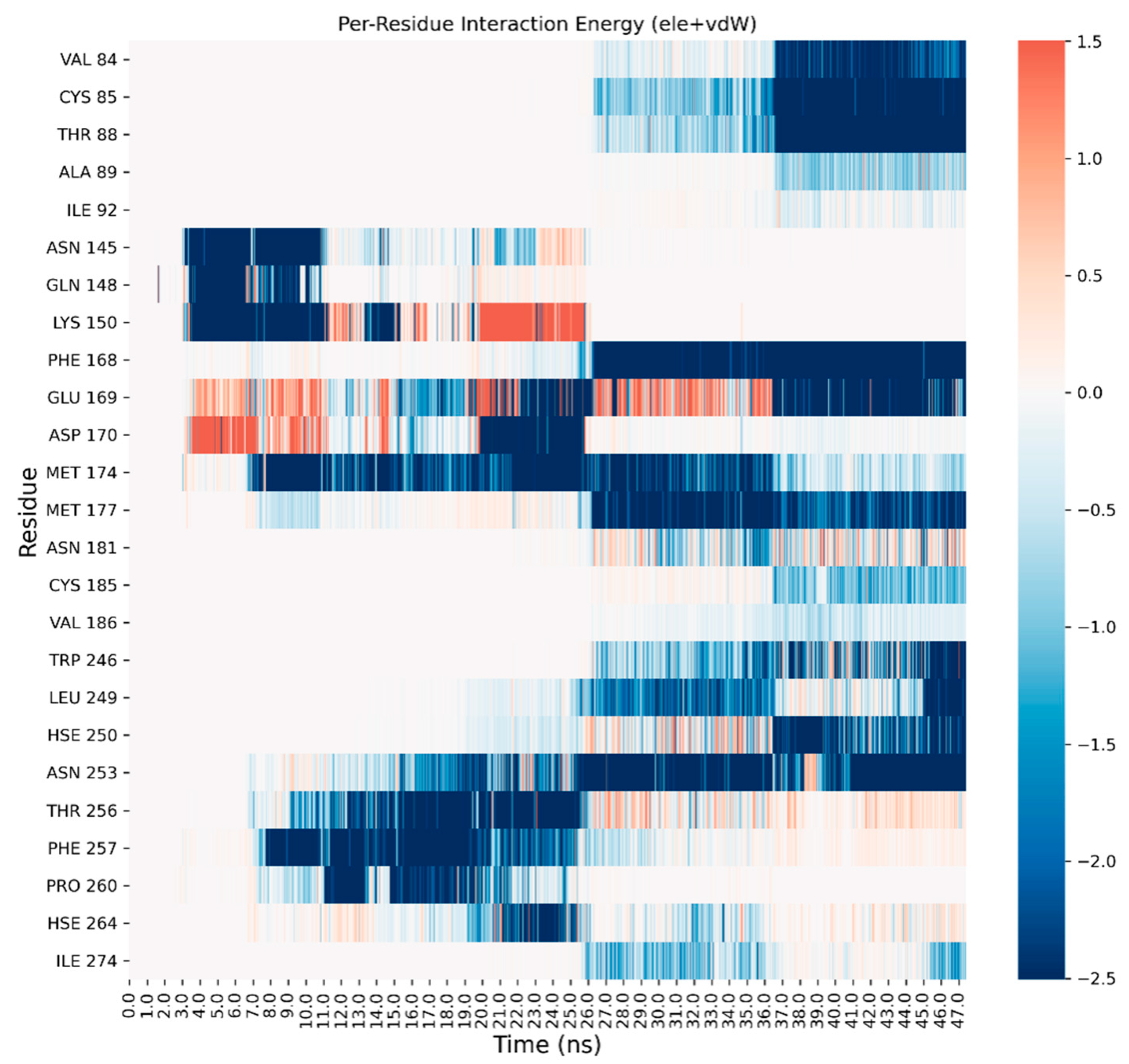
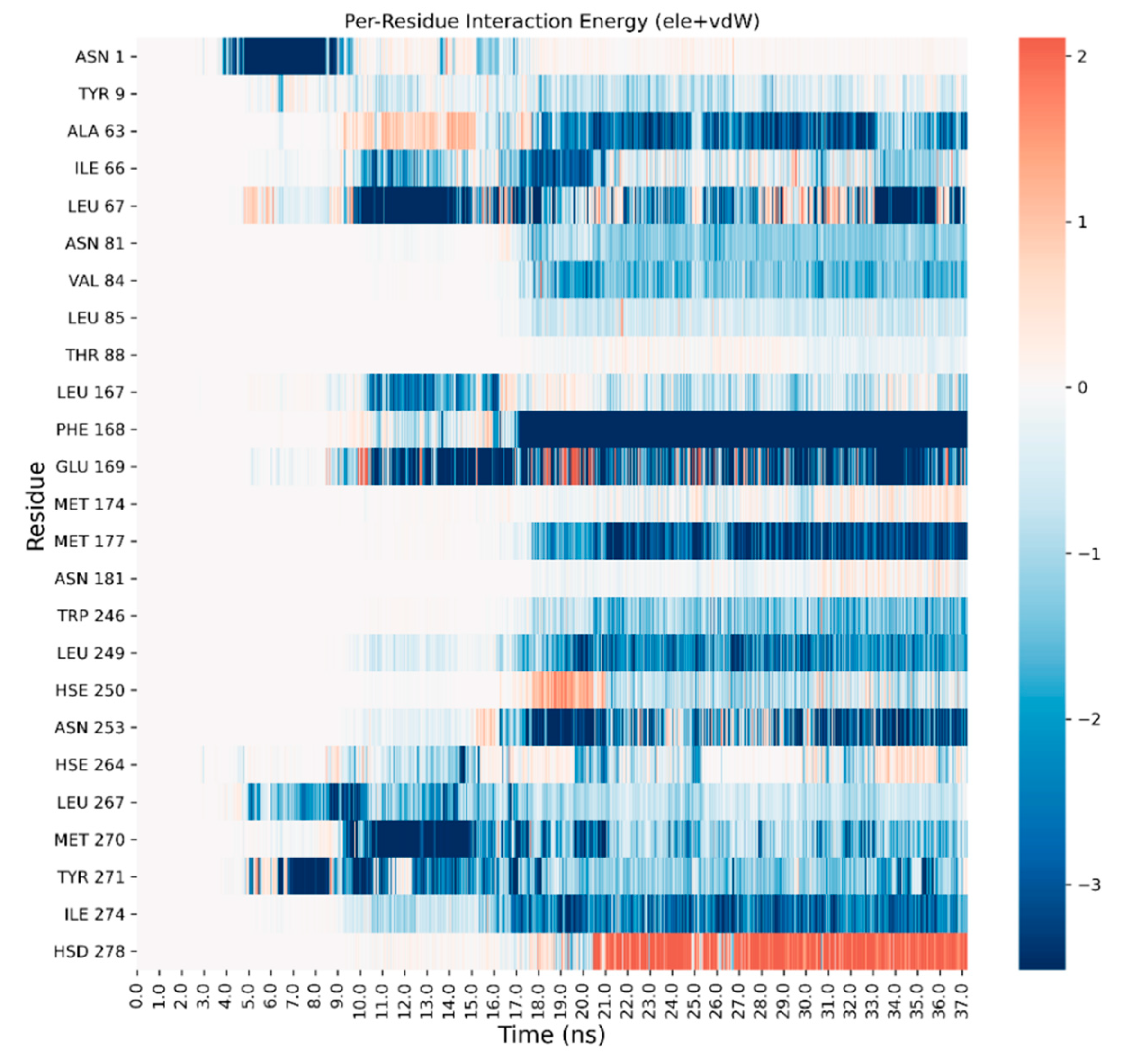
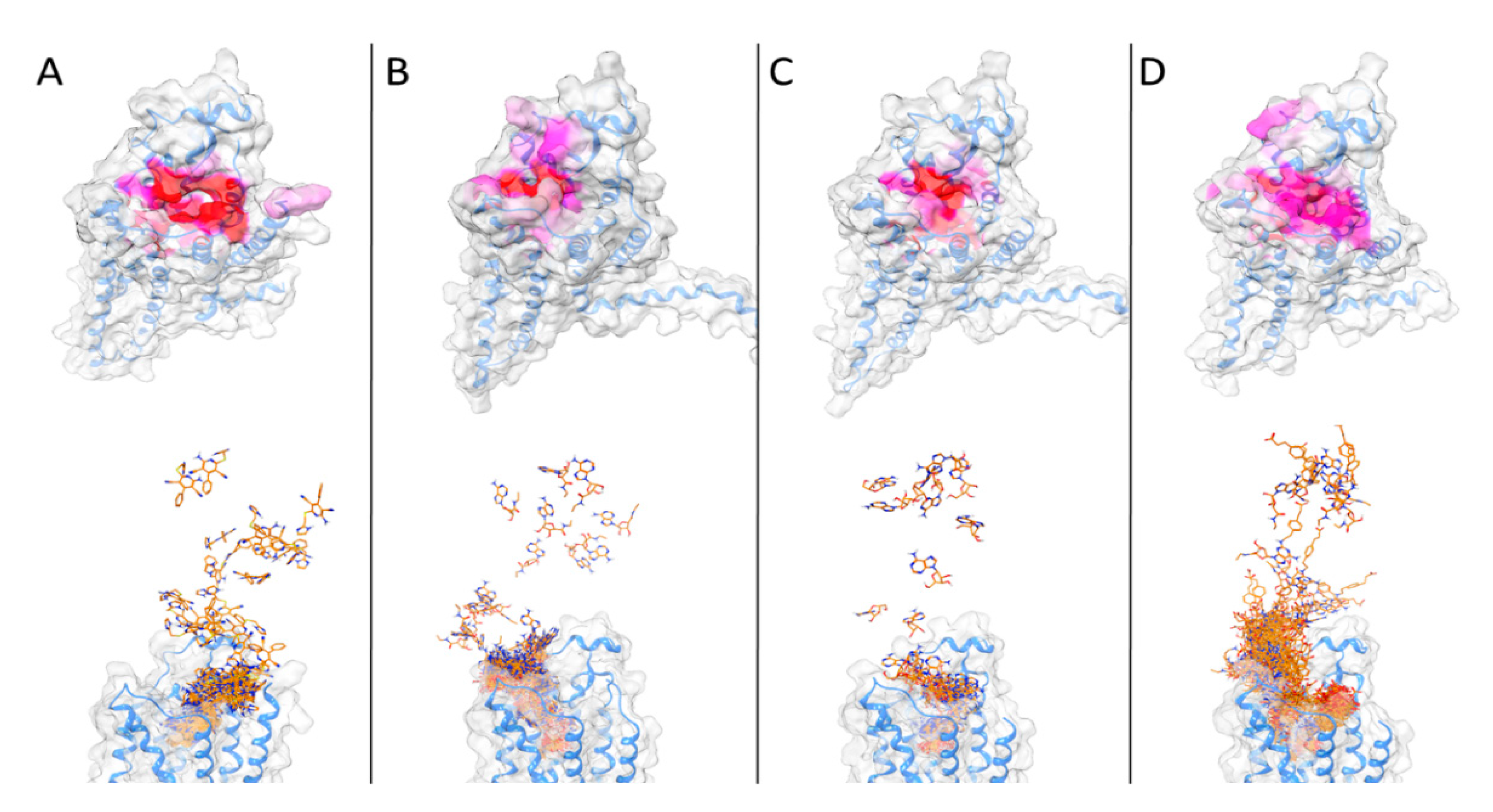
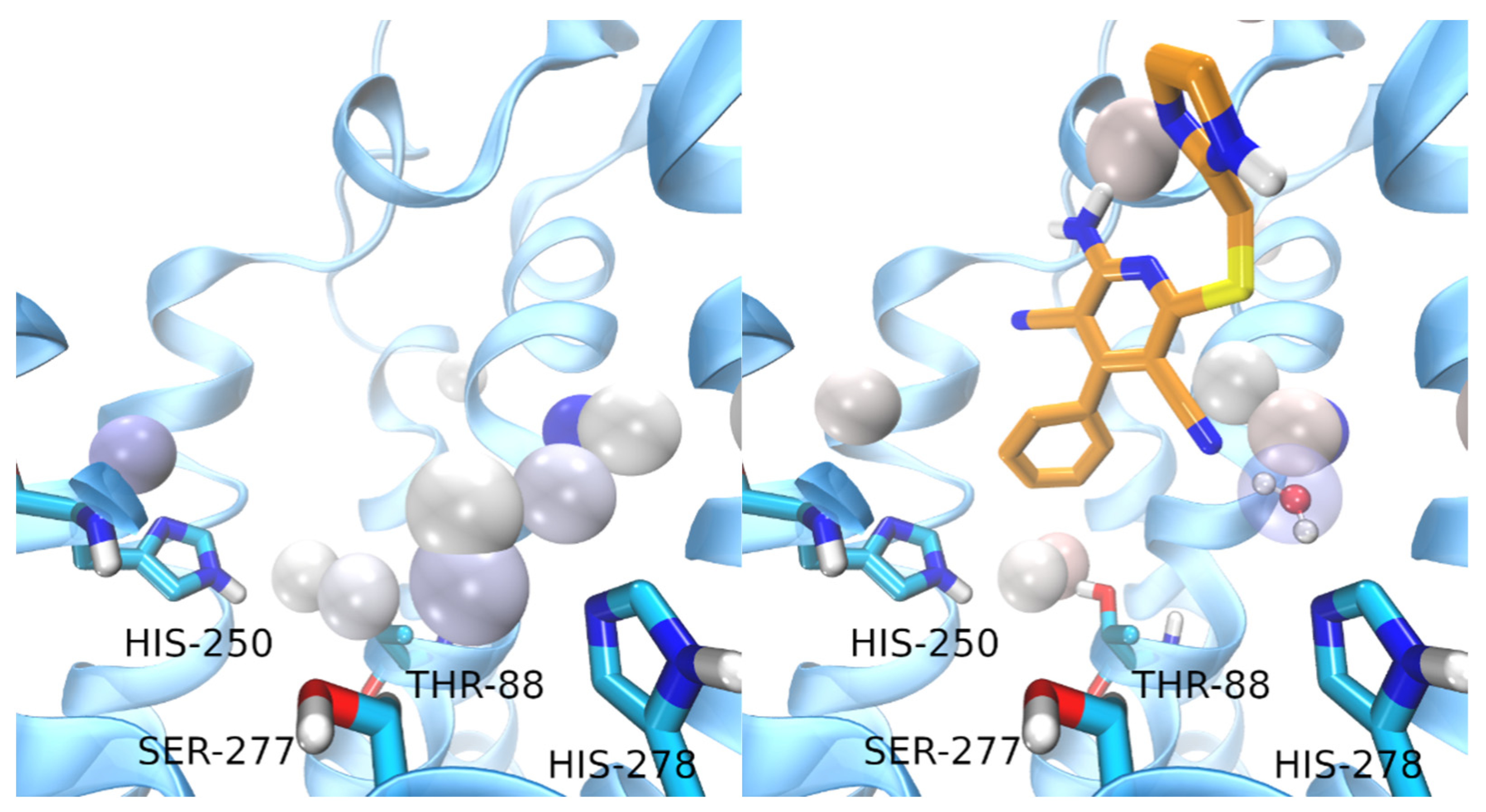
3. Results and Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Müller, C.E.; Jacobson, K. Recent developments in adenosine receptor ligands and their potential as novel drugs. Biochim. Biophys. Acta (BBA)-Biomembr. 2011, 1808, 1290–1308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.-F.; Eltzschig, H.K.; Fredholm, B.B. Adenosine receptors as drug targets—What are the challenges? Nat. Rev. Drug Discov. 2013, 12, 265–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navarro, G.; Borroto-Escuela, D.O.; Fuxe, K.; Franco, R. Purinergic signaling in Parkinson’s disease. Relevance for treatment. Neuropharmacology 2016, 104, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Shah, U.; Hodgson, R. Recent progress in the discovery of adenosine A(2A) receptor antagonists for the treatment of Parkinson’s disease. Curr. Opin. Drug Discov. Dev. 2010, 13, 466–480. [Google Scholar]
- Gao, Z.-G.; Jacobson, K.A. Purinergic Signaling in Mast Cell Degranulation and Asthma. Front. Pharmacol. 2017, 8, 947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vincenzi, F.; Pasquini, S.; Borea, P.A.; Varani, K. Targeting Adenosine Receptors: A Potential Pharmacological Avenue for Acute and Chronic Pain. Int. J. Mol. Sci. 2020, 21, 8710. [Google Scholar] [CrossRef]
- Fishman, P.; Bar-Yehuda, S.; Synowitz, M.; Powell, J.; Klotz, K.; Gessi, S.; Borea, P. Adenosine Receptors and Cancer. Nitric Oxide 2009, 193, 399–441. [Google Scholar]
- Mustafa, S.J.; Morrison, R.R.; Teng, B.; Pelleg, A. Adenosine Receptors and the Heart: Role in Regulation of Coronary Blood Flow and Cardiac Electrophysiology. Antiplatelet Agents 2009, 193, 161–188. [Google Scholar]
- Schmidt, J.; Ferk, P. Safety issues of compounds acting on adenosinergic signalling. J. Pharm. Pharmacol. 2017, 69, 790–806. [Google Scholar] [CrossRef] [Green Version]
- Jacobson, K.A.; Tosh, D.K.; Jain, S.; Gao, Z.-G. Historical and Current Adenosine Receptor Agonists in Preclinical and Clinical Development. Front. Cell. Neurosci. 2019, 13, 124. [Google Scholar] [CrossRef] [Green Version]
- Albrecht-Küpper, B.E.; Leineweber, K.; Nell, P.G. Partial adenosine A1 receptor agonists for cardiovascular therapies. Purinergic Signal. 2011, 8, 91–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soudijn, I.V.W.A.A.P.I.W.; Wijngaarden, I.; Ijzerman, A. Medicinal Chemistry of Adenosine A1 Receptor Ligands. Curr. Top. Med. Chem. 2003, 3, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Cristalli, G.; Lambertucci, C.; Marucci, G.; Volpini, R.; Ben, D.D. A2A Adenosine Receptor and its Modulators: Overview on a Druggable GPCR and on Structure-Activity Relationship Analysis and Binding Requirements of Agonists and Antagonists. Curr. Pharm. Des. 2008, 14, 1525–1552. [Google Scholar] [CrossRef] [PubMed]
- Jespers, W.; Schiedel, A.C.; Heitman, L.H.; Cooke, R.M.; Kleene, L.; van Westen, G.; Gloriam, D.E.; Müller, C.E.; Sotelo, E.; Gutiérrez-De-Terán, H. Structural Mapping of Adenosine Receptor Mutations: Ligand Binding and Signaling Mechanisms. Trends Pharmacol. Sci. 2018, 39, 75–89. [Google Scholar] [CrossRef] [Green Version]
- Lane, J.R.; Herenbrink, C.K.; van Westen, G.; Spoorendonk, J.A.; Hoffmann, C.; Ijzerman, A. A Novel Nonribose Agonist, LUF5834, Engages Residues That Are Distinct from Those of Adenosine-Like Ligands to Activate the Adenosine A2a Receptor. Mol. Pharmacol. 2012, 81, 475–487. [Google Scholar] [CrossRef] [Green Version]
- Amelia, T.; van Veldhoven, J.P.D.; Falsini, M.; Liu, R.; Heitman, L.H.; van Westen, G.J.P.; Segala, E.; Verdon, G.; Cheng, R.K.Y.; Cooke, R.M.; et al. Crystal Structure and Subsequent Ligand Design of a Nonriboside Partial Agonist Bound to the Adenosine A2A Receptor. J. Med. Chem. 2021, 64, 3827–3842. [Google Scholar] [CrossRef]
- Molecular Operating Environment (MOE), 2019.01; Chemical Computing Group ULC: Montreal, QC, Canada, 2021; Available online: https://www.chemcomp.com/Research-Citing_MOE.htm (accessed on 19 January 2021).
- Humphrey, W.; Dalke, A.; Schulten, K. Sartorius products. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Wallace, J.A.; Shen, J.K. Predicting pKa Values with Continuous Constant pH Molecular Dynamics. Methods Enzymol. 2009, 466, 455–475. [Google Scholar] [CrossRef]
- Wallace, J.A.; Wang, Y.; Shi, C.; Pastoor, K.J.; Nguyen, B.-L.; Xia, K.; Shen, J.K. Toward accurate prediction of pKa values for internal protein residues: The importance of conformational relaxation and desolvation energy. Proteins: Struct. Funct. Bioinform. 2011, 79, 3364–3373. [Google Scholar] [CrossRef]
- Huang, S.K.; Almurad, O.; Pejana, R.J.; Morrison, Z.A.; Pandey, A.; Picard, L.-P.; Nitz, M.; Sljoka, A.; Prosser, R.S. Allosteric modulation of the adenosine A2A receptor by cholesterol. eLife 2022, 11, e73901. [Google Scholar] [CrossRef] [PubMed]
- Harvey, M.; Giupponi, G.; De Fabritiis, G. ACEMD: Accelerating Biomolecular Dynamics in the Microsecond Time Scale. J. Chem. Theory Comput. 2009, 5, 1632–1639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brooks, B.R.; Brooks, C.L.; MacKerell, A.D.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef] [PubMed]
- CGenFF Home. Available online: https://cgenff.umaryland.edu/ (accessed on 14 December 2021).
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2009, 31, 671–690. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Yoluk, O.; MacKerell, A.D. FFParam: Standalone package for CHARMM additive and Drude polarizable force field parametrization of small molecules. J. Comput. Chem. 2020, 41, 958–970. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Berendsen, H.J.C.; Van Der Spoel, D.; Van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Sabbadin, D.; Moro, S. Supervised Molecular Dynamics (SuMD) as a Helpful Tool To Depict GPCR–Ligand Recognition Pathway in a Nanosecond Time Scale. J. Chem. Inf. Model. 2014, 54, 372–376. [Google Scholar] [CrossRef]
- Bakan, A.; Meireles, L.M.; Bahar, I. ProDy: Protein Dynamics Inferred from Theory and Experiments. Bioinform. 2011, 27, 1575–1577. [Google Scholar] [CrossRef] [Green Version]
- Michaud-Agrawal, N.; Denning, E.J.; Woolf, T.B.; Beckstein, O. MDAnalysis: A toolkit for the analysis of molecular dynamics simulations. J. Comput. Chem. 2011, 32, 2319–2327. [Google Scholar] [CrossRef] [Green Version]
- Gowers, R.J.; Linke, M.; Barnoud, J.; Reddy, T.J.E.; Melo, M.N.; Seyler, S.L.; Domanski, J.; Dotson, D.L.; Buchoux, S.; Kenney, I.M.; et al. MDAnalysis: A Python Package for the Rapid Analysis of Molecular Dynamics Simulations. In Proceedings of the 15th Python in Science Conference (SciPy 2016), Austin, TX, USA, 11–17 July 2016; pp. 98–105. [Google Scholar]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.C.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W.; et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef] [PubMed]
- Cuzzolin, A.; Deganutti, G.; Salmaso, V.; Sturlese, M.; Moro, S. AquaMMapS: An Alternative Tool to Monitor the Role of Water Molecules During Protein-Ligand Association. ChemMedChem 2018, 13, 522–531. [Google Scholar] [CrossRef] [Green Version]
- Sabbadin, D.; Ciancetta, A.; Deganutti, G.; Cuzzolin, A.; Moro, S. Exploring the recognition pathway at the human A2A adenosine receptor of the endogenous agonist adenosine using supervised molecular dynamics simulations. MedChemComm 2015, 6, 1081–1085. [Google Scholar] [CrossRef]
- Deganutti, G.; Welihinda, A.; Moro, S. Comparison of the Human A2AAdenosine Receptor Recognition by Adenosine and Inosine: New Insight from Supervised Molecular Dynamics Simulations. ChemMedChem 2017, 12, 1319–1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Q.; Van Rhee, A.M.; Kim, J.; Yehle, S.; Wess, J.; Jacobson, K.A. Hydrophilic side chains in the third and seventh transmembrane helical domains of human A2A adenosine receptors are required for ligand recognition. Mol. Pharmacol. 1996, 50, 512–521. [Google Scholar] [PubMed]
- Kim, J.; Wess, J.; van Rhee, A.M.; Schöneberg, T.; Jacobson, K.A. Site-directed Mutagenesis Identifies Residues Involved in Ligand Recognition in the Human A2a Adenosine Receptor. J. Biol. Chem. 1995, 270, 13987–13997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fredholm, B.B.; Ijzerman, A.P.; Jacobson, K.A.; Klotz, K.N.; Linden, J. International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol. Rev. 2001, 53, 527–552. [Google Scholar] [PubMed]
- Lopes, L.V.; Cunha, R.; Ribeiro, J. Cross talk between A(1) and A(2A) adenosine receptors in the hippocampus and cortex of young adult and old rats. J. Neurophysiol. 1999, 82, 3196–3203. [Google Scholar] [CrossRef]
- Cunha, R.A.; Correia-De-Sá, P.; Sebastião, A.M.; Ribeiro, J.A. Preferential activation of excitatory adenosine receptors at rat hippocampal and neuromuscular synapses by adenosine formed from released adenine nucleotides. J. Cereb. Blood Flow Metab. 1996, 119, 253–260. [Google Scholar] [CrossRef] [Green Version]
- Guo, D.; Mulder-Krieger, T.; Ijzerman, A.; Heitman, L.H. Functional efficacy of adenosine A2A receptor agonists is positively correlated to their receptor residence time. J. Cereb. Blood Flow Metab. 2012, 166, 1846–1859. [Google Scholar] [CrossRef] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bolcato, G.; Pavan, M.; Bassani, D.; Sturlese, M.; Moro, S. Ribose and Non-Ribose A2A Adenosine Receptor Agonists: Do They Share the Same Receptor Recognition Mechanism? Biomedicines 2022, 10, 515. https://doi.org/10.3390/biomedicines10020515
Bolcato G, Pavan M, Bassani D, Sturlese M, Moro S. Ribose and Non-Ribose A2A Adenosine Receptor Agonists: Do They Share the Same Receptor Recognition Mechanism? Biomedicines. 2022; 10(2):515. https://doi.org/10.3390/biomedicines10020515
Chicago/Turabian StyleBolcato, Giovanni, Matteo Pavan, Davide Bassani, Mattia Sturlese, and Stefano Moro. 2022. "Ribose and Non-Ribose A2A Adenosine Receptor Agonists: Do They Share the Same Receptor Recognition Mechanism?" Biomedicines 10, no. 2: 515. https://doi.org/10.3390/biomedicines10020515