
Cardiogenic Pulmonary Edema in Emergency Medicine
 , ,
, ,  , , ,
, , ,
Abstract
:Highlights
- Cardiogenic pulmonary edema is the most common cause of respiratory failure and results from increased cardiac filling pressure and alveolar-epithelial barrier breakdown due to factors like inflammation, leukocyte infiltration, procoagulant processes, and ion channel modification by reactive oxygen and nitrogen species.
- Rapid evaluation of the patient with cardiogenic pulmonary edema and appropriate treatment are necessary to reduce mortality and morbidity.
- Bedside tests, especially ultrasound, help us to quickly identify and to understand the need of amine support. Optimal therapy for patients with cardiogenic pulmonary edema hinges on precise phenotypic identification and etiological assessment, with early consideration of non-invasive ventilation and diuretics.
- Hypoperfusion necessitates inotropes and, at times, vasopressors, while individuals exhibiting persistent symptoms and diuretic resistance may benefit from vasodilators and additional therapeutic strategies, including beta-agonists, underscoring the need for further studies to explore medical interventions in mitigating pulmonary damage in CPE patients.
Abstract
1. Introduction
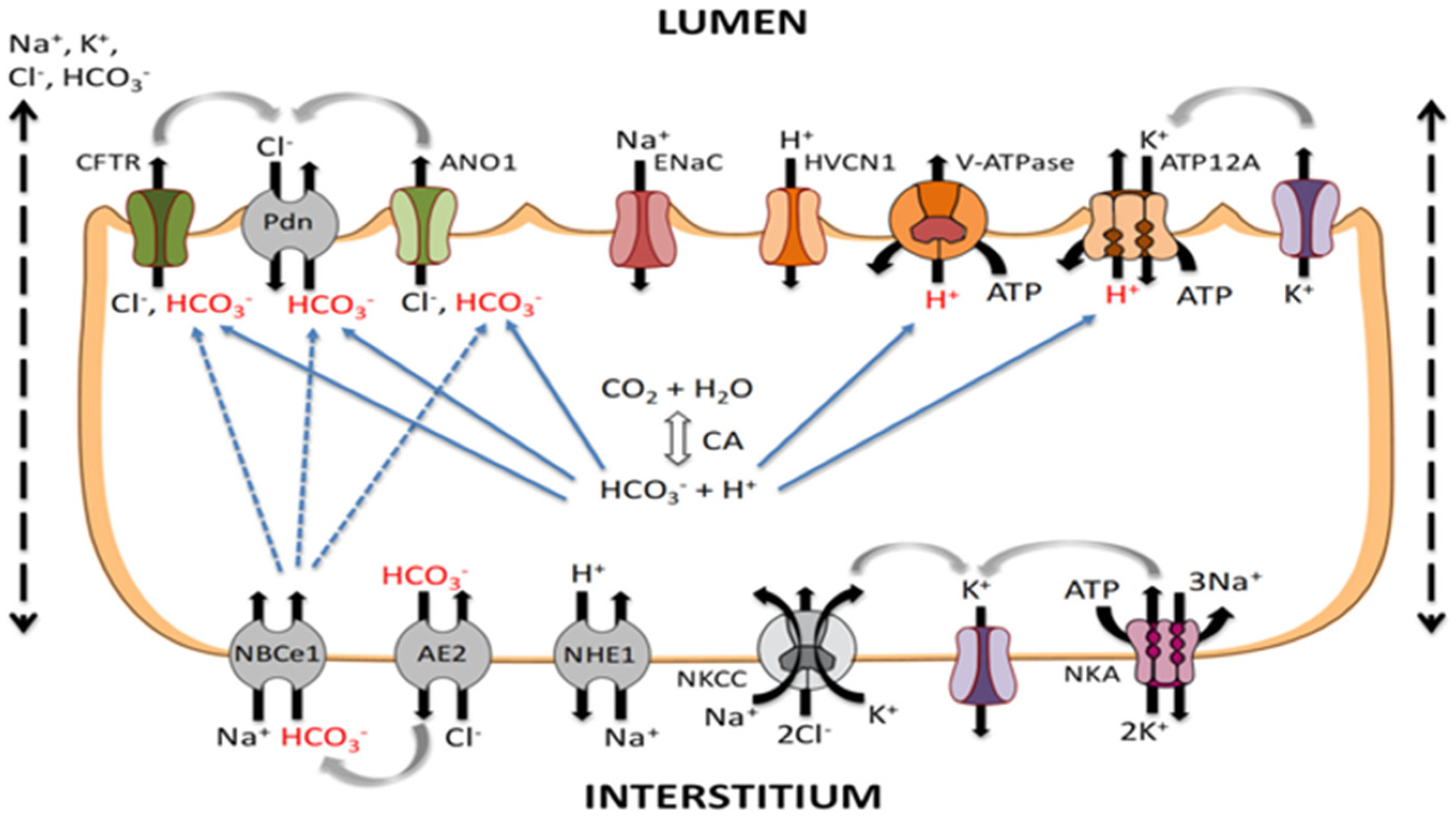
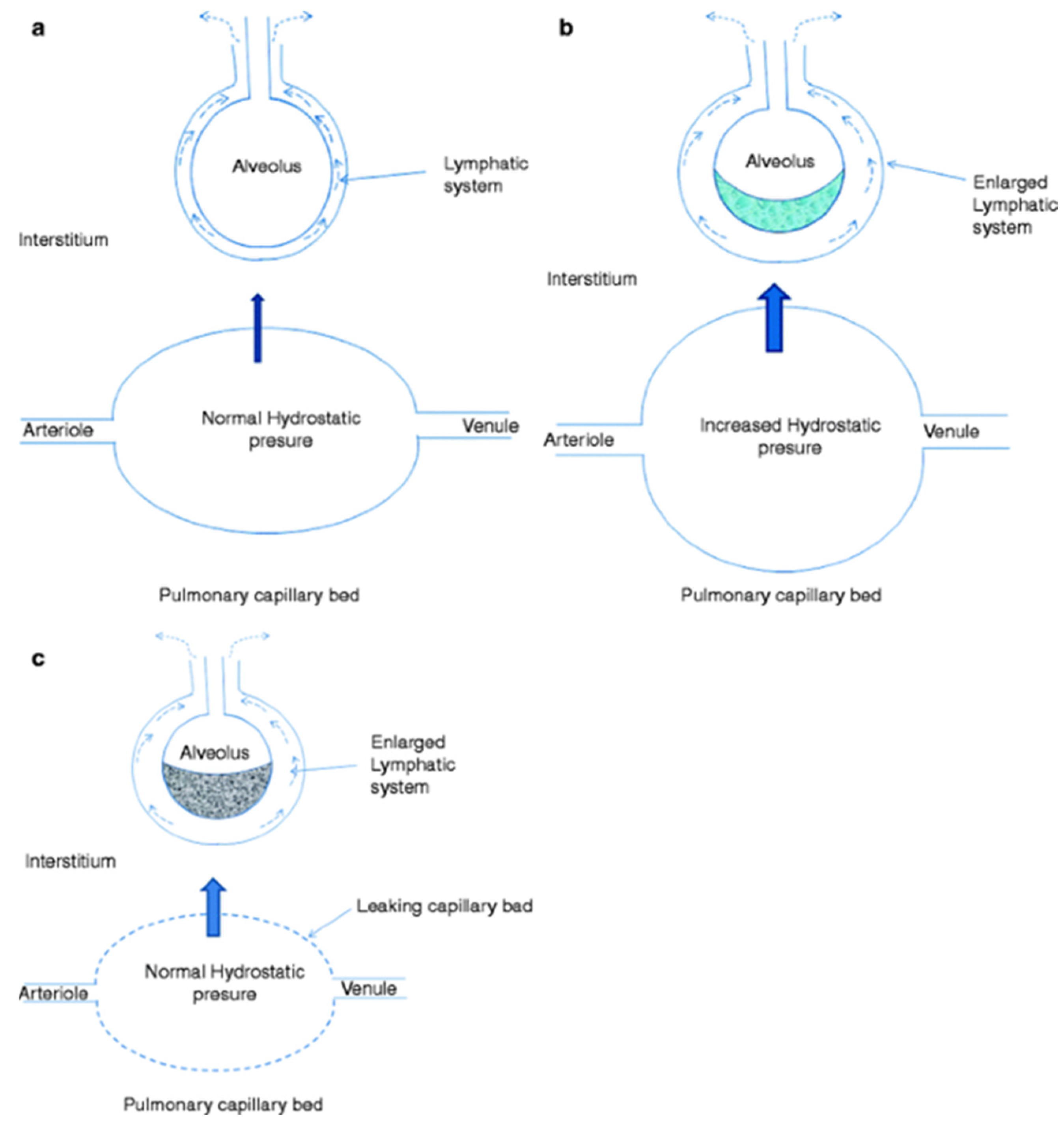
2. Pathophysiology of Pulmonary Edema
3. Etiology of Cardiogenic Pulmonary Edema
4. Clinical Manifestations and Diagnostic Process
4.1. History and Physical Examination
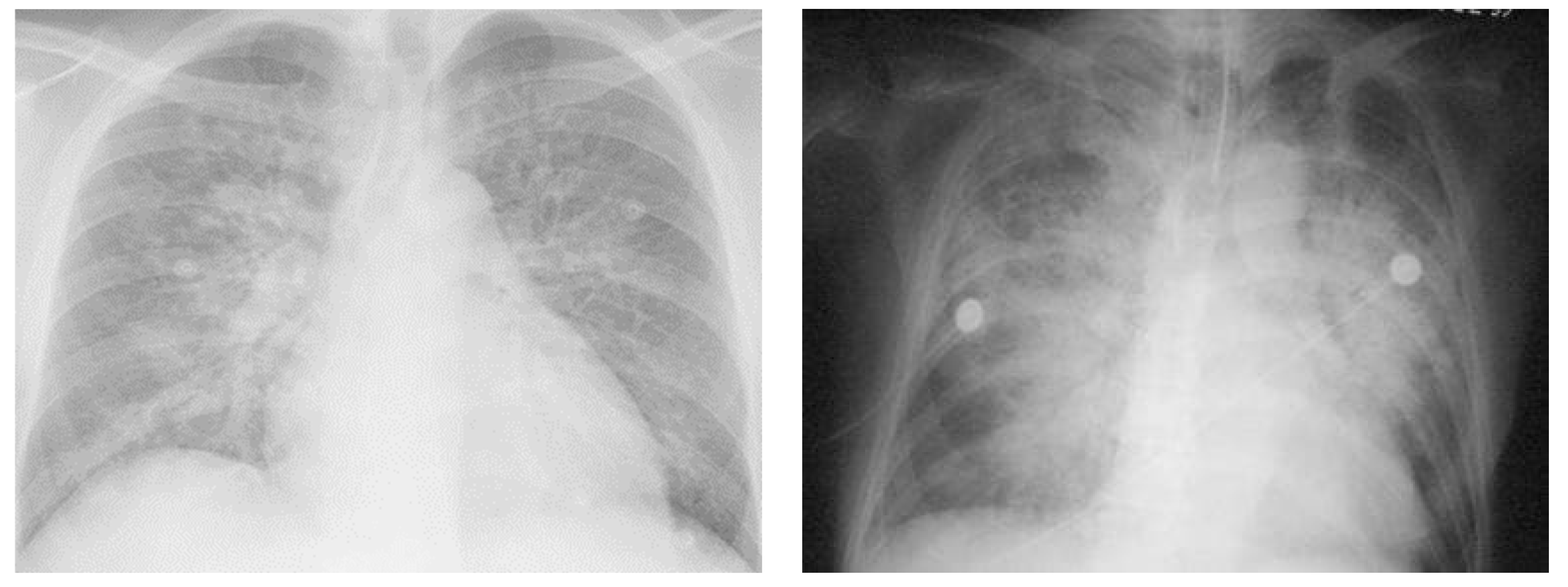
4.2. Tests Available for Diagnosing Pulmonary Edema
5. Treatment
5.1. Supplemental Oxygen and Assisted Ventilation
5.2. Diuretic Therapy
5.3. Ultrafiltration
5.4. Vasodilator Therapy
5.5. Management of Arrhythmias
5.6. Management of Hypotensive Patients
5.7. Additional Management Strategies
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gheorghiade, M.; Zannad, F.; Sopko, G.; Klein, L.; Piña, I.L.; Konstam, M.A.; Massie, B.M.; Roland, E.; Targum, S.; Collins, S.P.; et al. International Working Group on Acute Heart Failure Syndromes. Acute heart failure syndromes: Current state and framework for future research. Circulation 2005, 112, 3958–3968. [Google Scholar] [CrossRef] [PubMed]
- Nieminen, M.S.; Brutsaert, D.; Dickstein, K.; Drexler, H.; Follath, F.; Harjola, V.P.; Hochadel, M.; Komajda, M.; Lassus, J.; Lopez-Sendon, J.L.; et al. EuroHeart Survey Investigators; Heart Failure Association, European Society of Cardiology. EuroHeart Failure Survey II (EHFS II): A survey on hospitalized acute heart failure patients: Description of population. Eur. Heart J. 2006, 27, 2725–2736. [Google Scholar] [CrossRef] [PubMed]
- Crane, S.D. Epidemiology, treatment and outcome of acidotic, acute, cardiogenic pulmonary oedema presenting to an emergency department. Eur. J. Emerg. Med. 2002, 9, 320–324. [Google Scholar] [CrossRef] [PubMed]
- Wiener, R.S.; Moses, H.W.; Richeson, J.F.; Gatewood, R.P., Jr. Hospital and long-term survival of patients with acute pulmonary edema associated with coronary artery disease. Am. J. Cardiol. 1987, 60, 33–35. [Google Scholar] [CrossRef] [PubMed]
- Ware, L.B.; Matthay, M.A. Clinical practice. Acute pulmonary edema. N. Engl. J. Med. 2005, 353, 2788–2796. [Google Scholar] [CrossRef]
- Rocca, E.; Zanza, C.; Longhitano, Y.; Piccolella, F.; Romenskaya, T.; Racca, F.; Savioli, G.; Saviano, A.; Piccioni, A.; Mongodi, S. Lung Ultrasound in Critical Care and Emergency Medicine: Clinical Review. Adv. Respir. Med. 2023, 91, 203–223. [Google Scholar] [CrossRef]
- Gri, N.; Longhitano, Y.; Zanza, C.; Monticone, V.; Fuschi, D.; Piccioni, A.; Bellou, A.; Esposito, C.; Ceresa, I.F.; Savioli, G. Acute Oncologic Complications: Clinical-Therapeutic Management in Critical Care and Emergency Departments. Curr. Oncol. 2023, 30, 7315–7334. [Google Scholar] [CrossRef]
- Zanza, C.; Facelli, V.; Romenskaya, T.; Bottinelli, M.; Caputo, G.; Piccioni, A.; Franceschi, F.; Saviano, A.; Ojetti, V.; Savioli, G.; et al. Lactic Acidosis Related to Pharmacotherapy and Human Diseases. Pharmaceuticals 2022, 15, 1496. [Google Scholar] [CrossRef]
- Dobbe, L.; Rahman, R.; Elmassry, M.; Paz, P.; Nugent, K. Cardiogenic Pulmonary Edema. Am. J. Med. Sci. 2019, 358, 389–397. [Google Scholar] [CrossRef]
- Yancy, C.W.; Jessup, M.; Bozkurt, B.; Butler, J.; Casey, D.E., Jr.; Drazner, M.H.; Fonarow, G.C.; Geraci, S.A.; Horwich, T.; Januzzi, J.L.; et al. American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. 2013 ACCF/AHA guideline for the management of heart failure: A report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation 2013, 128, e240–e327. [Google Scholar] [CrossRef]
- Rochwerg, B.; Brochard, L.; Elliott, M.W.; Hess, D.; Hill, N.S.; Nava, S.; Navalesi, P.; Antonelli, M.; Brozek, J.; Conti, G.; et al. Official ERS/ATS clinical practice guidelines: Noninvasive ventilation for acute respiratory failure. Eur. Respir. J. 2017, 50, 1602426. [Google Scholar] [CrossRef] [PubMed]
- De Pasquale, C.G.; Arnolda, L.F.; Doyle, I.R.; Grant, R.L.; Aylward, P.E.; Bersten, A.D. Prolonged alveolocapillary barrier damage after acute cardiogenic pulmonary edema. Crit. Care Med. 2003, 31, 1060–1067. [Google Scholar] [CrossRef] [PubMed]
- Sliwa, K.; Woodiwiss, A.; Libhaber, E.; Zhanje, F.; Libhaber, C.; Motara, R.; Essop, R. C-reactive protein predicts response to pentoxifylline in patients with idiopathic dilated cardiomyopathy. Eur. J. Heart Fail. 2004, 6, 731–734. [Google Scholar] [CrossRef] [PubMed]
- Shaw, S.M.; Shah, M.K.; Williams, S.G.; Fildes, J.E. Immunological mechanisms of pentoxifylline in chronic heart failure. Eur. J. Heart Fail. 2009, 11, 113–118. [Google Scholar] [CrossRef]
- Herrero, R.; Sanchez, G. New insights into the mechanisms of pulmonary edema in acute lung injury. Ann. Transl. Med. 2018, 6, 32. [Google Scholar] [CrossRef]
- Iles, K.E.; Song, W.; Miller, D.W.; Dickinson, D.A.; Matalon, S. Reactive species and pulmonary edema. Expert Rev. Respir. Med. 2009, 3, 487–496. [Google Scholar] [CrossRef]
- Racca, F.; Longhitano, Y.; Zanza, C.; Draisci, G.; Stoia, P.A.; Gollo, E.; Maio, M.; Grattarola, C.; Astuto, M.; Vaschetto, R.; et al. Peri-Partum respiratory management in neuro-muscular disorders (IT-NEUMA-Pregn study): A proposal by an italian panel and a call for an international collaboration. Pulmonology 2023, S2531-0437. [Google Scholar] [CrossRef]
- Murray, J.F. Pulmonary edema: Pathophysiology and diagnosis. Int. J. Tuberc. Lung Dis. 2011, 15, 155–160. [Google Scholar]
- Szidon, J.P. Pathophysiology of the congested lung. Cardiol. Clin. 1989, 7, 39–48. [Google Scholar] [CrossRef]
- Gropper, M.A.; Wiener-Kronish, J.P.; Hashimoto, S. Acute cardiogenic pulmonary edema. Clin. Chest Med. 1994, 15, 501–515. [Google Scholar] [CrossRef]
- Günther, A.; Siebert, C.; Schmidt, R.; Ziegler, S.; Grimminger, F.; Yabut, M.; Temmesfeld, B.; Walmrath, D.; Morr, H.; Seeger, W. Surfactant alterations in severe pneumonia, acute respiratory distress syndrome, and cardiogenic lung edema. Am. J. Respir. Crit. Care Med. 1996, 153, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.C.; Zentner, M.D.; Deng, H.T.; Kim, K.J.; Wu, R.; Yang, P.C.; Ann, D.K. Oxidative stress disrupts glucocorticoid hormone-dependent transcription of the amiloride-sensitive epithelial sodium channel α-subunit in lung epithelial cells through ERK-dependent and thioredoxin-sensitive pathways. J. Biol. Chem. 2000, 24, 8600–8609. [Google Scholar] [CrossRef] [PubMed]
- Matthay, M.A.; Ware, L.B.; Zimmerman, G.A. The acute respiratory distress syndrome. J. Clin. Investig. 2012, 122, 2731–2740. [Google Scholar] [CrossRef] [PubMed]
- Zemans, R.L.; Matthay, M.A. Bench-to-bedside review: The role of the alveolar epithelium in the resolution of pulmonary edema in acute lung injury. Crit. Care 2004, 8, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Vivona, M.L.; Matthay, M.; Chabaud, M.B.; Friedlander, G.; Clerici, C. Hypoxia reduces alveolar epithelial sodium and fluid transport in rats: Reversal by β-adrenergic agonist treatment. Am. J. Respir. Cell Mol. Biol. 2001, 25, 554–561. [Google Scholar] [CrossRef] [PubMed]
- Roux, J.; Kawakatsu, H.; Gartland, B.; Pespeni, M.; Sheppard, D.; Matthay, M.A.; Canessa, C.M.; Pittet, J.F. Interleukin-1β decreases expression of the epithelial sodium channel α-subunit in alveolar epithelial cells via a p38 MAPK-dependent signaling pathway. J. Biol. Chem. 2005, 280, 18579–18589. [Google Scholar] [CrossRef] [PubMed]
- Herrero, R.; Kajikawa, O.; Matute-Bello, G.; Wang, Y.; Hagimoto, N.; Mongovin, S.; Wong, V.; Park, D.R.; Brot, N.; Heinecke, J.W.; et al. The biological activity of FasL in human and mouse lungs is determined by the structure of its stalk region. J. Clin. Investig. 2011, 121, 1174–1190. [Google Scholar] [CrossRef]
- Herrero, R.; Tanino, M.; Smith, L.S.; Kajikawa, O.; Wong, V.A.; Mongovin, S.; Matute-Bello, G.; Martin, T.R. The Fas/FasL pathway impairs the alveolar fluid clearance in mouse lungs. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 305, L377–L388. [Google Scholar] [CrossRef]
- Perl, M.; Lomas-Neira, J.; Chung, C.-S.; Ayala, A. Epithelial cell apoptosis and neutrophil recruitment in acute lung injury-a unifying hypothesis? What we have learned from small interfering RNAs. Mol. Med. 2008, 14, 465–475. [Google Scholar] [CrossRef]
- Elia, N.; Tapponnier, M.; Matthay, M.A.; Hamacher, J.; Pache, J.-C.; Bründler, M.-A.; Totsch, M.; De Baetselier, P.; Fransen, L.; Fukuda, N.; et al. Functional identification of the alveolar edema reabsorption activity of murine tumor necrosis factor-α. Am. J. Respir. Crit. Care Med. 2003, 168, 1043–1050. [Google Scholar] [CrossRef]
- Dagenais, A.; Fréchette, R.; Yamagata, Y.; Yamagata, T.; Carmel, J.-F.; Clermont, M.-E.; Brochiero, E.; Massé, C.; Berthiaume, Y. Downregulation of ENaC activity and expression by TNF-α in alveolar epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 286, L301–L311. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.S.; Choi, Y.H.; Kim, Y.S.; Baik, S.H.; Oh, Y.J.; Sheen, S.S.; Park, J.H.; Hwang, S.C.; Park, K.J. Evaluation of bronchoalveolar lavage fluid from ARDS patients with regard to apoptosis. Respir. Med. 2008, 102, 464–469. [Google Scholar] [CrossRef] [PubMed]
- Alwi, I. Diagnosis and management of cardiogenic pulmonary edema. Acta Medica Indones. 2010, 42, 176–184. [Google Scholar]
- Piper, S.E.; McDonagh, T.A. Chemotherapy-related Cardiomyopathy. Eur. Cardiol. 2015, 10, 19–24. [Google Scholar] [CrossRef]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. ESC Scientific Document Group. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. Heart J. 2021, 42, 3599–3726. [Google Scholar] [CrossRef]
- Al Deeb, M.; Barbic, S.; Featherstone, R.; Dankoff, J.; Barbic, D. Point-of-care ultrasonography for the diagnosis of acute cardiogenic pulmonary edema in patients presenting with acute dyspnea: A systematic review and meta-analysis. Acad. Emerg. Med. 2014, 21, 843–852. [Google Scholar] [CrossRef]
- Thomas, S.S.; Nohria, A. Hemodynamic classifications of acute heart failure and their clinical application—An update. Circ. J. 2012, 76, 278–286. [Google Scholar] [CrossRef]
- Gluecker, T.; Capasso, P.; Schnyder, P.; Gudinchet, F.; Schaller, M.D.; Revelly, J.P.; Chiolero, R.; Vock, P.; Wicky, S. Clinical and radiologic features of pulmonary edema. Radiographics 1999, 19, 1507–1531, discussion 1532–1533. [Google Scholar] [CrossRef]
- Attias, D.; Mansencal, N.; Auvert, B.; Vieillard-Baron, A.; Delos, A.; Lacombe, P.; N’Guetta, R.; Jardin, F.; Dubourg, O. Prevalence, characteristics, and outcomes of patients presenting with cardiogenic unilateral pulmonary edema. Circulation 2010, 122, 1109–1115. [Google Scholar] [CrossRef]
- Agricola, E.; Bove, T.; Oppizzi, M.; Marino, G.; Zangrillo, A.; Margonato, A.; Picano, E. “Ultrasound comet-tail images”: A marker of pulmonary edema: A comparative study with wedge pressure and extravascular lung water. Chest 2005, 127, 1690–1695. [Google Scholar] [CrossRef]
- Coiro, S.; Porot, G.; Rossignol, P.; Ambrosio, G.; Carluccio, E.; Tritto, I.; Huttin, O.; Lemoine, S.; Sadoul, N.; Donal, E.; et al. Prognostic value of pulmonary congestion assessed by lung ultrasound imaging during heart failure hospitalisation: A two-centre cohort study. Sci. Rep. 2016, 6, 39426, Erratum in Sci. Rep. 2017, 7, 43972. [Google Scholar] [CrossRef] [PubMed]
- Volpicelli, G.; Elbarbary, M.; Blaivas, M.; Lichtenstein, D.A.; Mathis, G.; Kirkpatrick, A.W.; Melniker, L.; Gargani, L.; Noble, V.E.; Via, G.; et al. International Liaison Committee on Lung Ultrasound (ILC-LUS) for International Consensus Conference on Lung Ultrasound (ICC-LUS). International evidence-based recommendations for point-of-care lung ultrasound. Intensive Care Med. 2012, 38, 577–591. [Google Scholar] [CrossRef] [PubMed]
- Maisel, A.S.; Krishnaswamy, P.; Nowak, R.M.; McCord, J.; Hollander, J.E.; Duc, P.; Omland, T.; Storrow, A.B.; Abraham, W.T.; Wu, A.H.; et al. Breathing Not Properly Multinational Study Investigators. Rapid measurement of B-type natriuretic peptide in the emergency diagnosis of heart failure. N. Engl. J. Med. 2002, 347, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Heart Failure Society of America; Lindenfeld, J.; Albert, N.M.; Boehmer, J.P.; Collins, S.P.; Ezekowitz, J.A.; Givertz, M.M.; Katz, S.D.; Klapholz, M.; Moser, D.K.; et al. HFSA 2010 Comprehensive Heart Failure Practice Guideline. J. Card. Fail. 2010, 16, e1–e2. [Google Scholar] [CrossRef] [PubMed]
- Hunt, S.A.; Abraham, W.T.; Chin, M.H.; Feldman, A.M.; Francis, G.S.; Ganiats, T.G.; Jessup, M.; Konstam, M.A.; Mancini, D.M.; Michl, K.; et al. 2009 focused update incorporated into the ACC/AHA 2005 Guidelines for the Diagnosis and Management of Heart Failure in Adults: A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines: Developed in collaboration with the International Society for Heart and Lung Transplantation. Circulation 2009, 119, e391–e479. [Google Scholar] [CrossRef]
- Park, J.H.; Balmain, S.; Berry, C.; Morton, J.J.; McMurray, J.J. Potentially detrimental cardiovascular effects of oxygen in patients with chronic left ventricular systolic dysfunction. Heart 2010, 96, 533–538. [Google Scholar] [CrossRef]
- Racca, F.; Geraci, C.; Cremascoli, L.; Ruvolo, D.; Piccolella, F.; Romenskaya, T.; Longhitano, Y.; Martuscelli, E.; Saviano, A.; Savioli, G.; et al. Invasive Mechanical Ventilation in Traumatic Brain Injured Patients with Acute Respiratory Failure. Rev. Recent Clin. Trials. 2023, 18, 3–11. [Google Scholar] [CrossRef]
- Williams, J.W., Jr.; Cox, C.E.; Hargett, C.W.; Gilstrap, D.L.; Castillo, C.E.; Govert, J.A.; Lugogo, N.L.; Coeytaux, R.R.; McCrory, D.C.; Hasselblad, V.; et al. Noninvasive Positive-Pressure Ventilation (NPPV) for Acute Respiratory Failure [Internet]; Report No.: 12-EHC089-EF; Agency for Healthcare Research and Quality (US): Rockville, MD, USA, 2012. [Google Scholar]
- Masip, J.; Roque, M.; Sánchez, B.; Fernández, R.; Subirana, M.; Expósito, J.A. Noninvasive ventilation in acute cardiogenic pulmonary edema: Systematic review and meta-analysis. JAMA 2005, 294, 3124–3130. [Google Scholar] [CrossRef]
- Winck, J.C.; Azevedo, L.F.; Costa-Pereira, A.; Antonelli, M.; Wyatt, J.C. Efficacy and safety of non-invasive ventilation in the treatment of acute cardiogenic pulmonary edema--a systematic review and meta-analysis. Crit. Care 2006, 10, R69. [Google Scholar] [CrossRef]
- Collins, S.P.; Mielniczuk, L.M.; Whittingham, H.A.; Boseley, M.E.; Schramm, D.R.; Storrow, A.B. The use of noninvasive ventilation in emergency department patients with acute cardiogenic pulmonary edema: A systematic review. Ann. Emerg. Med. 2006, 48, 260–269.e1-4. [Google Scholar] [CrossRef]
- Gray, A.; Goodacre, S.; Newby, D.E.; Masson, M.; Sampson, F.; Nicholl, J.; 3CPO Trialists. Noninvasive ventilation in acute cardiogenic pulmonary edema. N. Engl. J. Med. 2008, 359, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Weng, C.L.; Zhao, Y.T.; Liu, Q.H.; Fu, C.J.; Sun, F.; Ma, Y.L.; Chen, Y.W.; He, Q.Y. Meta-analysis: Noninvasive ventilation in acute cardiogenic pulmonary edema. Ann. Intern. Med. 2010, 152, 590–600. [Google Scholar] [CrossRef] [PubMed]
- Zanza, C.; Longhitano, Y.; Leo, M.; Romenskaya, T.; Franceschi, F.; Piccioni, A.; Pabon, I.M.; Santarelli, M.T.; Racca, F. Practical Review of Mechanical Ventilation in Adults and Children in the Operating Room and Emergency Department. Rev. Recent Clin. Trials 2022, 17, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Vital, F.M.; Ladeira, M.T.; Atallah, A.N. Non-invasive positive pressure ventilation (CPAP or bilevel NPPV) for cardiogenic pulmonary oedema. Cochrane Database Syst. Rev. 2013, CD005351, Update in Cochrane Database Syst. Rev. 2019, 4, CD005351. [Google Scholar] [CrossRef]
- Rusterholtz, T.; Kempf, J.; Berton, C.; Gayol, S.; Tournoud, C.; Zaehringer, M.; Jaeger, A.; Sauder, P. Noninvasive pressure support ventilation (NIPSV) with face mask in patients with acute cardiogenic pulmonary edema (ACPE). Intensive Care Med. 1999, 25, 21–28. [Google Scholar] [CrossRef]
- Liesching, T.; Nelson, D.L.; Cormier, K.L.; Sucov, A.; Short, K.; Warburton, R.; Hill, N.S. Randomized trial of bilevel versus continuous positive airway pressure for acute pulmonary edema. J. Emerg. Med. 2014, 46, 130–140. [Google Scholar] [CrossRef]
- Felker, G.M.; O’Connor, C.M.; Braunwald, E. Heart Failure Clinical Research Network Investigators. Loop diuretics in acute decompensated heart failure: Necessary? Evil? A necessary evil? Circ. Heart Fail. 2009, 2, 56–62. [Google Scholar] [CrossRef]
- Dikshit, K.; Vyden, J.K.; Forrester, J.S.; Chatterjee, K.; Prakash, R.; Swan, H.J. Renal and extrarenal hemodynamic effects of furosemide in congestive heart failure after acute myocardial infarction. N. Engl. J. Med. 1973, 288, 1087–1090. [Google Scholar] [CrossRef]
- Felker, G.M.; Lee, K.L.; Bull, D.A.; Redfield, M.M.; Stevenson, L.W.; Goldsmith, S.R.; LeWinter, M.M.; Deswal, A.; Rouleau, J.L.; Ofili, E.O.; et al. NHLBI Heart Failure Clinical Research Network. Diuretic strategies in patients with acute decompensated heart failure. N. Engl. J. Med. 2011, 364, 797–805. [Google Scholar] [CrossRef]
- Costanzo, M.R.; Guglin, M.E.; Saltzberg, M.T.; Jessup, M.L.; Bart, B.A.; Teerlink, J.R.; Jaski, B.E.; Fang, J.C.; Feller, E.D.; Haas, G.J.; et al. UNLOAD Trial Investigators. Ultrafiltration versus intravenous diuretics for patients hospitalized for acute decompensated heart failure. J. Am. Coll. Cardiol. 2007, 49, 675–683. [Google Scholar] [CrossRef]
- National Clinical Guideline Centre (UK). Acute Heart Failure: Diagnosing and Managing Acute Heart Failure in Adults; National Institute for Health and Care Excellence: London, UK, 2014. [Google Scholar]
- Hofmann, R.; Steinwender, C.; Kammler, J.; Kypta, A.; Wimmer, G.; Leisch, F. Intravenous amiodarone bolus for treatment of atrial fibrillation in patients with advanced congestive heart failure or cardiogenic shock. Wien. Klin. Wochenschr. 2004, 116, 744–749. [Google Scholar] [CrossRef] [PubMed]
- McMurray, J.J.; Adamopoulos, S.; Anker, S.D.; Auricchio, A.; Böhm, M.; Dickstein, K.; Falk, V.; Filippatos, G.; Fonseca, C.; Gomez-Sanchez, M.A.; et al. ESC Committee for Practice Guidelines. ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure 2012: The Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure 2012 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association (HFA) of the ESC. Eur. Heart J. 2012, 33, 1787–1847. [Google Scholar] [CrossRef] [PubMed]
- Tarvasmäki, T.; Lassus, J.; Varpula, M.; Sionis, A.; Sund, R.; Køber, L.; Spinar, J.; Parissis, J.; Banaszewski, M.; Silva Cardoso, J.; et al. CardShock study investigators. Current real-life use of vasopressors and inotropes in cardiogenic shock—Adrenaline use is associated with excess organ injury and mortality. Crit. Care 2016, 20, 208. [Google Scholar] [CrossRef] [PubMed]
- Léopold, V.; Gayat, E.; Pirracchio, R.; Spinar, J.; Parenica, J.; Tarvasmäki, T.; Lassus, J.; Harjola, V.P.; Champion, S.; Zannad, F.; et al. Epinephrine and short-term survival in cardiogenic shock: An individual data meta-analysis of 2583 patients. Intensive Care Med. 2018, 44, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Mebazaa, A.; Nieminen, M.S.; Packer, M.; Cohen-Solal, A.; Kleber, F.X.; Pocock, S.J.; Thakkar, R.; Padley, R.J.; Põder, P.; Kivikko, M. SURVIVE Investigators. Levosimendan vs dobutamine for patients with acute decompensated heart failure: The SURVIVE Randomized Trial. JAMA 2007, 297, 1883–1891. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W. beta2 adrenergic agonists in acute lung injury? The heart of the matter. Crit. Care 2009, 13, 1011. [Google Scholar] [CrossRef]
- Taylor, B.J.; Snyder, E.M.; Richert, M.L.; Wheatley, C.M.; Chase, S.C.; Olson, L.J.; Johnson, B.D. Effect of β2-adrenergic receptor stimulation on lung fluid in stable heart failure patients. J. Heart Lung Transplant. 2017, 36, 418–426. [Google Scholar] [CrossRef]
- Velez, M. Advances in contemporary medical management to treat patients with heart failure. Curr. Opin. Cardiol. 2023, 38, 136–142. [Google Scholar] [CrossRef]
- Cox, Z.L.; Collins, S.P.; Aaron, M.; Hernandez, G.A., III; Davidson, B.T.; Fowler, M.; Lindsell, C.J.; Frank, E.H., Jr.; A Jenkins, C.A. Efficacy and safety of dapagliflozin in acute heart failure: Rationale and design of the DICTATE-AHF trial. Am. Heart J. 2021, 232, 116–124. [Google Scholar] [CrossRef]
- Carballo, S.; Stirnemann, J.; Garin, N.; Darbellay Farhoumand, P.; Serratrice, J.; Carballo, D. Prognosis of patients eligible for dapagliflozin in acute heart failure. Eur. J. Clin. Investig. 2020, 50, e13245. [Google Scholar] [CrossRef]
- Longhitano, Y.; Zanza, C.; Romenskaya, T.; Saviano, A.; Persiano, T.; Leo, M.; Piccioni, A.; Betti, M.; Maconi, A.; Pindinello, I.; et al. Single-Breath Counting Test Predicts Non-Invasive Respiratory Support Requirements in Patients with COVID-19 Pneumonia. J. Clin. Med. 2021, 11, 179. [Google Scholar] [CrossRef] [PubMed]
- Zanza, C.; Romenskaya, T.; Zuliani, M.; Piccolella, F.; Bottinelli, M.; Caputo, G.; Rocca, E.; Maconi, A.; Savioli, G.; Longhitano, Y. Acute Traumatic Pain in the Emergency Department. Diseases 2023, 11, 45. [Google Scholar] [CrossRef] [PubMed]
- Copetti, R.; Soldati, G.; Copetti, P. Chest sonography: A useful tool to differentiate acute cardiogenic pulmonary edema from acute respiratory distress syndrome. Cardiovasc. Ultrasound 2008, 6, 16. [Google Scholar] [CrossRef] [PubMed]
- Savioli, G.; Ceresa, I.F.; Manzoni, F.; Ricevuti, G.; Bressan, M.A.; Oddone, E. Role of a Brief Intensive Observation Area with a Dedicated Team of Doctors in the Management of Acute Heart Failure Patients: A Retrospective Observational Study. Medicina 2020, 56, 251. [Google Scholar] [CrossRef]
- Ceresa, I.F.; Savioli, G.; Angeli, V.; Novelli, V.; Muzzi, A.; Grugnetti, G.; Cobianchi, L.; Manzoni, F.; Klersy, C.; Lago, P.; et al. Preparing for the Maximum Emergency with a Simulation: A Table-Top Test to Evaluate Bed Surge Capacity and Staff Compliance with Training. Open Access Emerg. Med. 2020, 12, 377–387. [Google Scholar] [CrossRef]
- Piccioni, A.; Valletta, F.; Franza, L.; Rosa, F.; Manca, F.; Zanza, C.; Savioli, G.; Gasbarrini, A.; Covino, M.; Franceschi, F. Evaluation of procalcitonin in hemorrhagic shock: A pilot study. Clin. Ter. 2023, 174, 432–435. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zanza, C.; Saglietti, F.; Tesauro, M.; Longhitano, Y.; Savioli, G.; Balzanelli, M.G.; Romenskaya, T.; Cofone, L.; Pindinello, I.; Racca, G.; et al. Cardiogenic Pulmonary Edema in Emergency Medicine. Adv. Respir. Med. 2023, 91, 445-463. https://doi.org/10.3390/arm91050034
Zanza C, Saglietti F, Tesauro M, Longhitano Y, Savioli G, Balzanelli MG, Romenskaya T, Cofone L, Pindinello I, Racca G, et al. Cardiogenic Pulmonary Edema in Emergency Medicine. Advances in Respiratory Medicine. 2023; 91(5):445-463. https://doi.org/10.3390/arm91050034
Chicago/Turabian StyleZanza, Christian, Francesco Saglietti, Manfredi Tesauro, Yaroslava Longhitano, Gabriele Savioli, Mario Giosuè Balzanelli, Tatsiana Romenskaya, Luigi Cofone, Ivano Pindinello, Giulia Racca, and et al. 2023. "Cardiogenic Pulmonary Edema in Emergency Medicine" Advances in Respiratory Medicine 91, no. 5: 445-463. https://doi.org/10.3390/arm91050034