
Unveiling the Reaction Mechanism of the Das/Chechik/Marek Synthesis of Stereodefined Quaternary Carbon Centers


1
FP-ENAS/Faculdade de Ciências da Saúde, Universidade Fernando Pessoa, Rua Carlos da Maia, 296, 4200-150 Porto, Portugal
2
UCIBIO@REQUIMTE, BioSIM, Departamento de Biomedicina, Faculdade de Medicina, Universidade do Porto, 4200-319 Porto, Portugal
*
Author to whom correspondence should be addressed.
Appl. Sci. 2021, 11(11), 5002; https://doi.org/10.3390/app11115002
Submission received: 13 May 2021
/
Revised: 26 May 2021
/
Accepted: 27 May 2021
/
Published: 28 May 2021
(This article belongs to the Special Issue The Application of Quantum Mechanics in Reactivity of Molecules II)
Abstract
:The reaction mechanism of the Cu+-catalyzed introduction of two all-carbon-substituted stereocenters in an ynamide system using a Grignard reagent, a zinc carbenoid, and an aldehyde, was investigated using density-functional theory. In contrast to the formation of an organocopper(I) compound and subsequent carbocupration reaction, previously postulated as the initial step, the reaction proved to instead proceed through an initial complexation of the substrate alkyne bond by the Cu+-catalyst, which primes this bond for reaction with the Grignard reagent. Subsequent addition of the zinc carbenoid then enables the nucleophilic attack on the incoming aldehyde, which is revealed as the rate-limiting step. Our computations have also identified the factors governing the regio- and setereoselectivity of this interesting reaction, and suggest possible paths for its further development.
1. Introduction
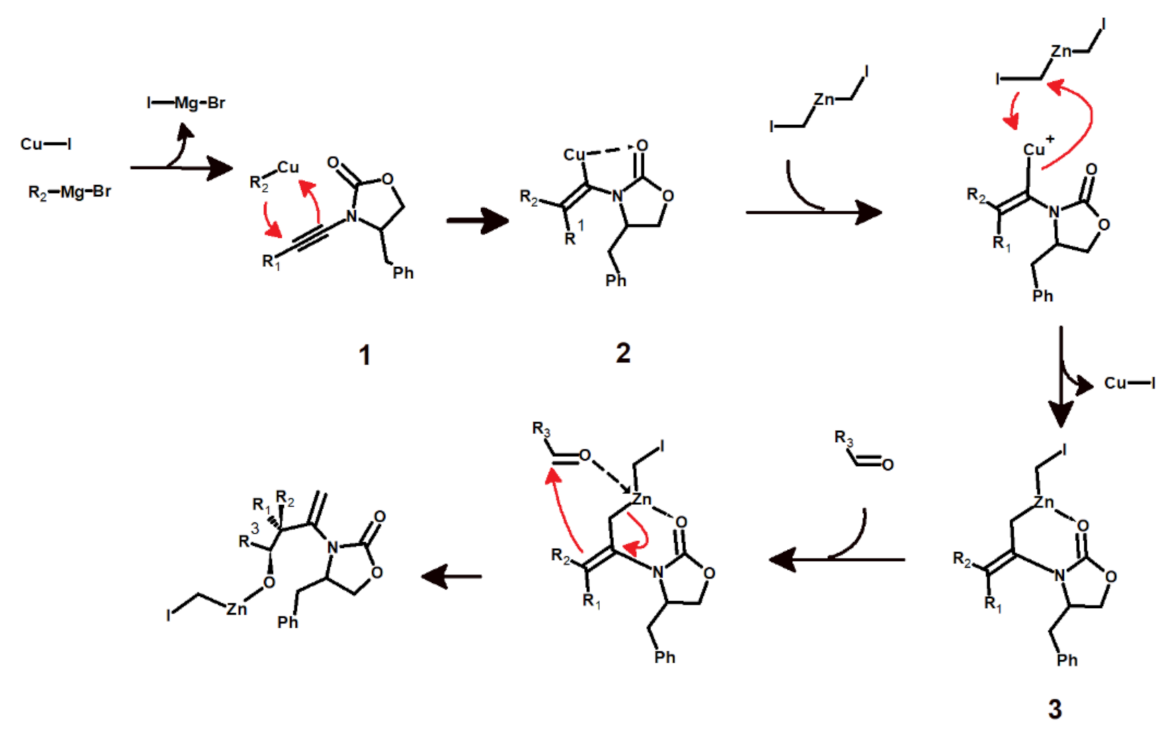
Ten years ago, Chechik, Das, and Marek developed a simple method to generate stereodefined quaternary all-carbon-substituted carbon centers in acyclic systems [1]. The strategy relies on the initial formation of a novel C–C bond through the addition of an organometallic compound to an ynamide 1, a reaction where the required stereocontrol is postulated to be provided by the complexation of the metal atom by the carbonyl moiety of the ynamide (Figure 1). Since the immediate addition of an aldehyde to provide the last substituent in the quaternary carbon is strongly disfavored, the carbometallated ynamide 2 is instead reacted with the Simmons–Smith–Furukawa zinc carbenoid (ICH2ZnCH2I), yielding a much more reactive allylzinc compound 3 which is susceptible to addition of a carbonyl compound to provide the last substituent at the quaternary center. This step is stereocontrolled, presumably through the formation of a Zimmerman–Traxler transition state (a six-membered ring transition state adopting a chair conformation) involving an interaction between the carbonyl oxygen and the zinc atom.
This reaction has been extensively cited since, and was subsequently modified to yield stereo-defined tri-substituted enolates [2]. In this contribution, we analyze the proposed reaction mechanism using computational methods. Our computations characterized the precise nature of the different transition states, identified the rate-determining step, and showed that, instead of the proposed pathway, the initial carbo-metalation event proceeds through the formation of a π-complex between CuI and the alkyne, followed by direct methylation of the alkyne bond by the unmodified Grignard reagent.
2. Materials and Methods
The reaction mechanism was investigated using the PBEPW91 functional [3,4], which has been shown to afford good geometric and/or energetic agreement with high-level CCSD(T) or MP2 benchmarking computations in similar Cu+-containing model systems [5,6]. All geometry optimizations were performed using auto-generated delocalized coordinates [7] with the Firefly quantum chemistry package [8], which is partially based on the GAMESS (US) code [9]. The SBKJ pseudo-potential (and associated basis set) [10] was used for Cu, Zn, Br, and I. A medium-sized basis set, 6-31G(d), was used for all other elements. Transition states were confirmed to contain a single imaginary frequency. IRC computations confirmed that the obtained transition states did connect the relevant reactant and product states. Zero-point and thermal effects on the free energies at 298.15 K were computed at the optimized geometries. DFT energies of the optimized geometries obtained with each density functional were then computed using the same functional using 6-311G(2d,p) for all elements except Cu and Zn (which used the s6-31G * basis set developed by Swart et al. [11] and the halogens Br and I, which used 6-311G(d,p). Unless otherwise noted, all energy values described in the text include solvation effects in tetrahydrofuran (ε = 7.6) computed using the Polarizable Continuum Model [12,13,14] implemented in Firefly, as well as dispersion and repulsion interactions with the continuum solvent, which were computed using the method developed by Amovili and Mennucci [15]. Average local ionization energies [16] and distribution of electrostatic potentials on the molecular surface [17] were computed using Multiwfn 3.6 [18]. To facilitate the exploration of a large number of possible pathways, we used a truncated model of the reagent, where R1 substituent on the alkyne is a simple methyl group and the bulky phenylmethyl substituent on the oxazolindinone ring was replaced by a methyl group. Numerical simulation of the kinetic behavior predicted by the relative energies of the pre-reactional complexes and transition states was performed using COPASI [19].
3. Results
3.1. Formation of Methylcopper(I)
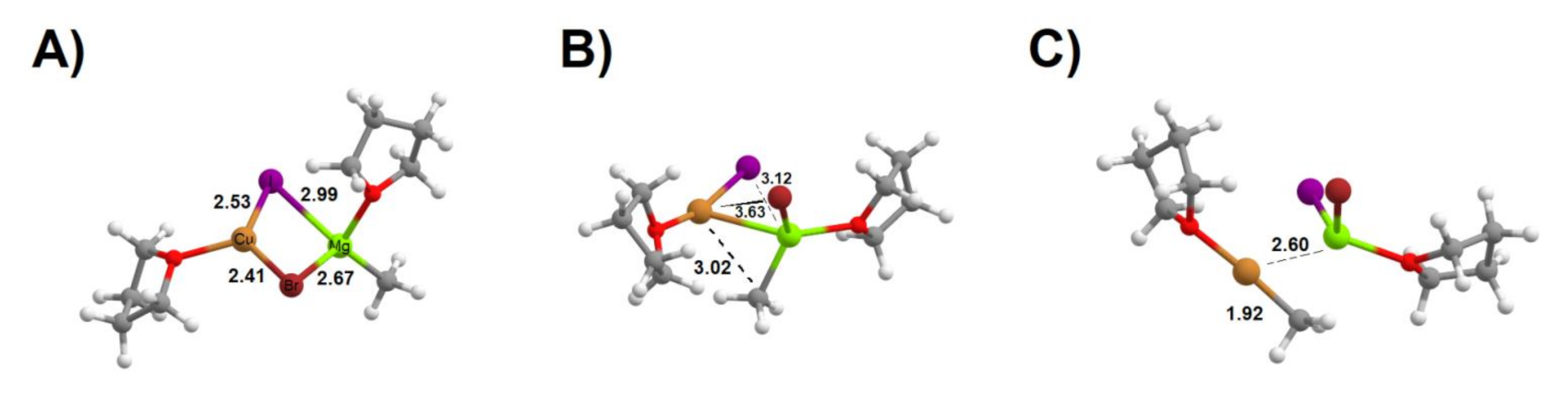
In the mechanism proposed be Das et al., the reaction is carried out in tetrahydrofuran (THF) solution. The ynamide first reacts with CuCH3 prepared in situ through the reaction of CuI with MgCH3Br. Our DFT computations showed that CuCH3 synthesis from THF-complexed CuI and CH3MgBr is exergonic by 11.8 kcal·mol−1 and that it proceeds readily with a barrier of only 9.6 kcal·mol−1. It begins through the formation of a pre-reactional complex between the THF-solvated CuI and CH3MgBr, where the metals and halogens form a quadrilateral arrangement with each halogen about equally bound to each metal and a metal–metal distance of 2.85 Å (Figure 2A). The ensuing electronic arrangement allows the Mg–CH3 bond to easily vibrate out of the plane formed by the metals and halogens and its distance from Cu to shorten from 4.64 Å away to 3.02 Å. At this point, the transition state is reached and the methyl group effortlessly breaks its bond to Mg and becomes solely bound to Cu (Figure 2B). The product of this step contains a CuCH3 loosely bound to the iodine, which then gets completely transferred to the Mg atom (Figure 2C) at a cost of 1.9 kcal·mol−1.
3.2. Carbocupration of the Alkyne Bond
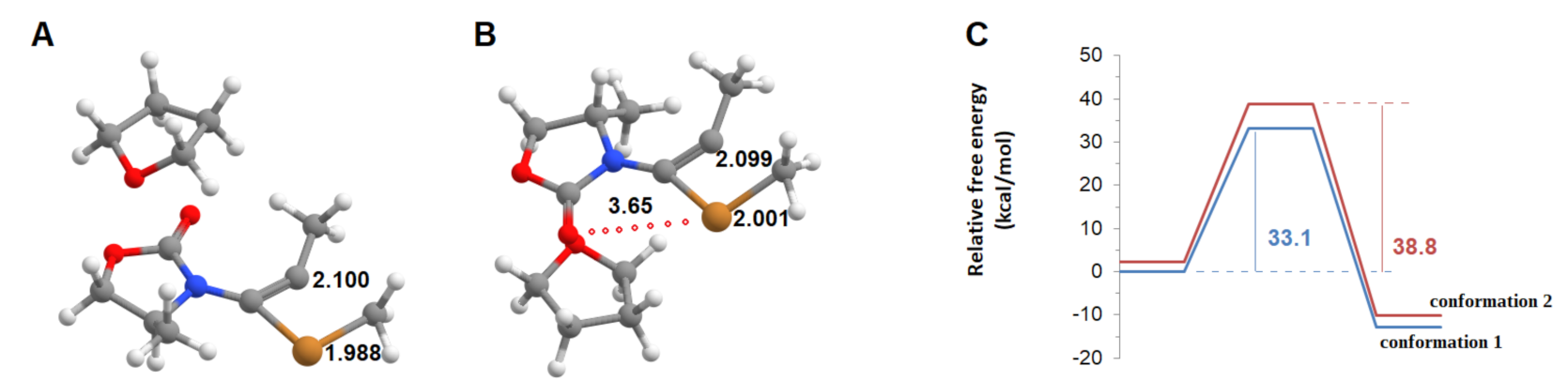
Addition of the CuCH3 to the ynamide triple bond may subsequently occur in two distinct ways, depending on the orientation of the Cu atom relative to the ynamide carbonyl (Figure 3). In conformation 1, no interaction between copper and the ynamide oxygen exists, whereas in conformation 2, the metal atom initially interacts with oxygen through a 2.72 Å bond. Conformation 1 is only slightly more stable (by 2.3 kcal·mol−1) than conformation 2. Methyl transfer from CuCH3 to the alkyne may occur from either of these conformations. In both cases, the transition state is reached when the methyl group lies 2.10 Å from the receiving carbon atom. Distances between the methyl group and the Cu atom hardly differ (by 0.01 Å) in the transition states arising from each conformation. Activation energies are, in both cases, extremely high (Figure 3C) and are incompatible with an experimentally viable mechanism, since they translate to reaction rates between 3.5 × 10−12 and 2.1 × 10−16 s−1 at 298 K. The addition of the methyl group to the ynamide must therefore follow a different mechanism.
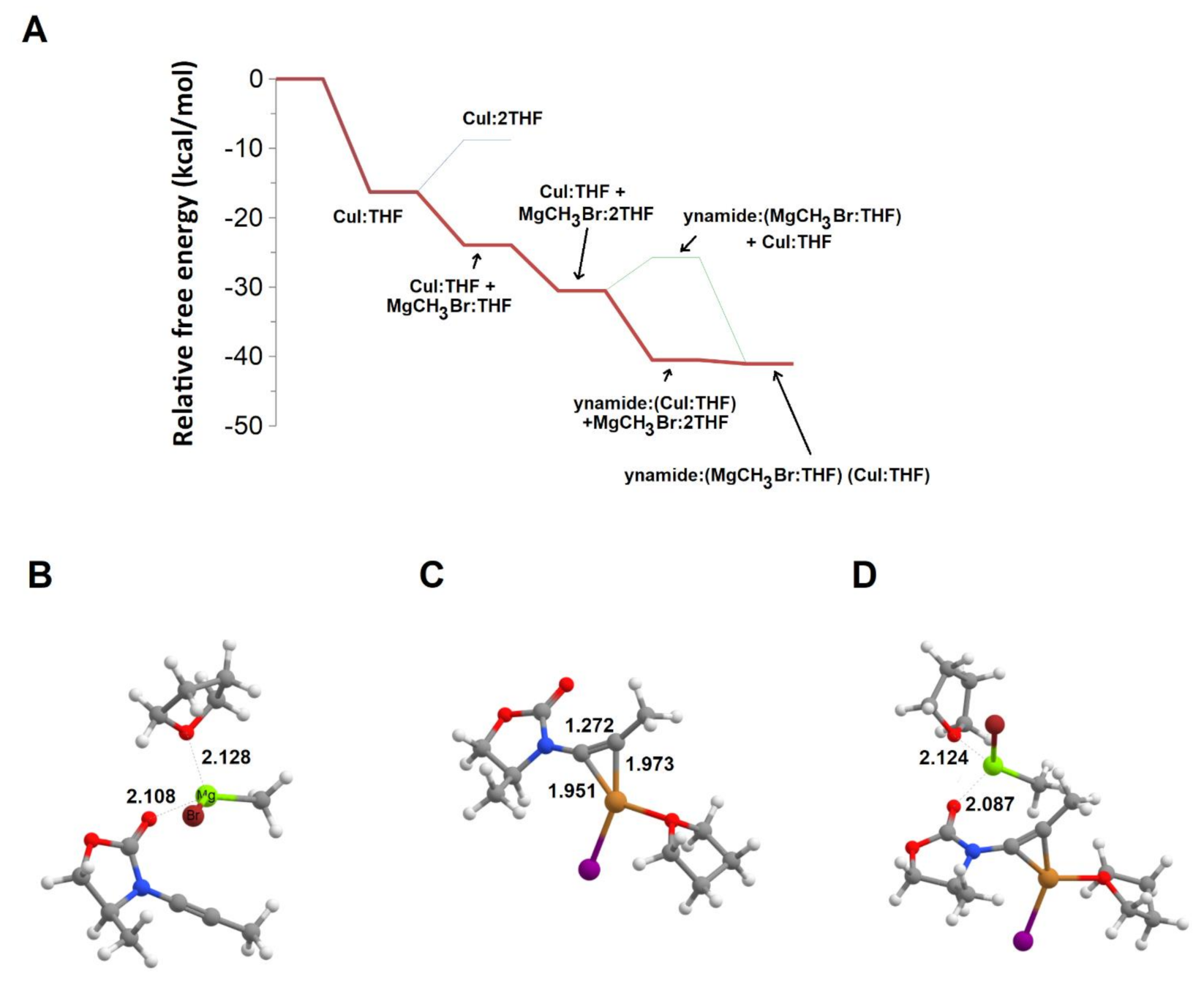
We therefore analyzed the pathways arising from direct complexation of the ynamide substrate with THF-solvated CuI or MgCH3Br. Analysis of the solvation energetics shows that the free energy change upon solvation of CuI by a single THF is favorable by 16.3 kcal·mol−1, and that the addition of a second THF molecule to the complex is strongly disfavored because the small decrease in electronic energy afforded by the formation of the new Cu–O bond is more than offset by the decrease of system entropy caused by the decrease of molecularity of the system (Figure 4A). CH3MgBr, in contrast, exists as a complex with two solvent molecules due to the comparable electronic energy decreases entailed by the formation of each of the Mg–O bonds, which are in both cases more than enough to offset the entropic losses. The complexation of MgCH3Br:2THF with the substrate carbonyl (Figure 4B) proceeds through the ejection of one of the metal-solvating THF molecules and therefore entails an increase in the entropy of the system. The reaction is, however, slightly disfavored, by 4.8 kcal·mol−1, showing that the interaction of MgCH3Br with the ynamide carbonyl is weaker than its interaction with the THF molecule that has been released. The π-complexation of Cu+ by the ynamine alkyne bond (Figure 4C), in contrast, is exergonic by 10.0 kcal·mol−1 and proceeds without a barrier. It is therefore much more likely for CuI to complex the ynamide than to undergo the metathesis described above to yield CuCH3. Subsequent addition of MgCH3Br to the Cu+-complexed ynamide (Figure 4D) then becomes exergonic (by 0.5 kcal·mol−1 for MgCH3Br:2THF and 7.1 kcal·mol−1 for MgCH3Br:THF) and yields the reactive ynamide: (CuI:THF):(MgCH3Br:THF) complex where direct transfer of the methyl group from the organomagnesium to the alkyne bond can occur. Interestingly, the energetics of the addition of CuI:THF and MgCH3Br:THF to the ynamide show that the process is cooperative: ynamide complexation by both metal compounds is exergonic by 17.1 kcal·mol−1, which is higher than the sum of each of the isolated additions of CuI:THF (10.0 kcal·mol−1) and MgCH3Br:THF (1.8 kcal·mol−1) to the bare ynamide. Interaction of each individual metal complex therefore facilitates the addition of the second complex, by triggering an electronic distribution that either facilitates reaction of CuI with the alkyne bond (in the case of an initial reaction with MgCH3Br) or else renders the carbonyl more nucleophilic to facilitate its reaction with Mg2+ (when CuI adds to the alkyne bond). Analysis of the geometry of the CuI-complexed (or Mg-complexed) ynamides (Figure 4B,C) suggests that this effect is at least partly due to the lower ability of the lone pair of the nitrogen losing the ability to resonate with the alkyne π-bonds in both situations: in the Cu+–alkyne complex, the π-bond which (in the free ynamide) partially overlaps with the lone pair orbital now interacts mostly with CuI, and the remaining alkyne π-bond lies almost perpendicular to the nitrogen lone pair orbital. The lone pair can therefore resonate more strongly with the carbonyl, leaving its oxygen atom more nucleophilic and enabling a better bond with the Mg2+. Conversely, in the Mg+-complexed ynamide this electron pair has partly delocalized into the N–C=O moiety, which decreases the HOMO–LUMO gap and facilitates subsequent attack by CuI.
3.3. Analysis of the Regioselectivity of Methyl Transfer
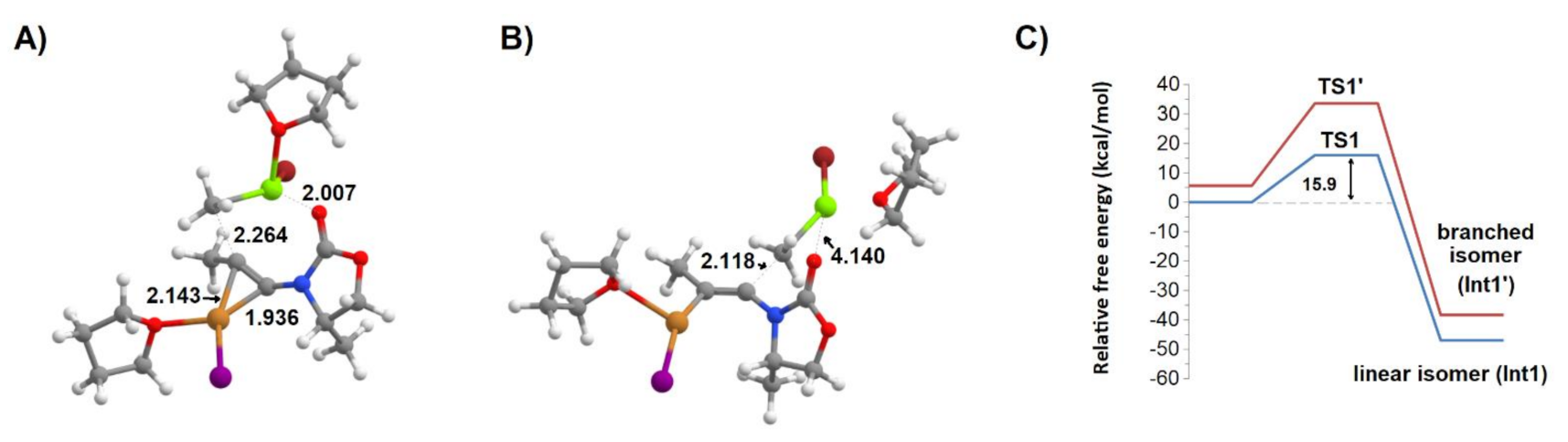
The methyl group may be transferred to either end of the alkyne bond. To maintain consistency with the nomenclature common in the field [20,21], we will refer to the methyl addition to the heteroatom-substituted alkyne carbon as “the branched reaction”, and refer to the methyl addition to the other end as “the linear reaction”. Analysis of the potential energy surface reveals that the transition state (TS1) leading to the experimentally-obtained “linear” intermediate Int1 (Figure 5A) is 17.6 kcal·mol−1 more stable than the transition state (TS1′) leading to the “branched” isomer (Int1′) (Figure 5B). The transition states differ most markedly in the length of the bonds between Cu+ and the alkyne bond atoms and in the presence or absence of a Mg2+–carbonyl bond (which is not compatible with the short distance between the organometallic methyl and the ring-proximal alkyne carbon atom in TS1′). The reaction products arising from these transition states differ in energy by a non-negligible amount (8.6 kcal·mol−1), in spite of having very similar Mg–O and Cu+–alkyne distances. This suggested that other factors govern the relative stabilities of the “linear” and “branched” isomers. Analysis of the energies of the different fragments in these intermediates (the organic moiety, CuI–THF and MgBr–THF) shows that most of the extra stability of the linear isomer is due to the higher stability of the linear organic moiety, rather than (as postulated earlier) on the existence of more favorable interactions between the organic portion and the metals in the linear isomer (Table 1). Those interactions are, however, crucial in the kinetic preference of the “linear” pathway over the pathway leading to the “branched” product.
3.4. Homologation of Intermediate 1 with ICH2ZnCH2I
Intermediate 1 is very crowded around the C=C double bond (Figure 6A). Addition of other substituents to this bond to generate the desired quaternary compound must therefore await the ejection of the MgBr moiety. This step is performed through the intervention of the THF solvent molecules, which will replace the carbonyl as Mg2+ ligands. In a system containing a truncated model of Intermediate 1 (where the CuI-complexed moiety has been removed to decrease computational cost) with four extra THF molecules, MgBr removal proceeds readily through simple elongation of the Mg2+–carbonyl bond from 2.18 Å to 2.66 Å. At this point (11.7 kcal·mol−1 above the initial state) the transition state is reached and the Mg2+-ynamide bond breaks, whereupon a hexagonal Mg2+Br:5THF species is released. This reaction is readily reversible because the end species is only 0.5 kcal·mol−1 more stable than the initial state.
The attack of this Mg-free intermediate (Int1) complexed by CuI by the Simmons–Smith–Furukawa zinc carbenoid (ICH2ZnCH2I) may proceed from different directions, which can be (roughly) defined by the position of the carbenoid relative to the nucleophilic C=O moiety on the oxazolidinone ring and by the dihedral angle between the C=C bond and the plane of the ring. Since this vast array of possibilities considerably complicates the description of the results, we will first analyze the formation of intermediate 2 through each of the attack vectors, and then discuss the possible routes of interconversion between the different forms of intermediate 2.
3.4.1. Attack through the Direction Opposite the Oxazolidinone Carbonyl
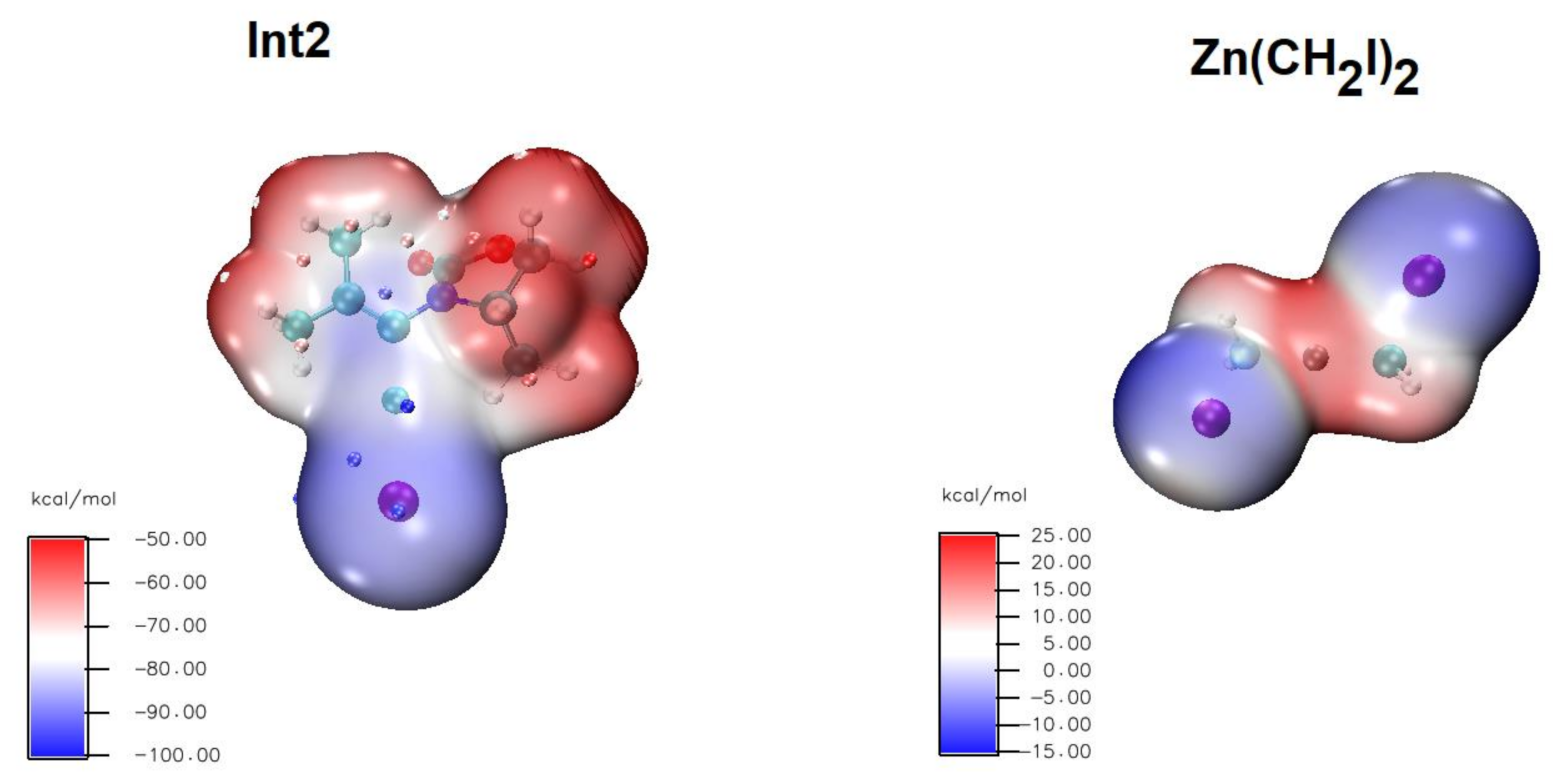
The pre-reactional complex arising from the attack through the face opposite the oxazolidinone carbonyl (Figure 6B) is appreciably more stable (by 10.5 kcal·mol−1) than the infinitely-separated reactants due to the electrostatic attraction between the positively-charged Zn2+ and the anionic intermediate 1. Surprisingly, our exploration of the subsequent potential energy surface showed that the CH2 transfer from the zinc carbenoid to the C=C bond in this case takes place through the direct intervention of Cu+ (Figure 6C). This preference can be rationalized by examination of the average local ionization energies (Figure 7A) of the Int2 intermediate, which feature an absolute minimum (i.e., highest nucleophilic character) on the Cu+, which will therefore be the location with the highest propensity to attack the relatively electrophilic CH2 group (Figure 7B) on ICH2ZnCH2I. In the transition state, located only 10.7 kcal·mol−1 above the pre-reactional complex, the bond order of the C–I bond has decreased to 0.45, and a half-bond has formed between the CH2 and Cu+ group. The collapse of this transition state is then effected through complete transfer of the loosely-bound iodine to the Zn2+ and formation of a new C–C bond between CH2 and the hetero-substituted carbon in the C=C double bond. This is accompanied by a partial migration of Cu+ to the other carbon of the double bond and by reduction of 0.5 in the total bond orders of each fragment. Analysis of Löwdin charge variations shows that the ZnICH2I moiety present in the product now contains 0.69 electrons more than in the beginning, of which 0.46 electrons originate in the Int2 moiety and 0.23 have come from the transferred CH2.
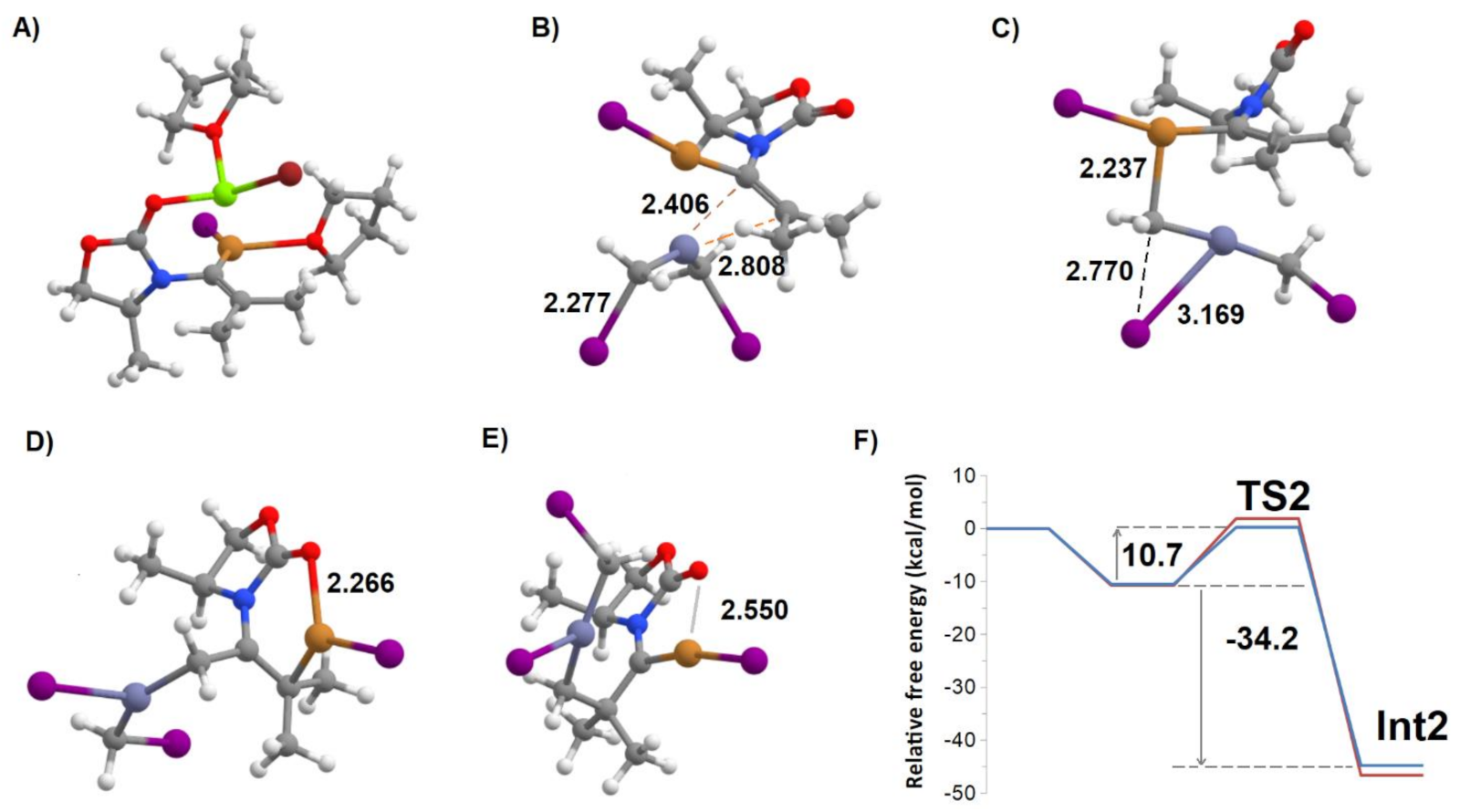
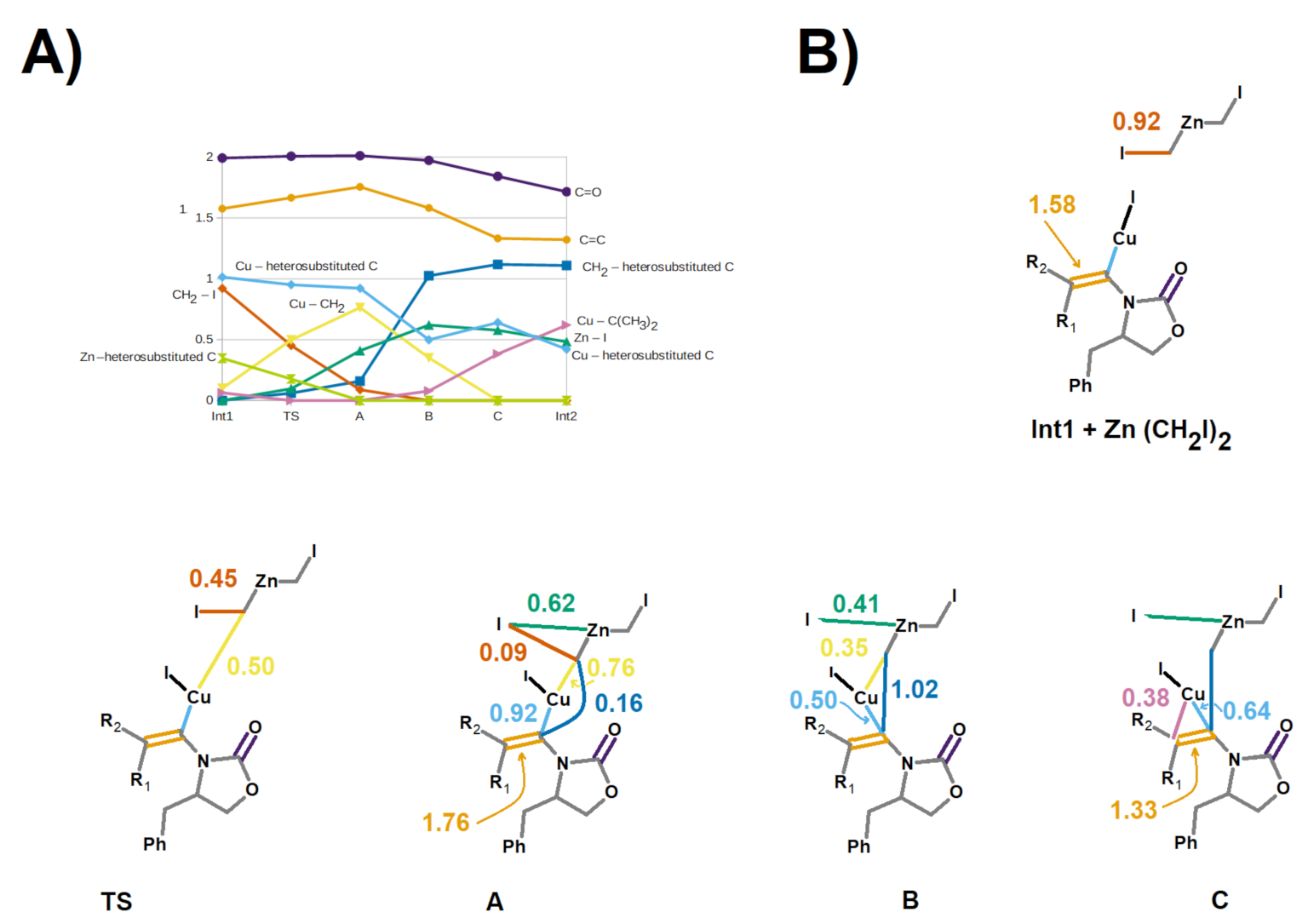
Examination of the evolution of the bond orders along the reaction coordinate leading from the transition state to Int2 (Figure 8) shows that this rearrangement occurs in three stages: first the bond order between CH2 and I decreases, concurrently with a simultaneous increase of the bond orders of the Cu–CH2 and Zn–I bonds. Then, as the CH2–I bond completely disappears, the bond orders of both the Cu–CH2 bond and the bond between Cu and the heterosubstituted ynamide carbon decrease in similar amounts to enable the formation of the bond between CH2 and the heterosubstituted carbon. This stage is accompanied by a decrease in the double-bond character of the formal C=C bond in ynamide. Finally, the remnants of the Cu–CH2 bond are replaced by the formation of a half-bond between Cu and the non-heterosubstituted carbon of the C=C bond, simultaneously with the further loss of some of its double-bond character. This complex electronic rearrangement occurs spontaneously without additional transition states, and yields a very stable intermediate (Int2) that lies 34.2 kcal·mol−1 below the pre-reactional complex. The alternative formation of an intermediate bearing the new CH2 group on the opposite carbon (Int2′) is thermodynamically much less favored, since Int2′ lies 22.3 kcal·mol−1 above Int2. Separate analysis of the energies of the individual fragments (Table 2) shows that the relative instability of Int2′ is not due to geometric constrains that affect the stabilization of the Zn-containing moiety by the oxazolidinone, but to the intrinsically higher energy of the organic moiety of Int2′ (compared to that of Int2).
Two products of the attack on Int1 are possible (Int2α and Int2β) depending on the direction of approach of the Simmons–Smith–Furukawa zinc carbenoid relative to the plane formed by the C=C bond and the coordinating CuI. Those attacks are not equally favored because in one of them (which we will designate as “attack from below the plane of the oxazolidine ring”, or “encumbered”) the attacking zinc carbenoid may clash with the substituent on the oxazolidinone ring (methyl, in our models, or the bulkier phenylmethyl in the experimental setup). Our computations reveal that, indeed, when the attack proceeds from below the ring plane, the steric interaction of the ring substituent with the zinc carbenoid increases the transition state energy by a modest amount (1.6 kcal·mol−1). Although the reaction is thermodynamically more favorable (by 1.8 kcal·mol−1) than in the attack from the direction above the ring, the high spontaneity of the reaction (exergonic by more than 34 kcal·mol−1 in both cases) entails that the reverse reactions will have too large an activation energy and the reaction outcome will therefore be kinetically-controlled (rather than thermodynamically-controlled), leading to a non-racemic mixture of the two products where the one arising from the attack of Int1 by the zinc compound from the “un-encumbered” direction (Int2α) will be predominant (Figure 6F).
3.4.2. Attack Proceeding through Zn Coordination by the Oxazolidinone Carbonyl
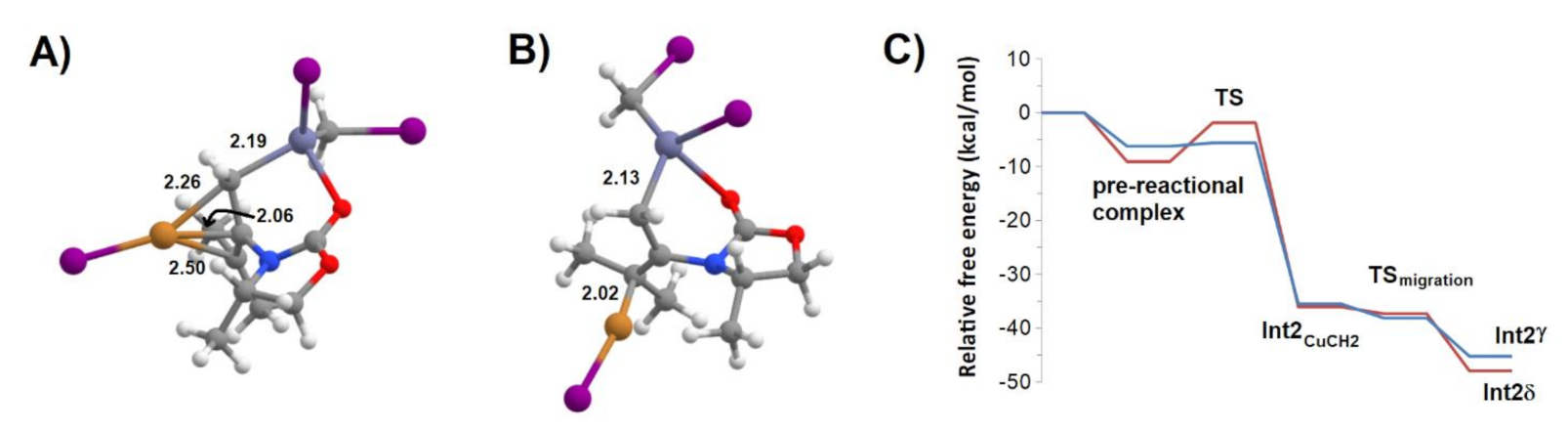
The pre-reactional complex arising from complexation of the zinc carbenoid species by the oxazolidinone ring (Figure 9A) is slightly less favored than the pre-reactional complex described in the previous section. It affords, however a more favorable path to the transfer of CH2 to Int1, with barriers as low as 0.6 kcal·mol−1 (when approaching from the direction above the plane of the ring) or 7.2 kcal·mol−1 (when approaching from the opposite direction). As in the path described in the previous section, CH2 is attacked by the excess electron density in Cu+, with concomitant weakening of the CH2–I bond and migration of this iodide to Zn (Figure 9B). However, in contrast to that path, subsequent CH2 transfer to the N-heterosubstituted carbon in the C=C bond is now accompanied by the detachment of Cu from this bond, and its reattachment to the transferred CH2 (Figure 9C). A facile Cu-migration from CH2 to the non-heterosubstituted carbon in the C=C bond (Figure 10) quickly converts this intermediate (Int2CuCH2) into Int2 isomers where (as in the Int2α and Int2β described above) Zn remains bound to the transferred CH2 group and Cu is bound to the C=C bonded atoms, and which can be distinguished from Int2α and Int2β by the existence of stabilizing interactions between Zn and the oxazolidinone ring. Numerical simulation of the kinetic behavior predicted by the relative energies of the pre-reactional complexes and transition states of their conversion into Int2 intermediates confirmed that, despite their slightly higher energy, the pre-reactional complexes where Zn2+ interacts with the carbonyl oxygen intermediates have a dominant kinetic role, because of the very small barriers of their associated transition states. Accordingly, the resulting products (Int2δ and Int2γ) will each be two to three times more abundant than Int2α, and Int2β (which arises from the most stable pre-reactional complex) will be residual due to its transition state having a larger energy than the one required for complete reagent separation (and subsequent recombination into the pre-reactive complexes leading to the other isomers).
3.5. Simultaneous Generation of Two Stereocenters through Addition of Aldehyde to Int2
Each of the four possible Int2 species may now perform a nucleophilic attack on an incoming aldehyde. This attack is aided by the Zn2+ moiety, which binds to the carbonyl in the incoming aldehyde, thereby increasing the electrophilicity of the carbonyl carbon atom and stabilizing the negative charge that accumulates on the carbonyl oxygen atom: in the absence of this coordination, the attack has an activation free energy of 32.2 kcal·mol−1, rather than the 18.8–27.9 kcal·mol−1 observed otherwise (Table 3).
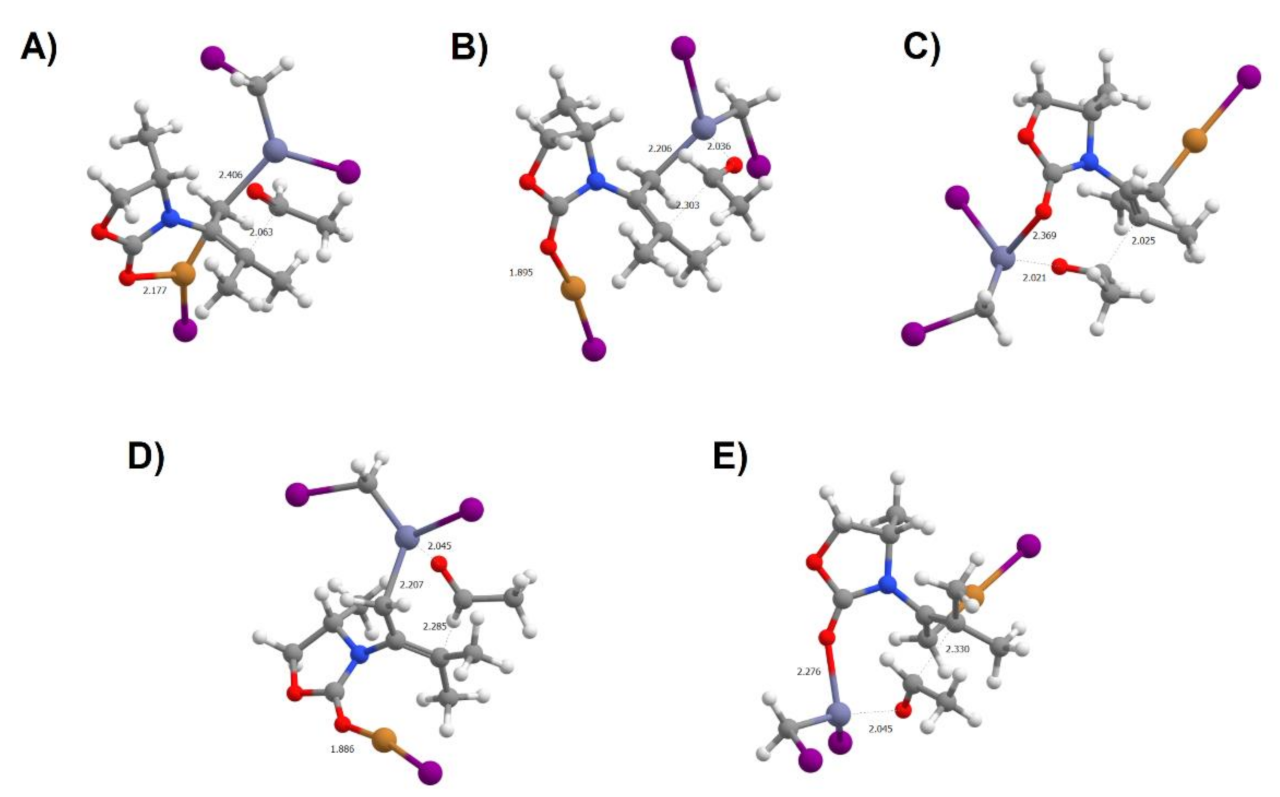
Interestingly, the position of the Cu+ ligand in the transition states arising from Int2δ and Int2γ is very similar to that in their Int2CuCH2 precursors (as shown in Figure 11, panels C and E), because complexation of the acetaldehyde carbonyl by the Zn2+ releases the Zn atom from the =CH2 group, and thereby favors the return of Cu+ to =CH2. Overall, the lowest activation energies are observed for the reactions starting from the abundant Int2γ and the (scarcer) Int2α, which lead to products with the (S) configuration in the carbon originally present on the alkyne. Different preferences are seen for the orientation of the acetaldehyde (and consequent configuration on the second stereocenter): the (S)(S) isomer is preferred by 0.5 kcal·mol−1 when the reaction proceeds from Int2α, whereas (S)(R) is preferred by 1.0 kcal·mol−1 when the reaction proceeds from Int2γ. This preference correlates with the orientation of the acetaldehyde relative to the substituents of the non-heterosubstituted alkyne bond carbon: the isomer where the hydrogen of acetaldehyde lies above them is consistently preferred due to diminished steric strain.
Production of the products bearing an (R) configuration on the stereocenter arising from the alkyne carbon is much less favorable than those with the opposite configuration (Table 3). The reasons for this behavior are varied: for the reactions starting from Int2β, the increase of the transition state energies by 5–7 kcal·mol−1 correlates with the higher proximity of the incoming acetaldehyde with the methyl substituent on the oxazolindinone ring (Figure 11D), whereas for the reactions starting from Int2δ the transition state energy is identical to that of the (favored) reactions starting from Int2α, and the large activation energies are instead due to the higher stability of Int2δ relative to the energy of the other isoforms.
4. Discussion
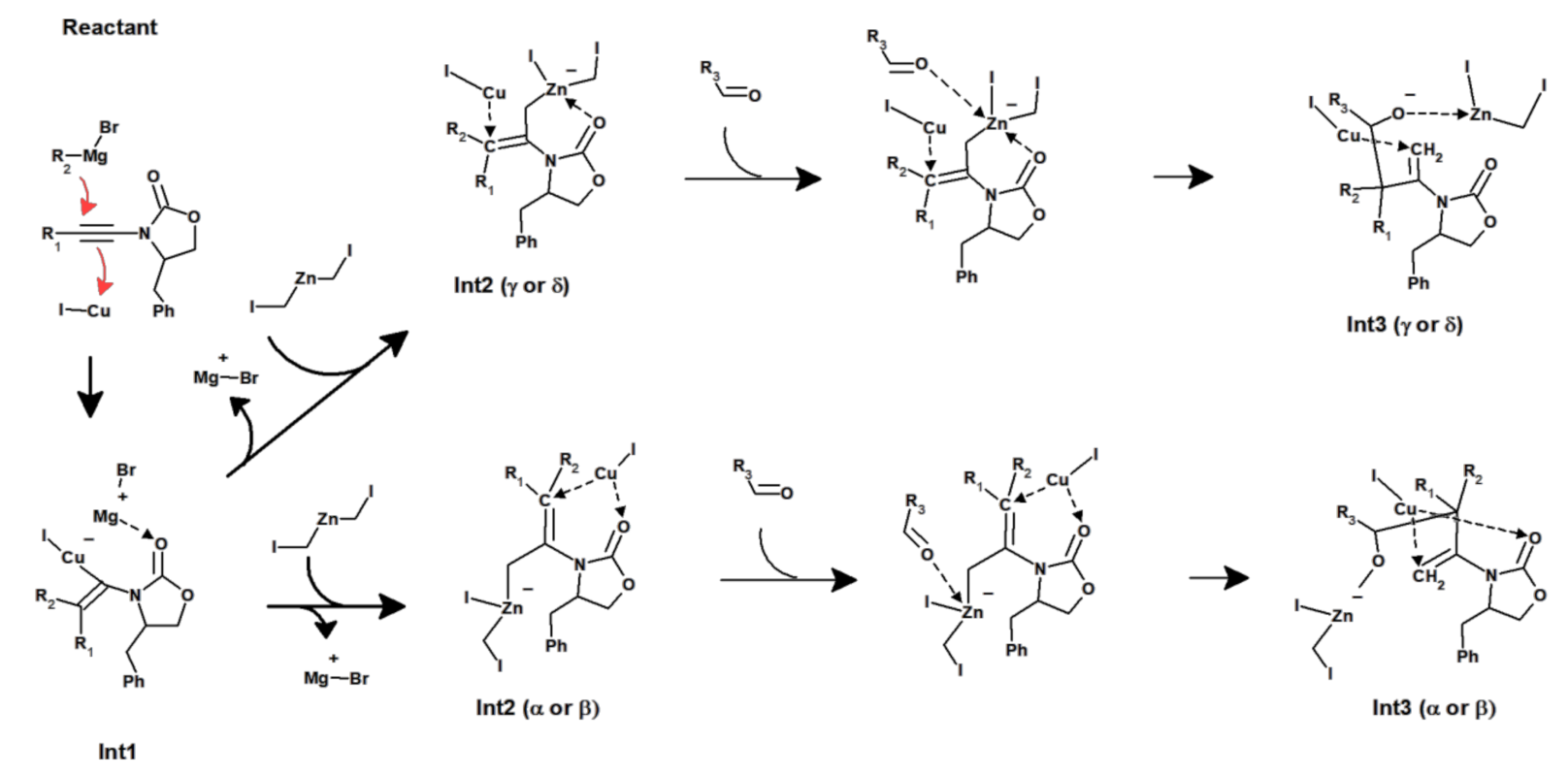
The mechanism arising from our density-functional studies (Figure 12) differs in important respects from the previously postulated reaction sequence. First, the transition state for the postulated carbocupration was shown to lie far above the energies compatible with measurable reaction rates, and the alkylation of the alkyne instead proceeds through direct alkylation of the Cu+-complexed alkyne by the Grignard reagent. Second, the removal of the MgBr+ moiety left behind by the Grignard reagent requires the active involvement of the solvent, which entails that eventual modifications of this method must retain the use of a solvent with sufficient Mg2+-chelating ability. Third, the reaction with the zinc carbenoid is not independent of the Cu+ catalyst but instead proceeds through methylene transfer to Cu+. This reaction step may, against expectation, occur either with (leading to Int2γ/δ) or without (leading to Int2α) the assistance of a Zn2+-carbonyl stabilizing interaction. Finally, the stereoselectivity of the carbonyl insertion does not arise from interactions between the aldehyde sidechains and the bulky substituent on the oxazolidinone ring: inspection of the transition states of the most favorable reactions (Figure 11B,C, and Supplementary Materials) shows that the aldehyde actually lies quite far from the oxazolidinone ring. Instead, in both cases (Int2α and Int2γ), the disfavored transition state is the one where the bulky substituent on the aldehyde lies immediately above both of the R1 and R2 substituents present on the other, newly formed, stereocenter. We expect these insights will prove useful for the continuing development of this important methodology and to further increase its scope and stereoselectivity.
Supplementary Materials
Coordinates of all molecules described in the text, as well as individual energetic data, are available online at https://www.mdpi.com/article/10.3390/app11115002/s1.
Author Contributions
C.E.P.B. and P.J.S. jointly performed the computations. P.J.S. further analyzed the results, conceived and designed the study, and wrote the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work has been financed by FEDER through Programa Operacional Factores de Competitividade–COMPETE and by Portuguese Funds through FCT–Fundação para a Ciência e a Tecnologia under project PTDC/QUI-QUI/111288/2009. This work was also supported by the Applied Molecular Biosciences Unit–UCIBIO, which is financed by national funds from FCT (UIDB/04378/2020).
Data Availability Statement
Full inputs and output files may be downloaded from figshare (DOI: 10.6084/m9.figshare.5481541, accessed date on 27 May 2021).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Das, J.P.; Chechik, H.; Marek, I.; Information, S.; Das, J.P.; Chechik, H.; Marek, I.; Stereocenters, Q.; Methods, G. A unique approach to aldol products for the creation of all-carbon quaternary stereocentres. Nat. Chem. 2009, 1, 128–132. [Google Scholar] [CrossRef] [PubMed]
- Minko, Y.; Pasco, M.; Lercher, L.; Botoshansky, M.; Marek, I. Forming all-carbon quaternary stereogenic centres in acyclic systems from alkynes. Nature 2012, 490, 522–526. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P. Unified theory of exchange and correlation beyond the local density approximation. In Electronic Structure of Solids ’91; Ziesche, P., Eschrig, H., Eds.; Akademie: Berlin, Germany, 1991; Volume 17, pp. 11–20. [Google Scholar]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernardo, C.E.P.; Bauman, N.P.; Piecuch, P.; Silva, P.J. Evaluation of density functional methods on the geometric and energetic descriptions of species involved in Cu+-promoted catalysis. J. Mol. Model. 2013, 19, 5457–5467. [Google Scholar] [CrossRef] [PubMed]
- Silva, P.J.; Bernardo, C.E.P. Influence of Alkyne and Azide Substituents on the Choice of the Reaction Mechanism of the Cu+-Catalyzed Addition of Azides to Iodoalkynes. J. Phys. Chem. A 2018, 122, 7497–7507. [Google Scholar] [CrossRef] [PubMed]
- Baker, J.; Kessi, A.; Delley, B. The generation and use of delocalized internal coordinates in geometry optimization. J. Chem. Phys. 1996, 105, 192–212. [Google Scholar] [CrossRef]
- Granovsky, A.A. Firefly 8.0.0. 2013. Available online: http://classic.chem.msu.su/gran/gamess/index.html (accessed on 1 December 2013).
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.; et al. General atomic and molecular electronic structure system. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Stevens, W.J.; Krauss, M.; Basch, H.; Jasien, P.G. Relativistic compact effective potentials and efficient, shared-exponent basis sets for the third-, fourth-, and fifth-row atoms. Can. J. Chem. 1992, 70, 612–630. [Google Scholar] [CrossRef]
- Swart, M.; Güell, M.; Luis, J.M.; Solà, M. Spin-state-corrected Gaussian-type orbital basis sets. J. Phys. Chem. A 2010, 114, 7191–7197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomasi, J.; Persico, M. Molecular Interactions in Solution: An Overview of Methods Based on Continuous Distributions of the Solvent. Chem. Rev. 1994, 94, 2027–2094. [Google Scholar] [CrossRef]
- Mennucci, B.; Tomasi, J. Continuum solvation models: A new approach to the problem of solute’s charge distribution and cavity boundaries. J. Chem. Phys. 1997, 106, 5151–5158. [Google Scholar] [CrossRef]
- Cossi, M.; Mennucci, B.; Pitarch, J.; Tomasi, J. Correction of cavity-induced errors in polarization charges of continuum solvation models. J. Comput. Chem. 1998, 19, 833–846. [Google Scholar] [CrossRef]
- Amovilli, C.; Mennucci, B. Self-consistent-field calculation of Pauli repulsion and dispersion contributions to the solvation free energy in the polarizable continuum model. J. Phys. Chem. B 1997, 5647, 1051–1057. [Google Scholar] [CrossRef]
- Sjoberg, P.; Murray, J.S.; Brinck, T.; Politzer, P. Average local ionization energies on the molecular surfaces of aromatic systems as guides to chemical reactivity. Can. J. Chem. 1990, 68, 1440–1443. [Google Scholar] [CrossRef] [Green Version]
- Lu, T.; Chen, F. Quantitative analysis of molecular surface based on improved Marching Tetrahedra algorithm. J. Mol. Graph. Model. 2012, 38, 314–323. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Mendes, P.; Hoops, S.; Sahle, S.; Gauges, R.; Dada, J.; Kummer, U. Computational modeling of biochemical networks using COPASI. Methods Mol. Biol. 2009, 500, 17–59. [Google Scholar] [PubMed]
- Chemla, F.; Ferreira, F. Carbocupration of Alkynes. In PATAI’S Chemistry of Functional Groups; John Wiley & Sons, Ltd.: Chichester, UK, 2011; pp. 527–584. ISBN 9780470682531. [Google Scholar]
- Basheer, A.; Marek, I. Recent advances in carbocupration of α-heterosubstituted alkynes. Beilstein J. Org. Chem. 2010, 6, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Reaction mechanism proposed by Das et al. (adapted from [1]).
Figure 1.
Reaction mechanism proposed by Das et al. (adapted from [1]).

Figure 2.
Formation of CuCH3 from CuI and MgCH3Br. (A) reactant state; (B) transition state; (C) separated products. Several important bond distances (in Å) are highlighted.
Figure 2.
Formation of CuCH3 from CuI and MgCH3Br. (A) reactant state; (B) transition state; (C) separated products. Several important bond distances (in Å) are highlighted.

Figure 3.
Carbocupration of the ynamide with THF-solvated CuCH3. (A) transition state (conformation 1); (B) transition state (conformation 2); (C) potential energy surface of the carbocupration reaction arising from conformation 1 (blue) and conformation 2 (red). Several important bond distances (in Å) are highlighted.
Figure 3.
Carbocupration of the ynamide with THF-solvated CuCH3. (A) transition state (conformation 1); (B) transition state (conformation 2); (C) potential energy surface of the carbocupration reaction arising from conformation 1 (blue) and conformation 2 (red). Several important bond distances (in Å) are highlighted.

Figure 4.
Addition of THF-solvated MgCH3Br or CuI to the ynamide. (A) potential energy surface (starting from infinitely separated THF, CuI, MgCH3Br, and ynamide) of the solvation of each metallic reagent and subsequent reactions with ynamide; (B) ynamide: (MgCH3Br:THF); (C) ynamide: (CuI:THF); (D) ynamide: (MgCH3Br:THF):(CuI:THF). Several important bond distances (in Å) are highlighted.
Figure 4.
Addition of THF-solvated MgCH3Br or CuI to the ynamide. (A) potential energy surface (starting from infinitely separated THF, CuI, MgCH3Br, and ynamide) of the solvation of each metallic reagent and subsequent reactions with ynamide; (B) ynamide: (MgCH3Br:THF); (C) ynamide: (CuI:THF); (D) ynamide: (MgCH3Br:THF):(CuI:THF). Several important bond distances (in Å) are highlighted.

Figure 5.
Regioselectivity of addition of THF-solvated MgCH3Br to CuI:ynamide. (A) transition state (TS1) of the formation of the linear isomer; (B) transition state (TS1’) of the formation of the branched isomer; (C) potential energy surface of the formation of each regioisomer. Several important bond distances (in Å) are highlighted.
Figure 5.
Regioselectivity of addition of THF-solvated MgCH3Br to CuI:ynamide. (A) transition state (TS1) of the formation of the linear isomer; (B) transition state (TS1’) of the formation of the branched isomer; (C) potential energy surface of the formation of each regioisomer. Several important bond distances (in Å) are highlighted.

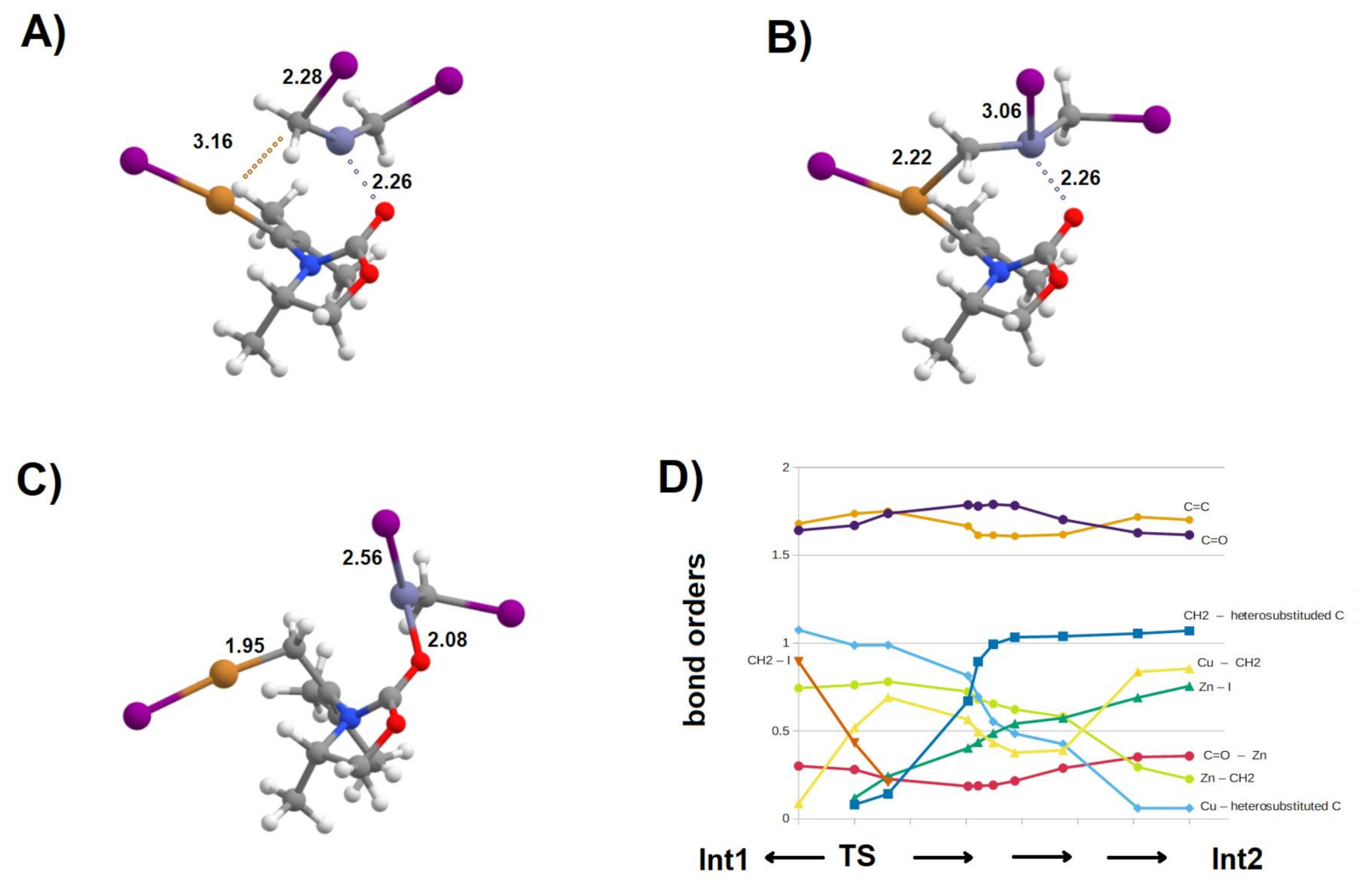
Figure 6.
Important structures in the pathway towards intermediate 2 (Int2). (A) Int1:(CuI:THF):(MgBr:THF); (B) Int1:CuI + ICH2ZnCH2I pre-reactional complex; (C) TS2; (D) Int2; (E) Int2′; (F) Potential energy surface of the zinc homologation of Mg-free Int1. The blue line refers to the attack from the “un-encumbered” direction (leading to isomer “α” of Int2), whereas the red line describes homologation from the “encumbered” reaction, leading to isomer “β“ of Int2.
Figure 6.
Important structures in the pathway towards intermediate 2 (Int2). (A) Int1:(CuI:THF):(MgBr:THF); (B) Int1:CuI + ICH2ZnCH2I pre-reactional complex; (C) TS2; (D) Int2; (E) Int2′; (F) Potential energy surface of the zinc homologation of Mg-free Int1. The blue line refers to the attack from the “un-encumbered” direction (leading to isomer “α” of Int2), whereas the red line describes homologation from the “encumbered” reaction, leading to isomer “β“ of Int2.

Figure 7.
Electrostatic potentials mapped on the density isosurfaces of Int2 (0.001 a.u.) and ICH2ZnCH2I (0.001 a.u.). For Int2, the minima (maxima) of the average localized ionization energy are shown as blue (red) dots superposed on the density isosurface.
Figure 7.
Electrostatic potentials mapped on the density isosurfaces of Int2 (0.001 a.u.) and ICH2ZnCH2I (0.001 a.u.). For Int2, the minima (maxima) of the average localized ionization energy are shown as blue (red) dots superposed on the density isosurface.

Figure 8.
Electronic rearrangements from Int1 to Int2. (A) Evolution of bond orders of key chemical bonds along the reaction coordinate; (B) Schematic structures along the reaction coordinate. Each bond is colored according to the palette in the graph in panel A. Structures A, B, and C are located (respectively) 2.5, 31.7, and 43.2 kcal·mol−1 below the transition state, on the reaction coordinate leading to Int2. Selected bond orders are shown.
Figure 8.
Electronic rearrangements from Int1 to Int2. (A) Evolution of bond orders of key chemical bonds along the reaction coordinate; (B) Schematic structures along the reaction coordinate. Each bond is colored according to the palette in the graph in panel A. Structures A, B, and C are located (respectively) 2.5, 31.7, and 43.2 kcal·mol−1 below the transition state, on the reaction coordinate leading to Int2. Selected bond orders are shown.

Figure 9.
Important structures in the pathway towards intermediate 2 (Int2CuCH2) arising from attack through the carbonyl side of the ring. (A) pre-reactional complex; (B) TS2; (C) Int2CuCH2; (D) changes in bond order on the pathway from pre-reactional complex to Int2CuCH2.
Figure 9.
Important structures in the pathway towards intermediate 2 (Int2CuCH2) arising from attack through the carbonyl side of the ring. (A) pre-reactional complex; (B) TS2; (C) Int2CuCH2; (D) changes in bond order on the pathway from pre-reactional complex to Int2CuCH2.

Figure 10.
Further structures on the pathway from Int2CuCH2 to Int2. (A) Cu migration transition state; (B) Int2γ; (C) potential energy surface of the pathway from Int1 to Int2CuCH2 to Int2. The blue line refers to the attack from above the ring plane (leading to isomer γ of Int2), and the red line to an attack below the plane of the ring (leading to isomer δ of Int2).
Figure 10.
Further structures on the pathway from Int2CuCH2 to Int2. (A) Cu migration transition state; (B) Int2γ; (C) potential energy surface of the pathway from Int1 to Int2CuCH2 to Int2. The blue line refers to the attack from above the ring plane (leading to isomer γ of Int2), and the red line to an attack below the plane of the ring (leading to isomer δ of Int2).

Figure 11.
Transition states leading to products with two stereocenters. (A) From Int2α to the (S)(S) product without Zn2+ complexation by carbonyl; (B) from Int2α to the (S)(R) product; (C) from Int2γ to the (S)(R) product; (D) from Int2β to the (R)(S) product; (E) from Int2δ to the (R)(S) product.
Figure 11.
Transition states leading to products with two stereocenters. (A) From Int2α to the (S)(S) product without Zn2+ complexation by carbonyl; (B) from Int2α to the (S)(R) product; (C) from Int2γ to the (S)(R) product; (D) from Int2β to the (R)(S) product; (E) from Int2δ to the (R)(S) product.

Figure 12.
Revised reaction mechanism.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Electronic energy (kcal·mol−1) differences (including solvation effects in THF) between the individual fragments of linear and branched isomers of intermediate 1, computed with the PBEPW91 density-functional. Computations of the energies of the individual fragments have been performed at the same geometry as that in the full molecule. Negative values denote preference for the “linear” over the “branched” isomer.
Table 1.
Electronic energy (kcal·mol−1) differences (including solvation effects in THF) between the individual fragments of linear and branched isomers of intermediate 1, computed with the PBEPW91 density-functional. Computations of the energies of the individual fragments have been performed at the same geometry as that in the full molecule. Negative values denote preference for the “linear” over the “branched” isomer.
| Reactant State | Transition State | Product | |
|---|---|---|---|
| Mg moiety | 0.6 | 5.8 | −2.0 |
| CuI moiety | 0.0 | −1.0 | 2.2 |
| Mg-organic moiety | −2.8 | −21.6 | −4.8 |
| CuI-organic moiety | −0.2 | −7.1 | −10.5 |
| organic moiety | −0.1 | 1.7 | −10.1 |
| Full complex | −3.0 | −17.7 | −10.3 |
Table 2.
Electronic energies including solvation effects in THF (in kcal·mol−1) of individual fragments of intermediates 2 and 2′, computed with the PBEPW91 density-functional. Computations of the energies of the individual fragments have been performed at the same geometry as that in the full molecule. The electron pair in the severed bond between the organic moiety and ZnI(CH2I) was divided symmetrically between the fragments.
Table 2.
Electronic energies including solvation effects in THF (in kcal·mol−1) of individual fragments of intermediates 2 and 2′, computed with the PBEPW91 density-functional. Computations of the energies of the individual fragments have been performed at the same geometry as that in the full molecule. The electron pair in the severed bond between the organic moiety and ZnI(CH2I) was divided symmetrically between the fragments.
| Int2 | Int2′ | Int2–Int2′ | |
|---|---|---|---|
| organic moiety | −348,955.2 | −348,892.7 | −62.5 |
| organic moiety + CuI | −478,913.8 | −478,885.5 | −28.3 |
| ZnI(CH2I) | −180,031.3 | −180,030.3 | −1.1 |
| Full molecule | −659,304.2 | −659,283.0 | −21.3 |
| interaction between ZnI(CH2I) and organic-CuI | −359.2 | −367.2 | 8.1 |
Table 3.
Activation free energies in THF (in kcal·mol−1) and key geometric parameters of the transition states linking the different Int2 isoforms to products with two quaternary stereocenters. The configuration at the stereocenter arising from the alkyne is shown first, followed by that of the stereocenter arising from the acetaldehyde. For the configuration assignment of the alkyne carbon, (where, for computational expediency, we had two identical methyl groups) we assigned the lowest priority to the methyl group originally present as the alkyne substituent and the second-lowest to the methyl group donated by the organometallic reagent.
Table 3.
Activation free energies in THF (in kcal·mol−1) and key geometric parameters of the transition states linking the different Int2 isoforms to products with two quaternary stereocenters. The configuration at the stereocenter arising from the alkyne is shown first, followed by that of the stereocenter arising from the acetaldehyde. For the configuration assignment of the alkyne carbon, (where, for computational expediency, we had two identical methyl groups) we assigned the lowest priority to the methyl group originally present as the alkyne substituent and the second-lowest to the methyl group donated by the organometallic reagent.
| Int2 Isoform Reacting with Acetaldehyde | Product Configuration | Activation Free Energy (kcal·mol−1) | C=O∙∙∙∙Zn Distance (Å) | C∙∙∙∙C=O Distance (Å) |
|---|---|---|---|---|
| Int2α | (S)(S) | 20.2 | 2.07 | 2.29 |
| Int2α | (S)(R) | 20.7 | 2.04 | 2.30 |
| Int2γ | (S)(S) | 19.8 | 2.02 | 2.06 |
| Int2γ | (S)(R) | 18.8 | 2.02 | 2.02 |
| Int2β | (R)(R) | 26.9 | 2.04 | 2.25 |
| Int2β | (R)(S) | 27.9 | 2.04 | 2.28 |
| Int2δ | (R)(R) | 24.9 | 2.05 | 2.32 |
| Int2δ | (R)(S) | 23.1 | 2.04 | 2.33 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Silva, P.J.; Bernardo, C.E.P. Unveiling the Reaction Mechanism of the Das/Chechik/Marek Synthesis of Stereodefined Quaternary Carbon Centers. Appl. Sci. 2021, 11, 5002. https://doi.org/10.3390/app11115002
AMA Style
Silva PJ, Bernardo CEP. Unveiling the Reaction Mechanism of the Das/Chechik/Marek Synthesis of Stereodefined Quaternary Carbon Centers. Applied Sciences. 2021; 11(11):5002. https://doi.org/10.3390/app11115002
Chicago/Turabian StyleSilva, Pedro J., and Carlos E. P. Bernardo. 2021. "Unveiling the Reaction Mechanism of the Das/Chechik/Marek Synthesis of Stereodefined Quaternary Carbon Centers" Applied Sciences 11, no. 11: 5002. https://doi.org/10.3390/app11115002
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.