
Hyperthermia Treatment as a Promising Anti-Cancer Strategy: Therapeutic Targets, Perspective Mechanisms and Synergistic Combinations in Experimental Approaches




Abstract
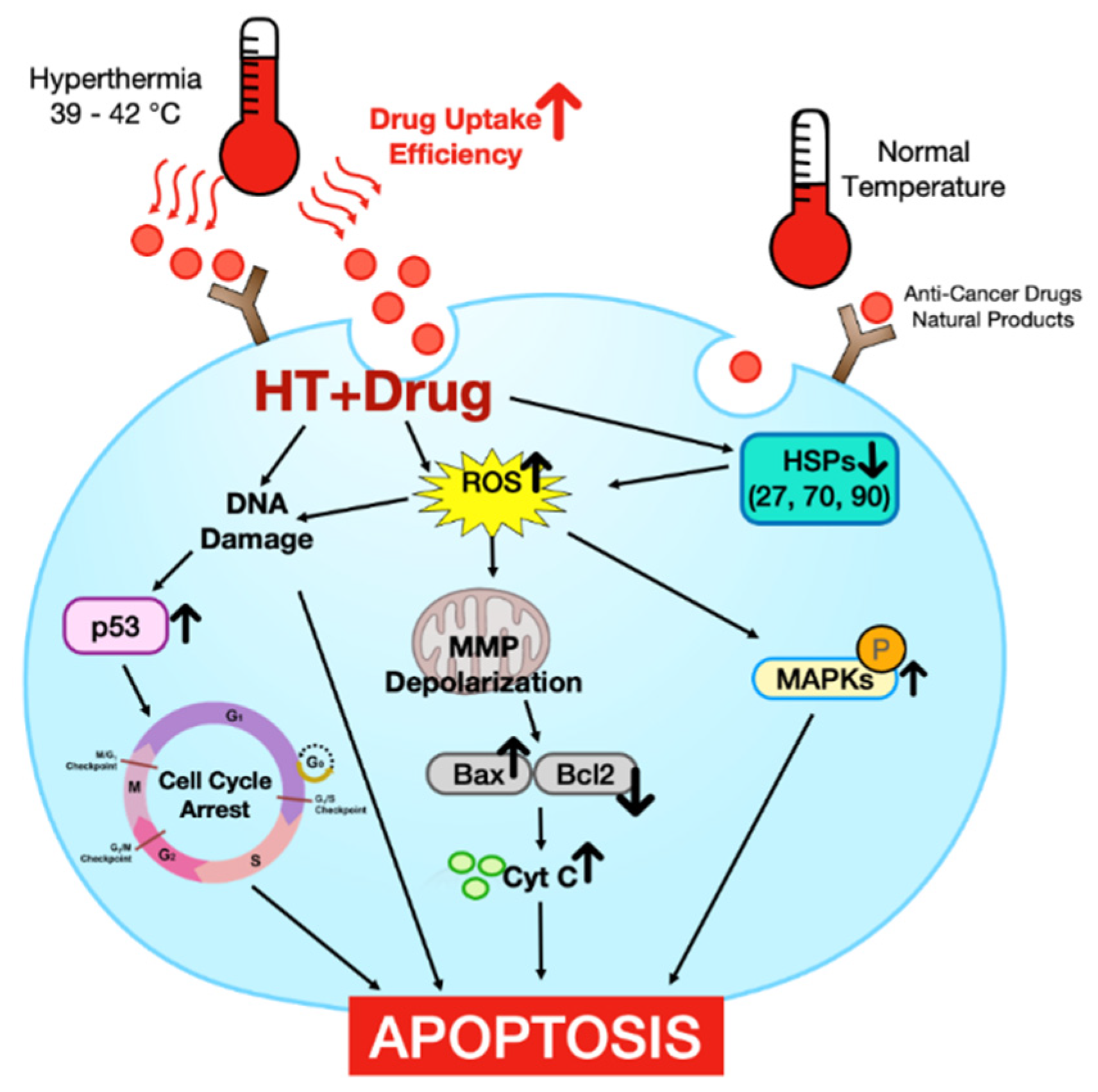
:1. Introduction
2. The Effect of HT Alone in Promoting Cell Death on Malignant Cells and the Mechanism
2.1. Reactive Oxygen Species (ROS) Production
2.2. Heat Shock Proteins (HSPs)
2.3. DNA Damage
2.4. Cell Cycle Arrest
2.5. Other Cellular Physiological Changes
2.5.1. Cytoskeletal Alterations
2.5.2. Change in Expression of Genes
2.5.3. Damage of Collagen Fiber
2.5.4. Cell Differentiation
2.5.5. Microvessel Damage
2.6. Regulation of Apoptosis Associated Transcription Factors and Proteins
2.6.1. Transcription Factors
2.6.2. Regulation of Anti/Pro-Apoptotic Protein
2.7. Activation of Caspase-3

{kind=link}
| 1-1. | |||
| HT | Cell Line and Observation Model | Molecular Mechanism | Ref |
| 43 °C, 40 min | Human tongue squamous cell carcinoma, Tca8113/in vitro | Apoptosis | [13] |
| 50–70 °C | Glioma/in vivo | Necrosis, Apoptosis | [14] |
| 42 °C, 4 h | Melanoma, HLA-A*0201+ Me275, Me290/in vitro | via up-regulation of both HSPs and tumor Ag expression | [15] |
| 1-2. | |||
| HT | Cell Line and Observation Model | Molecular Mechanism | Ref |
| 43 °C, 1 h | Human osteosarcoma, U-2/in vitro | Increasing ROS and caspase-3 activation, releasing of cytochrome c and ER stress | [16] |
| 1-3. | |||
| HT | Cell Line & Observation Model | Molecular Mechanism | Ref |
| 43 °C, 1 h | Spermatocytes/in vivo | Deregulated RNA (especially piRNA) metabolism | [17] |
| 43 °C, 1 h | Sarcomas/in vivo | Increased protein nitration/Decreased GSH levels and hsp 70 expression | [18] |
| 1-4. | |||
| HT | Cell Line and Observation Model | Molecular Mechanism | Ref |
| 42 °C, 1 h | Cervical carcinoma, SiHa, HeLa, C33A, Caski, C4I, HT3/Human prostate carcinoma, Du145, LNCaP, PC3/in vitro | E6 degradation enabling p53-dependent apoptosis and G2-phase arrest | [19] |
| 41–44 °C, 2 h | Human myeloid leukemia, TF-1, K562, HL-60/in vitro | Decreasing of telomerase activity | [20] |
| 42–48 °C, 30 min–2 h | NSLCLs, A549/in vitro | Inducing DNA damage, chromosomal damage and to inhibit DNA repair | [21] |
| 41 and 43 °C, 90 min | Myeloma/in vitro | Cell shrinkage, DNA fragmentation, karyorrhexis | [22] |
| 1-5. | |||
| HT | Cell Line and Observation Model | Molecular Mechanism | Ref |
| 45 °C, 30 min | Melanoma, B16-F10/in vitro | G0/G1 arrest | [23] |
| 1-6. | |||
| HT | Cell Line and Observation Model | Molecular Mechanism | Ref |
| 43.5 and 45 °C, 30 min | Lung cancer, H1299/in vitro | Cytoskeletal alterations | [24] |
| 43 °C, 1 h | Human neuroblastoma, SK-N-MC/in vitro | Loss of integrins from the cell surface, that blocks several physiological signaling pathways | [25] |
| 1-7. | |||
| HT | Cell Line and Observation Model | Molecular Mechanism | Ref |
| 43 °C, 1 h | Glioma BT4An tumor/in vivo | Changes of global gene expression | [26] |
| 41 °C, 30 min | Human lymphoma, U937/in vitro | Change in the expression of a large number of genes such as DNAJB1, HSPA1A, and HSPA1B | [27] |
| 1-8. | |||
| HT | Cell line and Observation Model | Molecular Mechanism | Ref |
| 40 and 42 °C, 1 h | Pancreatic cancer, Panc-1 and fibroblast, WI-38/in vitro | Affecting collagen fiber architecture and inducing apoptosis | [28] |
| 1-9. | |||
| HT | Cell Line and Observation Model | Molecular Mechanism | Ref |
| 43 °C (30, 60, 90 min),45 °C (20, 40, 60 min) | Erythroleukemia, K562/in vitro | Erythroid differentiation (inducing glycophorin A expression, hemoglobin synthesis) | [29] |
| 1-10. | |||
| HT | Cell Line and Observation Model | Molecular Mechanism | Ref |
| 43 °C, 30 min | Hepatoma, H22/in vivo | Apoptosis, Necrosis/Inadequate supply of nutrients, oxygen, and accumulation of acid | [30] |
| 1-11. | |||
| HT | Cell Line and Observation Model | Molecular Mechanism | Ref |
| 42 °C, 1 h | Human gastric cancer, MKN45/in vitro | Inhibiting the TNF-alpha-induced NF-kappaB activation | [31] |
| 43 °C, 1 h | Glioblastoma, C6/Human umbilical vein endothelial cells (HUVECs)/Human renal tubular epithelial cells, HK2/in vitro | Inhibiting proliferation and promoting apoptosis through the EGFR/STAT3 pathway | [32] |
| 42 °C, 0–4 h | Glioma, C6/in vitro | Stimulating TNF-α signaling to activate apoptosis, enhancing p38 MAPK expression and inhibiting the NF-κB pathway | [33] |
| 42 °C, 1–48 h | Hepatocellular carcinoma, HepG2, HUT/in vitro | Increased expression of Bax and down regulation of Bcl-2 and S100A4 genes | [34] |
| 43, 45, and 47 °C, 1 h | Human glioma, A172, T98G, U251MG, YKG-1/in vitro | A temperature-dependent AIF translocation that can cause apoptosis independent of p53 | [35] |
| 47 °C, 40 min | Human NSCLC, NCI-H1650/SCLC, NCI-H446/in vitro | Regulation of HIF-1a expression through AKT and ERK signaling pathways. | [12] |
| 44 °C, 1 h | Human prostate cancer, PC-3, LnCaP, DU-145/Murine prostate cancer, TRAMP-C2/in vitro | Inducing proteasome inhibition and loss of androgen receptor expression, abrogating AR expression, down-regulation of NF-kB | [36] |
| 42 °C, 1 h | Breast cancer, MDA-MB-231/in vitro | Aggregation-induced c-FLIP cytosolic depletion | [37] |
| 43, 45, and 47 °C, 30 min | Breast carcinoma cell, MCF-7/in vitro | Down-regulation of the expression of TGF-β1, EGF and MMPs, suppressing MMP-2/9 secretion and enzymatic activity | [38] |
| 45 °C, 40 min | Human cervical cancer, CaSki/in vitro | Apoptosis, Necrosis/Up-regulation of caspase-3 and Smac levels and down-regulation of anti-apoptotic Survivin, | [39] |
| 43 °C, 80 min | Human tongue squamous carcinoma, Tca8113/in vitro | Activation and translocation of PKC-δ | [40] |
| 44 °C, 40 min | Human squamous cell carcinoma (SAS) wild-type 53 and mutated-type 53/in vitro | Induction of caspase-3 activation and apoptosis in the wild type p53 and suppression of IL-12-related genes in the mutated p53 | [41] |
| 45 °C, 48 °C, 90 min | Human melanoma, A375/Squamous carcinoma, A431/in vitro | Apoptosis (45 °C), Necrosis (48 °C/Activation of caspase-3/7, ER stress and ER-mediated apoptosis | [42] |
| 43 °C, 3 h | Lung cancer, BEAS-2B and BZR-T33/in vitro | Increasing caspase-3 as a result of activation of cell-death membrane receptor, arrest of cells in the G2-Mphase of the cell cycle | [43] |
| 43 °C, 40 min | Esophageal cancer, EC109/in vitro | Inhibiting Survivin and XIAP and activating caspase 3 | [44] |
| 43 °C, 45 °C, 2 h | Human malignant melanoma, A375, A431/in vitro | low HT: induced extrinsic and intrinsic apoptotic pathways both of which activated caspase 6 only//high HT: mediated by the combined effects of caspases 3, 7 and 6 | [45] |
3. Combination of Hyperthermia and Anti-Cancer Drugs
3.1. Cisplatin
3.2. Cisplatin and 5-Fluorouracil
3.3. Cisplatin and Doxorubicin
3.4. Cisplatin and Ferucarbotran
3.5. Cisplatin + Sodium Arsenite
3.6. Paclitaxel and Cisplatin
3.7. Paclitaxel
3.8. Docetaxel
3.9. Doxorubicin
3.10. Bortezomib
3.11. Gemcitabine (GEM)
3.12. Mapatumumab
3.13. Cyclophosphamide (CTX)
3.14. CTX and Melatonin
3.15. Erlotinib
3.16. Macrosphelide (MS5)
3.17. Mafosfamide
3.18. Melphalan (Mel)
3.19. Methotrexate (MTX)
3.20. Mitomycin C (MMC)
3.21. Picibanil (OK-432)
3.22. TRAIL
3.23. Pelitinib
3.24. Pluronics
3.25. Ranpirnase
3.26. Sorafenib (SRF)
3.27. SurvivinT34A
3.28. Temozolomide
3.29. Tirapazamine (TPZ)
3.30. Toremifene
3.31. Trabectedin
3.32. Vinblastine
| 2.1. | ||||
| Compound | HT | Cell Line/Observation Model | Molecular Mechanism | Ref |
| Cisplatin | 43 °C, 1 h | Prostate cancer, PC-3, DU-145/in vitro | cleavage of caspase-3/activation of AMPKα-JNK and inhibition of Akt-mTOR-p70s6k signaling pathway | [48] |
| Cisplatin | 44 °C, 30 min | Human maxillary squamous cell carcinoma, cisplatin-resistant, IMC-3, IMC-3CR/in vitro | Increase of ROS production, repression of SESN1 | [49] |
| Cisplatin | 42–43 °C, 30 min | Ovarian cancer, SKOV3/in vitro, in vivo | Apoptosis | [50] |
| Cisplatin | 42.5 °C, 1 h | NSLCLs, A549, H460/in vitro | Pharmacokinetics change | [51] |
| Cisplatin | 44 °C, 30 min | Human maxillary carcinoma, IMC-3/in vitro | Cleavage of PARP | [52] |
| Carboplatin (CPt) | 42.5 °C, 1 h | Human retinoblastoma, WERI/in vitro | Cell cycle arrest (G2/M)/caspase 9 activation induced by the release of cytochrome C | [54] |
| Cisplatin and 5-fluorouracil | 42 °C | Orthotopic esophageal squamous cancer/in vitro, in vivo | Apoptosis | [56] |
| Cisplatin and doxorubicin | 42 °C, 1 h | Human Ovarian Carcinoma, OVCAR8/Colon cancer cell, HCT116/in vitro | DNA damage (blocking PARylation) | [58] |
| Cisplatin ferucarbotran (Resovist) | 42.5 °C, 20 min | Human oral cancer, HSC-3, OSC-19/in vitro | Cell cycle arrest (G2/M) | [60] |
| Sodium arsenite (NaAsO2) and cisplatin | 39 °C, 1 h | Ovarian cancer, A2780, CP70/in vitro | Cell cycle arrest (G2/M) | [62] |
| Paclitaxel and cisplatin | 43 °C, 1 h | Osteosarcoma, OS732, MG63/in vitro | Apoptosis (up-regulating Fas) | [63] |
| 2-2. | ||||
| Compound | HT | Cell Line/Observation Model | Molecular Mechanism | Ref |
| Paclitaxel | 41 °C, 2 h | Human breast cancer, MCF-7/in vitro | Apoptosis | [65] |
| Paclitaxel | 41.5, 43 °C, 2 h | Human breast, MCF-7/Ovarian cancer, SKOV-3/Hepatocellular carcinoma, HepG2/in vitro | Cell cycle arrest (G2/M) | [66] |
| Paclitaxel | 41 °C, 2 h | Human Breast cancer, MCF-7/in vitro | activation of caspase-7/Cell cycle arrest (G2/M) | [67] |
| Paclitaxel (PTX) | 42 °C, 2 h | Human larynx carcinoma, HEp2/in vitro | Cell cycle arrest (G2/M) | [68] |
| Taxol | 41.5 °C, 2 h | Cervical adenocarcinoma, HeLa/in vitro | Necrosis/Cell cycle arrest (G2/M) | [69] |
| Docetaxel | 41 °C, 2 h | Human breast cancer cell line MCF-7 and MDA-MB-453/in vitro | Down-regulation of proteins in the Bcl-2 family | [71] |
| 2-3. | ||||
| Compound | HT | Cell Line and Observation Model | Molecular Mechanism | Ref |
| Doxorubicin and GSH | 42 °C, 1 h | NSLCLs, A549/in vitro | Increase of the GSH-mediated release of doxorubicin | [73] |
| Doxorubicin | 42 °C | Squamous cell carcinoma, SCC-7/in vivo | Apoptosis | [74] |
| Doxorubicin | 42 °C, 2 h | Murine colorectal adenocarcinoma, C26/in vitro | upregulation of p53 | [75] |
| Doxorubicin | gold nanoshells 43 °C | Human hepatoma, BEL-7402/in vitro | MMP depolarization/DNA cross-linking and inhibition of DNA repair mechanisms | [76] |
| Doxorubicin | 42 °C | Human hepatoma, Hep3B/in vivo | Apoptosis, Necrosis | [77] |
| Doxorubicin | 43 °C, 1 h | Uterine cancer, MES-SA/in vitro | Apoptosis (overcoming P-gp regulated multidrug resistance) | [78] |
| Doxorubicin, IR-780 loaded polymeric prodrug micelles | IR-780, NIR imaging | Breast cancer, MCF-7, MCF-7, ADR/in vitro, in vivo | MMP depolarization | [79] |
| A water-responsive phospholipid-calcium-carbonate hybrid nanoparticle (PL/ACC-DOX&ICG) | 43 °C, 1 h, with PL/ACC-DOX&ICG upon NIR laser irradiation. | Breast Cancer, 4T1/in vivo, in vitro | Apoptosis | [80] |
| Pegylated liposomal doxorubicin | Focused ultrasound system | Murine breast cancer, 4T1-luc2/in vivo | Apoptosis (increased perfusion, vascular permeability and interstitial microconvection) | [82] |
| Liposomal doxorubicin | 42 °C, 30 min | Hepatocellular carcinoma, HepG2/in vivo, in vitro | MMP depolarization | [83] |
| BCM- doxorubicin (a block copolymer micelle (BCM) formulation (which may reduce toxicities of doxorubicin in a similar way to pegylated liposomal doxorubicin) | 42 °C, 1 h | Human ovarian cancer HEYA8, OV-90, SKOV3/in vitro, in vivo | Growth inhibition (Pharmacokinetics change) | [84] |
| Lipodox® (liposome-encapsulated doxorubicin) | 42 °C, 30 min | Murine colon carcinoma, CT26/in vivo, in vitro | enhancement of the uptake of liposomal drugs by enhancing phagocytic activity | [86] |
| Redox-responsive hyaluronic acid nanogels | the laser irradiation at 808 nm, 60 s | Human breast cancer, MCF-7, ADR/in vitro | Apoptosis (increased intracellular doxorubicin accumulation) | [87] |
| 2-4. | ||||
| Compound | HT | Cell Line and Observation Model | Molecular Mechanism | Ref |
| Bortezomib | 44 °C, 30 min | Mantle cell lymphoma, Jeko-1, Rec-1, Granta 519, HBL-2, NCEB-1/in vitro | inhibition of HSP27/70 | [90] |
| Bortezomib | 44 °C, 15 min | Human leukemic monocyte lymphoma, U937/in vitro | Apoptosis (increase cells underwent the late apoptosis stage) | [91] |
| Gemcitabine | 43 °C, 1 h | Human pancreatic carcinoma, AsPC-1, MIAPaCa-2/in vitro | Apoptosis, Necrosis (blocking the activation of NF-B) | [94] |
| Gemcitabine | 42 °C, 90 min | Pancreatic cancer, SW1990/in vitro | activation of ROS/JNK signaling pathway/Cell cycle arrest (S-phase) | [95] |
| Gemcitabine | 43 °C, 3 h | Human non-small-cell lung cancer, BZR-T33/in vitro | Cell cycle arrest (G2/M)/cleavage of caspase-3 | [96] |
| Gemcitabine and NucAnt | 43 °C, 1 h | Human pancreatic cancer, BxPC3, PANC-1/in vitro | Cell cycle arrest (S-phase)/activation of expression of p53, p21, BcL-2, PARP, Bax and H2AX | [97] |
| Gemcitabine-loaded TSL | 41 °C, 24 h | Adenocarcinoma, CT-26/in vivo, in vitro | cleavage of the caspse-3/7 and causing the fragmentation of chromatin DNA | [98] |
| Mapatumumab | 42 °C, 1 h | Human colorectal carcinoma, CX-1, HCT116/in vitro | Elevation of ROS level/JNK activation/MMP depolarization | [100] |
| Mapatumumab and Oxaliplatin | 37 °C, 3 h | Human Colon Cancer, CX-1/in vitro, in vivo | MMP depolarization/ROS production | [101] |
| Mapatumumab and Oxaliplatin | 42 °C, 1 h | Human colorectal carcinoma, CX-1, HCT116/in vitro | Activating JNK signaling pathway | [103] |
| Cyclophosphamide | 43 °C, 1 h | Glioblastoma-like tumor, BT4An/in vivo | Anti-angiogenisis (upregulation of TSP-1) | [106] |
| Cyclophosphamide and melatonin | 43.5 °C | Carcinosarcoma, Walker 256/in vivo | N/A | [108] |
| Erlotinib | NIR irradiation, 50 °C within 500 s | NSLCLs, A549, H1975/Prostate cancer, PC-9/in vitro, in vivo | Cell cycle arrest (G0/G1)/inhibiting the epidermal growth factor receptor (EGFR) tyrosine kinase | [110] |
| Macrosphelide | 41 °C, 20 min | Human lymphoma, U937/in vitro | increasing in ROS generation and caspase 3, 8 activation/down-regulation of Bcl-2 | [111] |
| Mafosfamide | 41.7 °C, 1 h | Pleural mesothelioma, MSTO-211H/in vitro | Necrosis (shifting cell death from apoptosis to necrosis) | [113] |
| Melphalan | 42 °C, 2 h | Human Ewing tumor, RDES/in vitro | activation of caspase-3 | [114] |
| Methotrexate | mild HT (42 °C) | Human hepatoma, Caco-2, SW480/in vitro | Cell cycle arrest (S phase) | [117] |
| Methotrexate | 40 °C–50 °C, 1 h | Hepatocellular carcinoma, HepG2/in vitro | enhancement of the uptake of liposomal drugs | [118] |
| Mitomycin C | 42 °C, 1 h | Human colon cancer, LS174T, LS180, HCT116, CX-1 cell/in vitro | Activation of JNK pathway induced mitochondria-dependent apoptotic pathway | [121] |
| Mitomycin C | 42 °C, 12 min | Bladder cancer cell, RT112 and T24/ex vivo | cytochrome C release/HR DNA damage repair capacity decrease | [122] |
| OK-432 | (pUST) | Colorectal adenocarcinoma, CT26-luc tumor cell/in vivo | Apoptosis, Necrosis(tissue inflammation induced necrosis) | [124] |
| Oxaliplatin and Melphalan | 42 °C, 1 h | Human colorectal cancer, CX-1/in vitro | MMP depolarization | [125] |
| Pelitinib | 42.5 °C, 4 h | NSLCLs, A549 cell/in vitro | up-regulation of ABCB1/ABCG2 | [126] |
| Pluronic L61 | 43 °C, 20 min | Rat colorectal adenocarcinoma, DHD, K12, TRb/in vivo | MMP depolarization | [127] |
| Ranpirnase | 40 °C, 24 h | Human lymphoblastoid, TK6/in vivo, in vitro | Apoptosis | [130] |
| Sorafenib, indocyanine | 785 nm irradiation for 10 min at 2 W/cm2 | Hepatocellular carcinoma, Huh7/in vitro, in vivo | producing ROS and activating caspase-9, 3 | [132] |
| SurvivinT34A | 42 °C, 1 h | Murine colorectal carcinoma, CT26/Murine melanoma, B16-F10, MethA/in vitro, in vivo | activation of p53/bound to Hsp90 and abrogating the cytoprotection of Hsp90 | [134] |
| Temozolomide | 43 °C, 15 min | Human melanoma, DM6/in vitro, in vivo | N/A | [136] |
| Tirapazamine | 40 °C, 1 h | human head and neck squamous cell carcinoma, SAS/in vitro | p53 independent apoptosis | [137] |
| Toremifene | 43.5 °C, 30 min | Breast cancer, MCF-7/in vitro | weakening Estrogen receptor expression/G0/G1-phase cells↑ and S-phase cells↓ | [139] |
| Trabectedin | 41.8 °C and 43 °C, 90 min | Human sarcoma cell osteosarcoma, U2OS/Liposarcoma, SW872/Synovial sarcoma, SW982/Ewing sarcoma, RD-ES/Leiomyosarcoma, SKUT-1/Human colorectal carcinoma, DLD1/in vitro | BRCA2 degradation and impairment of DNA homologous recombination repair | [141] |
| Vinblastine | 44 °C, 1 h | BT4An rat glioma/in vivo | Apoptosis (disturbing established neovasculature, and producing vascular shutdown) | [144] |
4. Natural Products
4.1. Reactive Oxygen Species Production
4.1.1. Baicalin
4.1.2. Epigallocatechin Gallate (EGCG) and Chlorogenic Acid (CGA)
4.1.3. Cinnamaldehyde (CNM)
4.1.4. Nonivamide
4.1.5. 5Z-7-Oxozeaenol (OZ)
4.1.6. Withaferin A (WA)
4.1.7. Ascorbic Acid (AscH2)
4.1.8. Docosahexaenoic Acid (DHA)
4.2. Regulation of Anti/Pro-Apoptotic Transcription Protein
4.2.1. ch282-5
4.2.2. Crocin
4.2.3. Perillyl Alcohol (POH)
4.2.4. 5-Aminolevulinic Acid (5-ALA)-Mediated PDT
4.2.5. Curcumin
4.3. Mitochondrial Membrane Potential (MMP) Depolarization
4.3.1. Furan-Fused Tetracyclic Synthesized Compounds (DFs)
4.3.2. Betulinic Acid (BA)
4.3.3. Curcumin and 5-FU
4.4. Cell Cycle Arrest
Arsenic Trioxide (As2O3)
4.5. Regulation of Heat Shock Response
4.5.1. Curcumin and Resveratrol
4.5.2. Quercetin
4.5.3. Quercetin and Tamoxifen
4.5.4. Quercetin + Lipopolysaccharide (LPS)
4.6. Other Mechanisms
4.6.1. β-Lapachone (β-Lap)
4.6.2. Enediyne
| 3-1. | ||||
| Compound | HT | Cell Line and Observation Model | Molecular Mechanism | Ref |
| Baicalin | 44 °C, 12 min | Myelomonocytic leukemia, U937/in vitro | Caspase activation/Bax and Noxa↑, Downregulation of antiapoptotic proteins/Bcl-2↓, MMP depolarization, increase of ROS, ER stress | [146] |
| Epigallocatechin gallate and chlorogenic acid | 10-cycles at 43.5–36 °C | Human pancreatic cancer, PANC-1/in vitro | Cell cycle arrest (G2/M)/the induction of the ROS-dependent mitochondria-mediated apoptosis | [148] |
| Cinnamaldehyde | 43 °C, 30 min | NSLCLs, A549/in vitro | ROS production and Mitogen-Activated Protein Kinase Family↑ | [150] |
| Cinnamaldehyde | 43 °C, 30 min | Renal adenocarcinoma, ACHN/in vitro | inhibition in HSP70 expression, Cell cycle arrest, increase of ROS | [151] |
| Nonivamide | 44 °C, 15 min | Human lymphoma, U937/in vitro | elevation of intracellular ROS/mitochondrial dysfunction/increased activation of JNK and p38 | [152] |
| 5Z-7-oxozeaenol | 44 °C, 10 min | Human T lymphoblast, Molt-4/in vitro | HSP70↓/p38and jnk↑/ROS production (ER stress-induced apoptosis) | [155] |
| Withaferin A | 44 °C, 30 min | Human cervical cancer, HeLa/in vitro | inducing JNK phosphorylation (p-JNK), and decreases in the phosphorylation of ERK (p-ERK) | [157] |
| Ascorbic acid | 42 °C, 15 min | Ehrlich ascites tumor, EAT/in vitro | Cell cycle arrest (G2/M)/H2O2 induced apoptosis | [159] |
| Docosahexaenoic acid | 44 °C, 10 min | Human myelomonocytic lymphoma, U937/in vitro | MMP depolarization (inducing phosphorylation of protein kinase C (PKC)-d) | [162] |
| 3-2. | ||||
| Compound | HT | Cell Line and Observation Model | Molecular Mechanism | Ref |
| Ch282-5 | 43 °C, 1 h | Melanoma, M21, B16F10/in vitro, in vivo | anti-apoptotic proteins of Bcl-2 and IAP family and activating/disturbing the mTOR/p70S6k signaling pathway/MAPKproteins(JNK and p38 MAPK) | [164] |
| Crocin | 43 °C, 2 h | Breast cancer, MDA-MB-468/in vitro | increasing the Bax↑Bcl2↓/HSP70, HSP90↓ | [167] |
| Perillyl alcohol | 43 °C, 1 h | Mammary carcinoma, SCK/in vitro | Cell cycle arrest (TGF-b induced G1 arrest)/p53 and p21 proteins | [170] |
| 5-aminolevulinic acid | 40–46 °C | Glioma, Human grade IV GBM cell line(ACBT)/in vitro | Apoptosis | [172] |
| Curcumin | 42 °C, 1 h | Murine Lewis lung carcinoma, MS-1/Endothelial LL/2/in vitro, in vivo | Apoptosis/angiogenesis↓ | [174] |
| 3.3. | ||||
| Compound | HT | Cell Line and Observation Model | Molecular Mechanism | Ref |
| Furan-fused tetracyclic compounds | 44 °C, 20 min | Human lymphoma, U937/in vitro | MMP depolarization/release of cytochrome c/activating caspase-3 and 8/expression of Fas | [175] |
| Betulinic acid | 42 °C, 2 h | Human melanoma, DB-1/in vivo | MMP depolarization | [177] |
| Curcumin and 5-Fluorouracil/magnetic nanoparticles encapsulated poly(D,L-lactic-co-glycolic acid) | 80 °C, 60 min, 120 min | Human breast adenocarcinoma, MCF7/in vitro | destabilizing the cytoskeleton and MMP depolarization | [179] |
| Arsenic trioxide | 43 °C, 30 min | Esophageal carcinoma, EC-1/in vitro | Cell cycle arrest G₂/M phase (and as the ratio of cells in G0/G1 and S phases decreased, cell death became more pronounced) | [182] |
| Curcumin and resveratrol | 42 °C, 30 min | Mice colon cancer, CT26/in vitro, in vivo | inducing apoptosis/HSP70↓/recruiting CD3+ T-cells and F4/80+ macrophages | [183] |
| Quercetin | 43 °C, 1 h | p53-negative prostatic adenocarcinoma, PC-3/Prostatic carcinoma, DU-145/in vivo, in vitro | antagonizing the expression of HSP72 | [184] |
| Quercetin | 42 °C, 1 h | Human myelogenous leukemia, K562/A, K562/in vitro | inhibition of the elevated protein expression and mRNA level of HSP70 and P-gp | [185] |
| Quercetin and tamoxifen | 42.5 °C, 1 h | Human melanoma, M10, M14, MNT1/in vitro | reducing heat shock protein-70 expression at both protein and mRNA levels | [189] |
| Quercetin (HSP70 inhibitor) + LPS | NIR light at 808 nm wavelength for 5 min, 40 °C, 35 min | Human thyroid duct carcinoma, TT/in vivo, in vitro | Apoptosis (enhancement cellular uptake and pronouncement tumor targeting ability) | [190] |
| β-lapachone | 42 °C, 1 h | Human osteosarcoma, HOS/in vitro | due to the heat-induced elevation of NQO1 activity | [191] |
| Enediyne | 42.5 °C, 1 h | Breast cancer, MDA-231/Melanoma, U-1/in vitro | inducing DSBs, and/or a reduction in DSB repair efficiency | [193] |
5. Conclusions and Future Direction
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| HT | hyperthermia |
| HSPs | heat shock proteins |
| ROS | reactive oxygen species |
| MMP | mitochondrial membrane potential |
| HTH | high temperature HT |
| HSP70 | 70-kDa heat shock protein |
| Bcl-2 | B-cell lymphoma-2 |
| Bcl-xL | B-cell lymphoma-extra large |
| Bax | BCL2 Associated X |
| ER | endoplasmic reticulum |
| HSF1 | heat shock factor 1 |
| ALA-PDT | 5-aminolevulinic acid-based photodynamic therapy |
| DUSPs | dual specificity phosphatases |
| MAPK | mitogen-activated protein kinase |
| TNF-α | tumor necrosis factor-alpha |
| NF-κB | nuclear factor-kappa B |
| AIF | apoptosis-inducing factor |
| NSCLCs | non-small cell lung carcinomas |
| SCLCs | small cell lung cancers |
| ERK | extracellular signal-regulated kinase |
| AR | androgen receptor |
| IκB | inhibitor of NF-κB |
| TRAIL | TNF-related apoptosis-inducing ligand |
| c-FLIP | cellular FLICE-inhibitory protein |
| TGF-b1 | transforming growth factor-beta 1 |
| VEGF | vascular endothelial growth factor |
| XIAP | X-linked inhibitor of apoptosis protein |
| ATF6 | Activating Transcription Factor 6 |
| gDNA | genomic DNA |
| mtDNA | mitochondrial DNA |
| PARP | poly (ADP-ribose) polymerase |
| AMPK | AMP-activated protein kinase |
| JNK | c-Jun N-terminal kinase |
| GNR | gold nanorod |
| MHT | mild HT |
| 5-FU | 5-Fluorouracil |
| RF | radiofrequency |
| AMF | alternating magnetic field |
| GSH | glutathione |
| HIFU-HT | pulsed high-intensity focused ultrasound-induced localized mild HT |
| mEHT | modulated electro hyperthermia |
| GNs | gold nanoshells |
| NIR | near-infrared |
| BLI | bioluminescene imaging |
| PL/ACC-DOX&ICG | phospholipid-calcium-carbonate hybrid nanoparticle loaded with doxorubicin and indocyanine green |
| MCTS | multicellular tumor spheroids |
| PLD | pegylated liposomal doxorubicin |
| ABC | ATP-binding cassette |
| BCM | block copolymer micelle |
| MCL | mantle cell lymphoma |
| PMBC | peripheral blood mononuclear cell |
| GEM | gemcitabine |
| N6L | NucAnt |
| CTX | Cyclophosphamide |
| EGFR | epidermal growth factor receptor |
| MoS2-SS-HA-Er | erlotinib-loaded MoS2 nanosheets functionalized with hyaluronic acid |
| MS5 | Macrosphelide |
| Mel | Melphalan |
| MTX | Methotrexate |
| SPION | superparamagnetic iron oxide nanoparticles |
| MMC | Mitomycin C |
| OK-432 | Picibanil |
| pUSHT | pulsed-wave ultrasound HT |
| IFNγ | interferon gamma |
| MDR | multidrug resistance |
| EO | ethylene oxide |
| PO | propylene oxide |
| SCID | severe combined immunodeficiency |
| SRF | sorafenib |
| SINP | SRF/ICG nanoparticles |
| survivinT-T34A | survivin’s threonine 34 to alanine |
| TPZ | Tirapazamine |
| DSB | double-strand breaks |
| EGCG | epigallocatechin gallate |
| CGA | chlorogenic acid |
| CNM | cinnamaldehyde |
| OZ | 5Z-7-oxozeaenol |
| TAK1 | transforming growth factor-β activated kinase 1 |
| WA | withaferin A |
| AscH2 | ascorbic acid |
| APPS | ascorbic acid-2-phosphate6-O-palmitate |
| APHD | Asc-2-phosphate-6-O-(2’-hexyl)decanoate |
| A6-P | 6-O-palmitoyl-Asc |
| VCIP | Asc-2,3,5,6-O-tetra-(2’-hexyl)decanoate |
| DHA | docosahexaenoic acid |
| PTP1B | protein tyrosine phosphatase 1B |
| PKC | protein kinase C |
| POH | perillyl alcohol |
| 5-ALA | 5-Aminolevulinic acid |
| DFs | furan-fused tetracyclic synthesized compounds |
| BA | betulinic acidg |
| TS | thymidylate synthase |
| As2O3 | arsenic trioxide |
| LPS | lipopolysaccharide |
| β-lap | β-lapachone |
| NQO1 | NAD(P)H:quinone oxidoreductase |
| ETC | electron transport chain |
References
- Hulvat, M.C. Cancer Incidence and Trends. Surg. Clin. N. Am. 2020, 100, 469–481. [Google Scholar] [CrossRef]
- Sankaranarayanan, R.; Swaminathan, R.; Jayant, K.; Brenner, H. An overview of cancer survival in Africa, Asia, the Caribbean and Central America: The case for investment in cancer health services. IARC Sci. Publ. 2011, 162, 257–291. [Google Scholar]
- Datta, N.R.; Jain, B.M.; Mathi, Z.; Datta, S.; Johari, S.; Singh, A.R.; Kalbande, P.; Kale, P.; Shivkumar, V.; Bodis, S. Hyperthermia: A Potential Game-Changer in the Management of Cancers in Low-Middle-Income Group Countries. Cancers 2022, 14, 315. [Google Scholar]
- Ahmed, K.; Tabuchi, Y.; Kondo, T. Hyperthermia: An effective strategy to induce apoptosis in cancer cells. Apoptosis 2015, 20, 1411–1419. [Google Scholar] [CrossRef] [PubMed]
- van der Zee, J.; Gonzalez, G.D. The Dutch Deep Hyperthermia Trial: Results in cervical cancer. Int. J. Hyperth. 2002, 18, 1–12. [Google Scholar] [CrossRef]
- Senturk, F.; Kocum, I.C.; Guler Ozturk, G. Stepwise implementation of a low-cost and portable radiofrequency hyperthermia system for in vitro/in vivo cancer studies. Instrum. Sci. Technol. 2021, 49, 629–641. [Google Scholar] [CrossRef]
- Montes-Robles, R.; Hernández, A.; Ibáñez, J.; Masot-Peris, R.; de la Torre, C.; Martínez-Máñez, R.; García-Breijo, E.; Fraile, R. Design of a low-cost equipment for optical hyperthermia. Sens. Actuators A Phys. 2017, 255, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, K.; Zaidi, S.F. Treating cancer with heat: Hyperthermia as promising strategy to enhance apoptosis. J. Pak. Med. Assoc. 2013, 63, 504–508. [Google Scholar] [PubMed]
- Kok, H.P.; Cressman, E.N.K.; Ceelen, W.; Brace, C.L.; Ivkov, R.; Grull, H.; Ter Haar, G.; Wust, P.; Crezee, J. Heating technology for malignant tumors: A review. Int. J. Hyperth. 2020, 37, 711–741. [Google Scholar] [CrossRef]
- Habash, R.W.Y. Therapeutic hyperthermia. Handb. Clin. Neurol. 2018, 157, 853–868. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, G.; Li, W.; He, X.; Xu, L. Radiofrequency ablation of advanced lung tumors: Imaging features, local control, and follow-up protocol. Int. J. Clin. Exp. Med. 2015, 8, 18137–18143. [Google Scholar] [PubMed]
- Wan, J.; Wu, W. Hyperthermia induced HIF-1a expression of lung cancer through AKT and ERK signaling pathways. J. Exp. Clin. Cancer Res. 2016, 35, 119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, W.; Bian, L.; Wang, N.; He, Y. Proteomic analysis of protein expression profiles during hyperthermia-induced apoptosis in Tca8113 cells. Oncol. Lett. 2013, 6, 135–143. [Google Scholar] [CrossRef]
- Takagi, H.; Azuma, K.; Tsuka, T.; Imagawa, T.; Osaki, T.; Okamoto, Y. Antitumor effects of high-temperature hyperthermia on a glioma rat model. Oncol. Lett. 2014, 7, 1007–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, H.; Cao, T.; Connolly, J.E.; Monnet, L.; Bennett, L.; Chapel, S.; Bagnis, C.; Mannoni, P.; Davoust, J.; Palucka, A.K.; et al. Hyperthermia enhances CTL cross-priming. J. Immunol. 2006, 176, 2134–2141. [Google Scholar] [CrossRef]
- Hou, C.H.; Lin, F.L.; Hou, S.M.; Liu, J.F. Hyperthermia induces apoptosis through endoplasmic reticulum and reactive oxygen species in human osteosarcoma cells. Int. J. Mol. Sci. 2014, 15, 17380–17395. [Google Scholar] [CrossRef] [Green Version]
- Kus-Liskiewicz, M.; Polanska, J.; Korfanty, J.; Olbryt, M.; Vydra, N.; Toma, A.; Widlak, W. Impact of heat shock transcription factor 1 on global gene expression profiles in cells which induce either cytoprotective or pro-apoptotic response following hyperthermia. BMC Genom. 2013, 14, 456. [Google Scholar] [CrossRef] [Green Version]
- Frank, J.; Lambert, C.; Biesalski, H.K.; Thews, O.; Vaupel, P.; Kelleher, D.K. Intensified oxidative and nitrosative stress following combined ALA-based photodynamic therapy and local hyperthermia in rat tumors. Int. J. Cancer 2003, 107, 941–948. [Google Scholar] [CrossRef]
- Oei, A.L.; van Leeuwen, C.M.; ten Cate, R.; Rodermond, H.M.; Buist, M.R.; Stalpers, L.J.; Crezee, J.; Kok, H.P.; Medema, J.P.; Franken, N.A. Hyperthermia Selectively Targets Human Papillomavirus in Cervical Tumors via p53-Dependent Apoptosis. Cancer Res. 2015, 75, 5120–5129. [Google Scholar] [CrossRef] [Green Version]
- Deezagi, A.; Manteghi, S.; Khosravani, P.; Vaseli-Hagh, N.; Soheili, Z.S. Induced apoptosis by mild hyperthermia occurs via telomerase inhibition on the three human myeloid leukemia cell lines: TF-1, K562, and HL-60. Leuk Lymphoma 2009, 50, 1519–1527. [Google Scholar] [CrossRef]
- Speit, G.; Schutz, P. Hyperthermia-induced genotoxic effects in human A549 cells. Mutat. Res. 2013, 747–748, 1–5. [Google Scholar] [CrossRef]
- Barni, S.; Pontiggia, P.; Bertone, V.; Vaccarone, R.; Silvotti, M.G.; Pontiggia, E.; Mathe, G. Hyperthermia-induced cell death by apoptosis in myeloma cells. Biomed. Pharmacother. 2001, 55, 170–173. [Google Scholar] [CrossRef] [PubMed]
- Garcia, M.P.; Cavalheiro, J.R.; Fernandes, M.H. Acute and long-term effects of hyperthermia in B16-F10 melanoma cells. PLoS ONE 2012, 7, e35489. [Google Scholar] [CrossRef]
- Pawlik, A.; Nowak, J.M.; Grzanka, D.; Gackowska, L.; Michalkiewicz, J.; Grzanka, A. Hyperthermia induces cytoskeletal alterations and mitotic catastrophe in p53-deficient H1299 lung cancer cells. Acta Histochem. 2013, 115, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Luchetti, F.; Mannello, F.; Canonico, B.; Battistelli, M.; Burattini, S.; Falcieri, E.; Papa, S. Integrin and cytoskeleton behaviour in human neuroblastoma cells during hyperthermia-related apoptosis. Apoptosis 2004, 9, 635–648. [Google Scholar] [CrossRef] [PubMed]
- Borkamo, E.D.; Dahl, O.; Bruland, O.; Fluge, O. Global gene expression analyses reveal changes in biological processes after hyperthermia in a rat glioma model. Int. J. Hyperth. 2008, 24, 425–441. [Google Scholar] [CrossRef]
- Furusawa, Y.; Tabuchi, Y.; Takasaki, I.; Wada, S.; Ohtsuka, K.; Kondo, T. Gene networks involved in apoptosis induced by hyperthermia in human lymphoma U937 cells. Cell Biol. Int. 2009, 33, 1253–1262. [Google Scholar] [CrossRef]
- Piehler, S.; Wucherpfennig, L.; Tansi, F.L.; Berndt, A.; Quaas, R.; Teichgraeber, U.; Hilger, I. Hyperthermia affects collagen fiber architecture and induces apoptosis in pancreatic and fibroblast tumor hetero-spheroids in vitro. Nanomedicine 2020, 28, 102183. [Google Scholar] [CrossRef]
- Sharif-Khatibi, L.; Kariminia, A.; Khoei, S.; Goliaei, B. Hyperthermia induces differentiation without apoptosis in permissive temperatures in human erythroleukaemia cells. Int. J. Hyperth. 2007, 23, 645–655. [Google Scholar] [CrossRef]
- Li, K.; Shen, S.Q.; Xiong, C.L. Microvessel damage may play an important role in tumoricidal effect for murine h(22) hepatoma cells with hyperthermia in vivo. J. Surg. Res. 2008, 145, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Kokura, S.; Yoshida, N.; Ueda, M.; Imamoto, E.; Ishikawa, T.; Takagi, T.; Naito, Y.; Okanoue, T.; Yoshikawa, T. Hyperthermia enhances tumor necrosis factor alpha-induced apoptosis of a human gastric cancer cell line. Cancer Lett. 2003, 201, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.D.; Zhang, Y.; Dong, T.X.; Xu, Y.T.; Zhang, W.; An, T.T.; Liu, P.F.; Yang, X.H. Hyperthermia with different temperatures inhibits proliferation and promotes apoptosis through the EGFR/STAT3 pathway in C6 rat glioma cells. Mol. Med. Rep. 2017, 16, 9401–9408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.C.; Zhang, Y.; Chen, H.Y.; Li, X.L.; Qin, L.J.; Li, Y.J.; Zhang, H.Y.; Wang, S. Hyperthermia promotes apoptosis and suppresses invasion in C6 rat glioma cells. Asian Pac. J. Cancer Prev. 2012, 13, 3239–3245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basile, A.; Biziato, D.; Sherbet, G.V.; Comi, P.; Cajone, F. Hyperthermia inhibits cell proliferation and induces apoptosis: Relative signaling status of P53, S100A4, and Notch in heat sensitive and resistant cell lines. J. Cell Biochem. 2008, 103, 212–220. [Google Scholar] [CrossRef]
- Fukami, T.; Nakasu, S.; Baba, K.; Nakajima, M.; Matsuda, M. Hyperthermia induces translocation of apoptosis-inducing factor (AIF) and apoptosis in human glioma cell lines. J. Neurooncol. 2004, 70, 319–331. [Google Scholar] [CrossRef]
- Pajonk, F.; van Ophoven, A.; McBride, W.H. Hyperthermia-induced proteasome inhibition and loss of androgen receptor expression in human prostate cancer cells. Cancer Res. 2005, 65, 4836–4843. [Google Scholar] [CrossRef] [Green Version]
- Morle, A.; Garrido, C.; Micheau, O. Hyperthermia restores apoptosis induced by death receptors through aggregation-induced c-FLIP cytosolic depletion. Cell Death Dis. 2015, 6, e1633. [Google Scholar] [CrossRef] [Green Version]
- Xie, X.; Shao, X.; Gao, F.; Jin, H.; Zhou, J.; Du, L.; Zhang, Y.; Ouyang, W.; Wang, X.; Zhao, L.; et al. Effect of hyperthermia on invasion ability and TGF-beta1 expression of breast carcinoma MCF-7 cells. Oncol. Rep. 2011, 25, 1573–1579. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Wang, X.; Du, L.; Zhao, L.; Lei, F.; Ouyang, W.; Zhang, Y.; Liao, Y.; Tang, J. Effect of hyperthermia on the apoptosis and proliferation of CaSki cells. Mol. Med. Rep. 2011, 4, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Huang, W.; Lin, Y.; Bian, L.; He, Y. Identification of proteins interacting with protein kinase C-delta in hyperthermia-induced apoptosis and thermotolerance of Tca8113 cells. Mol. Med. Rep. 2015, 12, 3821–3828. [Google Scholar] [CrossRef]
- Yasumoto, J.; Kirita, T.; Takahashi, A.; Ohnishi, K.; Imai, Y.; Yuki, K.; Ohnishi, T. Apoptosis-related gene expression after hyperthermia in human tongue squamous cell carcinoma cells harboring wild-type or mutated-type p53. Cancer Lett. 2004, 204, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Shellman, Y.G.; Howe, W.R.; Miller, L.A.; Goldstein, N.B.; Pacheco, T.R.; Mahajan, R.L.; LaRue, S.M.; Norris, D.A. Hyperthermia induces endoplasmic reticulum-mediated apoptosis in melanoma and non-melanoma skin cancer cells. J. Investig. Dermatol. 2008, 128, 949–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vertrees, R.A.; Das, G.C.; Coscio, A.M.; Xie, J.; Zwischenberger, J.B.; Boor, P.J. A mechanism of hyperthermia-induced apoptosis in ras-transformed lung cells. Mol. Carcinog. 2005, 44, 111–121. [Google Scholar] [CrossRef]
- Qin, S.; Xu, C.; Li, S.; Wang, X.; Sun, X.; Wang, P.; Zhang, B.; Ren, H. Hyperthermia induces apoptosis by targeting Survivin in esophageal cancer. Oncol. Rep. 2015, 34, 2656–2664. [Google Scholar] [CrossRef] [PubMed]
- Mantso, T.; Vasileiadis, S.; Anestopoulos, I.; Voulgaridou, G.P.; Lampri, E.; Botaitis, S.; Kontomanolis, E.N.; Simopoulos, C.; Goussetis, G.; Franco, R.; et al. Hyperthermia induces therapeutic effectiveness and potentiates adjuvant therapy with non-targeted and targeted drugs in an in vitro model of human malignant melanoma. Sci. Rep. 2018, 8, 10724. [Google Scholar] [CrossRef] [PubMed]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, S. Cisplatin: The first metal based anticancer drug. Bioorg. Chem. 2019, 88, 102925. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.F.; Yan, X.M.; Lan, B.; Lei, Y.R.; Li, X.H.; Gao, S.; Guo, Y.F.; Guo, F. Molecular mechanisms of synergistic induction of apoptosis by the combination therapy with hyperthermia and cisplatin in prostate cancer cells. Biochem. Biophys. Res. Commun. 2016, 479, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Narita, N.; Ito, Y.; Takabayashi, T.; Okamoto, M.; Imoto, Y.; Ogi, K.; Tokunaga, T.; Matsumoto, H.; Fujieda, S. Suppression of SESN1 reduces cisplatin and hyperthermia resistance through increasing reactive oxygen species (ROS) in human maxillary cancer cells. Int. J. Hyperth. 2018, 35, 269–278. [Google Scholar] [CrossRef]
- Mehtala, J.G.; Torregrosa-Allen, S.; Elzey, B.D.; Jeon, M.; Kim, C.; Wei, A. Synergistic effects of cisplatin chemotherapy and gold nanorod-mediated hyperthermia on ovarian cancer cells and tumors. Nanomedicine 2014, 9, 1939–1955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dou, Y.N.; Dunne, M.; Huang, H.; McKee, T.; Chang, M.C.; Jaffray, D.A.; Allen, C. Thermosensitive liposomal cisplatin in combination with local hyperthermia results in tumor growth delay and changes in tumor microenvironment in xenograft models of lung carcinoma. J. Drug Target. 2016, 24, 865–877. [Google Scholar] [CrossRef] [PubMed]
- Ohtsubo, T.; Igawa, H.; Saito, T.; Matsumoto, H.; Park, H.; Song, C.W.; Kano, E.; Saito, H. Enhancement of cell killing by induction of apoptosis after treatment with mild hyperthermia at 42 degrees C and cisplatin. Radiat. Res. 2001, 156, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Ho, G.Y.; Woodward, N.; Coward, J.I. Cisplatin versus carboplatin: Comparative review of therapeutic management in solid malignancies. Crit. Rev. Oncol. Hematol. 2016, 102, 37–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, E.K.; Park, S.R.; Lee, J.H.; Chung, H.S.; Ahn, H.E.; Rhee, Y.H.; Lim, B.U.; Park, H.J. Induction of apoptosis by carboplatin and hyperthermia alone or combined in WERI human retinoblastoma cells. Int. J. Hyperth. 2003, 19, 431–443. [Google Scholar] [CrossRef]
- Longley, D.B.; Harkin, D.P.; Johnston, P.G. 5-fluorouracil: Mechanisms of action and clinical strategies. Nat. Rev. Cancer 2003, 3, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Zhang, F.; Bai, Z.; Wang, J.; Qiu, L.; Li, Y.; Meng, Y.; Valji, K.; Yang, X. Orthotopic Esophageal Cancers: Intraesophageal Hyperthermia-enhanced Direct Chemotherapy in Rats. Radiology 2017, 282, 103–112. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Lu, L.; Yan, S.; Yi, H.; Yao, H.; Wu, D.; He, G.; Tao, X.; Deng, X. Autophagy and doxorubicin resistance in cancer. Anti-Cancer Drugs 2018, 29, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Schaaf, L.; Schwab, M.; Ulmer, C.; Heine, S.; Murdter, T.E.; Schmid, J.O.; Sauer, G.; Aulitzky, W.E.; van der Kuip, H. Hyperthermia Synergizes with Chemotherapy by Inhibiting PARP1-Dependent DNA Replication Arrest. Cancer Res. 2016, 76, 2868–2875. [Google Scholar] [CrossRef] [Green Version]
- Araya, T.; Kasahara, K.; Nishikawa, S.; Kimura, H.; Sone, T.; Nagae, H.; Ikehata, Y.; Nagano, I.; Fujimura, M. Antitumor effects of inductive hyperthermia using magnetic ferucarbotran nanoparticles on human lung cancer xenografts in nude mice. OncoTargets Ther. 2013, 6, 237–242. [Google Scholar] [CrossRef] [Green Version]
- Sato, I.; Umemura, M.; Mitsudo, K.; Kioi, M.; Nakashima, H.; Iwai, T.; Feng, X.; Oda, K.; Miyajima, A.; Makino, A.; et al. Hyperthermia generated with ferucarbotran (Resovist(R)) in an alternating magnetic field enhances cisplatin-induced apoptosis of cultured human oral cancer cells. J. Physiol. Sci. 2014, 64, 177–183. [Google Scholar] [CrossRef]
- Muenyi, C.S.; Pinhas, A.R.; Fan, T.W.; Brock, G.N.; Helm, C.W.; States, J.C. Sodium arsenite +/- hyperthermia sensitizes p53-expressing human ovarian cancer cells to cisplatin by modulating platinum-DNA damage responses. Toxicol. Sci. 2012, 127, 139–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muenyi, C.S.; Trivedi, A.P.; Helm, C.W.; States, J.C. Cisplatin plus sodium arsenite and hyperthermia induces pseudo-G1 associated apoptotic cell death in ovarian cancer cells. Toxicol. Sci. 2014, 139, 74–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, T.; Gong, W.; Li, X.; Zou, C.; Jiang, G.; Li, X.; Feng, D. Enhancement of osteosarcoma cell sensitivity to cisplatin using paclitaxel in the presence of hyperthermia. Int. J. Hyperth. 2013, 29, 248–255. [Google Scholar] [CrossRef]
- Abu Samaan, T.M.; Samec, M.; Liskova, A.; Kubatka, P.; Busselberg, D. Paclitaxel’s Mechanistic and Clinical Effects on Breast Cancer. Biomolecules 2019, 9, 789. [Google Scholar] [CrossRef] [Green Version]
- Liao, S.; Hu, X.; Liu, Z.; Lin, Y.; Liang, R.; Zhang, Y.; Li, Q.; Li, Y.; Liao, X. Synergistic action of microwave-induced mild hyperthermia and paclitaxel in inducing apoptosis in the human breast cancer cell line MCF-7. Oncol. Lett. 2019, 17, 603–615. [Google Scholar] [CrossRef]
- Michalakis, J.; Georgatos, S.D.; de Bree, E.; Polioudaki, H.; Romanos, J.; Georgoulias, V.; Tsiftsis, D.D.; Theodoropoulos, P.A. Short-term exposure of cancer cells to micromolar doses of paclitaxel, with or without hyperthermia, induces long-term inhibition of cell proliferation and cell death in vitro. Ann. Surg. Oncol. 2007, 14, 1220–1228. [Google Scholar] [CrossRef]
- Lin, Y.; Liu, Z.; Li, Y.; Liao, X.; Liao, S.; Cen, S.; Yang, L.; Wei, J.; Hu, X. Short-term hyperthermia promotes the sensitivity of MCF-7 human breast cancer cells to paclitaxel. Biol. Pharm. Bull. 2013, 36, 376–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giovinazzi, S.; Bellapu, D.; Morozov, V.M.; Ishov, A.M. Targeting mitotic exit with hyperthermia or APC/C inhibition to increase paclitaxel efficacy. Cell Cycle 2013, 12, 2598–2607. [Google Scholar] [CrossRef] [Green Version]
- Michalakis, J.; Georgatos, S.D.; Romanos, J.; Koutala, H.; Georgoulias, V.; Tsiftsis, D.; Theodoropoulos, P.A. Micromolar taxol, with or without hyperthermia, induces mitotic catastrophe and cell necrosis in HeLa cells. Cancer Chemother. Pharm. 2005, 56, 615–622. [Google Scholar] [CrossRef]
- Varnai, R.; Koskinen, L.M.; Mantyla, L.E.; Szabo, I.; FitzGerald, L.M.; Sipeky, C. Pharmacogenomic Biomarkers in Docetaxel Treatment of Prostate Cancer: From Discovery to Implementation. Genes 2019, 10, 599. [Google Scholar] [CrossRef] [Green Version]
- Lv, F.; Yu, Y.; Zhang, B.; Liang, D.; Li, Z.M.; You, W. Inhibitory effects of mild hyperthermia plus docetaxel therapy on ER(+/-) breast cancer cells and action mechanisms. J. Huazhong Univ. Sci. Technol. Med. Sci. 2013, 33, 870–876. [Google Scholar] [CrossRef]
- Meredith, A.M.; Dass, C.R. Increasing role of the cancer chemotherapeutic doxorubicin in cellular metabolism. J. Pharm. Pharm. 2016, 68, 729–741. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Kim, S.; Choi, B.H.; Park, M.T.; Lee, J.; Jeong, S.Y.; Choi, E.K.; Lim, B.U.; Kim, C.; Park, H.J. Hyperthermia improves therapeutic efficacy of doxorubicin carried by mesoporous silica nanocontainers in human lung cancer cells. Int. J. Hyperth. 2011, 27, 698–707. [Google Scholar] [CrossRef]
- Chae, S.Y.; Kim, Y.S.; Park, M.J.; Yang, J.; Park, H.; Namgung, M.S.; Rhim, H.; Lim, H.K. High-intensity focused ultrasound-induced, localized mild hyperthermia to enhance anti-cancer efficacy of systemic doxorubicin: An experimental study. Ultrasound Med. Biol. 2014, 40, 1554–1563. [Google Scholar] [CrossRef]
- Vancsik, T.; Forika, G.; Balogh, A.; Kiss, E.; Krenacs, T. Modulated electro-hyperthermia induced p53 driven apoptosis and cell cycle arrest additively support doxorubicin chemotherapy of colorectal cancer in vitro. Cancer Med. 2019, 8, 4292–4303. [Google Scholar] [CrossRef] [Green Version]
- Liang, Z.; Li, X.; Xie, Y.; Liu, S. ‘Smart’ gold nanoshells for combined cancer chemotherapy and hyperthermia. Biomed. Mater. 2014, 9, 025012. [Google Scholar] [CrossRef]
- Jeon, M.J.; Ahn, C.H.; Kim, H.; Chung, I.J.; Jung, S.; Kim, Y.H.; Youn, H.; Chung, J.W.; Kim, Y.I. The intratumoral administration of ferucarbotran conjugated with doxorubicin improved therapeutic effect by magnetic hyperthermia combined with pharmacotherapy in a hepatocellular carcinoma model. J. Exp. Clin. Cancer Res. 2014, 33, 57. [Google Scholar] [CrossRef]
- Tang, Y.; McGoron, A.J. Increasing the rate of heating: A potential therapeutic approach for achieving synergistic tumour killing in combined hyperthermia and chemotherapy. Int. J. Hyperth. 2013, 29, 145–155. [Google Scholar] [CrossRef]
- Li, Z.; Wang, H.; Chen, Y.; Wang, Y.; Li, H.; Han, H.; Chen, T.; Jin, Q.; Ji, J. pH- and NIR Light-Responsive Polymeric Prodrug Micelles for Hyperthermia-Assisted Site-Specific Chemotherapy to Reverse Drug Resistance in Cancer Treatment. Small 2016, 12, 2731–2740. [Google Scholar] [CrossRef]
- Liu, X.; Wang, C.; Ma, H.; Yu, F.; Hu, F.; Yuan, H. Water-Responsive Hybrid Nanoparticles Codelivering ICG and DOX Effectively Treat Breast Cancer via Hyperthermia-aided DOX Functionality and Drug Penetration. Adv. Healthc. Mater. 2019, 8, e1801486. [Google Scholar] [CrossRef]
- Gabizon, A.A.; Patil, Y.; La-Beck, N.M. New insights and evolving role of pegylated liposomal doxorubicin in cancer therapy. Drug Resist. Updates 2016, 29, 90–106. [Google Scholar] [CrossRef]
- Wu, S.K.; Chiang, C.F.; Hsu, Y.H.; Lin, T.H.; Liou, H.C.; Fu, W.M.; Lin, W.L. Short-time focused ultrasound hyperthermia enhances liposomal doxorubicin delivery and antitumor efficacy for brain metastasis of breast cancer. Int. J. Nanomed. 2014, 9, 4485–4494. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Wang, J.; Jin, Y.; Zhang, F.; Yang, X. Intratumoral radiofrequency hyperthermia-enhanced chemotherapy of liposomal doxorubicin on hepatocellular carcinoma. Am. J. Transl. Res. 2018, 10, 3619–3627. [Google Scholar]
- Eetezadi, S.; De Souza, R.; Vythilingam, M.; Lessa Cataldi, R.; Allen, C. Effects of Doxorubicin Delivery Systems and Mild Hyperthermia on Tissue Penetration in 3D Cell Culture Models of Ovarian Cancer Residual Disease. Mol. Pharm. 2015, 12, 3973–3985. [Google Scholar] [CrossRef]
- Zhao, M.; Ding, X.F.; Shen, J.Y.; Zhang, X.P.; Ding, X.W.; Xu, B. Use of liposomal doxorubicin for adjuvant chemotherapy of breast cancer in clinical practice. J. Zhejiang Univ. Sci. B 2017, 18, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Tsang, Y.W.; Chi, K.H.; Huang, C.C.; Chi, M.S.; Chiang, H.C.; Yang, K.L.; Li, W.T.; Wang, Y.S. Modulated electro-hyperthermia-enhanced liposomal drug uptake by cancer cells. Int. J. Nanomed. 2019, 14, 1269–1279. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Xu, Q.; Li, X.; Zhang, P.; Zhao, X.; Wang, Y. Redox-responsive hyaluronic acid nanogels for hyperthermia- assisted chemotherapy to overcome multidrug resistance. Carbohydr. Polym. 2019, 203, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.R.C.; Abdul-Majeed, S.; Cael, B.; Barta, S.K. Clinical Pharmacokinetics and Pharmacodynamics of Bortezomib. Clin. Pharm. 2019, 58, 157–168. [Google Scholar] [CrossRef]
- Cengiz Seval, G.; Beksac, M. The safety of bortezomib for the treatment of multiple myeloma. Expert Opin. Drug Saf. 2018, 17, 953–962. [Google Scholar] [CrossRef] [PubMed]
- Milani, V.; Lorenz, M.; Weinkauf, M.; Rieken, M.; Pastore, A.; Dreyling, M.; Issels, R. Combination of hyperthermia and bortezomib results in additive killing in mantle cell lymphoma cells. Int. J. Hyperth. 2009, 25, 262–272. [Google Scholar] [CrossRef]
- Saliev, T.; Feril, L.B., Jr.; Begimbetova, D.; Baiskhanova, D.; Klodzinskyi, A.; Bobrova, X.; Aipov, R.; Baltabayeva, T.; Tachibana, K. Hyperthermia enhances bortezomib-induced apoptosis in human white blood cancer cells. J. Therm. Biol. 2017, 67, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Schlack, K.; Boegemann, M.; Steinestel, J.; Schrader, A.J.; Krabbe, L.M. The safety and efficacy of gemcitabine for the treatment of bladder cancer. Expert Rev. Anticancer Ther. 2016, 16, 255–271. [Google Scholar] [CrossRef] [PubMed]
- Bami, H.; Goodman, C.; Boldt, G.; Vincent, M. Gemcitabine-induced pseudocellulitis: A case report and review of the literature. Curr. Oncol. 2019, 26, e703–e706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adachi, S.; Kokura, S.; Okayama, T.; Ishikawa, T.; Takagi, T.; Handa, O.; Naito, Y.; Yoshikawa, T. Effect of hyperthermia combined with gemcitabine on apoptotic cell death in cultured human pancreatic cancer cell lines. Int. J. Hyperth. 2009, 25, 210–219. [Google Scholar] [CrossRef]
- Jin, H.; Zhao, Y.; Yang, J.; Zhang, X.; Ma, S. Hyperthermia enhances the sensitivity of pancreatic cancer SW1990 cells to gemcitabine through ROS/JNK signaling. Oncol. Lett. 2018, 16, 6742–6748. [Google Scholar] [CrossRef] [Green Version]
- Vertrees, R.A.; Das, G.C.; Popov, V.L.; Coscio, A.M.; Goodwin, T.J.; Logrono, R.; Zwischenberger, J.B.; Boor, P.J. Synergistic interaction of hyperthermia and Gemcitabine in lung cancer. Cancer Biol. Ther. 2005, 4, 1144–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanhaji, M.; Goring, J.; Couleaud, P.; Aires, A.; Cortajarena, A.L.; Courty, J.; Prina-Mello, A.; Stapf, M.; Ludwig, R.; Volkov, Y.; et al. The phenotype of target pancreatic cancer cells influences cell death by magnetic hyperthermia with nanoparticles carrying gemicitabine and the pseudo-peptide NucAnt. Nanomedicine 2019, 20, 101983. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.K.; Shin, D.H.; Choi, M.H.; Kim, J.S. Enhanced antitumor efficacy of gemcitabine-loaded temperature-sensitive liposome by hyperthermia in tumor-bearing mice. Drug Dev. Ind. Pharm. 2014, 40, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Moretto, P.; Hotte, S.J. Targeting apoptosis: Preclinical and early clinical experience with mapatumumab, an agonist monoclonal antibody targeting TRAIL-R1. Expert Opin. Investig. Drugs 2009, 18, 311–325. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Kim, H.C.; Kim, S.Y.; Basse, P.; Park, B.H.; Lee, B.C.; Lee, Y.J. Hyperthermia-enhanced TRAIL- and mapatumumab-induced apoptotic death is mediated through mitochondria in human colon cancer cells. J. Cell Biochem. 2012, 113, 1547–1558. [Google Scholar] [CrossRef] [Green Version]
- Song, X.; Kim, S.Y.; Lee, Y.J. The role of Bcl-xL in synergistic induction of apoptosis by mapatumumab and oxaliplatin in combination with hyperthermia on human colon cancer. Mol. Cancer Res. 2012, 10, 1567–1579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogers, B.B.; Cuddahy, T.; Briscella, C.; Ross, N.; Olszanski, A.J.; Denlinger, C.S. Oxaliplatin: Detection and Management of Hypersensitivity Reactions. Clin. J. Oncol. Nurs. 2019, 23, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Kim, S.Y.; Lee, Y.J. Evidence for two modes of synergistic induction of apoptosis by mapatumumab and oxaliplatin in combination with hyperthermia in human colon cancer cells. PLoS ONE 2013, 8, e73654. [Google Scholar] [CrossRef] [Green Version]
- Ahlmann, M.; Hempel, G. The effect of cyclophosphamide on the immune system: Implications for clinical cancer therapy. Cancer Chemother. Pharm. 2016, 78, 661–671. [Google Scholar] [CrossRef]
- Taraboletti, G.; Rusnati, M.; Ragona, L.; Colombo, G. Targeting tumor angiogenesis with TSP-1-based compounds: Rational design of antiangiogenic mimetics of endogenous inhibitors. Oncotarget 2010, 1, 662–673. [Google Scholar] [CrossRef] [PubMed]
- Borkamo, E.D.; Fluge, O.; Mella, O.; Akslen, L.A.; Bruland, O.; Dahl, O. Hyperthermia improves the antitumour effect of metronomic cyclophosphamide in a rat transplantable brain tumour. Radiother. Oncol. 2008, 86, 435–442. [Google Scholar] [CrossRef]
- Emadi, A.; Jones, R.J.; Brodsky, R.A. Cyclophosphamide and cancer: Golden anniversary. Nat. Rev. Clin. Oncol. 2009, 6, 638–647. [Google Scholar] [CrossRef] [PubMed]
- Lushnikova, E.L.; Ovsyanko, E.V.; Nepomnyashchikh, L.M.; Efremov, A.V.; Morozov, D.V. Proliferation of Walker 256 carcinosarcoma cells: Effect of whole-body hyperthermia and antitumor agents. Bull. Exp. Biol. Med. 2011, 152, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Kaveh, S.; Ebrahimi, P.; Rezapour, A.; Mozafari, M.; Sayehmiri, K. Bevacizumab and erlotinib versus bevacizumab for colorectal cancer treatment: Systematic review and meta-analysis. Int. J. Clin. Pharm. 2019, 41, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Zhang, D.; Liu, J.; Wang, J.; Lu, Y.; Zheng, J.; Li, B.; Jia, L. Functionalized MoS2-erlotinib produces hyperthermia under NIR. J. Nanobiotechnol. 2019, 17, 76. [Google Scholar] [CrossRef]
- Ahmed, K.; Zhao, Q.L.; Matsuya, Y.; Yu, D.Y.; Salunga, T.L.; Nemoto, H.; Kondo, T. Enhancement of macrosphelide-induced apoptosis by mild hyperthermia. Int. J. Hyperth. 2007, 23, 353–361. [Google Scholar] [CrossRef]
- Pette, M.; Gold, R.; Pette, D.F.; Hartung, H.P.; Toyka, K.V. Mafosfamide induces DNA fragmentation and apoptosis in human T-lymphocytes. A possible mechanism of its immunosuppressive action. Immunopharmacology 1995, 30, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Ehlers, E.M.; Kuhnel, W.; Wiedemann, G.J. Hyperthermia and mafosfamide in a human-derived malignant pleural mesothelioma cell line. J. Cancer Res. Clin. Oncol. 2002, 128, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Krause, C.; Kluttermann, K.; Mauz-Korholz, C. Molecular mechanisms and gene regulation of melphalan- and hyperthermia-induced apoptosis in Ewing sarcoma cells. Anticancer Res. 2008, 28, 2585–2593. [Google Scholar] [PubMed]
- Rihacek, M.; Pilatova, K.; Sterba, J.; Pilny, R.; Valik, D. New Indings in Methotrexate Pharmacology-Diagnostic Possibilities and Impact on Clinical Care. Klin. Onkol. 2015, 28, 163–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bedoui, Y.; Guillot, X.; Selambarom, J.; Guiraud, P.; Giry, C.; Jaffar-Bandjee, M.C.; Ralandison, S.; Gasque, P. Methotrexate an Old Drug with New Tricks. Int. J. Mol. Sci. 2019, 20, 5023. [Google Scholar] [CrossRef] [Green Version]
- Costa Lima, S.A.; Gaspar, A.; Reis, S.; Duraes, L. Multifunctional nanospheres for co-delivery of methotrexate and mild hyperthermia to colon cancer cells. Mater. Sci. Eng. C 2017, 75, 1420–1426. [Google Scholar] [CrossRef]
- Li, Y.Q.; Xu, M.; Dhawan, U.; Liu, W.C.; Wu, K.T.; Liu, X.R.; Lin, C.; Zhao, G.; Wu, Y.C.; Chung, R.J. Iron-gold alloy nanoparticles serve as a cornerstone in hyperthermia-mediated controlled drug release for cancer therapy. Int. J. Nanomed. 2018, 13, 5499–5509. [Google Scholar] [CrossRef] [Green Version]
- Di Stasi, S.M.; Verri, C.; Celestino, F.; De Carlo, F.; Pagliarulo, V. Intravesical electro-osmotic administration of mitomycin C. Urologia 2016, 83, 18–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimou, A.; Syrigos, K.N.; Saif, M.W. Is there a role for mitomycin C in metastatic colorectal cancer? Expert Opin. Investig. Drugs 2010, 19, 723–735. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Lee, D.H.; Song, X.; Bartlett, D.L.; Kwon, Y.T.; Lee, Y.J. Role of Bcl-xL/Beclin-1 in synergistic apoptotic effects of secretory TRAIL-armed adenovirus in combination with mitomycin C and hyperthermia on colon cancer cells. Apoptosis 2014, 19, 1603–1615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van den Tempel, N.; Naipal, K.A.T.; Raams, A.; van Gent, D.C.; Franckena, M.; Boormans, J.L.; Kanaar, R. Ex vivo assays to predict enhanced chemosensitization by hyperthermia in urothelial cancer of the bladder. PLoS ONE 2018, 13, e0209101. [Google Scholar] [CrossRef]
- Lam, S.C.; Yuen, H.K.L. Medical and sclerosing agents in the treatment of orbital lymphatic malformations: What’s new? Curr. Opin. Ophthalmol. 2019, 30, 380–385. [Google Scholar] [CrossRef]
- Li, T.C.; Liu, C.C.; Lee, Y.Z.; Hsu, Y.H.; Chiang, C.F.; Miaw, S.C.; Lin, W.L. Combination Therapy of Pulsed-Wave Ultrasound Hyperthermia and Immunostimulant OK-432 Enhances Systemic Antitumor Immunity for Cancer Treatment. Int. J. Radiat. Oncol. Biol. Phys. 2020, 108, 140–149. [Google Scholar] [CrossRef]
- Yoo, J.; Lee, Y.J. Effect of hyperthermia and chemotherapeutic agents on TRAIL-induced cell death in human colon cancer cells. J. Cell Biochem. 2008, 103, 98–109. [Google Scholar] [CrossRef] [PubMed]
- To, K.K.; Poon, D.C.; Wei, Y.; Wang, F.; Lin, G.; Fu, L. Pelitinib (EKB-569) targets the up-regulation of ABCB1 and ABCG2 induced by hyperthermia to eradicate lung cancer. Br. J. Pharm. 2015, 172, 4089–4106. [Google Scholar] [CrossRef] [Green Version]
- Krupka, T.M.; Exner, A.A. Structural parameters governing activity of Pluronic triblock copolymers in hyperthermia cancer therapy. Int. J. Hyperth. 2011, 27, 663–671. [Google Scholar] [CrossRef] [Green Version]
- Lee, I. Ranpirnase (Onconase), a cytotoxic amphibian ribonuclease, manipulates tumour physiological parameters as a selective killer and a potential enhancer for chemotherapy and radiation in cancer therapy. Expert Opin. Biol. Ther. 2008, 8, 813–827. [Google Scholar] [CrossRef] [PubMed]
- Nasu, M.; Carbone, M.; Gaudino, G.; Ly, B.H.; Bertino, P.; Shimizu, D.; Morris, P.; Pass, H.I.; Yang, H. Ranpirnase Interferes with NF-kappaB Pathway and MMP9 Activity, Inhibiting Malignant Mesothelioma Cell Invasiveness and Xenograft Growth. Genes Cancer 2011, 2, 576–584. [Google Scholar] [CrossRef] [PubMed]
- Halicka, H.D.; Ardelt, B.; Shogen, K.; Darzynkiewicz, Z. Mild hyperthermia predisposes tumor cells to undergo apoptosis upon treatment with onconase. Int. J. Oncol. 2007, 30, 841–847. [Google Scholar] [PubMed] [Green Version]
- Keating, G.M. Sorafenib: A Review in Hepatocellular Carcinoma. Target. Oncol. 2017, 12, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Wang, C.; Sun, J.; Sun, L.; Wan, J.; Wang, S.; Gu, D.; Yu, C.; Yang, C.; He, J.; et al. Self-Assembled and Self-Monitored Sorafenib/Indocyanine Green Nanodrug with Synergistic Antitumor Activity Mediated by Hyperthermia and Reactive Oxygen Species-Induced Apoptosis. ACS Appl. Mater. Interfaces 2019, 11, 43996–44006. [Google Scholar] [CrossRef] [PubMed]
- Aspe, J.R.; Wall, N.R. Survivin-T34A: Molecular mechanism and therapeutic potential. OncoTargets Ther. 2010, 3, 247–254. [Google Scholar] [CrossRef]
- Li, Z.M.; Zhao, Y.W.; Zhao, C.J.; Zhang, X.P.; Chen, L.J.; Wei, Y.Q.; Yang, H.S. Hyperthermia increases the therapeutic efficacy of survivinT34A in mouse tumor models. Cancer Biol. Ther. 2011, 12, 523–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aasland, D.; Gotzinger, L.; Hauck, L.; Berte, N.; Meyer, J.; Effenberger, M.; Schneider, S.; Reuber, E.E.; Roos, W.P.; Tomicic, M.T.; et al. Temozolomide Induces Senescence and Repression of DNA Repair Pathways in Glioblastoma Cells via Activation of ATR-CHK1, p21, and NF-kappaB. Cancer Res. 2019, 79, 99–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, S.H.; Ueno, T.; Yoshimoto, Y.; Yoo, J.S.; Abdel-Wahab, O.I.; Abdel-Wahab, Z.; Chu, E.; Pruitt, S.K.; Friedman, H.S.; Dewhirst, M.W.; et al. Optimizing a novel regional chemotherapeutic agent against melanoma: Hyperthermia-induced enhancement of temozolomide cytotoxicity. Clin. Cancer Res. 2006, 12, 289–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masunaga, S.; Ono, K.; Takahashi, A.; Ohnishi, K.; Ohnishi, T.; Suzuki, M.; Nagata, K.; Kinashi, Y.; Nagasawa, H.; Uto, Y.; et al. Usefulness of combined treatment with mild temperature hyperthermia and/or tirapazamine in the treatment of solid tumors: Its independence of p53 status. Cancer Sci. 2003, 94, 125–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maenpaa, J.U.; Ala-Fossi, S.L. Toremifene in postmenopausal breast cancer. Efficacy, safety and cost. Drugs Aging 1997, 11, 261–270. [Google Scholar] [CrossRef]
- Kanaya, Y.; Doihara, H.; Shiroma, K.; Ogasawara, Y.; Date, H. Effect of combined therapy with the antiestrogen agent toremifene and local hyperthermia on breast cancer cells implanted in nude mice. Surg. Today 2008, 38, 911–920. [Google Scholar] [CrossRef] [Green Version]
- D’Incalci, M. Trabectedin mechanism of action: What’s new? Future Oncol. 2013, 9, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Harnicek, D.; Kampmann, E.; Lauber, K.; Hennel, R.; Cardoso Martins, A.S.; Guo, Y.; Belka, C.; Mortl, S.; Gallmeier, E.; Kanaar, R.; et al. Hyperthermia adds to trabectedin effectiveness and thermal enhancement is associated with BRCA2 degradation and impairment of DNA homologous recombination repair. Int. J. Cancer 2016, 139, 467–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haque, A.; Rahman, M.A.; Faizi, M.S.H.; Khan, M.S. Next Generation Antineoplastic Agents: A Review on Structurally Modified Vinblastine (VBL) Analogues. Curr. Med. Chem. 2018, 25, 1650–1662. [Google Scholar] [CrossRef] [PubMed]
- Gourmelon, C.; Bourien, H.; Augereau, P.; Patsouris, A.; Frenel, J.S.; Campone, M. Vinflunine for the treatment of breast cancer. Expert Opin. Pharm. 2016, 17, 1817–1823. [Google Scholar] [CrossRef]
- Eikesdal, H.P.; Bjerkvig, R.; Dahl, O. Vinblastine and hyperthermia target the neovasculature in BT(4)AN rat gliomas: Therapeutic implications of the vascular phenotype. Int. J. Radiat. Oncol. Biol. Phys. 2001, 51, 535–544. [Google Scholar] [CrossRef]
- Wan, D.; Ouyang, H. Baicalin induces apoptosis in human osteosarcoma cell through ROS-mediated mitochondrial pathway. Nat. Prod. Res. 2018, 32, 1996–2000. [Google Scholar] [CrossRef] [PubMed]
- Zakki, S.A.; Cui, Z.G.; Sun, L.; Feng, Q.W.; Li, M.L.; Inadera, H. Baicalin Augments Hyperthermia-Induced Apoptosis in U937 Cells and Modulates the MAPK Pathway via ROS Generation. Cell Physiol. Biochem. 2018, 45, 2444–2460. [Google Scholar] [CrossRef]
- Zhou, C.G.; Hui, L.M.; Luo, J.M. Epigallocatechin gallate inhibits the proliferation and induces apoptosis of multiple myeloma cells via inactivating EZH2. Eur. Rev. Med. Pharm. Sci 2018, 22, 2093–2098. [Google Scholar] [CrossRef]
- Lu, C.H.; Chen, W.T.; Hsieh, C.H.; Kuo, Y.Y.; Chao, C.Y. Thermal cycling-hyperthermia in combination with polyphenols, epigallocatechin gallate and chlorogenic acid, exerts synergistic anticancer effect against human pancreatic cancer PANC-1 cells. PLoS ONE 2019, 14, e0217676. [Google Scholar] [CrossRef] [Green Version]
- Mei, J.; Ma, J.; Xu, Y.; Wang, Y.; Hu, M.; Ma, F.; Qin, Z.; Xue, R.; Tao, N. Cinnamaldehyde Treatment of Prostate Cancer-Associated Fibroblasts Prevents Their Inhibitory Effect on T Cells Through Toll-Like Receptor 4. Drug Des. Dev. Ther. 2020, 14, 3363–3372. [Google Scholar] [CrossRef]
- Park, J.; Baek, S.H. Combination Therapy with Cinnamaldehyde and Hyperthermia Induces Apoptosis of A549 Non-Small Cell Lung Carcinoma Cells via Regulation of Reactive Oxygen Species and Mitogen-Activated Protein Kinase Family. Int. J. Mol. Sci. 2020, 21, 6229. [Google Scholar] [CrossRef]
- Ahn, C.R.; Park, J.; Kim, J.E.; Ahn, K.S.; Kim, Y.W.; Jeong, M.; Kim, H.J.; Park, S.H.; Baek, S.H. Cinnamaldehyde and Hyperthermia Co-Treatment Synergistically Induces ROS-Mediated Apoptosis in ACHN Renal Cell Carcinoma Cells. Biomedicines 2020, 8, 357. [Google Scholar] [CrossRef]
- Sun, L.; Cui, Z.G.; Zakki, S.A.; Feng, Q.W.; Li, M.L.; Inadera, H. Mechanistic study of nonivamide enhancement of hyperthermia-induced apoptosis in U937 cells. Free Radic. Biol. Med. 2018, 120, 147–159. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Powell, F.; Larsen, N.A.; Lai, Z.; Byth, K.F.; Read, J.; Gu, R.F.; Roth, M.; Toader, D.; Saeh, J.C.; et al. Mechanism and in vitro pharmacology of TAK1 inhibition by (5Z)-7-Oxozeaenol. ACS Chem. Biol. 2013, 8, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Acuna, U.M.; Wittwer, J.; Ayers, S.; Pearce, C.J.; Oberlies, N.H.; EJ, D.E.B. Effects of (5Z)-7-oxozeaenol on the oxidative pathway of cancer cells. Anticancer Res. 2012, 32, 2665–2671. [Google Scholar] [PubMed]
- Li, P.; Zhao, Q.L.; Jawaid, P.; Rehman, M.U.; Ahmed, K.; Sakurai, H.; Kondo, T. Enhancement of hyperthermia-induced apoptosis by 5Z-7-oxozeaenol, a TAK1 inhibitor, in Molt-4 cells. Int. J. Hyperth. 2017, 33, 411–418. [Google Scholar] [CrossRef]
- Hahm, E.R.; Kim, S.H.; Singh, K.B.; Singh, K.; Singh, S.V. A Comprehensive Review and Perspective on Anticancer Mechanisms of Withaferin A in Breast Cancer. Cancer Prev. Res. 2020, 13, 721–734. [Google Scholar] [CrossRef]
- Cui, Z.G.; Piao, J.L.; Rehman, M.U.; Ogawa, R.; Li, P.; Zhao, Q.L.; Kondo, T.; Inadera, H. Molecular mechanisms of hyperthermia-induced apoptosis enhanced by withaferin A. Eur. J. Pharm. 2014, 723, 99–107. [Google Scholar] [CrossRef]
- Blaszczak, W.; Barczak, W.; Masternak, J.; Kopczynski, P.; Zhitkovich, A.; Rubis, B. Vitamin C as a Modulator of the Response to Cancer Therapy. Molecules 2019, 24, 453. [Google Scholar] [CrossRef] [Green Version]
- Asada, R.; Kageyama, K.; Tanaka, H.; Kimura, M.; Saitoh, Y.; Miwa, N. Carcinostatic effects of diverse ascorbate derivatives in comparison with aliphatic chain moiety structures: Promotion by combined hyperthermia and reduced cytotoxicity to normal cells. Oncol. Lett. 2012, 3, 1042–1046. [Google Scholar] [CrossRef] [PubMed]
- Kuban-Jankowska, A.; Gorska-Ponikowska, M.; Sahu, K.K.; Kostrzewa, T.; Wozniak, M.; Tuszynski, J. Docosahexaenoic Acid Inhibits PTP1B Phosphatase and the Viability of MCF-7 Breast Cancer Cells. Nutrients 2019, 11, 2554. [Google Scholar] [CrossRef] [Green Version]
- Song, E.A.; Kim, H. Docosahexaenoic Acid Induces Oxidative DNA Damage and Apoptosis, and Enhances the Chemosensitivity of Cancer Cells. Int. J. Mol. Sci. 2016, 17, 1257. [Google Scholar] [CrossRef] [Green Version]
- Cui, Z.G.; Piao, J.L.; Kondo, T.; Ogawa, R.; Tsuneyama, K.; Zhao, Q.L.; Feril, L.B., Jr.; Inadera, H. Molecular mechanisms of hyperthermia-induced apoptosis enhanced by docosahexaenoic acid: Implication for cancer therapy. Chem. Biol. Interact. 2014, 215, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Delbridge, A.R.; Strasser, A. The BCL-2 protein family, BH3-mimetics and cancer therapy. Cell Death Differ. 2015, 22, 1071–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Z.; Lan, B.; Peng, B.; Wang, X.; Zhang, G.; Li, X.; Guo, F. Combination therapy with BH3 mimetic and hyperthermia tends to be more effective on anti-melanoma treatment. Biochem. Biophys. Res. Commun. 2018, 503, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Veisi, A.; Akbari, G.; Mard, S.A.; Badfar, G.; Zarezade, V.; Mirshekar, M.A. Role of crocin in several cancer cell lines: An updated review. Iran. J. Basic Med. Sci. 2020, 23, 3–12. [Google Scholar] [CrossRef]
- Hosseinzadeh, H.; Mehri, S.; Heshmati, A.; Ramezani, M.; Sahebkar, A.; Abnous, K. Proteomic screening of molecular targets of crocin. DARU J. Pharm. Sci. 2014, 22, 5. [Google Scholar] [CrossRef] [Green Version]
- Mostafavinia, S.E.; Khorashadizadeh, M.; Hoshyar, R. Antiproliferative and Proapoptotic Effects of Crocin Combined with Hyperthermia on Human Breast Cancer Cells. DNA Cell Biol. 2016, 35, 340–347. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.C.; Fonseca, C.O.; Schonthal, A.H. Preclinical development and clinical use of perillyl alcohol for chemoprevention and cancer therapy. Am. J. Cancer Res. 2015, 5, 1580–1593. [Google Scholar] [PubMed]
- Chen, T.C.; da Fonseca, C.O.; Schonthal, A.H. Intranasal Perillyl Alcohol for Glioma Therapy: Molecular Mechanisms and Clinical Development. Int. J. Mol. Sci. 2018, 19, 3905. [Google Scholar] [CrossRef] [Green Version]
- Ahn, K.J.; Lee, C.K.; Choi, E.K.; Griffin, R.; Song, C.W.; Park, H.J. Cytotoxicity of perillyl alcohol against cancer cells is potentiated by hyperthermia. Int. J. Radiat. Oncol. Biol. Phys. 2003, 57, 813–819. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K. 5-Aminolevulinic acid-mediated photodynamic therapy for bladder cancer. Int. J. Urol. 2017, 24, 97–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirschberg, H.; Sun, C.H.; Tromberg, B.J.; Yeh, A.T.; Madsen, S.J. Enhanced cytotoxic effects of 5-aminolevulinic acid-mediated photodynamic therapy by concurrent hyperthermia in glioma spheroids. J. Neurooncol. 2004, 70, 289–299. [Google Scholar] [CrossRef] [Green Version]
- Giordano, A.; Tommonaro, G. Curcumin and Cancer. Nutrients 2019, 11, 2376. [Google Scholar] [CrossRef] [Green Version]
- Tang, J.C.; Shi, H.S.; Wan, L.Q.; Wang, Y.S.; Wei, Y.Q. Enhanced antitumor effect of curcumin liposomes with local hyperthermia in the LL/2 model. Asian Pac. J. Cancer Prev. 2013, 14, 2307–2310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, D.Y.; Matsuya, Y.; Zhao, Q.L.; Ahmed, K.; Wei, Z.L.; Nemoto, H.; Kondo, T. Enhancement of hyperthermia-induced apoptosis by a new synthesized class of furan-fused tetracyclic compounds. Apoptosis 2007, 12, 1523–1532. [Google Scholar] [CrossRef]
- Zhang, X.; Hu, J.; Chen, Y. Betulinic acid and the pharmacological effects of tumor suppression (Review). Mol. Med. Rep. 2016, 14, 4489–4495. [Google Scholar] [CrossRef] [Green Version]
- Wachsberger, P.R.; Burd, R.; Wahl, M.L.; Leeper, D.B. Betulinic acid sensitization of low pH adapted human melanoma cells to hyperthermia. Int. J. Hyperth. 2002, 18, 153–164. [Google Scholar] [CrossRef]
- Wei, Y.; Yang, P.; Cao, S.; Zhao, L. The combination of curcumin and 5-fluorouracil in cancer therapy. Arch. Pharm. Res. 2018, 41, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, S.; Girija, A.R.; Nagaoka, Y.; Iwai, S.; Suzuki, M.; Kizhikkilot, V.; Yoshida, Y.; Maekawa, T.; Nair, S.D. Curcumin and 5-fluorouracil-loaded, folate- and transferrin-decorated polymeric magnetic nanoformulation: A synergistic cancer therapeutic approach, accelerated by magnetic hyperthermia. Int. J. Nanomed. 2014, 9, 437–459. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Pan, D.; Yang, H.; Huang, J.; He, Z.; Li, H.; Li, D. Effects of arsenic trioxide combined with platinum drugs in treatment of cervical cancer: A protocol for systematic review and meta-analysis of randomized controlled trials. Medicine 2020, 99, e22950. [Google Scholar] [CrossRef] [PubMed]
- Hoonjan, M.; Jadhav, V.; Bhatt, P. Arsenic trioxide: Insights into its evolution to an anticancer agent. J. Biol. Inorg. Chem. 2018, 23, 313–329. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.H.; Liang, H.J.; Zhang, Q.Q.; Li, S.Q.; Li, X.R.; Huo, X.Q.; Yang, Q.H.; Li, W.W.; Gu, J.F.; Hua, Q.L.; et al. Radiosensitivity enhancement by arsenic trioxide in conjunction with hyperthermia in the EC-1 esophageal carcinoma cell line. Asian Pac. J. Cancer Prev. 2012, 13, 1693–1697. [Google Scholar] [CrossRef]
- Kuo, I.M.; Lee, J.J.; Wang, Y.S.; Chiang, H.C.; Huang, C.C.; Hsieh, P.J.; Han, W.; Ke, C.H.; Liao, A.T.C.; Lin, C.S. Potential enhancement of host immunity and anti-tumor efficacy of nanoscale curcumin and resveratrol in colorectal cancers by modulated electro- hyperthermia. BMC Cancer 2020, 20, 603. [Google Scholar] [CrossRef]
- Asea, A.; Ara, G.; Teicher, B.A.; Stevenson, M.A.; Calderwood, S.K. Effects of the flavonoid drug quercetin on the response of human prostate tumours to hyperthermia in vitro and in vivo. Int. J. Hyperth. 2001, 17, 347–356. [Google Scholar] [CrossRef]
- Shen, J.; Zhang, W.; Wu, J.; Zhu, Y. The synergistic reversal effect of multidrug resistance by quercetin and hyperthermia in doxorubicin-resistant human myelogenous leukemia cells. Int. J. Hyperth. 2008, 24, 151–159. [Google Scholar] [CrossRef]
- Hansen, R.K.; Oesterreich, S.; Lemieux, P.; Sarge, K.D.; Fuqua, S.A. Quercetin inhibits heat shock protein induction but not heat shock factor DNA-binding in human breast carcinoma cells. Biochem. Biophys. Res. Commun. 1997, 239, 851–856. [Google Scholar] [CrossRef]
- Osborne, C.K. Tamoxifen in the treatment of breast cancer. N. Engl. J. Med. 1998, 339, 1609–1618. [Google Scholar] [CrossRef] [PubMed]
- O’Brian, C.A.; Liskamp, R.M.; Solomon, D.H.; Weinstein, I.B. Inhibition of protein kinase C by tamoxifen. Cancer Res. 1985, 45, 2462–2465. [Google Scholar] [PubMed]
- Piantelli, M.; Tatone, D.; Castrilli, G.; Savini, F.; Maggiano, N.; Larocca, L.M.; Ranelletti, F.O.; Natali, P.G. Quercetin and tamoxifen sensitize human melanoma cells to hyperthermia. Melanoma Res. 2001, 11, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Zhang, M.; Fu, Q.; Li, J.; Sun, H. Targeted near infrared hyperthermia combined with immune stimulation for optimized therapeutic efficacy in thyroid cancer treatment. Oncotarget 2016, 7, 6878–6890. [Google Scholar] [CrossRef] [Green Version]
- Hori, T.; Kondo, T.; Lee, H.; Song, C.W.; Park, H.J. Hyperthermia enhances the effect of beta-lapachone to cause gammaH2AX formations and cell death in human osteosarcoma cells. Int. J. Hyperth. 2011, 27, 53–62. [Google Scholar] [CrossRef]
- Shao, R.G. Pharmacology and therapeutic applications of enediyne antitumor antibiotics. Curr. Mol. Pharm. 2008, 1, 50–60. [Google Scholar] [CrossRef]
- Garrett, J.E.; Metzger, E.; Schmitt, K.; Soto, S.; Northern, S.; Kryah, L.; Irfan, M.; Rice, S.; Brown, M.; Zaleski, J.M.; et al. Enhancement of Cytotoxicity of Enediyne Compounds by Hyperthermia: Effects of Various Metal Complexes on Tumor Cells. Radiat. Res. 2020, 193, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Slimen, I.B.; Najar, T.; Ghram, A.; Dabbebi, H.; Ben Mrad, M.; Abdrabbah, M. Reactive oxygen species, heat stress and oxidative-induced mitochondrial damage. A review. Int. J. Hyperth. 2014, 30, 513–523. [Google Scholar] [CrossRef]
- Eppink, B.; Krawczyk, P.M.; Stap, J.; Kanaar, R. Hyperthermia-induced DNA repair deficiency suggests novel therapeutic anti-cancer strategies. Int. J. Hyperth. 2012, 28, 509–517. [Google Scholar] [CrossRef]
- Krawczyk, P.M.; Eppink, B.; Essers, J.; Stap, J.; Rodermond, H.; Odijk, H.; Zelensky, A.; van Bree, C.; Stalpers, L.J.; Buist, M.R.; et al. Mild hyperthermia inhibits homologous recombination, induces BRCA2 degradation, and sensitizes cancer cells to poly (ADP-ribose) polymerase-1 inhibition. Proc. Natl. Acad. Sci. USA 2011, 108, 9851–9856. [Google Scholar] [CrossRef] [Green Version]
- Srinivas, U.S.; Tan, B.W.Q.; Vellayappan, B.A.; Jeyasekharan, A.D. ROS and the DNA damage response in cancer. Redox Biol. 2019, 25, 101084. [Google Scholar] [CrossRef]
- Chen, F.; Rezavi, R.; Wang, C.C.; Harrison, L.E. Proteasome inhibition potentiates the cytotoxic effects of hyperthermia in HT-29 colon cancer cells through inhibition of heat shock protein 27. Oncology 2007, 73, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Burns, T.F. Targeting Heat Shock Proteins in Cancer: A Promising Therapeutic Approach. Int. J. Mol. Sci. 2017, 18, 1978. [Google Scholar] [CrossRef] [Green Version]
- Gilchrist, R.K.; Medal, R.; Shorey, W.D.; Hanselman, R.C.; Parrott, J.C.; Taylor, C.B. Selective inductive heating of lymph nodes. Ann. Surg. 1957, 146, 596–606. [Google Scholar] [CrossRef]
- Jordan, A.; Wust, P.; Fahling, H.; John, W.; Hinz, A.; Felix, R. Inductive heating of ferrimagnetic particles and magnetic fluids: Physical evaluation of their potential for hyperthermia. Int. J. Hyperth. 1993, 9, 51–68. [Google Scholar] [CrossRef]
- Ito, A.; Kuga, Y.; Honda, H.; Kikkawa, H.; Horiuchi, A.; Watanabe, Y.; Kobayashi, T. Magnetite nanoparticle-loaded anti-HER2 immunoliposomes for combination of antibody therapy with hyperthermia. Cancer Lett. 2004, 212, 167–175. [Google Scholar] [CrossRef]
- DeNardo, S.J.; DeNardo, G.L.; Miers, L.A.; Natarajan, A.; Foreman, A.R.; Gruettner, C.; Adamson, G.N.; Ivkov, R. Development of tumor targeting bioprobes ((111)In-chimeric L6 monoclonal antibody nanoparticles) for alternating magnetic field cancer therapy. Clin Cancer Res. 2005, 11 Pt 2, 7087s–7092s. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeNardo, S.J.; DeNardo, G.L.; Natarajan, A.; Miers, L.A.; Foreman, A.R.; Gruettner, C.; Adamson, G.N.; Ivkov, R. Thermal dosimetry predictive of efficacy of 111In-ChL6 nanoparticle AMF—Induced thermoablative therapy for human breast cancer in mice. J. Nucl. Med. 2007, 48, 437–444. [Google Scholar]
- Kennedy, L.C.; Bickford, L.R.; Lewinski, N.A.; Coughlin, A.J.; Hu, Y.; Day, E.S.; West, J.L.; Drezek, R.A. A new era for cancer treatment: Gold-nanoparticle-mediated thermal therapies. Small 2011, 7, 169–183. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, R.L.; Zhang, M.; Diagaradjane, P.; Peddibhotla, S.; Contreras, A.; Hilsenbeck, S.G.; Woodward, W.A.; Krishnan, S.; Chang, J.C.; Rosen, J.M. Thermal enhancement with optically activated gold nanoshells sensitizes breast cancer stem cells to radiation therapy. Sci. Transl. Med. 2010, 2, 55ra79. [Google Scholar] [CrossRef] [Green Version]
- Gad, S.C.; Sharp, K.L.; Montgomery, C.; Payne, J.D.; Goodrich, G.P. Evaluation of the toxicity of intravenous delivery of auroshell particles (gold-silica nanoshells). Int. J. Toxicol. 2012, 31, 584–594. [Google Scholar] [CrossRef] [PubMed]
- Jang, B.; Kim, Y.S.; Choi, Y. Effects of gold nanorod concentration on the depth-related temperature increase during hyperthermic ablation. Small 2011, 7, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Kaur, P.; Aliru, M.L.; Chadha, A.S.; Asea, A.; Krishnan, S. Hyperthermia using nanoparticles—Promises and pitfalls. Int. J. Hyperth. 2016, 32, 76–88. [Google Scholar] [CrossRef]

| Mechanism | HT | HT + Anti-Cancer Drug | HT + Natural Product |
|---|---|---|---|
| ROS | 1 | 6 | 7 |
| HSP | 2 | 2 | 4 |
| DNA damage | 4 | 5 | 1 |
| MMP depolarization | 8 | 3 | |
| Cell cycle arrest | 2 | 12 | 1 |
| Pharmacokinetics change | 7 | ||
| transcription factor | 6 | 5 | |
| Regulation of apoptotic protein | 9 | 7 | 3 |
| Cellular physiological changes | 6 | ||
| Apoptosis (Unknown mechanism) | 11 | 2 | |
| Anti-angiogenesis | 1 | ||
| Necrosis | 5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yi, G.Y.; Kim, M.J.; Kim, H.I.; Park, J.; Baek, S.H. Hyperthermia Treatment as a Promising Anti-Cancer Strategy: Therapeutic Targets, Perspective Mechanisms and Synergistic Combinations in Experimental Approaches. Antioxidants 2022, 11, 625. https://doi.org/10.3390/antiox11040625
Yi GY, Kim MJ, Kim HI, Park J, Baek SH. Hyperthermia Treatment as a Promising Anti-Cancer Strategy: Therapeutic Targets, Perspective Mechanisms and Synergistic Combinations in Experimental Approaches. Antioxidants. 2022; 11(4):625. https://doi.org/10.3390/antiox11040625
Chicago/Turabian StyleYi, Ga Yeong, Min Ju Kim, Hyo In Kim, Jinbong Park, and Seung Ho Baek. 2022. "Hyperthermia Treatment as a Promising Anti-Cancer Strategy: Therapeutic Targets, Perspective Mechanisms and Synergistic Combinations in Experimental Approaches" Antioxidants 11, no. 4: 625. https://doi.org/10.3390/antiox11040625