Dibasic Derivatives of Phenylcarbamic Acid as Prospective Antibacterial Agents Interacting with Cytoplasmic Membrane

 ,
,
Abstract
:1. Introduction
2. Results and Discussion
2.1. Chemistry
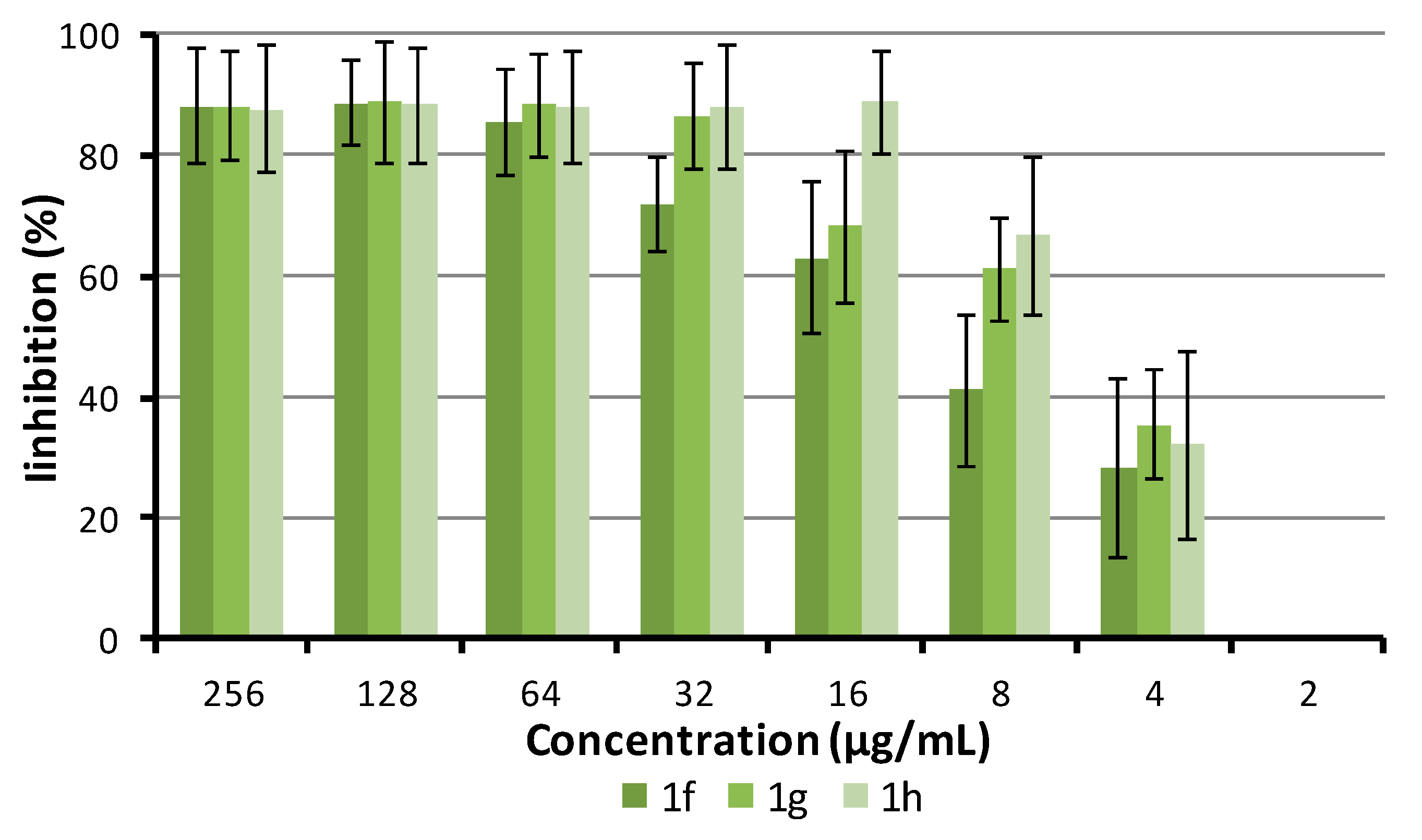
2.2. Antibacterial Activity
2.3. In Vitro Antiproliferative Assay
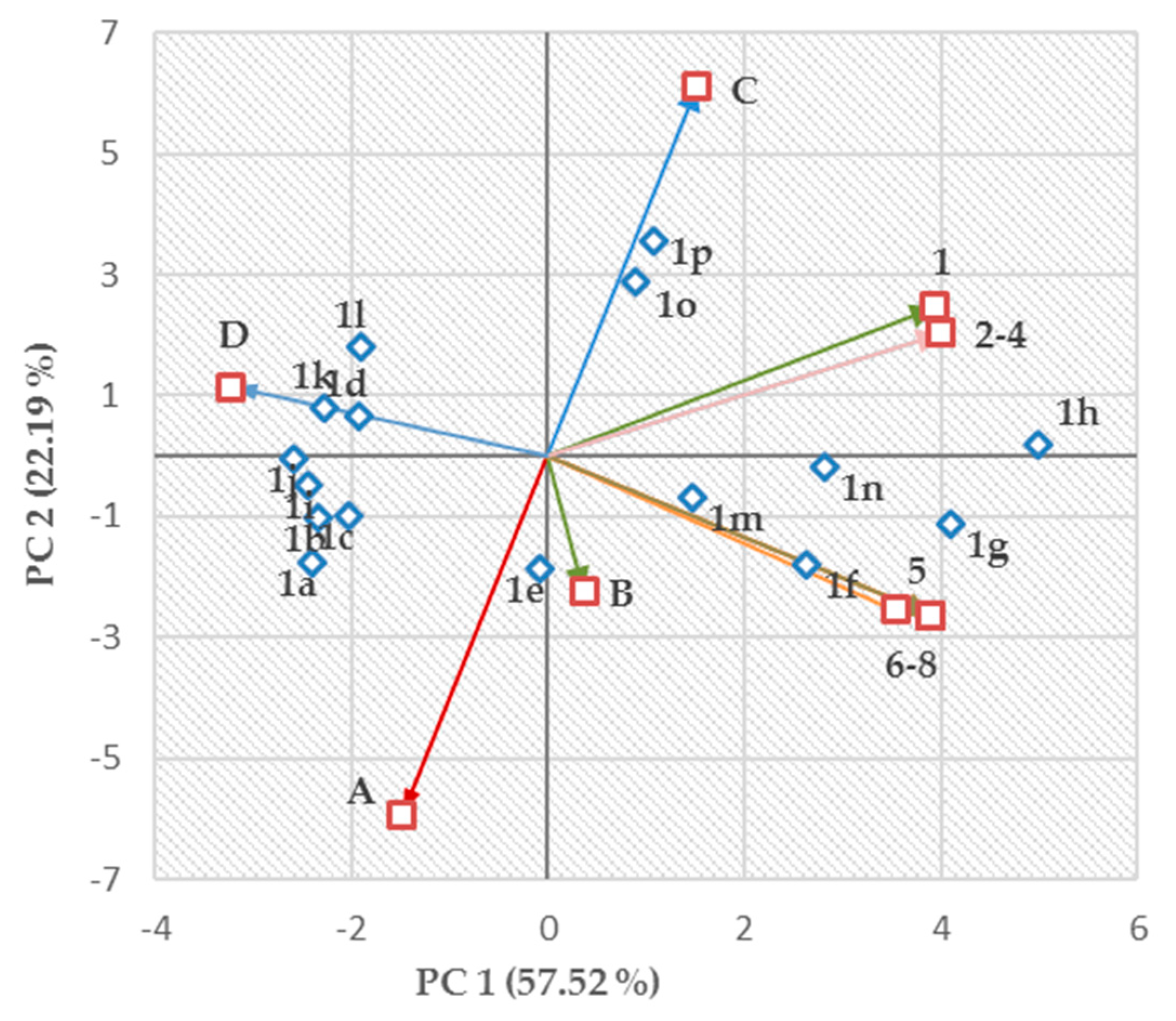
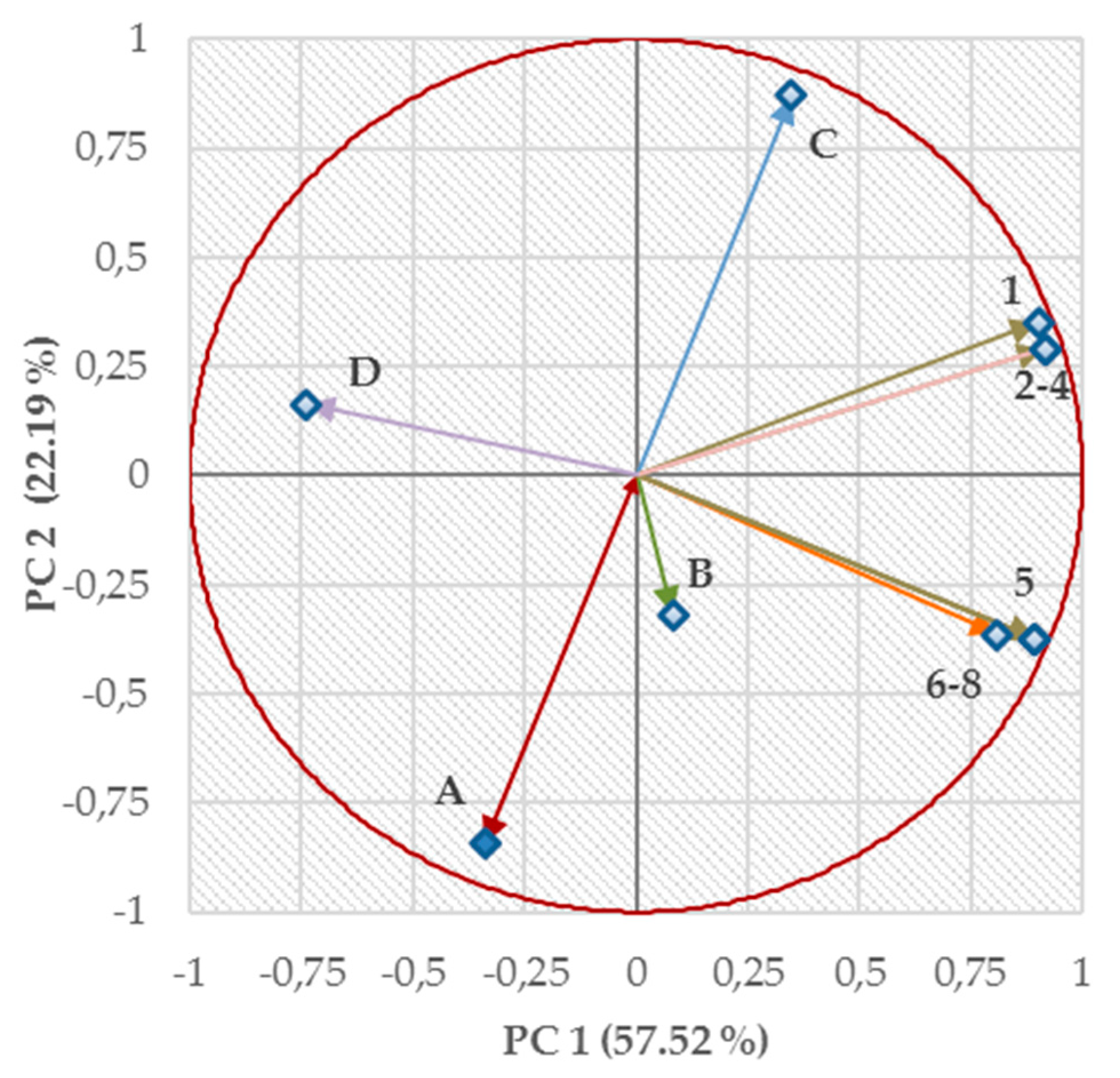
2.4. Structure–Activity Relationships
2.5. Advanced Antimicrobial Evaluation
2.5.1. Synergistic Effect
2.5.2. Time-Kill Studies
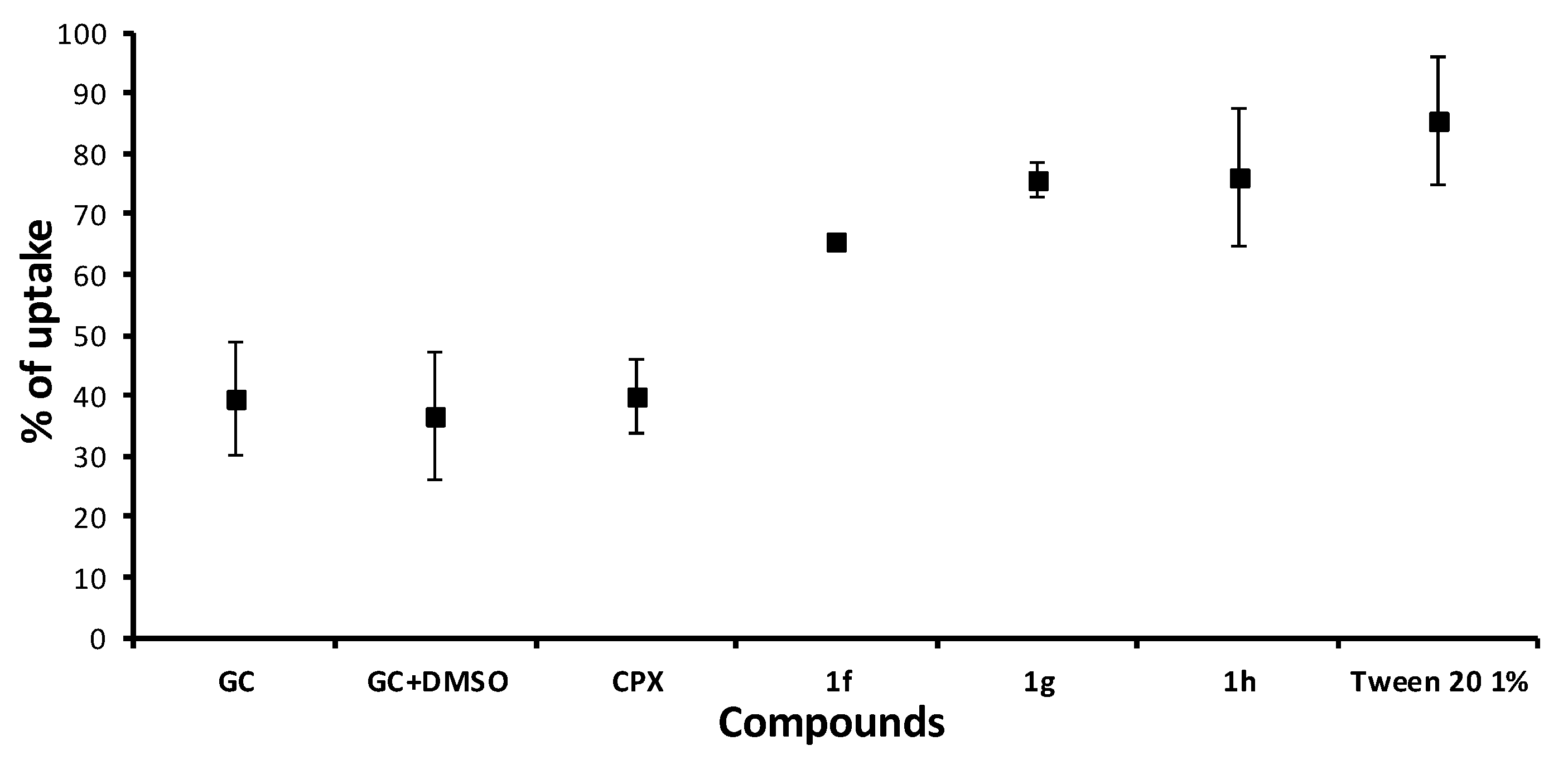
2.5.3. Crystal Violet Uptake
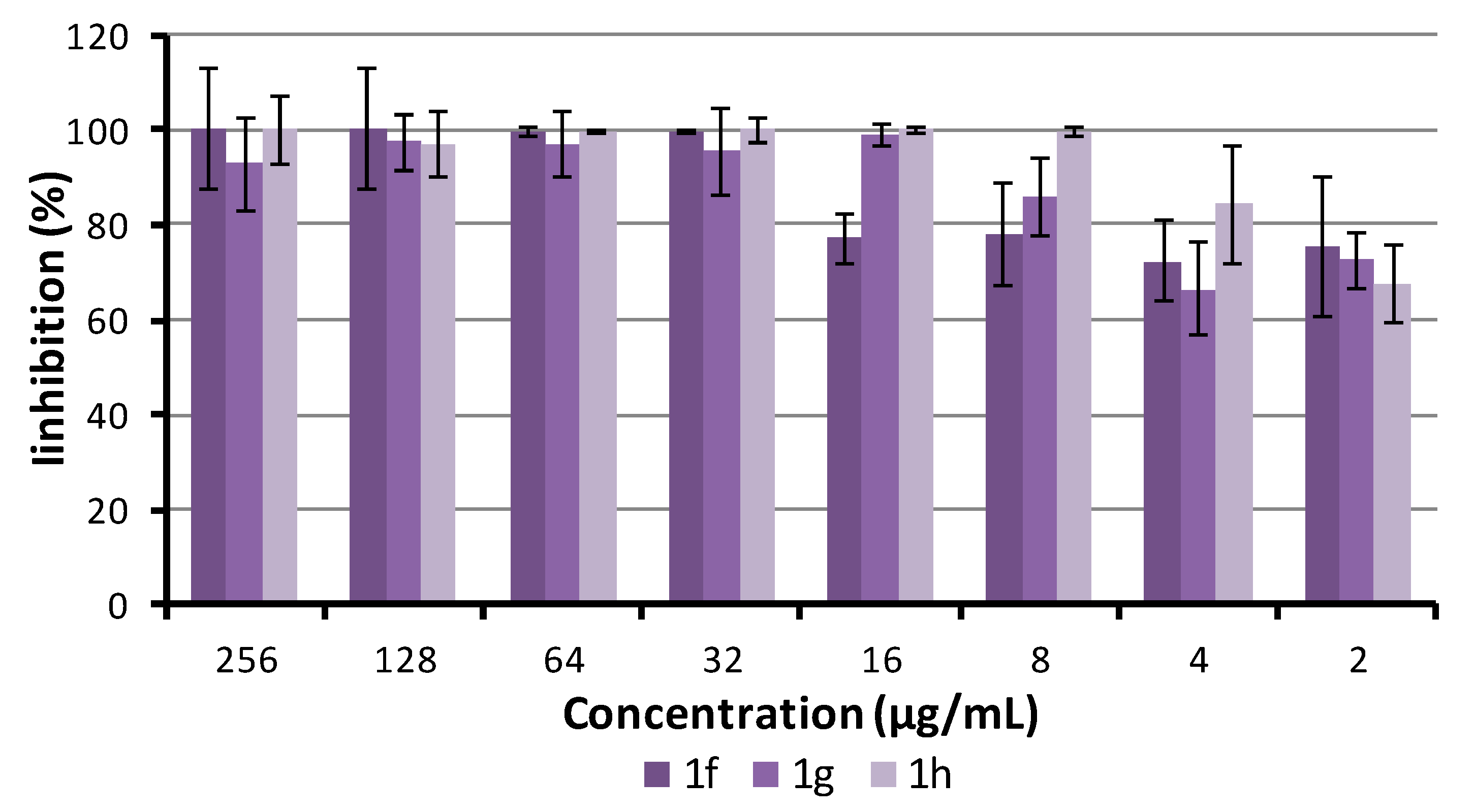
2.5.4. Antibiofilm Activity
3. Materials and Methods
3.1. Chemistry
3.2. Minimum Inhibitory Concentration Determination
3.3. Synergy Effect with Clinically Used Drugs
3.4. Time-Kill Study
3.5. Crystal Violet Uptake
3.6. Biofilm Inhibition Assay
3.7. Biofilm Susceptibility to Compounds
3.8. Calculations and Statistical Analyses
3.9. In Vitro Antiproliferative Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Dastidar, S.G.; Kristiansen, J.E.; Molnar, J.; Amaral, L. Role of phenothiazines and structurally similar compounds of plant origin in the fight against infections by drug resistant bacteria. Antibiotics 2013, 2, 58–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamad, M.; Al-Marzooq, F.; Orive, G.; Al-Tel, T.H. Superbugs but no drugs: Steps in averting a post-antibiotic era. Drug Discov. Today 2019, 24, 2225–2228. [Google Scholar] [CrossRef] [PubMed]
- Kristiansen, J.E.; Amaral, L. The potential management of resistant infections with non-antibiotics. J. Antimicrob. Chemoth. 1997, 40, 319–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amaral, L.; Lorian, V. Effects of chlorpromazine on the cell envelope proteins of Escherichia coli. Antimicrob. Agents Chemother. 1991, 35, 1923–1924. [Google Scholar] [CrossRef] [Green Version]
- Chattopadhyay, D.; Das, S.K.; Patra, A.R.; Bhattacharya, S.K. Non-Antibiotics—An alternative for microbial resistance: Scope and hope. In New Strategies Combating Bacterial Infection; Ahmad, I., Aqil, F., Eds.; Wiley-VCH: Weinhiem, Germany, 2009; pp. 89–125. [Google Scholar]
- Schmidt, R.M.; Rosenkranz, H.S. Antimicrobial activity of local anaesthetics: Lidocaine and procaine. J. Infect. Dis. 1970, 121, 596–607. [Google Scholar] [CrossRef]
- de Silva, S.S.; Carvalho, J.W.P.; Aires, C.P.; Nitschke, M. Disruption of Staphylococcus aureus biofilms using rhamnolipid biosurfactants. J. Dairy Sci. 2017, 100, 7864–7873. [Google Scholar] [CrossRef] [Green Version]
- Sriram, M.I.; Kalishwaralal, K.; Deepak, V.; Gracerosepat, R.; Srisakthi, K.; Gurunathan, S. Biofilm inhibition and antimicrobial action of lipopeptide biosurfactant produced by heavy metal tolerant strain Bacillus cereus NK1. Colloids Surf. B. Biointerfaces 2011, 85, 174–181. [Google Scholar] [CrossRef]
- Meylheuc, T.; Van Oss, C.J.; Bellon-Fontaine, M.N. Adsorption of biosurfactant on solid surfaces and consequences regarding the bioadhesion of Listeria monocytogenes LO28. J. Appl. Microbiol. 2001, 91, 822–832. [Google Scholar] [CrossRef] [Green Version]
- Wieczorek, D.; Dobrowolski, A.; Staszak, K.; Kwasniewska, D.; Dubyk, P. Surface and antimicrobial activity of sulfobetaines. J. Surfactants Deterg. 2016, 19, 813–822. [Google Scholar] [CrossRef]
- Rewak-Soroczynska, J.; Paluch, E.; Siebert, A.; Szalkiewicz, K.; Oblak, E. Biological activity of glycine and alanine derivatives of quaternary ammonium salts (QASs) against micro-organisms. Lett. Appl. Microbiol. 2019, 69, 212–220. [Google Scholar] [CrossRef]
- Aydin, O.N.; Eyigor, M.; Aydin, N. Antimicrobial activity of ropivacaine and other local anaesthetics. Eur. J. Anaesthesiol. 2001, 18, 687–694. [Google Scholar] [CrossRef] [PubMed]
- Mullin, G.S.; Rubinfeld, R.S. The antibacterial activity of topical anesthetics. Cornea 1997, 16, 662–665. [Google Scholar] [CrossRef] [PubMed]
- Kesici, S.; Demirci, M.; Kesici, U. Bacterial inhibition efficiency of prilocaine and bupivacaine. Int. Wound J. 2019, 16, 1185–1189. [Google Scholar] [CrossRef] [PubMed]
- Pina-Vaz, C.; Rodrigues, A.G.; Sansonetty, F.; Martinez-De-Oliveira, J.; Fonseca, A.F.; Mardh, P.A. Antifungal activity of local anesthetics against Candida species. Infect. Dis. Obstet. Gynecol. 2000, 8, 124–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srisatjaluk, R.L.; Klongnoi, B.; Wongsirichat, N. Antimicrobial effect of topical local anesthetic spray on oral microflora. J. Dent. Anesth. Pain. Med. 2016, 16, 17–24. [Google Scholar] [CrossRef] [Green Version]
- Johnson, S.M.; Saint, J.; Barbara, E.; Dine, A.P. Local anesthetics as antimicrobial agents: A review. Surg. Infect. 2008, 9, 205–213. [Google Scholar] [CrossRef] [Green Version]
- Mutlu, E. In vitro investigation of the antibacterial effects of lidocaine and bupivacaine alone and in combinations with fentanyl. Turk. Klin. J. Med. Sci. 2018, 38, 334–339. [Google Scholar] [CrossRef] [Green Version]
- Kramer, A.; Sorgatz, K.; Hoppe, H.; Meyer, F.U. Bacteriostatic and antiseptic efficacy of selected anaesthetics individually and in combination with an antiseptic mouthwash. Hygiene Medizin 1994, 19, 527–534. [Google Scholar]
- Razavi, B.M.; Fazly-Bazzaz, B.S. A review and new insights to antimicrobial action of local anesthetics. Eur. J. Clin. Microbiol. Infect. Dis. 2019, 38, 991–1002. [Google Scholar] [CrossRef]
- Abdelli, F.; Jardak, M.; Elloumi, J.; Stien, D.; Cherif, S.; Mnif, S.; Aifa, S. Antibacterial, anti-adherent and cytotoxic activities of surfactin (s) from a lipolytic strain Bacillus safensis F4. Biodegradation 2019, 30, 287–300. [Google Scholar] [CrossRef]
- Lewis, K. Riddle of biofilm resistance. Antimicrob. Agents Chemother. 2001, 45, 999–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamal, M.; Ahmad, W.; Andleeb, S.; Jalil, F.; Imran, M.; Nawaz, M.A.; Kamil, M.A. Bacterial biofilm and associated infections. J. Chin. Med. Assoc. 2018, 81, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Scott VanEpps, J.; Younger, J.G. Implantable device related infection. Shock 2016, 46, 597–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azevedo, M.M.; Cobrado, L.; Silva-Dias, A.; Pina-Vaz, C.; Rodrigues, A.G. Prevention and eradication of biofilm in medical indwelling devices. In Frontiers in Clinical Drug Research—Anti Infectives; Atta-ur-Rahman, Ed.; Bentham Science Publishers: Sharjah, UAE, 2017; pp. 204–232. [Google Scholar]
- Khatoon, Z.; McTiernan, C.D.; Suuronen, E.J.; Mah, T.F.; Alarcona, E.I. Bacterial biofilm formation on implantable devices and approaches to its treatment and prevention. Heliyon 2018, 4, e01067. [Google Scholar] [CrossRef] [Green Version]
- Mandakhalikar, K.D.; Chua, R.R.; Tambyah, P.A. New technologies for prevention of catheter associated urinary tract infection. Curr. Treat. Options Infect. Dis. 2016, 8, 24–41. [Google Scholar] [CrossRef]
- Gominet, M.; Compain, F.; Beloin, C.; Lebeaux, D. Central venous catheters and biofilms: Where do we stand in 2017? APMIS 2017, 125, 365–375. [Google Scholar] [CrossRef] [Green Version]
- Menezes, F.G.; Correa, L.; Medina-Pestana, J.O.; Aguiar, W.F.; Camargo, L.F.A. A randomized clinical trial comparing Nitrofurazone-coated and uncoated urinary catheters in kidney transplant recipients: Results from a pilot study. Transpl. Infect. Dis. 2019, 21, e13031. [Google Scholar] [CrossRef]
- Doherty, W. Instillagel: An anaesthetic antiseptic gel for use in catheterization. Br. J. Nurs. 1999, 8, 109–112. [Google Scholar] [CrossRef]
- Wilson, M.C.R. Biofilm and other causes of pain in catheterization. Br. J. Community Nurs. 2009, 14, 102–113. [Google Scholar] [CrossRef]
- Lai, N.M.; Chaiyakunapruk, N.; Lai, N.A.; O’Riordan, E.; Pau, W.S.C.; Saint, S. Catheter impregnation, coating or bonding for reducing central venous catheter-related infections in adults. Cochrane Database Syst. Rev. 2016, 3, CD007878. [Google Scholar] [CrossRef]
- Chong, H.Y.; Lai, N.M.; Apisarnthanarak, A.; Chaiyakunapruk, N. Comparative efficacy of antimicrobial central venous catheters in reducing catheter-related bloodstream infections in adults: Abridged cochrane systematic review and network meta-analysis. Clin. Infect. Dis. 2017, 64, S131–S140. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Tong, H.; Liu, H.; Wang, Y.; Wang, R.; Gao, H.; Yu, P.; Lv, Y.; Chen, S.; Wang, G.; et al. Effectiveness of antimicrobial-coated central venous catheters for preventing catheter-related blood-stream infections with the implementation of bundles: A systematic review and network meta-analysis. Ann. Intensive Care 2018, 8, 71. [Google Scholar] [CrossRef] [PubMed]
- Dang, F.P.; Li, H.J.; Tian, J.H. Comparative efficacy of 13 antimicrobial dressings and different securement devices in reducing catheter-related bloodstream infections: A Bayesian network meta-analysis. Medicine 2019, 98, e14940. [Google Scholar] [CrossRef] [PubMed]
- Yeung, S.S.T.; Loshak, H. Coated and Uncoated Central Venous Catheters: A Review of Comparative Clinical Effectiveness and Safety; CADTH Rapid Response Report: Summary with Critical Appraisal; Canadian Agency for Drugs and Technologies in Health: Ottawa, ON, Canada, January 2019.
- Majeed, A.; Sagar, F.; Latif, A.; Hassan, H.; Iftikhar, A.; Darouiche, R.O.; Mohajer, M.A. Does antimicrobial coating and impregnation of urinary catheters prevent catheter-associated urinary tract infection? A review of clinical and preclinical studies. Expert Rev. Med. Devices. 2019, 16, 809–820. [Google Scholar] [CrossRef] [PubMed]
- Al-Qahtani, M.; Safan, A.; Jassim, G.; Abadla, S. Efficacy of anti-microbial catheters in preventing catheter associated urinary tract infections in hospitalized patients: A review on recent updates. J. Infect. Public Health 2019, 12, 760–766. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, C.; Costa, F.; Pirttila, A.M.; Tejesvi, M.V.; Cristina, M.; Martins, L. Prevention of urinary catheter-associated infections by coating antimicrobial peptides from crowberry endophytes. Sci. Rep. 2019, 9, 10753. [Google Scholar] [CrossRef] [Green Version]
- Andersen, M.J.; Flores-Mireles, A.L. Urinary catheter coating modifications: The race against catheter-associated infections. Coatings 2020, 10, 23. [Google Scholar] [CrossRef] [Green Version]
- Otto, M. Staphylococcal biofilms. Curr. Top. Microbiol. Immunol. 2008, 322, 207–228. [Google Scholar]
- Malheiro, J.; Simoes, M. Antimicrobial resistance of biofilms in medical devices. In Biofilms and Implantable Medical Devices; Deng, Y., Lv, W., Eds.; Woodhead Publishing & Elsevier: Duxford, UK, 2017; pp. 97–113. [Google Scholar]
- Larsen, T.; Fiehn, N.E. Resistance of Streptococcus sanguis biofilms to antimicrobial agents. APMIS 1996, 104, 280–284. [Google Scholar] [CrossRef]
- Olsen, I. Biofilm-specific antibiotic tolerance and resistance. Eur. J. Clin. Microbiol. Infect. Dis. 2015, 34, 877–886. [Google Scholar] [CrossRef]
- Pace, J.L.; Rupp, M.E.; Finch, R.G. Biofilms, Infection, and Antimicrobial Therapy, 1st ed.; CRC Press, Taylor & Francis Group: Boca Raton, FL, USA, 2006. [Google Scholar]
- Vetas, D.; Dimitropoulou, E.; Mitropoulou, G.; Kourkoutas, Y.; Giaouris, E. Disinfection efficiencies of sage and spearmint essential oils against planktonic and biofilm Staphylococcus aureus cells in comparison with sodium hypochlorite. Int. J. Food Microbiol. 2017, 257, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Kwiecinska-Pirog, J.; Bogiel, T.; Gospodarek, E. Effects of ceftazidime and ciprofloxacin on biofilm formation in Proteus mirabilis rods. J. Antibiot. 2013, 66, 593–597. [Google Scholar] [CrossRef] [PubMed]
- Jampilek, J. Design and discovery of new antibacterial agents: Advances, perspectives, challenges. Curr. Med. Chem. 2018, 25, 4972–5006. [Google Scholar] [CrossRef] [PubMed]
- Singha, P.; Locklin, J.; Handa, H. A review of the recent advances in antimicrobial coatings for urinary catheters. Acta Biomater. 2017, 50, 20–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walters, M.C.; Roe, F.; Bugnicourt, A.; Franklin, M.J.; Stewart, P.S. Contributions of antibiotic penetration, oxygen limitation, and low metabolic activity to tolerance of Pseudomonas aeruginosa biofilms to ciprofloxacin and tobramycin. Antimicrob. Agents Chemother. 2003, 47, 317–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, C.W.; Mah, T.F. Molecular mechanisms of biofilm-based antibiotic resistance and tolerance in pathogenic bacteria. FEMS Microbiol. Rev. 2017, 41, 276–301. [Google Scholar] [CrossRef]
- Borrielo, G.; Richards, L.; Ehrlich, G.D.; Stewart, P.S. Arginine or nitrate enhances antibiotic susceptibility of Pseudomonas aeruginosa in biofilms. Antimicrob. Agents Chemother. 2006, 50, 382–384. [Google Scholar] [CrossRef] [Green Version]
- Van Ecker, H.; Sass, A.; Bazzini, S.; De Roy, K.; Udine, C.; Messiaen, T.; Coenye, T. Biofilm-grown Burkholderia cepacia complex cells survive antibiotic treatment by avoiding production of reactive oxygen species. PLoS ONE 2013, 8, 58943. [Google Scholar] [CrossRef] [Green Version]
- Malik, I.; Csollei, J.; Solovic, I.; Pospisilova, S.; Michnova, H.; Jampilek, J.; Cizek, A.; Kapustikova, I.; Curillova, J.; Pechacova, M.; et al. Dibasic derivatives of phenylcarbamic acid against mycobacterial strains: Old drugs and new tricks? Molecules 2018, 23, 2493. [Google Scholar] [CrossRef] [Green Version]
- Csollei, J.; Buciova, L.; Cizmarik, J.; Kopacova, L. Studies of local anaesthetics CXII. Preparation and activity of dibasic alkylesters of 2-, and 3-alkoxy-substituted phenylcarbamic acids. Ceskoslov. Farm 1993, 42, 127–129. [Google Scholar]
- Oravcova, V.; Zurek, L.; Townsend, A.; Clark, A.B.; Ellis, J.C.; Cizek, A. American crows as carriers of vancomycin-resistant enterococci with vanA gene. Environ. Microbiol. 2014, 16, 939–949. [Google Scholar] [CrossRef] [PubMed]
- Zadrazilova, I.; Pospisilova, S.; Masarikova, M.; Imramovsky, A.; Monreal-Ferriz, J.; Vinsova, J.; Cizek, A.; Jampilek, J. Salicylanilide carbamates: Promising antibacterial agents with high in vitro activity against methicillin-resistant Staphylococcus aureus (MRSA). Eur. J. Pharm. Sci. 2015, 77, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Tengler, J.; Kapustikova, I.; Pesko, M.; Govender, R.; Keltosova, S.; Mokry, P.; Kollar, P.; O´Mahony, J.; Coffey, A.; Kralova, K.; et al. Synthesis and biological evaluation of 2-hydroxy-3-[(2-aryloxyethyl)amino]propyl 4-[(alkoxycarbonyl)amino]benzoates. Sci. World J. 2013, 2013, 274570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imramovsky, A.; Pesko, M.; Kralova, K.; Vejsova, M.; Stolarikova, J.; Vinsova, J.; Jampilek, J. Investigating spectrum of biological activity of 4- and 5-chloro-2-hydroxy-N-[2-(arylamino)-1-alkyl-2-oxoethyl]- benzamides. Molecules 2011, 16, 2414–2430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pauk, K.; Zadrazilova, I.; Imramovsky, A.; Vinsova, J.; Pokorna, M.; Masarikova, M.; Cizek, A.; Jampilek, J. New derivatives of salicylamides: Preparation and antimicrobial activity against various bacterial species. Bioorg. Med. Chem. 2013, 21, 6574–6581. [Google Scholar] [CrossRef]
- Pere, P.; Lindgren, L.; Vaara, M. Poor antibacterial effect of ropivacaine: Comparison with bupivacaine. Anesthesiology 1999, 91, 884–886. [Google Scholar] [CrossRef] [Green Version]
- ROCHE. Cell Proliferation Reagent WST-1. Roche Diagnostics GmbH: Mannheim, Germany, 2011. Available online: https://www.sigmaaldrich.com/content/dam/sigma-aldrich/docs/Roche/Bulletin/1/cellprorobul.pdf (accessed on 24 January 2020).
- Kos, J.; Nevin, E.; Soral, M.; Kushkevych, I.; Gonec, T.; Bobal, P.; Kollar, P.; Coffey, A.; O´Mahony, J.; Liptaj, T.; et al. Synthesis and antimycobacterial properties of ring-substituted 6-hydroxynaphthalene- 2-carboxanilides. Bioorg. Med. Chem. 2015, 23, 2035–2043. [Google Scholar] [CrossRef]
- Lim, E.J.; Yoon, Y.J.; Heo, J.; Lee, T.H.; Kim, Y.H. Ciprofloxacin enhances TRAIL-induced apoptosis in lung cancer cells by upregulating the expression and protein stability of death receptors through CHOP expression. Int. J. Mol. Sci. 2018, 19, 3187. [Google Scholar] [CrossRef] [Green Version]
- Suffness, M.; Douros, J. Current status of the NCI plant and animal product program. J. Nat. Prod. 1982, 45, 1–14. [Google Scholar] [CrossRef]
- Abdi, H.; Williams, L.J. Principal component analysis. WIREs Comp. Stat. 2010, 2, 433–459. [Google Scholar] [CrossRef]
- van den Berg, R.A.; Hoefsloot, H.C.; Westerhuis, J.A.; Smilde, A.K.; van der Werf, M.J. Centering, scaling, and transformations: Improving the biological information content of metabolomics data. BMC Genom. 2006, 7, 142–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bro, R.; Smilde, A.K. Principal component analysis. Anal. Methods 2014, 6, 2812–2831. [Google Scholar] [CrossRef] [Green Version]
- Eriksson, L.; Jaworska, J.; Worth, A.P.; Cronin, M.T.; McDowell, R.M.; Gramatica, P. Methods for reliability and uncertainty assessment and for applicability evaluations of classification- and regression-based QSARs. Environ. Health Perspect. 2003, 111, 1361–1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, D.A. Stopping rules in principal components analysis: A comparison of heuristical and statistical approaches. Ecology 1993, 74, 2204–2214. [Google Scholar] [CrossRef]
- Hauke, J.; Kossowski, T. Comparison of values of Pearson’s and Spearman’s correlation coefficients on the same sets of data. Quaest. Geograph. 2011, 30, 87–93. [Google Scholar] [CrossRef] [Green Version]
- Schwalbe, R.; Steele-Moore, L.; Goodwin, A.C. Antimicrobial Susceptibility Testing Protocols, 1st ed.; CRC Press: Boca Raton, FL, USA, 2007. [Google Scholar]
- Bonapace, C.R.; Bosso, J.A.; Friedrich, L.V.; White, R.L. Comparison of methods of interpretation of checkerboard synergy testing. Diagn. Microbiol. Infect. Dis. 2002, 44, 363–366. [Google Scholar] [CrossRef]
- Breukink, E.; De Kruijff, B. Lipid II as a target for antibiotics. Nat. Rev. Drug Discov. 2006, 5, 321–323. [Google Scholar] [CrossRef]
- Tipper, D.J.; Strominger, J.L. Mechanism of action of penicillins: A proposal based on their structural similarity to acyl-D-alanyl-D-alanine. Proc. Natl. Acad. Sci. USA 1965, 54, 1133–1141. [Google Scholar] [CrossRef] [Green Version]
- Arias, C.A.; Murray, B.E. The rise of the Enterococcus: Beyond vancomycin resistance. Nat. Rev. Microbiol. 2012, 10, 266–278. [Google Scholar] [CrossRef] [Green Version]
- Stapleton, P.D.; Taylor, P.W. Methicillin resistance in Staphylococcus aureus: Mechanisms and modulation. Sci. Prog. 2002, 85, 57–72. [Google Scholar] [CrossRef]
- Cetinkaya, Y.; Falk, P.; Mayhall, C.G. Vancomycin-resistant enterococci. Clin. Microbiol. Rev. 2000, 13, 686–707. [Google Scholar] [CrossRef] [PubMed]
- Cha, J.O.; Park, Y.K.; Lee, Y.S.; Chung, G.T. In vitro biofilm formation and bactericidal activities of methicillin-resistant Staphylococcus aureus clones prevalent in Korea. Diagn. Microbiol. Infect. Dis. 2011, 70, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Devi, K.P.; Nisha, S.A.; Sakthivel, R.; Pandian, S.K. Eugenol (an essential oil of clove) acts as an antibacterial agent against Salmonella typhi by disrupting the cellular membrane. J. Ethnopharmacol. 2010, 130, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Pospisilova, S.; Kos, J.; Michnova, H.; Kapustikova, I.; Strharsky, T.; Oravec, M.; Moricz, A.M.; Bakonyi, J.; Kauerova, T.; Kollar, P.; et al. Synthesis and spectrum of biological activities of novel N-arylcinnamamides. Int. J. Mol. Sci. 2018, 19, 2318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clinical and Laboratory Standards Institute. Performance Standards for Antimicrobial Susceptibility Testing; The 8th Informational Supplement Document; CLSI: Wayne, PA, USA, 2012; M100-S22. [Google Scholar]
- Ringner, M. What is principal component analysis? Nat. Biotechnol. 2008, 26, 303–304. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||||
|---|---|---|---|---|---|---|
| Comp. | R | X | Y | log kw | γ (N/m) | log ε2 (Ch-T) |
| 1a | 2-OC4H9 | A | C | 3.7688 | 0.06464 | 4.19 |
| 1b | 2-OC5H11 | A | C | 4.0454 | 0.06366 | 4.05 |
| 1c | 2-OC6H13 | A | C | 4.6049 | 0.06222 | 4.52 |
| 1d | 2-OC7H15 | A | C | 4.9487 | 0.05985 | 4.08 |
| 1e | 3-OC4H9 | A | D | 4.0258 | 0.06316 | 4.24 |
| 1f | 3-OC5H11 | A | D | 4.6722 | 0.06285 | 4.27 |
| 1g | 3-OC6H13 | A | D | 4.9446 | 0.06105 | 4.13 |
| 1h | 3-OC7H15 | A | D | 5.5384 | 0.05786 | 4.27 |
| 1i | 2-OC4H9 | B | C | 4.4679 | 0.06302 | 4.08 |
| 1j | 2-OC5H11 | B | C | 4.8466 | 0.06206 | 4.22 |
| 1k | 2-OC6H13 | B | C | 5.2359 | 0.06065 | 4.10 |
| 1l | 2-OC7H15 | B | C | 5.8966 | 0.05853 | 4.14 |
| 1m | 3-OC4H9 | B | D | 4.7099 | 0.06298 | 4.09 |
| 1n | 3-OC5H11 | B | D | 5.2087 | 0.06154 | 4.18 |
| 1o | 3-OC6H13 | B | D | 5.6569 | 0.05925 | 4.01 |
| 1p | 3-OC7H15 | B | D | 6.1749 | 0.05692 | 4.20 |
| Comp. | MICs (µg/mL (µM)) | Tox IC50 (μM) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| SA | MRSA1 | MRSA2 | MRSA3 | EF | VRE1 | VRE2 | VRE3 | CA | ||
| 1a | >256 (>427) | >256 (>427) | >256 (>427) | >256 (>427) | >256 (>427) | >256 (>427) | >256 (>427) | >256 (>427) | >128 (>213) | 10.80 ± 1.11 |
| 1b | >256 (>417) | >256 (>417) | >256 (>417) | >256 (>417) | >256 (>417) | >256 (>417) | >256 (>417) | >256 (>417) | >128 (>208) | 12.53 ± 1.22 |
| 1c | >256 (>407) | >256 (>407) | >256 (>407) | >256 (>407) | >256 (>407) | >256 (>407) | >256 (>407) | >256 (>407) | >128 (>203) | 15.42 ± 0.22 |
| 1d | >256 (>398) | >256 (>398) | >256 (>398) | >256 (>398) | >256 (>398) | >256 (>398) | >256 (>398) | >256 (>398) | >128 (199) | 16.32 ± 0.70 |
| 1e | 64 (129) | 64 (129) | 64 (129) | 64 (129) | 128 (259) | 32 (65.1) | 64 (129) | 32 (65.1) | >128 (>259) | 4.20 ± 0.79 |
| 1f | 16 (31.6) | 16 (31.6) | 32 (63.2) | 16 (31.6) | 32 (63.2) | 16 (31.6) | 16 (31.6) | 16 (31.6) | 128 (252) | 4.19 ± 0.54 |
| 1g | 8 (15.4) | 8 (15.4) | 16 (30.8) | 8 (15.4) | 16 (30.8) | 8 (15.4) | 8 (15.4) | 8 (15.4) | 16 (30.8) | 3.50 ± 0.31 |
| 1h | 8 (15.0) | 8 (15.0) | 8 (15.0) | 8 (15.0) | 8 (15.0) | 4 (7.51) | 4 (7.51) | 4 (7.51) | 8 (15.0) | 3.67 ± 0.01 |
| 1i | >256 (>407) | >256 (>407) | >256 (>407) | >256 (>407) | >256 (>407) | >256 (>407) | >256 (>407) | >256 (>407) | >128 (>203) | 23.06 ± 0.95 |
| 1j | >256 (>398) | >256 (>398) | >256 (>398) | >256 (>398) | >256 (>398) | >256 (>398) | >256 (>398) | >256 (>398) | >128 (>199) | 26.45 ± 1.91 |
| 1k | >256 (>390) | >256 (>390) | >256 (>390) | >256 (>390) | >256 (>390) | >256 (>390) | >256 (>390) | >256 (>390) | >128 (>195) | 31.24 ± 2.24 |
| 1l | >256 (>382) | >256 (>382) | >256 (>382) | >256 (>382) | >256 (>382) | >256 (>382) | >256 (>382) | >256 (>382) | >128 (>191) | 29.75 ± 0.14 |
| 1m | 128 (245) | 32 (61.5) | 64 (122) | 64 (122) | 32 (61.5) | 32 (61.5) | 64 (122) | 16 (30.7) | >128 (>245) | 4.90 ± 0.19 |
| 1n | 128 (239) | 64 (119) | 128 (239) | 256 (478) | 16 (29.9) | 32 (59.9) | 64 (119) | 16 (29.9) | >128 (>239) | 4.99 ± 0.99 |
| 1o | >256 (>466) | 128 (233) | >256 (>466) | >256 (>466) | 16 (29.2) | 32 (58.3) | 256 (466) | 8 (14.6) | >128 (>233) | 4.71 ± 0.07 |
| 1p | >256 (>455) | 256 (455) | >256 (>455) | >256 (>455) | 8 (14.2) | 64 (113) | 256 (455) | 16 (28.4) | >128 (>227) | 6.32 ± 0.58 |
| AMP | 2 (5.72) | 16 (45.8) | >16 (>45.8) | >16 (>45.8) | 4 (11.5) | 4 (11.5) | 4 (11.5) | 2 (5.72) | – | >30 |
| CPX | 0.5 (1.51) | >16 (>45.8) | >16 (>45.8) | >16 (>45.8) | 1 (3.02) | 1 (3.02) | 1 (3.02) | 64 (193) | – | 0.71 ± 0.09 |
| VAN | 1 (0.69) | 2 (1.38) | 1 (0.69) | 1 (0.69) | – | 512 (353) | 512 (353) | 1024 (706) | – | >30 |
| 5-FC | – | – | – | – | – | – | – | – | 1 (7.75) | – |
| CMP | – | – | – | – | – | – | – | – | – | 0.20 ± 0.07 |
| SDS | 128 (443) | 128 (443) | 64 (222) | 64 (222) | 128 (443) | 256 (887) | 128 (443) | 128 (443) | – | 148.43 ± 8.42 |
| CRB | 0.25 (0.59) | 0.25 (0.59) | 0.5 (1.18) | 2 (4.73) | 0.5 (1.18) | 0.5 (1.18) | 0.25 (0.59) | 0.5 (1.18) | – | 1.73 ± 0.08 |
| CTC | 0.25 (0.63) | 0.25 (0.63) | 0.5 (1.26) | 1 (2.53) | 1 (2.53) | 0.5 (1.26) | 0.5 (1.26) | 0.5 (1.26) | – | 1.29 ± 0.07 |
| PRC | >256 (>1083) | >256 (>1083) | >256 (>1083) | >256 (>1083) | >256 (>1083) | >256 (>1083) | >256 (>1083) | >256 (>1083) | – | >300 |
| TRC | >256 (>1030) | >256 (>1030) | >256 (>1030) | >256 (>1030) | >256 (>1030) | >256 (>1030) | >256 (>1030) | >256 (>1030) | – | >300 |
| TEC | >256 (>968) | >256 (>968) | >256 (>968) | >256 (>968) | >256 (>968) | >256 (>968) | >256 (>968) | >256 (>968) | – | 224.13 ± 6.52 |
| Isolate | Combination (MIC (μg/mL)) | FIC Index | Comb. Effect (MICs (μg/mL)) |
|---|---|---|---|
| MRSA 63718 | Comp. 1g + CPX (16/16) | 1.000 | IND |
| Comp. 1h + CPX (8/16) | 0.531 | ADD 0.25/0.032 | |
| Comp. 1g + OXA (8/512) | 0.500 | SYN 2/128 | |
| Comp. 1h +OXA (4/512) | 0.562 | ADD 2/32; 1/256 | |
| MRSA SA 3202 | Comp. 1g + CPX (16/16) | 1.000 | IND |
| Comp. 1h + CPX (8/16) | 1.000 | IND | |
| Comp. 1g + OXA (8/512) | 1.000 | IND | |
| Comp. 1h+ OXA (8/512) | 0.750 | ADD 4/128 | |
| VRE 342B | Comp. 1g + VAN (8/1024) | 0.625 | ADD 4/128 |
| Comp. 1h + VAN (8/1024) | 0.500 | SYN 2/256 | |
| Comp. 1g + GEN (8/32) | 0.750 | ADD 4/8 | |
| Comp. 1h+ GEN (8/32) | 1.000 | IND | |
| VRE 368 | Comp. 1g + VAN (8/512) | 0.750 | ADD 4/128 |
| Comp. 1h + VAN (4/512) | 0.750 | ADD 2/256 | |
| Comp. 1g + GEN (8/64) | 1.000 | IND | |
| Comp. 1h+ GEN (2/128) | 1.016 | IND |
| Strain | Conc. | Log10 Difference in CFU/mL from Inoculum | |||
|---|---|---|---|---|---|
| 4 h | 6 h | 8 h | 24 h | ||
| SA | 1 × MIC | 1.54 | 1.98 | 2.33 | 2.74 |
| 2 × MIC | 0.87 | 0.98 | 1.26 | 2.24 | |
| 4 × MIC | −0.26 | −0.04 | 0.10 | 1.85 | |
| MRSA1 | 1 × MIC | 0.00 | 0.66 | 1.00 | 1.99 |
| 2 × MIC | −1.33 | −0.93 | −0.26 | 1.29 | |
| 4 × MIC | −2.57 * | −1.99 * | −1.26 * | 1.21 | |
| EF | 1 × MIC | −1.63 | −1.01 | −0.61 | 0.27 |
| 2 × MIC | −3.37 * | −3.43 * | −2.68 | 1.04 | |
| 4 × MIC | −5.62 * | −5.14 * | −5.92 * | −0.31 | |
| VRE1 | 1 × MIC | −0.14 | 0.97 | 1.01 | 1.11 |
| 2 × MIC | −0.36 | −1.89 | −0.64 | 1.51 | |
| 4 × MIC | −0.92 | −1.00 | −1.09 | 0.77 | |
| VRE2 | 1 × MIC | −2.00 | 0.29 | 0.25 | −0.16 |
| 2 × MIC | −2.34 | −1.97 | −1.01 | 0.32 | |
| 4 × MIC | −2.72 * | −2.71 * | −2.45 | 0.97 | |
| VRE3 | 1 × MIC | −0.09 | −0.29 | 0.19 | 2.41 |
| 2 × MIC | −0.75 | −0.74 | 0.56 | 0.43 | |
| 4 × MIC | −1.85 | −1.85 | −2.15 | −2.27 | |
| Strain | Conc. | Log10 Difference in CFU/mL from Inoculum | |||
|---|---|---|---|---|---|
| 4 h | 6 h | 8 h | 24 h | ||
| SA | 1 × MIC | 0.08 | 0.31 | 0.50 | 2.07 |
| 2 × MIC | 0.05 | 0.10 | 0.05 | 2.01 | |
| 4 × MIC | −0.25 | −0.13 | −0.08 | 1.93 | |
| MRSA1 | 1 × MIC | −0.73 * | −0.11 | 0.98 | 1.58 |
| 2 × MIC | −0.92 * | −0.42 * | −0.13 | 1.62 | |
| 4 × MIC | −2.48 * | −2.40 * | −2.08 * | 1.35 | |
| EF | 1 × MIC | −0.64 | 0.16 | 0.24 | 0.34 |
| 2 × MIC | −1.87 | −1.36 | −0.86 | 0.42 | |
| 4 × MIC | −2.57 * | −2.23 * | −1.62 | 0.49 | |
| VRE1 | 1 × MIC | 0.07 | 1.08 | −0.08 | 1.42 |
| 2 × MIC | 0.09 | 0.41 | −0.02 | 0.47 | |
| 4 × MIC | −0.78 | −0.99 | −0.66 | 0.85 | |
| VRE2 | 1 × MIC | 0.46 | 0.71 | 1.25 | 1.13 |
| 2 × MIC | 0.28 | 0.54 | −0.37 | 0.66 | |
| 4 × MIC | −0.32 | 0.05 | 0.22 | 0.37 | |
| VRE3 | 1 × MIC | −0.70 | 0.08 | −0.05 | 0.92 |
| 2 × MIC | −1.81 | −1.31 | −0.81 | 0.95 | |
| 4 × MIC | −1.75 * | −0.76 | −0.76 | 1.08 | |
| Strain | Conc. | Log10 Difference in CFU/mL from Inoculum | |||
|---|---|---|---|---|---|
| 4 h | 6 h | 8 h | 24 h | ||
| SA | 1 × MIC | −0.37 | −0.65 | −0.84 | 2.90 |
| 2 × MIC | −0.26 | −0.59 | −1.12 | −3.67 | |
| 4 × MIC | −0.27 | −0.38 | −1.18 | −5.47 | |
| MRSA1 | 1 × MIC | −1.00 | −1.50 | −1.76 | 0.55 |
| 2 × MIC | −0.91 | −1.43 | −1.97 | −5.18 | |
| 4 × MIC | −1.31 | −1.75 | −3.89 | −5.19 | |
| Sample | (µg/mL) | ||
|---|---|---|---|
| MIC | MBIC80 | MBEC80 | |
| 1f | 16 | 32 | 64 |
| 1g | 8 | 8 | 16 |
| 1h | 8 | 8 | 16 |
| SDS | 128 | 32 | 128 |
| CRB | 0.25 | 0.25 | 64 |
| CTC | 0.25 | 0.25 | >256 |
| PRC | >256 | >256 | >256 |
| TRC | >256 | >256 | >256 |
| TEC | >256 | >256 | >256 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pospíšilová, Š.; Malík, I.; Bezouskova, K.; Kauerova, T.; Kollar, P.; Csöllei, J.; Oravec, M.; Cizek, A.; Jampilek, J. Dibasic Derivatives of Phenylcarbamic Acid as Prospective Antibacterial Agents Interacting with Cytoplasmic Membrane. Antibiotics 2020, 9, 64. https://doi.org/10.3390/antibiotics9020064
Pospíšilová Š, Malík I, Bezouskova K, Kauerova T, Kollar P, Csöllei J, Oravec M, Cizek A, Jampilek J. Dibasic Derivatives of Phenylcarbamic Acid as Prospective Antibacterial Agents Interacting with Cytoplasmic Membrane. Antibiotics. 2020; 9(2):64. https://doi.org/10.3390/antibiotics9020064
Chicago/Turabian StylePospíšilová, Šárka, Ivan Malík, Kristyna Bezouskova, Tereza Kauerova, Peter Kollar, Jozef Csöllei, Michal Oravec, Alois Cizek, and Josef Jampilek. 2020. "Dibasic Derivatives of Phenylcarbamic Acid as Prospective Antibacterial Agents Interacting with Cytoplasmic Membrane" Antibiotics 9, no. 2: 64. https://doi.org/10.3390/antibiotics9020064