
Discovery of Resistance Genes in Rye by Targeted Long-Read Sequencing and Association Genetics

 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Collection of Danish Puccinia Recondita f. sp. Secalis Samples, Their Multiplication and Single Pustule Isolation
2.2. Plant Materials and DNA Extraction
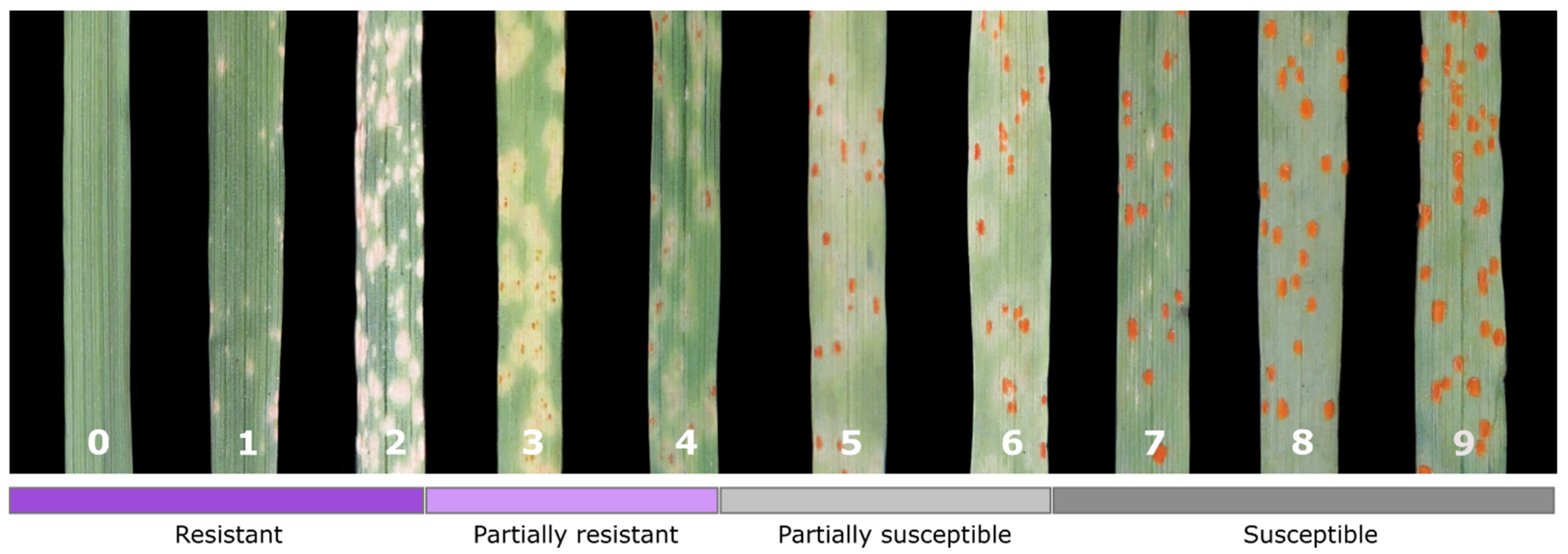
2.3. Phenotyping for Pathogen Resistance
2.4. Molecular Marker Resource and SNP Genotyping
2.5. Data Analysis
2.6. Genome-Wide Association Study
2.7. In Silico Test of the Bait Library
2.8. Phylogenetic Analysis and Pairwise Selection of Restorer Lines
2.9. Single-Molecule Real-Time High-Fidelity Resistance Gene Enrichment Sequencing
2.10. De Novo Assembly and NLR Annotation
2.11. In Vitro Test of the Bait Library
2.12. K-Mer Presence/Absence Matrix
2.13. Association Genetics RenSeq Analysis
2.14. Characterization of the Candidate Leaf Rust Resistance Gene Pr3
2.15. Graphical Editing
3. Results
3.1. Phenotyping of Rye Breeding Lines for Resistance to Leaf Rust
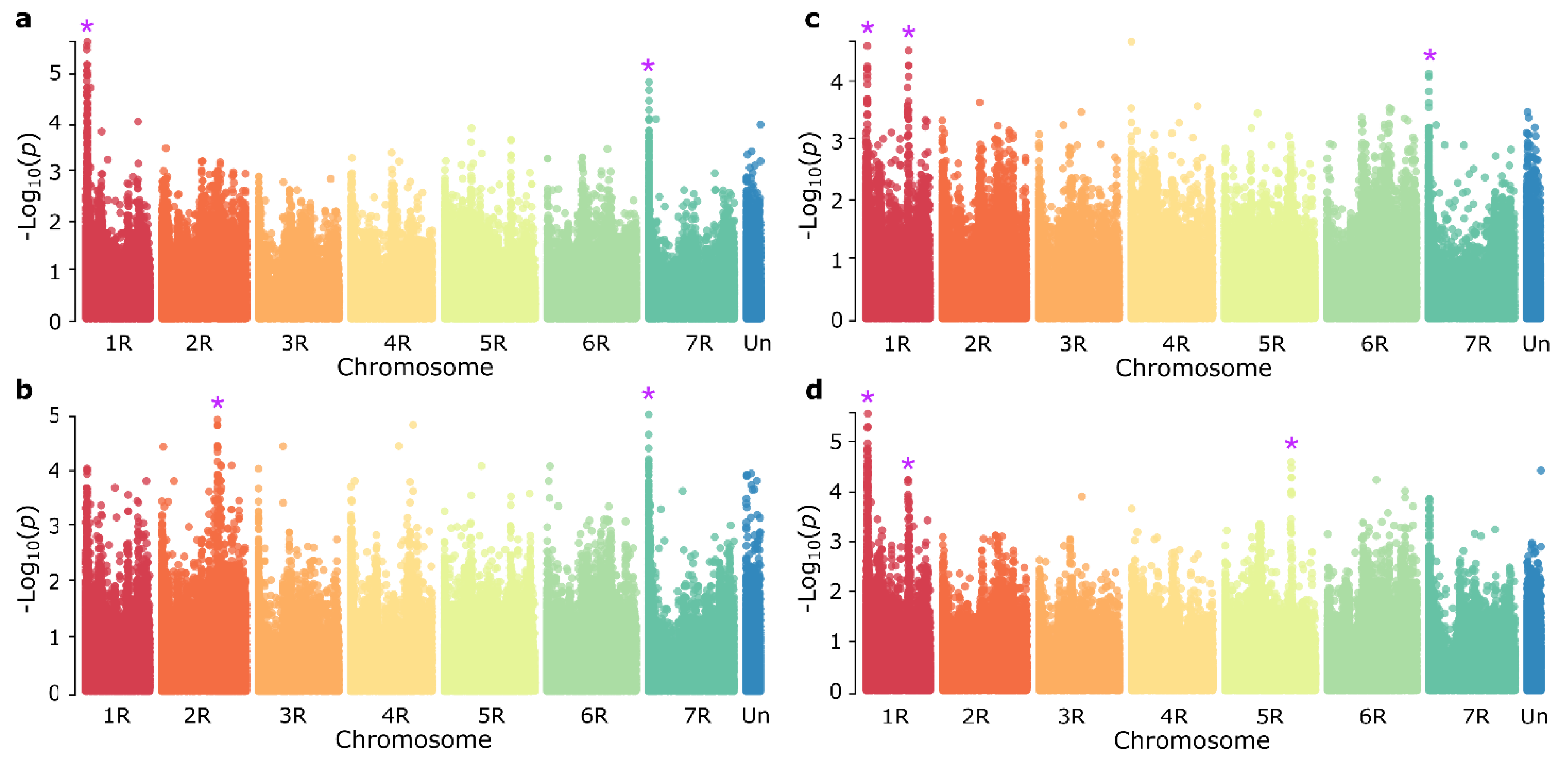
3.2. Genome-Wide Association Study
3.3. In Silico Test of 600 K Triticeae NLR Bait-Library for Rye
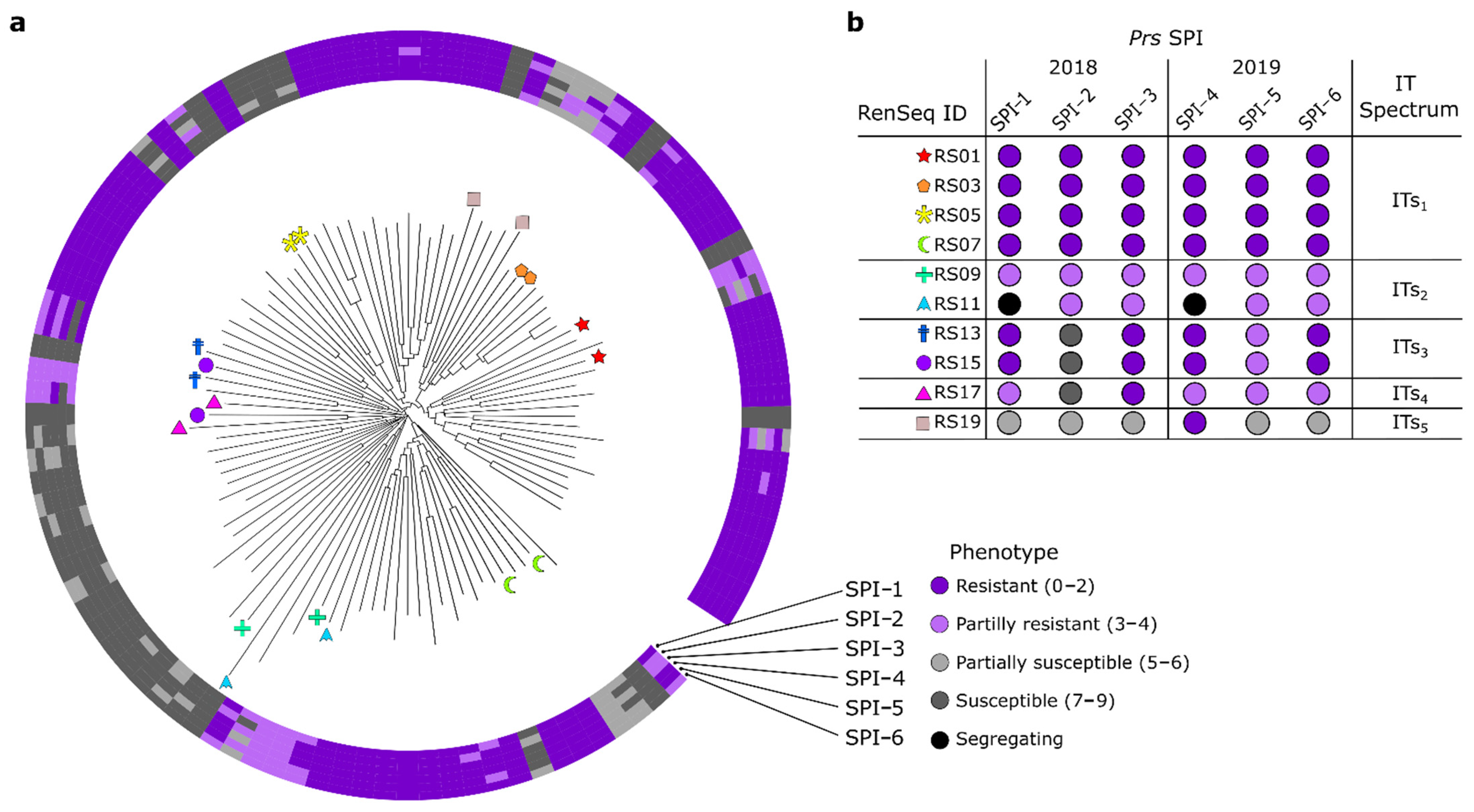
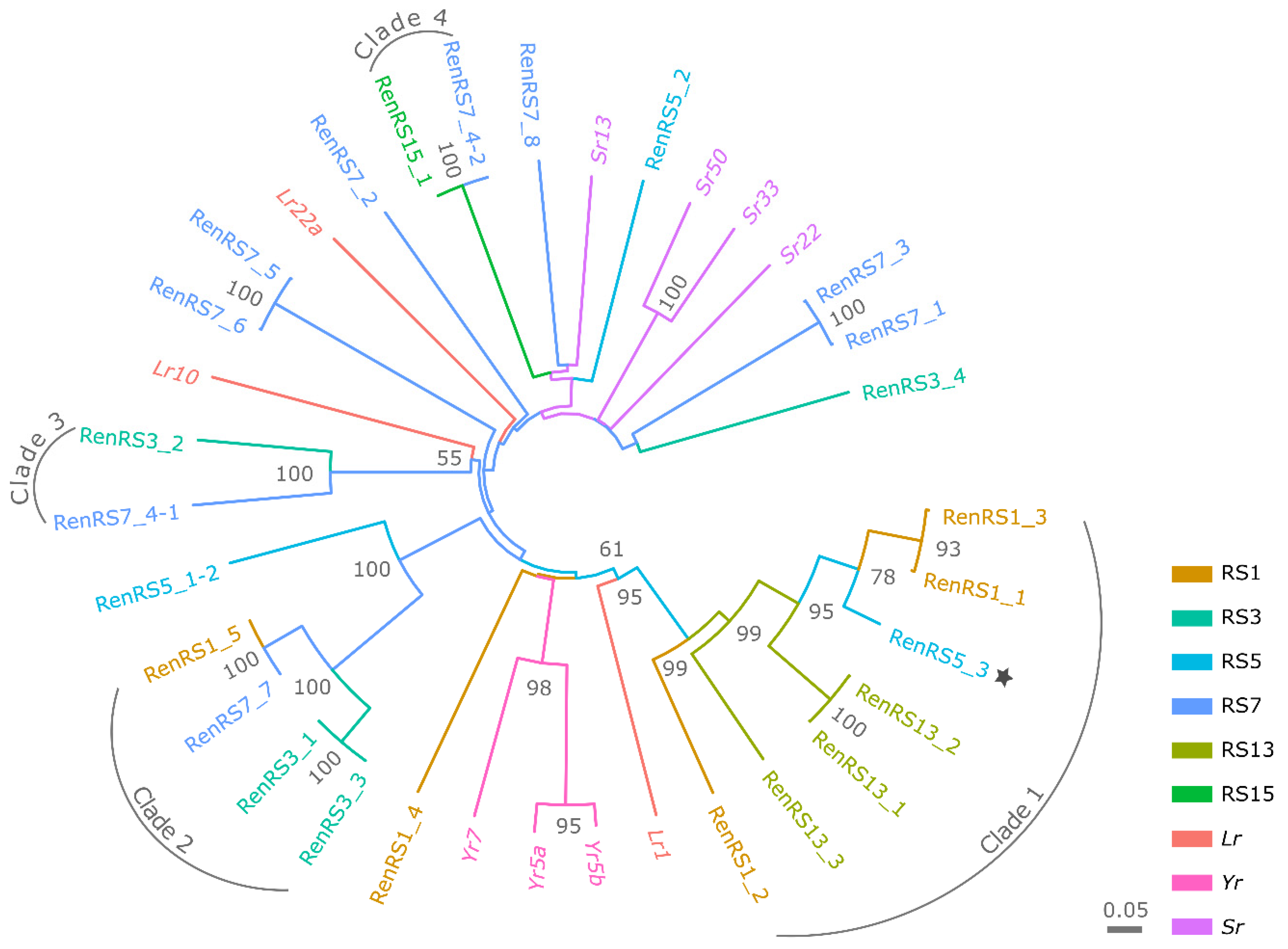
3.4. Phylogenetic Analysis and Pairwise Selection of Restorer Lines
3.5. SMRT RenSeq, De Novo Assembly and NLR Annotation
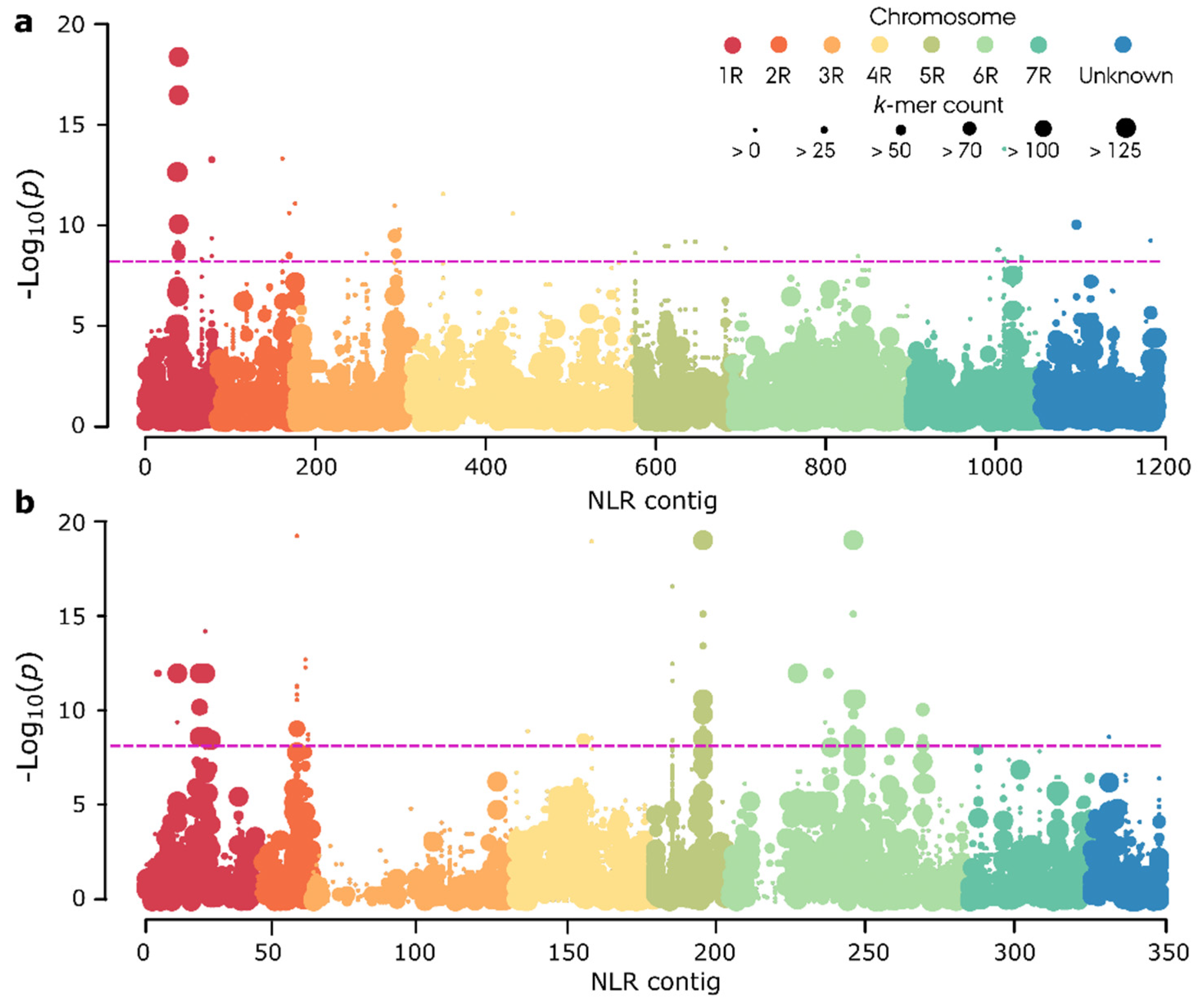
3.6. K-Mer-Based Association Genetics RenSeq (AgRenSeq) Analysis
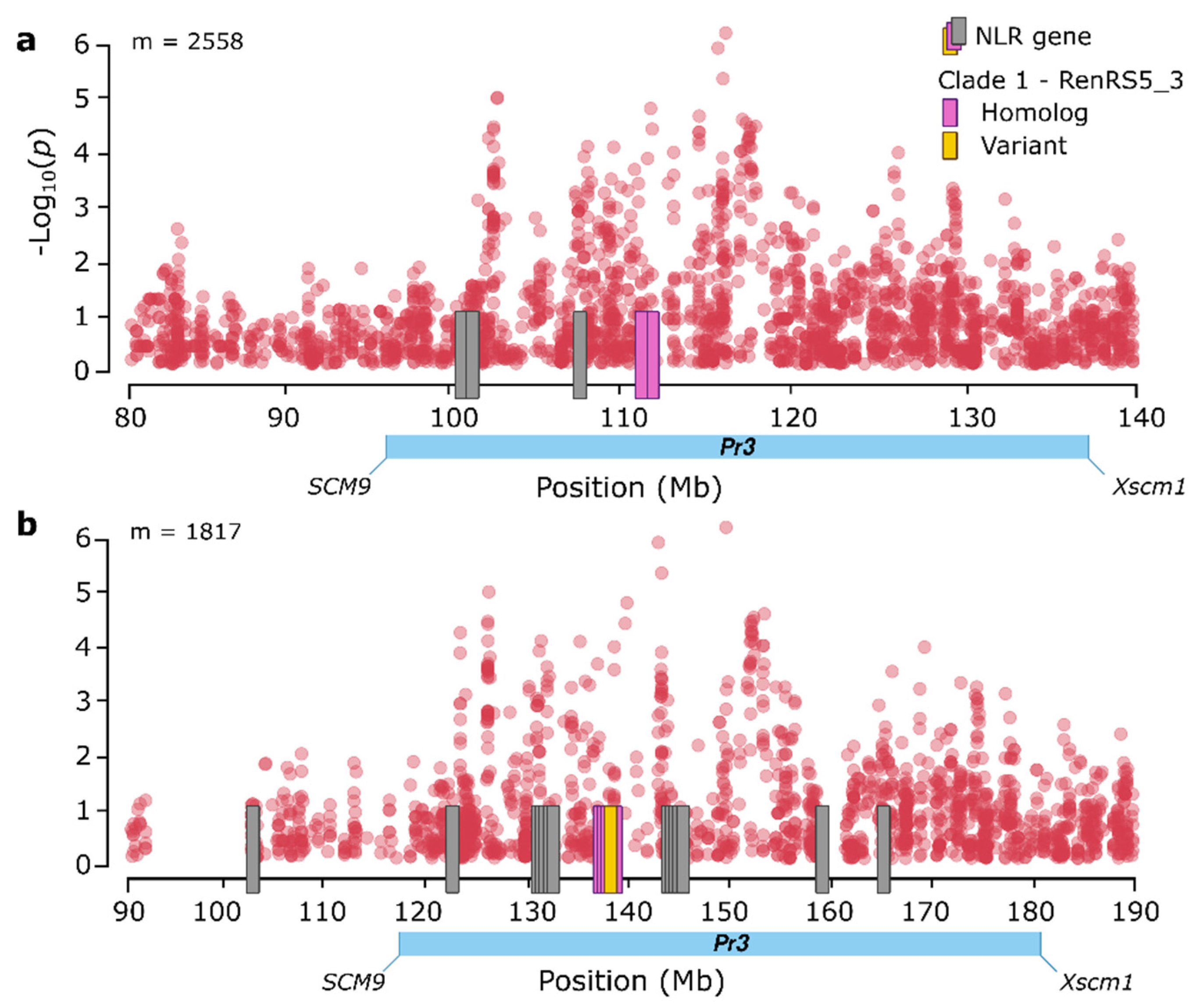
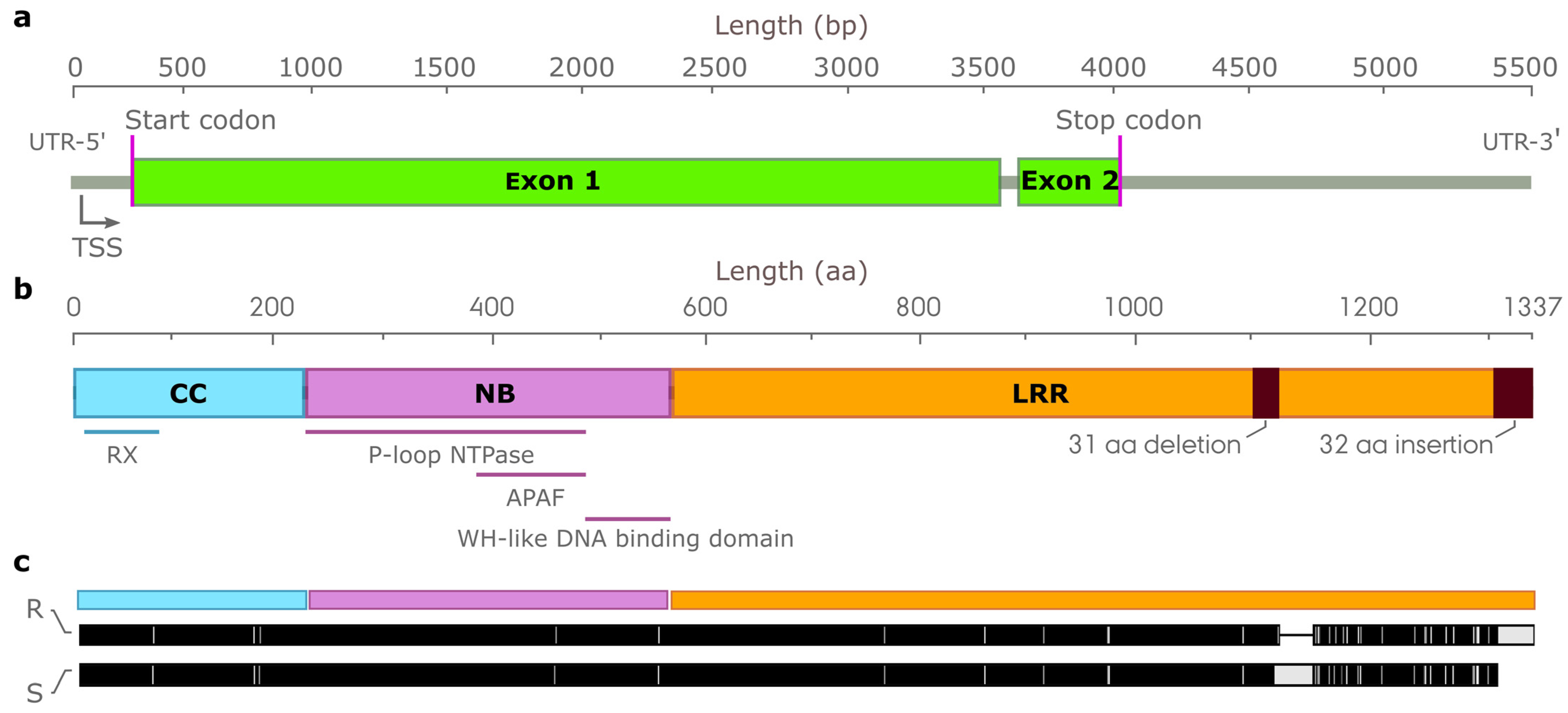
3.7. Characterization of Candidate Genes Conferring Resistance to Leaf Rust Resistance
3.8. Characterization of Candidate NLR Genes Conferring Resistance to Leaf Rust on Chromosome Arm 1RS
4. Discussion
4.1. Leaf Rust Resistance Genes in Restorer Germplasm
4.2. Test of NLR Capture by the Bait Library
4.3. K-Mer Association Genetics with SMRT RenSeq Data
4.4. Co-Discovery of a Candidate Pr Gene on Chromosome Arm 1RS
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Miedaner, T.; Sperling, U. Effect of leaf rust and yield components of winter rye hybrids and assessment of quantitative resistance. J. Phytopathol. 1995, 143, 725–730. [Google Scholar] [CrossRef]
- Miedaner, T.; Gey, A.-K.M.; Sperling, U.; Geiger, H.H. Quantitative-genetic analysis of leaf-rust resistance in seedling and adult-plant stages of inbred lines and their testcrosses in winter rye. Plant Breed. 2002, 121, 475–479. [Google Scholar] [CrossRef]
- Sortsinfo. The Danish Official Trial Records. Available online: https://sortinfo.dk/ (accessed on 10 February 2021).
- Miedaner, T.; Klocke, B.; Flath, K.; Geiger, H.H.; Weber, W.E. Diversity, spatial variation, and temporal dynamics of virulences in the German leaf rust (Puccinia recondita f. sp. secalis) population in winter rye. Eur. J. Plant Pathol. 2011, 132, 23–35. [Google Scholar] [CrossRef]
- Anikster, Y.; Bushnell, W.; Roelfs, A.; Eilam, T.; Manisterski, J. Puccinia recondita causing leaf rust on cultivated wheats, wild wheats, and rye. Can. J. Bot. 1997, 75, 2082–2096. [Google Scholar] [CrossRef]
- Hanzlik, K.; Gerowitt, B. Occurrence and distribution of important weed species in German winter oilseed rape fields. J. Plant Dis. Prot. 2012, 119, 107–120. [Google Scholar] [CrossRef]
- De Mol, F.; Von Redwitz, C.; Gerowitt, B. Weed species composition of maize fields in G ermany is influenced by site and crop sequence. Weed Res. 2015, 55, 574–585. [Google Scholar] [CrossRef]
- Andreasen, C.; Stryhn, H. Increasing weed flora in Danish beet, pea and winter barley fields. Crop Prot. 2012, 36, 11–17. [Google Scholar] [CrossRef]
- Figueroa, M.; Dodds, P.N.; Henningsen, E.C. Evolution of virulence in rust fungi-multiple solutions to one problem. Curr. Opin. Plant Biol. 2020, 56, 20–27. [Google Scholar] [CrossRef]
- Kolmer, J.A.; Ordonez, M.E.; German, S.; Morgounov, A.; Pretorius, Z.; Visser, B.; Goyeau, H.; Anikster, Y.; Acevedo, M. Multilocus genotypes of the wheat leaf rust fungus Puccinia triticina in worldwide regions indicate past and current long-distance migration. Phytopathology 2019, 109, 1453–1463. [Google Scholar] [CrossRef] [Green Version]
- Hovmoller, M.S.; Yahyaoui, A.H.; Milus, E.A.; Justesen, A.F. Rapid global spread of two aggressive strains of a wheat rust fungus. Mol. Ecol. 2008, 17, 3818–3826. [Google Scholar] [CrossRef]
- Nelson, R.; Wiesner-Hanks, T.; Wisser, R.; Balint-Kurti, P. Navigating complexity to breed disease-resistant crops. Nat. Rev. Genet. 2018, 19, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Wehling, P.; Linz, A.; Hackauf, B.; Roux, S.R.; Ruge, B.; Klocke, B. Leaf-rust resistance in rye (Secale cereale L.). 1. Genetic analysis and mapping of resistance genes Pr1 and Pr2. Theor. Appl. Genet. 2003, 107, 432–438. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.R.; Hackauf, B.; Linz, A.; Ruge, B.; Klocke, B.; Wehling, P. Leaf-rust resistance in rye (Secale cereale L.). 2. Genetic analysis and mapping of resistance genes Pr3, Pr4, and Pr5. Theor. Appl. Genet. 2004, 110, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Vendelbo, N.M.; Mahmood, K.; Sarup, P.; Orabi, J.; Hovmøller, M.; Justesen, A.F.; Kristensen, P.S.; Jahoor, A. Discovery of a novel leaf rust (Puccinia recondita) resistance in rye (Secale cereale L.) Using Association Genomics. Cells 2021, 11, 64. [Google Scholar] [CrossRef]
- McIntosh, R.A.; Frieb, B.; Jiang, J.; Gill, B.S. Cytogenetical studies in wheat XVI. Chromosomal location of a new gene for resistance to leaf rust in a Japanese wheat-rye translocation line. Euphytica 1995, 82, 141–147. [Google Scholar] [CrossRef]
- Friebe, B.; Jiang, J.; Raupp, W.J.; McIntosh, R.A.; Gill, B.S. Characterization of wheat-alien translocations conferring resistance to diseases and pests: Current status. Euphytica 1996, 91, 59–87. [Google Scholar] [CrossRef]
- Kourelis, J.; Van Der Hoorn, R.A. Defended to the nines: 25 years of resistance gene cloning identifies nine mechanisms for R protein function. Plant Cell 2018, 30, 285–299. [Google Scholar] [CrossRef] [Green Version]
- Qi, D.; DeYoung, B.J.; Innes, R.W. Structure-function analysis of the coiled-coil and leucine-rich repeat domains of the RPS5 disease resistance protein. Plant Physiol. 2012, 158, 1819–1832. [Google Scholar] [CrossRef] [Green Version]
- Monino-Lopez, D.; Nijenhuis, M.; Kodde, L.; Kamoun, S.; Salehian, H.; Schentsnyi, K.; Stam, R.; Lokossou, A.; Abd-El-Haliem, A.; Visser, R.G. Allelic variants of the NLR protein Rpi-chc1 differentially recognize members of the Phytophthora infestans PexRD12/31 effector superfamily through the leucine-rich repeat domain. Plant J. 2021, 107, 182–197. [Google Scholar] [CrossRef]
- Williams, S.J.; Sornaraj, P.; de Courcy-Ireland, E.; Menz, R.I.; Kobe, B.; Ellis, J.G.; Dodds, P.N.; Anderson, P.A. An autoactive mutant of the M flax rust resistance protein has a preference for binding ATP, whereas wild-type M protein binds ADP. Mol. Plant-Microbe Interact. 2011, 24, 897–906. [Google Scholar] [CrossRef] [Green Version]
- Maekawa, T.; Cheng, W.; Spiridon, L.N.; Töller, A.; Lukasik, E.; Saijo, Y.; Liu, P.; Shen, Q.-H.; Micluta, M.A.; Somssich, I.E. Coiled-coil domain-dependent homodimerization of intracellular barley immune receptors defines a minimal functional module for triggering cell death. Cell Host Microbe 2011, 9, 187–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabanus-Wallace, M.T.; Hackauf, B.; Mascher, M.; Lux, T.; Wicker, T.; Gundlach, H.; Báez, M.; Houben, A.; Mayer, K.F.X.; Guo, L.; et al. Chromosome-scale genome assembly provides insights into rye biology, evolution and agronomic potential. Nat. Genet. 2021, 53, 564–573. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Wang, L.; Yang, J.; He, H.; Jin, H.; Li, X.; Ren, T.; Ren, Z.; Li, F.; Han, X.; et al. A high-quality genome assembly highlights rye genomic characteristics and agronomically important genes. Nat. Genet. 2021, 53, 574–584. [Google Scholar] [CrossRef] [PubMed]
- Miedaner, T.; Korzun, V. Marker-assisted selection for disease resistance in wheat and barley breeding. Phytopathology 2012, 102, 560–566. [Google Scholar] [CrossRef] [Green Version]
- Beukert, U.; Thorwarth, P.; Zhao, Y.; Longin, C.F.H.; Serfling, A.; Ordon, F.; Reif, J.C. Comparing the potential of marker-assisted selection and genomic prediction for improving rust resistance in hybrid wheat. Front. Plant Sci. 2020, 11, 1650. [Google Scholar] [CrossRef]
- Liu, R.; Lu, J.; Zhou, M.; Zheng, S.; Liu, Z.; Zhang, C.; Du, M.; Wang, M.; Li, Y.; Wu, Y. Developing stripe rust resistant wheat (Triticum aestivum L.) lines with gene pyramiding strategy and marker-assisted selection. Genet. Res. Crop Evol. 2020, 67, 381–391. [Google Scholar] [CrossRef]
- Hickey, L.T.; Germán, S.E.; Pereyra, S.A.; Diaz, J.E.; Ziems, L.A.; Fowler, R.A.; Platz, G.J.; Franckowiak, J.D.; Dieters, M.J. Speed breeding for multiple disease resistance in barley. Euphytica 2017, 213, 1–14. [Google Scholar] [CrossRef]
- Ghosh, S.; Watson, A.; Gonzalez-Navarro, O.E.; Ramirez-Gonzalez, R.H.; Yanes, L.; Mendoza-Suárez, M.; Simmonds, J.; Wells, R.; Rayner, T.; Green, P. Speed breeding in growth chambers and glasshouses for crop breeding and model plant research. Nat. Protoc. 2018, 13, 2944–2963. [Google Scholar] [CrossRef] [Green Version]
- Bajgain, P.; Rouse, M.N.; Tsilo, T.J.; Macharia, G.K.; Bhavani, S.; Jin, Y.; Anderson, J.A. Nested association mapping of stem rust resistance in wheat using genotyping by sequencing. PLoS ONE 2016, 11, e0155760. [Google Scholar] [CrossRef] [Green Version]
- Rollar, S.; Serfling, A.; Geyer, M.; Hartl, L.; Mohler, V.; Ordon, F. QTL mapping of adult plant and seedling resistance to leaf rust (Puccinia triticina Eriks.) in a multiparent advanced generation intercross (MAGIC) wheat population. Theor. Appl. Genet. 2021, 134, 37–51. [Google Scholar] [CrossRef]
- Alqudah, A.M.; Sallam, A.; Baenziger, P.S.; Börner, A. GWAS: Fast-forwarding gene identification and characterization in temperate Cereals: Lessons from Barley—A review. J. Adv. Res. 2020, 22, 119–135. [Google Scholar] [CrossRef] [PubMed]
- Korte, A.; Farlow, A. The advantages and limitations of trait analysis with GWAS: A review. Plant Methods 2013, 9, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Florence, J.; Vernaldi, S.; Maekawa, T. Evolution and conservation of plant NLR functions. Front. Immunol. 2013, 4, 297:1–297:16. [Google Scholar] [CrossRef] [Green Version]
- Arora, S.; Steuernagel, B.; Gaurav, K.; Chandramohan, S.; Long, Y.; Matny, O.; Johnson, R.; Enk, J.; Periyannan, S.; Singh, N.; et al. Resistance gene cloning from a wild crop relative by sequence capture and association genetics. Nat. Biotechnol. 2019, 37, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Dong, O.X.; Ronald, P.C. Genetic engineering for disease resistance in plants: Recent progress and future perspectives. Plant Physiol. 2019, 180, 26–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jupe, F.; Witek, K.; Verweij, W.; Sliwka, J.; Pritchard, L.; Etherington, G.J.; Maclean, D.; Cock, P.J.; Leggett, R.M.; Bryan, G.J.; et al. Resistance gene enrichment sequencing (RenSeq) enables reannotation of the NB-LRR gene family from sequenced plant genomes and rapid mapping of resistance loci in segregating populations. Plant J. 2013, 76, 530–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witek, K.; Jupe, F.; Witek, A.I.; Baker, D.; Clark, M.D.; Jones, J.D. Accelerated cloning of a potato late blight-resistance gene using RenSeq and SMRT sequencing. Nat. Biotechnol. 2016, 34, 656–660. [Google Scholar] [CrossRef] [Green Version]
- Xing, L.; Hu, P.; Liu, J.; Witek, K.; Zhou, S.; Xu, J.; Zhou, W.; Gao, L.; Huang, Z.; Zhang, R. Pm21 from Haynaldia villosa encodes a CC-NBS-LRR protein conferring powdery mildew resistance in wheat. Mol. Plant 2018, 11, 874–878. [Google Scholar] [CrossRef] [Green Version]
- Wenger, A.M.; Peluso, P.; Rowell, W.J.; Chang, P.-C.; Hall, R.J.; Concepcion, G.T.; Ebler, J.; Fungtammasan, A.; Kolesnikov, A.; Olson, N.D. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat. Biotechnol. 2019, 37, 1155–1162. [Google Scholar] [CrossRef]
- Van de Weyer, A.L.; Monteiro, F.; Furzer, O.J.; Nishimura, M.T.; Cevik, V.; Witek, K.; Jones, J.D.G.; Dangl, J.L.; Weigel, D.; Bemm, F. A species-wide inventory of NLR genes and alleles in Arabidopsis thaliana. Cell 2019, 178, 1260–1272.e1214. [Google Scholar] [CrossRef] [Green Version]
- Steuernagel, B.; Periyannan, S.K.; Hernandez-Pinzon, I.; Witek, K.; Rouse, M.N.; Yu, G.; Hatta, A.; Ayliffe, M.; Bariana, H.; Jones, J.D.; et al. Rapid cloning of disease-resistance genes in plants using mutagenesis and sequence capture. Nat. Biotechnol. 2016, 34, 652–655. [Google Scholar] [CrossRef] [PubMed]
- Seong, K.; Seo, E.; Witek, K.; Li, M.; Staskawicz, B. Evolution of NLR resistance genes with noncanonical N-terminal domains in wild tomato species. New Phytol. 2020, 227, 1530–1543. [Google Scholar] [CrossRef] [PubMed]
- Barbey, C.R.; Lee, S.; Verma, S.; Bird, K.A.; Yocca, A.E.; Edger, P.P.; Knapp, S.J.; Whitaker, V.M.; Folta, K.M. Disease resistance genetics and genomics in octoploid strawberry. G3 Genes Genomes Genet. 2019, 9, 3315–3332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- USDA. United States Department of Agriculture: Wheat and Barley DNA Extraction Protocol (96-Well Plate Format). Available online: https://www.ars.usda.gov/ARSUserFiles/60701500/SmallGrainsGenotypingLaboratory/Protocols/wheat%20and%20barleyDNA%20extraction_original.pdf (accessed on 7 July 2021).
- Pallotta, M.A.; Warner, P.; Fox, R.L.; Kuchel, H.; Jefferies, S.J.; Langridge, P. Marker assisted wheat breeding in the southern region of Australia. In Proceedings of the Tenth International Wheat Genetics Symposium; Istituto Sperimentale per la Cerealicoltura: Puglia, Italy, 2003; pp. 789–791. [Google Scholar]
- McNeal, F.H.; Konzak, C.F.; Smith, E.P.; Tate, W.S.; Russell, T.S. A Uniform System for Recording and Processing Cereal Research Data; No. REP-10904; CIMMYT: El Batan, Mexico, 1971. [Google Scholar]
- Hovmoller, M.S.; Rodriguez-Algaba, J.; Thach, T.; Sorensen, C.K. Race typing of Puccinia striiformis on wheat. In Wheat Rust Diseases: Methods and Protocols; Humana Press: New York, NY, USA, 2017; pp. 29–40. [Google Scholar]
- Thach, T.; Ali, S.; Justesen, A.; Rodriguez-Algaba, J.; Hovmøller, M. Recovery and virulence phenotyping of the historic ‘Stubbs collection’ of the yellow rust fungus Puccinia striiformis from wheat. Ann. Appl. Biol. 2015, 167, 314–326. [Google Scholar] [CrossRef]
- Bauer, E.; Schmutzer, T.; Barilar, I.; Mascher, M.; Gundlach, H.; Martis, M.M.; Twardziok, S.O.; Hackauf, B.; Gordillo, A.; Wilde, P.; et al. Towards a whole-genome sequence for rye (Secale cereale L.). Plant J. 2017, 89, 853–869. [Google Scholar] [CrossRef] [Green Version]
- Vendelbo, N.M.; Mahmood, K.; Sarup, P.; Orabi, J.; Kristensen, P.S.; Jahoor, A. Discovery of a novel powdery mildew (Blumeria graminis) resistance locus in rye (Secale cereale L.). Sci. Rep. 2021, 11, 1–15. [Google Scholar]
- RStudio Team. Rstudio: Integrated Development for R. RStudio, Inc., Boston. Available online: http://www.rstudio.com (accessed on 2 August 2021).
- R Core Team. R A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria. Available online: https://www.R-project.org/ (accessed on 8 February 2021).
- Lipka, A.E.; Tian, F.; Wang, Q.; Peiffer, J.; Li, M.; Bradbury, P.J.; Gore, M.A.; Buckler, E.S.; Zhang, Z. GAPIT: Genome association and prediction integrated tool. Bioinformatics 2012, 28, 2397–2399. [Google Scholar] [CrossRef] [Green Version]
- NCBI. National Center for Biotechnology Information. Available online: https://www.ncbi.nlm.nih.gov (accessed on 13 June 2021).
- Paradis, E.; Schliep, K. ape 5.0: An environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 2019, 35, 526–528. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, 256–259. [Google Scholar] [CrossRef] [Green Version]
- Nurk, S.; Walenz, B.P.; Rhie, A.; Vollger, M.R.; Logsdon, G.A.; Grothe, R.; Miga, K.H.; Eichler, E.E.; Phillippy, A.M.; Koren, S. HiCanu: Accurate assembly of segmental duplications, satellites, and allelic variants from high-fidelity long reads. Genome Res. 2020, 30, 1291–1305. [Google Scholar] [CrossRef]
- Steuernagel, B.; Jupe, F.; Witek, K.; Jones, J.D.; Wulff, B.B. NLR-parser: Rapid annotation of plant NLR complements. Bioinformatics 2015, 31, 1665–1667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steuernagel, B.; Witek, K.; Krattinger, S.G.; Ramirez-Gonzalez, R.H.; Schoonbeek, H.J.; Yu, G.; Baggs, E.; Witek, A.I.; Yadav, I.; Krasileva, K.V.; et al. The NLR-annotator tool enables annotation of the intracellular immune receptor repertoire. Plant Physiol. 2020, 183, 468–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wickham, H. ggplot2. Wiley Interdiscip. Rev. Comput. Stat. 2011, 3, 180–185. [Google Scholar] [CrossRef]
- Marcais, G.; Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 2011, 27, 764–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- GeneiousPrime. Geneious Prime (v. 2020.2.3). 2020. Available online: https://www.geneious.com (accessed on 11 December 2021).
- Stanke, M.; Steinkamp, R.; Waack, S.; Morgenstern, B. AUGUSTUS: A web server for gene finding in eukaryotes. Nucleic Acids Res. 2004, 32, W309–W312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, P.; Binns, D.; Chang, H.Y.; Fraser, M.; Li, W.; McAnulla, C.; Hunter, S. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [Green Version]
- Martin, E.C.; Sukarta, O.C.; Spiridon, L.; Grigore, L.G.; Constantinescu, V.; Tacutu, R.; Goverse, A.; Petrescu, A.-J. LRRpredictor—A new LRR motif detection method for irregular motifs of plant NLR proteins using an ensemble of classifiers. Genes 2020, 11, 286. [Google Scholar] [CrossRef] [Green Version]
- Toparslan, E.; Karabag, K.; Bilge, U. A workflow with R: Phylogenetic analyses and visualizations using mitochondrial cytochrome b gene sequences. PLoS ONE 2020, 15, e0243927. [Google Scholar] [CrossRef]
- Yu, G. Using ggtree to visualize data on tree-like structures. Curr. Protoc. Bioinform. 2020, 69, e96. [Google Scholar] [CrossRef]
- UniProt, C. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, 480–489. [Google Scholar] [CrossRef]
- Mago, R.; Miah, H.; Lawrence, G.J.; Wellings, C.R.; Spielmeyer, W.; Bariana, H.S.; McIntosh, R.A.; Pryor, A.J.; Ellis, J.G. High-resolution mapping and mutation analysis separate the rust resistance genes Sr31, Lr26 and Yr9 on the short arm of rye chromosome 1. Theor. Appl. Genet. 2005, 112, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Gaikpa, D.S.; Kessel, B.; Presterl, T.; Ouzunova, M.; Galiano-Carneiro, A.L.; Mayer, M.; Melchinger, A.E.; Schön, C.-C.; Miedaner, T. Exploiting genetic diversity in two European maize landraces for improving Gibberella ear rot resistance using genomic tools. Theor. Appl. Genet. 2021, 134, 793–805. [Google Scholar] [CrossRef] [PubMed]
- Vendelbo, N.M.; Sarup, P.; Orabi, J.; Kristensen, P.S.; Jahoor, A. Genetic structure of a germplasm for hybrid breeding in rye (Secale cereale L.). PLoS ONE 2020, 15, e0239541. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; San Vicente, F.; Huang, K.; Dhliwayo, T.; Costich, D.E.; Semagn, K.; Sudha, N.; Olsen, M.; Prasanna, B.M.; Zhang, X.; et al. Molecular characterization of CIMMYT maize inbred lines with genotyping-by-sequencing SNPs. Theor. Appl. Genet. 2016, 129, 753–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibson, G. Rare and common variants: Twenty arguments. Nat. Rev. Genet. 2012, 13, 135–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sucher, J.; Boni, R.; Yang, P.; Rogowsky, P.; Buchner, H.; Kastner, C.; Kumlehn, J.; Krattinger, S.G.; Keller, B. The durable wheat disease resistance gene Lr34 confers common rust and northern corn leaf blight resistance in maize. Plant Biotechnol. J. 2017, 15, 489–496. [Google Scholar] [CrossRef]
- Xie, J.; Guo, G.; Wang, Y.; Hu, T.; Wang, L.; Li, J.; Qiu, D.; Li, Y.; Wu, Q.; Lu, P. A rare single nucleotide variant in Pm5e confers powdery mildew resistance in common wheat. New Phytol. 2020, 228, 1011–1026. [Google Scholar] [CrossRef]
- Halterman, D.A.; Wise, R.P. A single-amino acid substitution in the sixth leucine-rich repeat of barley MLA6 and MLA13 alleviates dependence on RAR1 for disease resistance signaling. Plant J. 2004, 38, 215–226. [Google Scholar] [CrossRef]
- Anderson, P.A.; Lawrence, G.J.; Morrish, B.C.; Ayliffe, M.A.; Finnegan, E.J.; Ellis, J.G. Inactivation of the flax rust resistance gene M associated with loss of a repeated unit within the leucine-rich repeat coding region. Plant Cell 1997, 9, 641–651. [Google Scholar]
- Barragan, A.C.; Weigel, D. Plant NLR diversity: The known unknowns of Pan-NLRomes. Plant Cell 2021, 33, 814–831. [Google Scholar] [CrossRef]
- Wei, F.; Gobelman-Werner, K.; Morroll, S.M.; Kurth, J.; Mao, L.; Wing, R.; Leister, D.; Schulze-Lefert, P.; Wise, R.P. The Mla (powdery mildew) resistance cluster is associated with three NBS-LRR gene families and suppressed recombination within a 240-kb DNA interval on chromosome 5S (1HS) of barley. Genetics 1999, 153, 1929–1948. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.; Dickinson, M.; Balint-Kurti, P.; Dixon, M.; Jones, J. Two complex resistance loci revealed in tomato by classical and RFLP mapping of the Cf-2, Cf-4, Cf-5, and Cf-9 genes for resistance to Cladosporium fulvum. Mol. Plant Microbe Interact. 1993, 6, 348. [Google Scholar] [CrossRef]
- Smith, S.M.; Pryor, A.J.; Hulbert, S.H. Allelic and haplotypic diversity at the rp1 rust resistance locus of maize. Genetics 2004, 167, 1939–1947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richter, T.E.; Pryor, T.J.; Bennetzen, J.L.; Hulbert, S.H. New rust resistance specificities associated with recombination in the Rp1 complex in maize. Genetics 1995, 141, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Lukaszewski, A.J. Frequency of 1RS. 1AL and 1RS. 1BL translocations in United States wheats. Crop Sci. 1990, 30, 1151–1153. [Google Scholar] [CrossRef]
- Qiu, L.; Wang, H.; Li, Y.; Wang, W.; Liu, Y.; Mu, J.; Geng, M.; Guo, W.; Hu, Z.; Ma, J. Fine mapping of the wheat leaf rust resistance gene LrLC10 (Lr13) and validation of its co-segregation markers. Front. Plant Sci. 2020, 11, 470. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, H.; Nyamesorto, B.; Luo, Y.; Mu, X.; Wang, F.; Kang, Z.; Lagudah, E.; Huang, L. A new mode of NPR1 action via an NB-ARC–NPR1 fusion protein negatively regulates the defence response in wheat to stem rust pathogen. New Phytol. 2020, 228, 959–972. [Google Scholar] [CrossRef]
- Zhu, H.; Li, C.; Gao, C. Applications of CRISPR–Cas in agriculture and plant biotechnology. Nat. Rev. Mol. Cell Biol. 2020, 21, 661–677. [Google Scholar] [CrossRef]
- Chen, S.; Rouse, M.N.; Zhang, W.; Zhang, X.; Guo, Y.; Briggs, J.; Dubcovsky, J. Wheat gene Sr60 encodes a protein with two putative kinase domains that confers resistance to stem rust. New Phytol. 2020, 225, 948–959. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chromosome | Position (Mb) | Marker ID | −Log10(p) | Phentypic Variance Explained (%) |
|---|---|---|---|---|
| 1RS | 115.55 | AX-99251803 | 6.11 | 13.1 |
| 1RL | 625.54 | AX-99805135 | 4.50 | 9.3 |
| 2RL | 818.90 | AX-99478491 | 4.90 | 9.6 |
| 5RL | 770.21 | AX-99776626 | 4.49 | 9.3 |
| 7RS | 26.93 | AX-99684185 | 4.83 | 12.1 |
| Clade | NLR Contigs | Assemblies | Anchoring Position in Lo7 | ||
|---|---|---|---|---|---|
| Resistant | SPI-Resistant | Chromosome Arm | Position (Mb) | ||
| 1 | 7 | RS1, RS3, RS5 | RS13 | 1RS | 111.15 |
| 2 | 4 | RS1, RS3, RS7 | 5RL | 792.53 | |
| 3 | 2 | RS3, RS7 | NA | NA | |
| 4 | 2 | RS7 | RS15 | 5RL | 807.97 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vendelbo, N.M.; Mahmood, K.; Steuernagel, B.; Wulff, B.B.H.; Sarup, P.; Hovmøller, M.S.; Justesen, A.F.; Kristensen, P.S.; Orabi, J.; Jahoor, A. Discovery of Resistance Genes in Rye by Targeted Long-Read Sequencing and Association Genetics. Cells 2022, 11, 1273. https://doi.org/10.3390/cells11081273
Vendelbo NM, Mahmood K, Steuernagel B, Wulff BBH, Sarup P, Hovmøller MS, Justesen AF, Kristensen PS, Orabi J, Jahoor A. Discovery of Resistance Genes in Rye by Targeted Long-Read Sequencing and Association Genetics. Cells. 2022; 11(8):1273. https://doi.org/10.3390/cells11081273
Chicago/Turabian StyleVendelbo, Nikolaj M., Khalid Mahmood, Burkhard Steuernagel, Brande B. H. Wulff, Pernille Sarup, Mogens S. Hovmøller, Annemarie Fejer Justesen, Peter S. Kristensen, Jihad Orabi, and Ahmed Jahoor. 2022. "Discovery of Resistance Genes in Rye by Targeted Long-Read Sequencing and Association Genetics" Cells 11, no. 8: 1273. https://doi.org/10.3390/cells11081273