De novo Transcriptome Assembly of Myllocerinus aurolineatus Voss in Tea Plants
 Xin Xie
Xin Xie Junmei Jiang1†
Junmei Jiang1†  Meiqing Chen
Meiqing Chen- 1Key Laboratory of Agricultural Microbiology, College of Agriculture, Guizhou University, Guiyang, China
- 2State Key Laboratory Breeding Base of Green Pesticide and Agricultural Bioengineering, Key Laboratory of Green Pesticide and Agricultural Bioengineering, Ministry of Education, Guizhou University, Guiyang, China
Myllocerinus aurolineatus Voss is a species of the insecta class in the arthropod. In this study, we first observed and identified M. aurolineatus Voss in tea plants in Guizhou, China, where it caused severe quantity and quality losses in tea plants. Knowledge on M. aurolineatus Voss genome is inadequate, especially for biological or functional research. We performed the first transcriptome sequencing by using the Illumina Hiseq™ technique on M. aurolineatus Voss. Over 55.9 million high-quality paired-end reads were generated and assembled into 69,439 unigenes using the Trinity short read software, resulting in a cluster of 1,207 bp of the N50 length. A total of 69,439 genes were predicted by BLAST to known proteins in the NCBI database and were distributed into Gene Ontology (20,190), eukaryotic complete genomes (12,488), and the Kyoto Encyclopedia of Genes and Genomes (3,170). We also identified 96,790 single-nucleotide polymorphisms and 13,121 simple sequence repeats in these unigenes. Our transcriptome data provide a useful resource for future functional studies of M. aurolineatus Voss for dispersal control in tea plants.
Introduction
Tea plant Camellia sinensis is a major economic crop in China and accounts for half of the total plantation area worldwide (Li et al., 2017). Many nutritional ingredients are produced in tea shoots, which are used as raw materials for commercial tea processing (Comblain et al., 2016). However, various tea plant pests jeopardize the quantity and quality of tea, such as the tea leaf beetle, one of the most serious pests affecting tea plants in China, Vietnam, Indonesia, Japan, and other tea-producing countries in Asia (Sun et al., 2010; Yang et al., 2013; Roy and Muraleedharan, 2014). Here, we reported a leaf beetle, Myllocerinus aurolineatus Voss in tea plant in Guizhou province, China. M. aurolineatus was first reported in China in 1991, where the species emerged in tea plants (Zhang, 1991).
M. aurolineatus is an exceptionally destructive and productive pest that shows strong tolerance and adaptability against environment challenges, including those with pesticide treatment, extreme temperatures, and volatile compound attractions (Sun et al., 2010, 2017). In Meitan district, Guizhou, M. aurolineatus pupates from March to April, then larvae become adult and begin to mate from June to July. Finally, the outbreak of this pest will be from July to August. Pesticide resistance, the diapause process, and other biological flexibilities are moderated by series molecular mechanisms (Casida, 2017; Jugulam and Gill, 2018; Zhang et al., 2018). However, lack of bona fide reference regarding gene annotation and genomic information restricts our research on the molecular mechanism of these physiological processes for exploring new management approaches for this species. To date, only 157 nucleotide sequences including 51 expressed sequences (EST) have been deposited in GenBank for this beetle. In addition, only few genetic markers have been reported for M. aurolineatus (Ma et al., 2014; Mukhopadhyay et al., 2016).
De novo transcriptome assemblies constitute a valid technology to study reference-free genomes in non-model organisms and provided abundant sequence and gene expression information for further functional research (Gayral et al., 2013; Smith-Unna et al., 2016). With the development of sequencing technologies, such as Illumina and 454 Life Sciences, genomic information in model and non-model organisms have been broadly uncovered along with numerous genes involved in biotic and abiotic stresses, pesticide resistance, developmental pathways, diapause transitions, and hormone regulations by homology blast with related organisms (Ekblom and Galindo, 2011; Tarrant et al., 2016).
In this study, Illumina Hiseq™ technique was used for de novo transcriptome assembly and analysis of M. aurolineatus, a new reported tea plant pest. Sequences were performed with a BLAST search to the known proteins of the NCBI database and the CDS fragments were predicted. Gene Ontology (GO), euKaryotic Ortholog Groups (KOG), and the Kyoto Encyclopedia of Genes and Genomes (KEGG) were annotated. Furthermore, we used these data to generate simple sequence repeat (SSR), and single-nucleotide polymorphism (SNP) markers, which will be potential resources for trait mapping. We believe that the transcriptome assembly of M. aurolineatus obtained in our study represents crucial resources for research into molecular pathway, gene function, metabolic regulation associated with pesticide resistance, and biocontrol areas.
Materials and Methods
M. aurolineatus Collection
In August, the mature M. aurolineatus were captured from the top leaves of the 20-year tea plant in Meitan District, Guizhou, China. Three beetles were kept in liquid nitrogen as a group and subsequently used for transcriptome sequencing.
Nucleic Acid Isolation
Total RNA was extracted using the Trizol kit (Sangon, China) according to the manufacturer's protocol, and treated with RNase-free DNase I to remove genomic DNA contamination. RNA integrity was evaluated with a 1.0% agarose gel. Thereafter, RNA quality and quantity were assessed using a Nanodrop (Thermo, USA) and an Agilent 2100 Bioanalyzer (Agilent Technologies, USA). The high-quality RNA samples were subsequently submitted for library preparation and sequencing.
Library Preparation and Sequencing
A total of 2 μg of RNA sample was used as input material for the RNA sample preparations. Sequencing libraries were generated using VAHTSTM mRNA-seq V2 Library Prep Kit for Illumina® following the manufacturer's protocols. First-strand cDNA was synthesized using random hexamer primer and M-MuLV Reverse Transcriptase. Second-strand cDNA synthesis was subsequently performed using DNA polymerase I and RNase H. Remaining overhangs were converted into blunt ends via exonuclease/polymerase activities. To select cDNA fragments of preferentially 150–200 bp in length, the library fragments were purified with AMPure XP system (Beckman Coulter, Beverly, USA). Then, 3 μl of USER Enzyme (NEB, USA) was utilized with size-selected, adaptor-ligated cDNA at 37°C for 15 min followed by 5 min at 95°C in PCR cycler. Finally, PCR products were purified (AMPure XP system) and the library quality was assessed on the Agilent Bioanalyzer 2100 system. The libraries were then quantified and pooled. Paired-end sequencing of the library was performed on the HiSeq XTen sequencers (Illumina, USA).
Data Assessment and Quality Control
FastQC (version 0.11.2) was used for evaluating the quality of sequenced data. Raw reads were filtered by Trimmomatic (version 0.36) according to five steps: (1) removing adaptor sequence; (2) removing low quality bases from reads 3′ to 5′ (Q < 20); (3) removing low-quality bases from reads 5′ to 3′ (Q < 20); (4) using a sliding window method to remove the base value <20 of reads tail (window size is 5 bp); (5) removing reads with reads length <35 nt and its pairing reads. The remaining clean data were used for further analysis.
Transcriptome Assembly and Gene Annotation
The remaining clean reads were de novo assembled into transcripts using Trinity short read software (version 2.0.6) (parameter: min_kmer_cov 2). Transcripts with a minimum length of 200 bp were clustered to minimize redundancy. For each cluster (representing the transcriptional complexity for the same gene), the longest sequence was preserved and designated as a unigene. Unigenes were blasted against the NCBI non-redundant protein database, SwissProt, TrEMBL, Conserved Domain Database (CDD), Protein family (Pfam), and KOG databases (E-value < 1e−5). According to the priority order of the best aligned results, the Unigene ORF (open reading frame) was determined using the ORF-predictor server. At the same time, TransDecoder (version 3.0.1) was used to predict the CDS sequences of the un-aligned Unigenes. GO functional annotation information was obtained according to transcript annotation results of SwissProt and TrEMBL. KEGG Automatic Annotation Server (version 2.1) was used for the KEGG annotation.
Results and Discussion
M. aurolineatus Identification
M. aurolineatus were captured in the tea plant in Meitan District, Guizhou, China, and the morphological character was described as follows: body, gray black; head with two longitudinal lines yellow pale, with metallic shimmer in dorsal view; antennae geniculate, with scapus straight and thin, expanded in apical three flagella; pronotum with four longitudinal lines yellow pale; elytra with several yellow pale long patches, with black traverse patches in the center in dorsal view (Figure 1).

Figure 1. The morphological characteristics of the adult M. aurolineatus on tea leaves.
Illumina Sequencing and De novo Assembly
We used the Illumina Hiseq™ platform to sequence the constructed cDNA library of M. aurolineatus and obtained 55,915,882 raw reads. To acquire clean reads, 3,121,732 of the low-quality reads and adapter sequences were trimmed by using FastQC software, thus generating 52,794,150 clean reads from the M. aurolineatus transcriptomes. The Q20 and Q30 percentages (sequencing error rate < 0.1%) of M. aurolineatus were 98.23 and 93.87%, respectively (Table 1). The average read length was 146.72 bp, and the percentage of GC content was 40.00% (Table 1). Subsequently, 69,439 unigenes were assembled from the high-quality reads with an average length of 669.28 bp and an N50 of 1,207 bp and N90 of 253 bp. Unigenes with a length of more than 500 and 1,000 bp were 22,395 and 11,856 (Table 2). The length and GC content frequency distributions of all unigenes are shown in Table 2 and Figure 2.
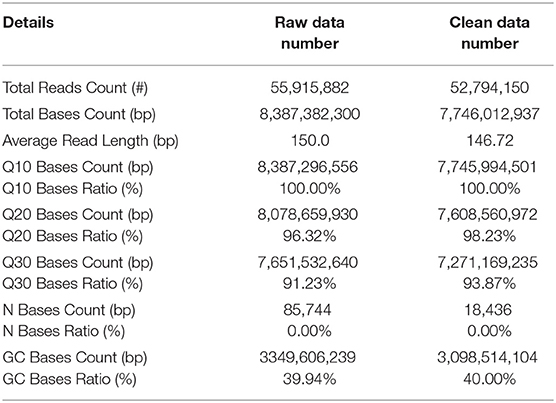
Table 1. Summary of the M. aurolineatus transcriptome.

Table 2. Transcriptome assembly results.
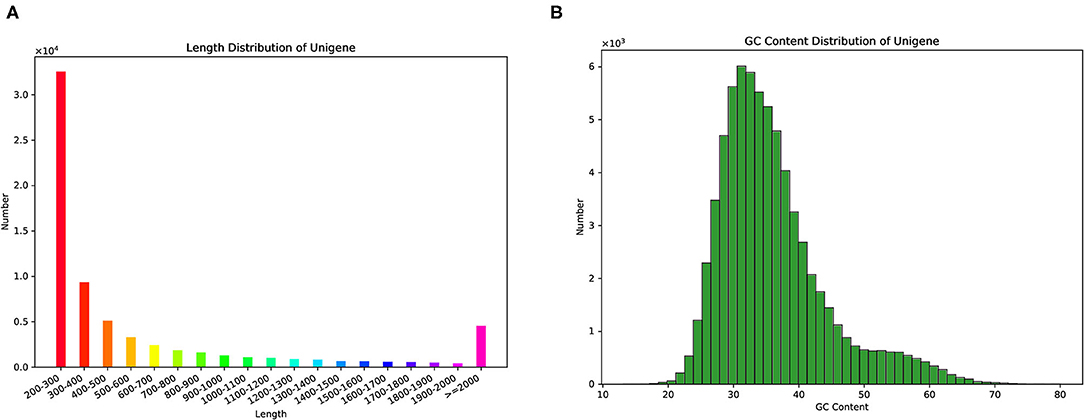
Figure 2. The length and GC content frequency distributions of all unigenes. (A) The length distributions; (B) GC content frequency distributions.
Annotation of Predicted Proteins
To annotate the proteins, we employed the BLASTx program for blasting against NT (NCBI nucleotide sequences), NR (NCBI non-redundant protein sequences), COG (Clusters of Orthologous Groups of proteins), KOG (euKaryotic Ortholog Groups), Swiss-Prot (a manually annotated and reviewed protein sequence database), TrEMBL, Pfam, CDD, GO, and KEGG databases. Of the M. aurolineatus transcripts, 69,439 unigenes were annotated in the above databases, including 23,491 unigenes (33.83%) in the NR database and 17,040 unigenes (24.54%) in the SwissProt database. Moreover, 26,351 unigenes (37.95%) were annotated in at least one database (Table 3). These results were similar to other Arthropoda species, such as 10,218 unigenes were identified in Dendroctonus ponderosae (Regniere and Bentz, 2007), 4,251 in Tribolium castaneum (Li et al., 2008), 1,207 in Papilio xuthus (Zhang and Zhang, 2017), and 353 in Acyrthosiphon pisum (Bishnoi and Singla, 2017). A total of 43.56% annotated unigenes were matched from D. ponderosae according to the proportion on the distribution of best-matched species, followed by 18.12, 5.15, 1.51, and 1.27% sequences that matched sequences from T. castaneum, P. xuthus, A. pisum, and Oryctes borbonicus, respectively. The remaining 30.39% of sequences were matched from other species (Figure 3).
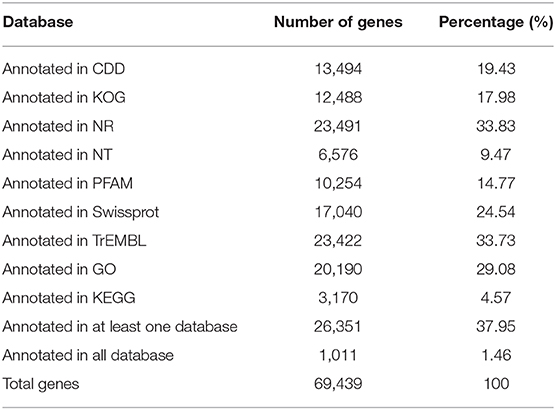
Table 3. Percentage of annotated genes.
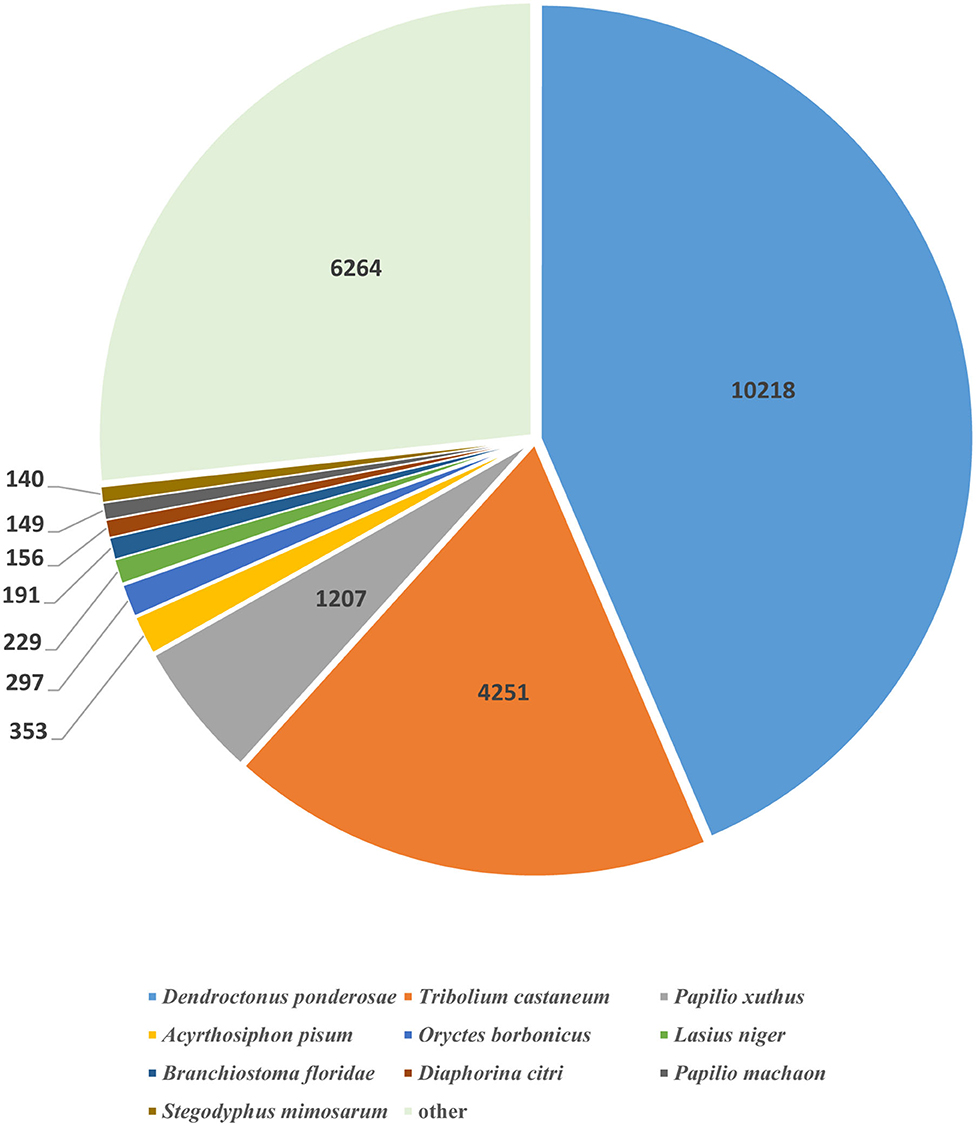
Figure 3. Species distribution of top BLASTx results. The pie chart shows the species distribution of unigenes top BLASTx results against the NR protein database with a cutoff E value <1e−3.
GO, KOG, and KEGG Classifications of Unigenes
Cluster groups of the unigenes were calculated against GO, KOG, and the KEGG databases using BLASTx search with a cutoff E value of 10−5. A total of 29.08% unigenes (20,190) were annotated in these three databases (Table 3).
As an international standardized gene functional classification system, GO determines the functional description of genes and their products (Tarrant et al., 2016). Three independent classifications, namely, “biological process,” “cellular component,” and “molecular function,” are included in this process. As shown in Figure 4 and Supplementary Table 1, 20,190 (29.08%) unigenes were assigned to the main GO classifications, including 71 sub-classifications. Within the molecular function group, binding (GO:0005488) and catalytic activity (GO:0003824) processes were the main proportions with 12,042 and 9,471 unigenes, respectively. In the biological process, cellular (GO:0009987) and metabolic processes (GO:0008152) were the largest two groups including 13,156 and 10,939 unigenes, respectively. Cell (GO:0005623, 13,874 unigenes) and cell part (GO:0044464, 13,837 unigenes) represented the majority of processes in the cellular component category.
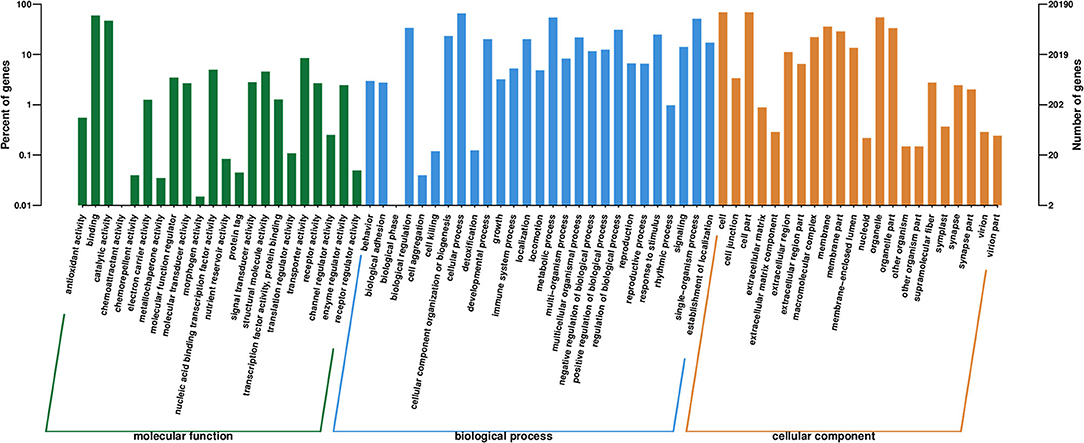
Figure 4. Gene Ontology (GO) categories of the unigenes. Distribution of the GO categories assigned to the M. aurolineatus transcriptome. Unique transcripts (unigenes) were annotated in three categories: cellular components, molecular functions, and biological process.
KOG annotation is widely used to cluster the protein functions in many species (Moon et al., 2008; Kuzniar et al., 2009). In our study, a total of 12,488 unigenes were clustered into 26 functional categories (Figure 5). Among the 26 KOG categories, the cluster for “General function prediction only” (1,731, 13.86%) was the largest group, followed by “Signal transduction mechanisms” (1,653, 13.24%), “Post-translational modification, protein turnover, chaperones” (1,205, 9.65%), and “Transcription” (996, 7.98%). Only few unigenes were assigned under the “Nuclear structure,” “Cell motility,” and “Defense mechanisms” groups (Figure 5 and Supplementary Table 2).
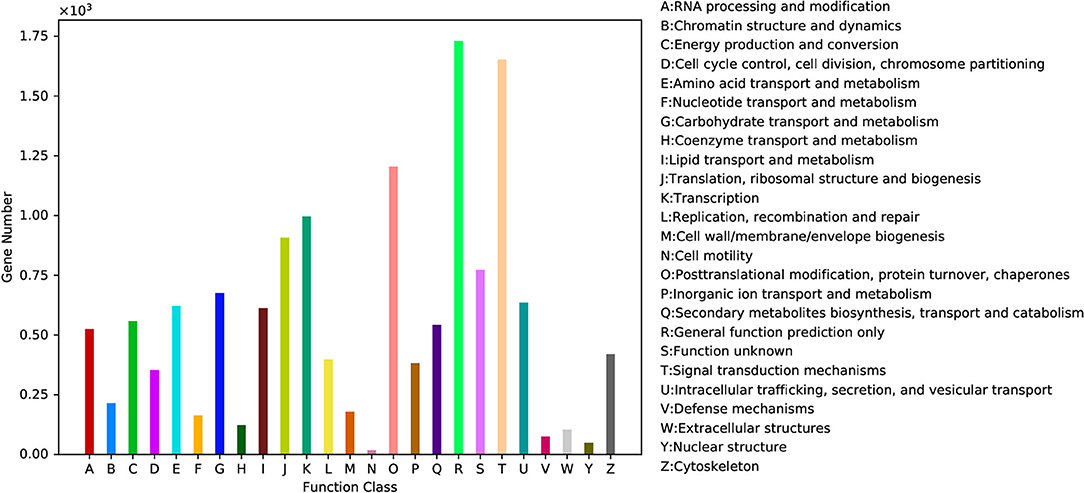
Figure 5. Cluster of KOG classification of the unigenes.
To further identify the predicted proteins' biological pathways, the KEGG pathway database was employed to annotate the unigenes by analyzing the gene functions systematically and integrating the chemical and genomic information (Du et al., 2016; Kanehisa et al., 2016). In our results, 3,170 unigenes were assigned and annotated to 279 different biological pathways (Supplementary Table 3). “Signal transduction” (667 unigenes) was the most enriched pathway, followed by “Translation” (415 unigenes), “Transport and catabolism” (364 unigenes), and “Folding sorting and degradation” (329 unigenes) (Figure 6).
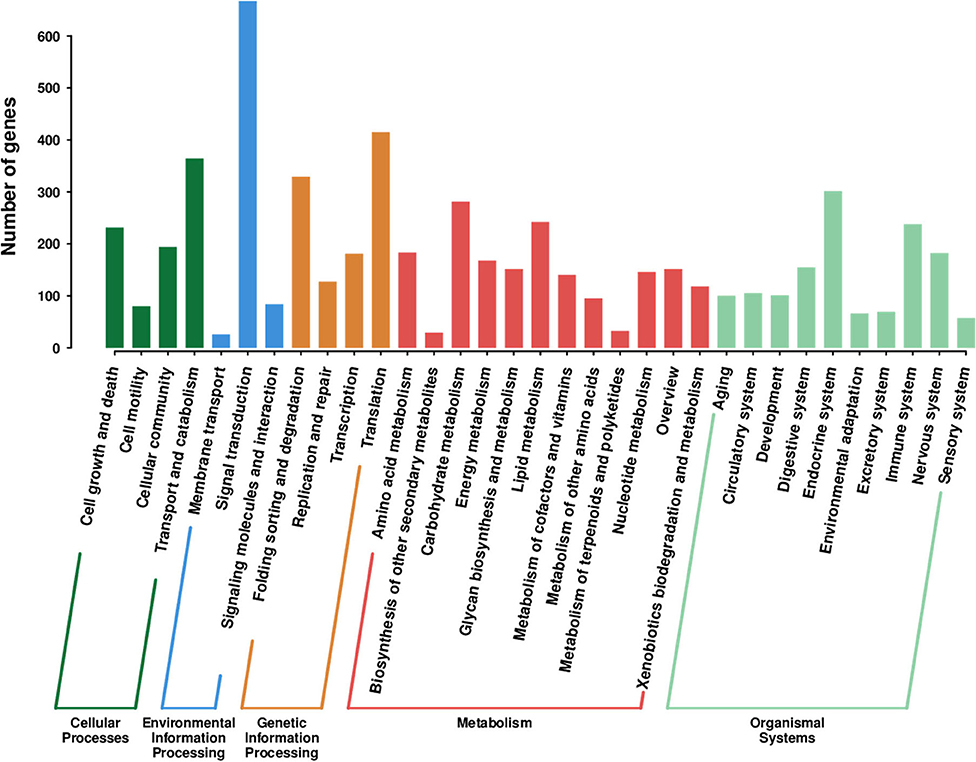
Figure 6. KEGG classifications of unigenes of M. aurolineatus.
CDS Prediction
BLAST alignment was used to search the coding sequences (CDS) in NR, SwissProt, and TrEMBL databases, and the remaining undefined sequences were assigned by using TransDecoder software. A total of 39,876 CDS sequences were predicted in our data (Supplementary Tables 4, 5). After transcript combination, the gene coverage was searched by BEDTools software. From our data, 63,340 genes can be detected in the 90–100% gene distribution rate, followed by 3,314 genes in the 80–90% gene distribution rate, 1,538 genes in the 70–80% gene distribution rate, 867 genes in the 60–70% gene distribution rate, and only few genes in the gene distribution rate under 50% (Supplementary Figure 1).
SSR Discovery
SSRs or microsatellite sequences, represented by 2–6 base pairs in length of core sequences, are polymorphic loci present in genomic DNA (Powell et al., 1996). To date, SSRs have been used to genotype numerous species for functional analysis (Kantety et al., 2002). Our results obtained 13,848 SSRs, including 9.40% dinucleotide repeats, 10.32% trinucleotide repeats, and 1.05% tetra/penta/hexanucleotide repeats. A total of 1,355 SSRs (9.78%) with more than 15 base pairs in length and 1,525 SSRs (11.01%) with <15 base pairs in length were found (Figure 7). Among the dinucleotide repeat motifs, (AT/AT)n (855 SSRs) and (AG/CT)n (271 SSRs) were the most common types and significantly outnumbered the other types of dinucleotide repeat motifs. Among the trinucleotide repeat motifs, (AAT/ATT)n (547 SSRs) and (ACC/GGT)n (197 SSRs) were the most common types (Figure 7). A total of 6,512 unique sequences with microsatellites flanking sequences on both sides of the microsatellites were obtained after removing the microsatellites that lacked sufficient flanking sequences for primer design, which allows for the design of primers for genotyping.
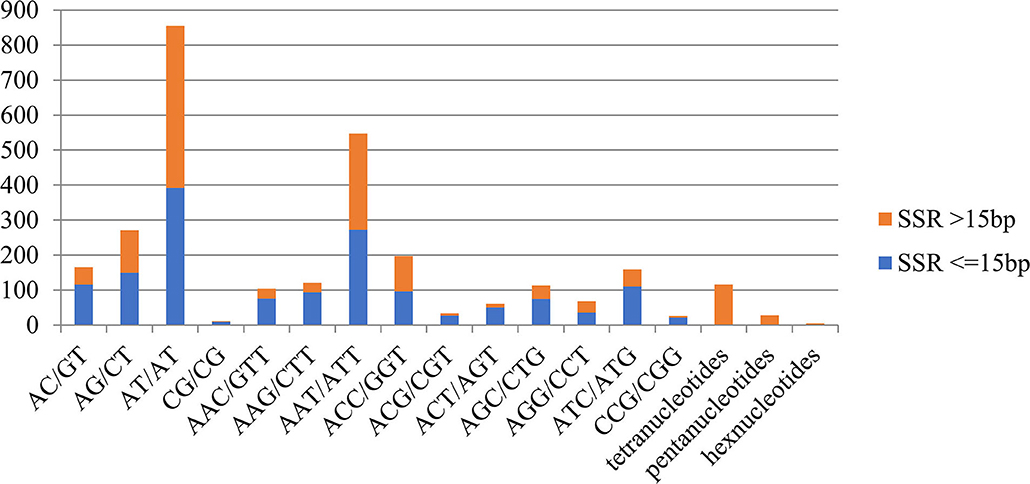
Figure 7. Distribution of simple sequence repeat (SSR) nucleotide classes among different nucleotide types found in the transcriptome of M. aurolineatus.
SNP Identification
The type of variation in the genome was commonly analyzed by using SNPs (Williams et al., 2010; Gimode et al., 2016). SNPs were generated by aligning sequences used for contig assembly. We obtained 96,790 SNPs after removing those that had a base mutation frequency lower than 1% (Supplementary Table 6). The proportions of transition substitutions of A->G, T->C, G->A, and C->T were 16.95% (16,408), 16.50% (15,970), 15.94% (15,430), and 15.82% (15,313), respectively (Figure 8). A total transition:transversion ratio was 1.87:1. A small proportion of transversion-type SNPs and a large proportion of transition-type SNPs exist because of the differences in the numbers of hydrogen bonds and base structures between different bases.
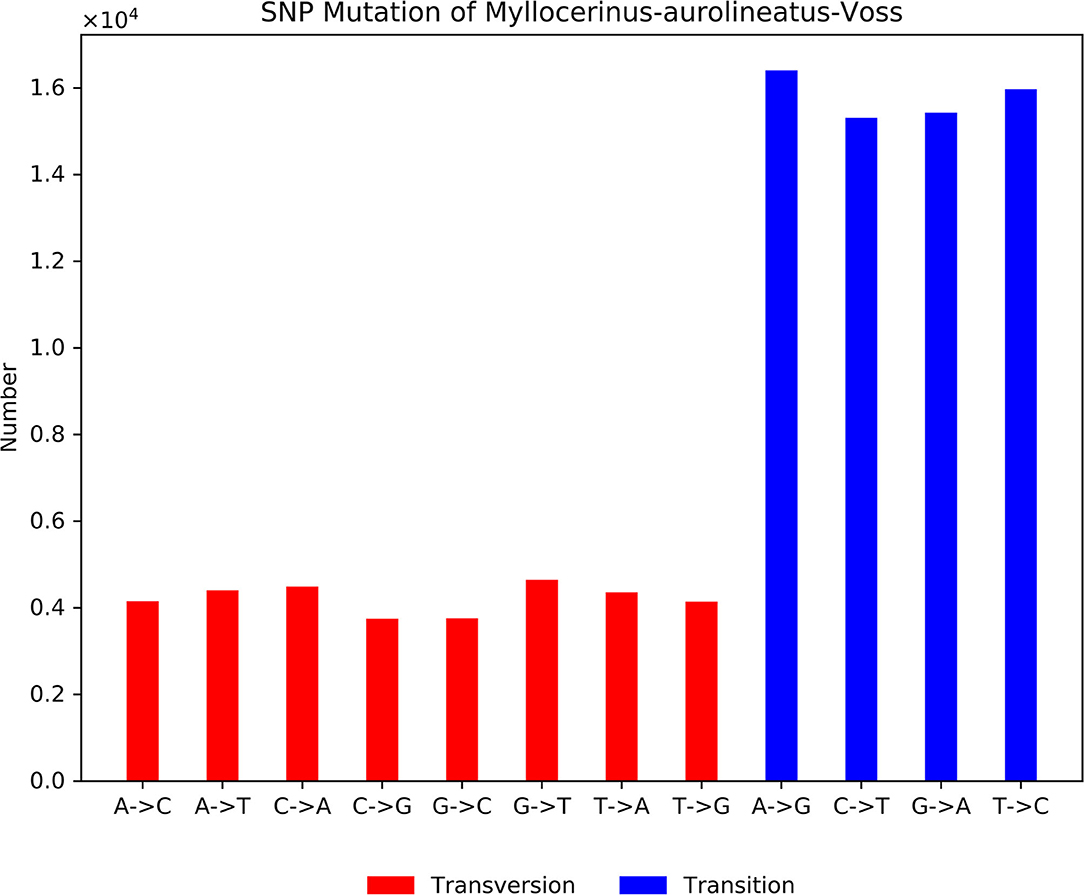
Figure 8. Classification of SNPs identified in the M. aurolineatus.
Conclusion
In the present study, we identified a new pest in tea plants in Guizhou, China and first reported the transcriptome of M. aurolineatus Voss by using de novo assembly technology with next-generation sequencing. We identified 69,439 unigenes, which constitute an important database for future studies on M. aurolineatus gene function analysis. Furthermore, we annotated these unigenes in the GO, KOG, and KEGG databases. In addition, to explore the molecular biology research of M. aurolineatus, we predicted many SSRs and SNPs that will provide a basal foundation for genetic analysis.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: NCBI BioProject ID: PRJNA694293.
Author Contributions
XL: conceptualization. XX: methodology. MC: software. XX: validation. JJ and MH: formal analysis. JJ: investigation. LJ: resources. XX and JJ: writing—original draft preparation. XL: writing—review and editing and project administration. XL and XX: funding acquisition. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Key Research and Development Program of China (Grant No. 2016YFD0200902), the Advanced Programs of Guizhou Province for the Returned Overseas Scholars [(2018)02], the Science and Technology Foundation of Guizhou Province [(2019)2408], the Project of Guizhou Province [No. (2018)5781], and the Ph.D. Funding of Guizhou University (No. 201754).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We sincerely thank the reviewer for the constructive criticisms and valuable comments.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fsufs.2021.631990/full#supplementary-material
Supplementary Figure 1. Distribution of gene's coverage of M. aurolineatus.
Supplementary Table 1. Gene Ontology (GO) classification of proteins in biological process, cellular component, and molecular function process.
Supplementary Table 2. euKaryotic Ortholog Groups (KOG) functional categories of M. aurolineatus proteins.
Supplementary Table 3. The annotation of identified unigenes in different biological pathways.
Supplementary Table 4. The coding sequences (CDS) prediction of identified unigenes.
Supplementary Table 5. The peptide prediction of identified unigenes.
Supplementary Table 6. Single nucleotide polymorphism (SNP) identification of identified unigenes.
References
Bishnoi, R., and Singla, D. (2017). APMicroDB: a microsatellite database of Acyrthosiphon pisum. Genom Data 12, 111–115. doi: 10.1016/j.gdata.2017.03.014
Casida, J. E. (2017). Pesticide interactions: mechanisms, benefits, and risks. J. Agric. Food Chem. 65, 4553–4561. doi: 10.1021/acs.jafc.7b01813
Comblain, F., Dubuc, J. E., Lambert, C., Sanchez, C., Lesponne, I., Serisier, S., et al. (2016). Identification of targets of a new nutritional mixture for osteoarthritis management composed by curcuminoids extract, hydrolyzed collagen and green tea extract. PLoS ONE 11:e0156902. doi: 10.1371/journal.pone.0156902
Du, J., Li, M., Yuan, Z., Guo, M., Song, J., Xie, X., et al. (2016). A decision analysis model for KEGG pathway analysis. BMC Bioinformatics 17:407. doi: 10.1186/s12859-016-1285-1
Ekblom, R., and Galindo, J. (2011). Applications of next generation sequencing in molecular ecology of non-model organisms. Heredity 107, 1–15. doi: 10.1038/hdy.2010.152
Gayral, P., Melo-Ferreira, J., Glemin, S., Bierne, N., Carneiro, M., Nabholz, B., et al. (2013). Reference-free population genomics from next-generation transcriptome data and the vertebrate-invertebrate gap. PLoS Genet. 9:e1003457. doi: 10.1371/journal.pgen.1003457
Gimode, D., Odeny, D. A., de Villiers, E. P., Wanyonyi, S., Dida, M. M., Mneney, E. E., et al. (2016). Identification of SNP and SSR markers in finger millet using next generation sequencing technologies. PLoS ONE 11:e0159437. doi: 10.1371/journal.pone.0159437
Jugulam, M., and Gill, B. S. (2018). Molecular cytogenetics to characterize mechanisms of gene duplication in pesticide resistance. Pest Manag. Sci. 74, 22–29. doi: 10.1002/ps.4665
Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M., and Tanabe, M. (2016). KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 44, 457–462. doi: 10.1093/nar/gkv1070
Kantety, R. V., La Rota, M., Matthews, D. E., and Sorrells, M. E. (2002). Data mining for simple sequence repeats in expressed sequence tags from barley, maize, rice, sorghum and wheat. Plant Mol. Biol. 48, 501–510. doi: 10.1023/A:1014875206165
Kuzniar, A., Lin, K., He, Y., Nijveen, H., Pongor, S., and Leunissen, J. A. (2009). ProGMap: an integrated annotation resource for protein orthology. Nucleic Acids Res. 37, 428–434. doi: 10.1093/nar/gkp462
Li, B., Predel, R., Neupert, S., Hauser, F., Tanaka, Y., Cazzamali, G., et al. (2008). Genomics, transcriptomics, and peptidomics of neuropeptides and protein hormones in the red flour beetle Tribolium castaneum. Genome Res. 18, 113–122. doi: 10.1101/gr.6714008
Li, H., Huang, W., Wang, G. L., Wang, W. L., Cui, X., and Zhuang, J. (2017). Transcriptomic analysis of the biosynthesis, recycling, and distribution of ascorbic acid during leaf development in tea plant (Camellia sinensis (L.) O. Kuntze). Sci Rep. 7:46212. doi: 10.1038/srep46212
Ma, J. Q., Yao, M. Z., Ma, C. L., Wang, X. C., Jin, J. Q., Wang, X. M., et al. (2014). Construction of a SSR-based genetic map and identification of QTLs for catechins content in tea plant (Camellia sinensis). PLoS ONE 9:e93131. doi: 10.1371/journal.pone.0093131
Moon, E. K., Chung, D. I., Hong, Y. C., Ahn, T. I., and Kong, H. H. (2008). Acanthamoeba castellanii: gene profile of encystation by ESTs analysis and KOG assignment. Exp. Parasitol. 119, 111–116. doi: 10.1016/j.exppara.2008.01.001
Mukhopadhyay, M., Mondal, T. K., and Chand, P. K. (2016). Biotechnological advances in tea (Camellia sinensis [L.] O. Kuntze): a review. Plant Cell Rep. 35, 255–287. doi: 10.1007/s00299-015-1884-8
Powell, W., Machray, G. C., and Provan, J. (1996). Polymorphism revealed by simple sequence repeats. Trends Plant Sci. 1, 215–222. doi: 10.1016/S1360-1385(96)86898-0
Regniere, J., and Bentz, B. (2007). Modeling cold tolerance in the mountain pine beetle, Dendroctonus ponderosae. J. Insect Physiol. 53, 559–572. doi: 10.1016/j.jinsphys.2007.02.007
Roy, S., and Muraleedharan, N. (2014). Microbial management of arthropod pests of tea: current state and prospects. Appl. Microbiol. Biotechnol. 98, 5375–5386. doi: 10.1007/s00253-014-5749-9
Smith-Unna, R., Boursnell, C., Patro, R., Hibberd, J. M., and Kelly, S. (2016). TransRate: reference-free quality assessment of de novo transcriptome assemblies. Genome Res. 26, 1134–1144. doi: 10.1101/gr.196469.115
Sun, X., Zhang, X., Wu, G., Li, X., Liu, F., Xin, Z., et al. (2017). n-Pentacosane Acts as both Contact and Volatile Pheromone in the tea Weevil, Myllocerinus aurolineatus. J. Chem. Ecol. 43, 557–562. doi: 10.1007/s10886-017-0857-5
Sun, X. L., Wang, G. C., Cai, X. M., Jin, S., Gao, Y., and Chen, Z. M. (2010). The tea weevil, Myllocerinus aurolineatus, is attracted to volatiles induced by conspecifics. J. Chem. Ecol. 36, 388–395. doi: 10.1007/s10886-010-9771-9
Tarrant, A. M., Baumgartner, M. F., Lysiak, N. S., Altin, D., Storseth, T. R., and Hansen, B. H. (2016). Transcriptional Profiling of Metabolic Transitions during Development and Diapause Preparation in the Copepod Calanus finmarchicus. Integr. Comp. Biol. 56, 1157–1169. doi: 10.1093/icb/icw060
Williams, L. M., Ma, X., Boyko, A. R., Bustamante, C. D., and Oleksiak, M. F. (2010). SNP identification, verification, and utility for population genetics in a non-model genus. BMC Genet. 11:32. doi: 10.1186/1471-2156-11-32
Yang, Z. W., Duan, X. N., Jin, S., Li, X. W., Chen, Z. M., Ren, B. Z., et al. (2013). Regurgitant derived from the tea geometrid Ectropis obliqua suppresses wound-induced polyphenol oxidases activity in tea plants. J. Chem. Ecol. 39, 744–751. doi: 10.1007/s10886-013-0296-x
Zhang, H. Z., Li, Y. Y., An, T., Huang, F. X., Wang, M. Q., Liu, C. X., et al. (2018). Comparative Transcriptome and iTRAQ Proteome Analyses Reveal the Mechanisms of Diapause in Aphidius gifuensis Ashmead (Hymenoptera: Aphidiidae). Front. Physiol. 9:1697. doi: 10.3389/fphys.2018.01697
Keywords: Myllocerinus aurolineatus Voss, transcriptome, pest, tea (Camellia sinensis), identification of pests
Citation: Xie X, Jiang J, Chen M, Huang M, Jin L and Li X (2021) De novo Transcriptome Assembly of Myllocerinus aurolineatus Voss in Tea Plants. Front. Sustain. Food Syst. 5:631990. doi: 10.3389/fsufs.2021.631990
Received: 21 November 2020; Accepted: 25 January 2021;
Published: 01 March 2021.
Edited by:
Wei Li, Institute of Plant Protection (CAAS), ChinaReviewed by:
Xueqing Geng, Shanghai Jiao Tong University, ChinaQun En Liu, China National Rice Research Institute (CAAS), China
Zhiqiang Li, Institute of Plant Protection (CAAS), China
Copyright © 2021 Xie, Jiang, Chen, Huang, Jin and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiangyang Li, xyli1@gzu.edu.cn
†These authors have contributed equally to this work