Profiling Non-motor Symptoms in Monogenic Parkinson’s Disease
 Xinyao Liu1,2
Xinyao Liu1,2  Weidong Le1,2,3*
Weidong Le1,2,3*- 1Liaoning Provincial Center for Clinical Research on Neurological Diseases, The First Affiliated Hospital, Dalian Medical University, Dalian, China
- 2Liaoning Provincial Key Laboratory for Research on the Pathogenic Mechanisms of Neurological Diseases, The First Affiliated Hospital, Dalian Medical University, Dalian, China
- 3Institute of Neurology, Sichuan Academy of Medical Sciences-Sichuan Provincial Hospital, Chengdu, China
Parkinson’s disease (PD) is the second most common neurodegenerative disease in the elder population, pathologically characterized by the progressive loss of dopaminergic neurons in the substantia nigra. While the precise mechanisms underlying the pathogenesis of PD remain unknown, various genetic factors have been proved to be associated with PD. To date, at least 23 loci and 19 disease-causing genes for PD have been identified. Although monogenic (often familial) cases account for less than 5% of all PD patients, exploring the phenotypes of monogenic PD can help us understand the disease pathogenesis and progression. Primary motor symptoms are important for PD diagnosis but only detectable at a relatively late stage. Despite typical motor symptoms, various non-motor symptoms (NMS) including sensory complaints, mental disorders, autonomic dysfunction, and sleep disturbances also have negative impacts on the quality of life in PD patients and pose major challenges for disease management. NMS is common in all stages of the PD course. NMS can occur long before the onset of PD motor symptoms or can present in the middle or late stage of the disease accompanied by motor symptoms. Therefore, the profiling and characterization of NMS in monogenic PD may help the diagnosis and differential diagnosis of PD, which thereby can execute early intervention to delay the disease progression. In this review, we summarize the characteristics, clinical phenotypes, especially the NMS of monogenic PD patients carrying mutations of SNCA, LRRK2, VPS35, Parkin, PINK1, DJ-1, and GBA. The clinical implications of this linkage between NMS and PD-related genes are also discussed.
Introduction
Parkinson’s disease (PD) is the second most common neurodegenerative disease in the elderly population worldwide. In the 15 most populous countries in the world, more than 4 million people over the age of 50 have PD, and this number is expected to double by 2030 (Dorsey et al., 2007). PD reduces patients’ quality of daily life and causes a heavy economic burden on patients, their families, and the whole society (Meissner et al., 2011). Various pathological mechanisms, such as aging, neuroinflammation, abnormal protein aggregation, mitochondrial dysfunction, oxidative stress, and environmental neurotoxins, have been reported to be associated with the progressive loss of dopaminergic neurons in the substantia nigra of PD brain. However, to date, the exact molecular mechanisms underlying the loss of these neurons remain elusive (Hirsch et al., 2013).
The clinical manifestations of PD involve motor and non-motor symptoms (NMS). Motor symptoms include bradykinesia, rigidity, resting tremor, postural and gait instability, etc., while NMS consists of cognitive decline, autonomic dysfunction, depression, anxiety, sleep disorder, and olfactory impairment (Kalia and Lang, 2015). Since the identification of the first PD-related gene SNCA in 1997 (Singleton et al., 2003), at least 23 loci and 19 disease-causing genes for PD have been identified (Deng et al., 2018). According to the recommendations of the Movement Disorder Society Nomenclature of Genetic Movement Disorders, the confirmed forms of monogenic PD with a PARK designation can be categorized into those presenting with: (1) classical PD; (2) early-onset PD but clinically similar to nongenetic PD; and (3) atypical parkinsonism (Marras et al., 2016b). In general, autosomal dominant forms are more frequently associated with a phenotype overlapping idiopathic PD (iPD), and autosomal recessive forms with young-onset parkinsonism similar to iPD or parkinsonism with atypical features (Marras et al., 2016b).
NMS usually occurs years to decades before motor symptoms, indicating the PD pathological process may initiate long before the presence of clinical motor symptoms (Pfeiffer, 2016). At present, most studies in PD are still focusing on the improvement of diagnosis and treatment of motor symptoms. The identification of NMS in PD is of significance for the early management of PD. Prompt identification and treatment of these NMS may improve patients’ quality of life (Huang et al., 2019; Seppi et al., 2019). It is worth noting that relatively few studies have specifically targeted the NMS in monogenic PD. Therefore, this review will summarize the characteristics, clinical phenotypes, especially the NMS of monogenic PD patients bearing mutations in various PD-related genes including SNCA, the leucine-rich repeat kinase 2 (LRRK2), vacuolar protein sorting 35 (VPS35), Parkin, PTEN-induced putative kinase 1 (PINK1), DJ-1 and glucocerebrosidase (GBA). The clinical implications of the linkage between NMS and PD-related genes are also discussed.
PARK1/PARK4-SNCA
In the discovery of monogenic forms of PD, SNCA is the first identified and is likely the most intensively investigated gene, although SNCA mutations are extremely rare in PD patients (up to 1% in different populations) (Singleton et al., 2003). Misfolded α-synuclein is a major constituent of Lewy bodies and Lewy neurites, which are pathological hallmarks of PD. The contribution of α-synuclein to PD is well established, but the exact molecular mechanisms remain obscure (Hirsch et al., 2013).
Compared with iPD, PD patients carrying SNCA mutations are characterized as early-onset, rapid progression, good levodopa response, and obvious NMS (Kasten and Klein, 2013). According to the statistical analysis of 146 SNCA mutation carriers, the proportion of male patients is 54%. Most patients develop symptoms after the age of 40 (Trinh et al., 2018). Gene multiplication seems to be more common in the European and Asian populations (Kasten and Klein, 2013). Patients carrying SNCA mutations often experience bradykinesia and rigidity, while only about 30% of mutation carriers report resting tremor and postural instability (Kasten et al., 2017).
As for the NMS features of SNCA monogenic PD, cognitive decline is the most common symptom, followed by depression, autonomic dysfunction, and other psychotic manifestations (Kasten et al., 2017; Chen et al., 2020). The longitudinal clinical assessments at a 2-year follow-up study conducted by Papadimitriou et al. (2016) have shown that the prominent NMS includes olfactory, autonomic, and cognitive dysfunctions in A53T symptomatic and asymptomatic carriers. SNCA multiplication might have a gene dosage effect concerning dementia. Fuchs et al. (2007) reported a Swedish family with parkinsonism due to duplication of the SNCA gene. Farrer et al. (2004) found early-onset PD and dementia due to triplication of the SNCA gene in a Swedish-American family. The proband began to develop the disease at the age of 31, and the disease progressed rapidly with tremor, stiffness, and bradykinesia; at the age of 45, visual hallucinations and auditory hallucinations and delusions appeared; later, the patient developed an intellectual disability and severe dementia, then died at the age of 52 (Farrer et al., 2004). Dan et al. (2016) showed that SNCA Rep-1 is related to depression in PD through the study of the genotype and depression performance in 1,134 Chinese PD patients. Recently, Rotter et al. (2019) reported that SNCA mRNA expression was positively correlated with the severity of depressive symptoms. The evaluation of dysautonomia including cardiovascular dysfunction was reported in six SNCA mutations and was confirmed on neuroimaging and/or neuropathology (Singleton et al., 2004). Both missense mutations, as well as α-synuclein multiplications, have been associated with various degrees of dysautonomia, including in asymptomatic carriers, preceding the onset of motor symptoms (Singleton et al., 2004; Kara et al., 2014). In familial PD with multiplied SNCA, although the triplications lead to an earlier and more severe phenotype, SNCA duplication may sometimes represent variable degrees of autonomic dysfunction (such as orthostatic hypotension, urinary incontinence, severe constipation, etc) (Kruger et al., 2001). Another study showed that hyposmia is associated with rapid eye movement sleep behavior disorder (RBD) in PD patients carrying SNCA mutations (Li et al., 2017). A recent study by Carmona-Abellan et al. (2019) found that E46K-SNCA carriers had moderate to severe p-α-synuclein deposits and small-fiber neurodegeneration including nerve fascicles and glands in epidermal and dermal layers. The severity of the latter skin abnormalities in E46K-SNCA was correlated with sudomotor dysfunction in hands (Carmona-Abellan et al., 2019). Tambasco et al. (2016) summarized the NMS in SNCA point mutations (PMc), duplication (Dc), and triplication (Tc) carriers. They found that among the reported 137 cases, depression is more common in Tc (69.0%) and Dc (35.0%) than in PMc (13.2%) (Tambasco et al., 2016). The frequency of anxiety (Tc 3.4%, Dc 5.0%, and PMc 5.9%) and sleep disturbances (Tc 20.7%, Dc 17.3%, and PMc 10.3%), usually presenting as RBD, manifest with a similar frequency for all 3 groups (Tambasco et al., 2016). Also, 69% of Tc, 42.5% Dc, and 25% of PMc have cognitive impairment (Tambasco et al., 2016). Furthermore, psychosis is more frequent among patients with Tc (69.0%) than those with either Dc (40%) or PMc (7.3%) (Tambasco et al., 2016). Gastrointestinal tract disturbances and urinary dysfunctions are more frequent in Tc (24.1%) than in Dc (12.5%) and PMc (10.3%) (Tambasco et al., 2016). Postural hypotension is present in 37.9% of Tc, 25% of Dc, and 17.6% of PMc in general; but the E46K and G51D PMc have high incidences of postural hypotension and falls, 44.4% and 37.5%, respectively. Hyposmia is less common (6.9%-22.2%) in PD patients carrying SNCA mutations (Tambasco et al., 2016).
PARK8-LRRK2
In autosomal dominant PD, mutations in the LRRK2 gene are the most common cause of monogenic PD (Paisán-Ruiz et al., 2004). Autosomal dominant missense mutations in the LRRK2 gene account for 1% to 2% of all PD cases, a higher proportion among Ashkenazi Jews and North African Berbers (Paisán-Ruiz et al., 2013). Currently, the detailed function and pathogenicity of LRRK2 in PD are not fully understood. Rab GTPase family members are cellular physiological substrates of LRRK2 kinase (Jeong et al., 2018). Studies of the relationship between LRRK2 and Rab protein members Rab7, Rab5B, and Rab29 have demonstrated that LRRK2 pathogenic mutations dysregulate vesicle trafficking (Shin et al., 2008; Dodson et al., 2012; MacLeod et al., 2013). Many Rab proteins including Rab3A/B/C/D, Rab8A/B, Rab10, Rab12, Rab35, Rab43, Rab5B/C, and Rab29 are the substrates of LRRK2 kinase (Steger et al., 2017). LRRK2-mediated Rab35 phosphorylation positively regulates α-synuclein propagation (Bae et al., 2018). LRRK2 also affects mitochondrial morphology and function and regulates autophagy, both etiologic factors in PD (Rosenbusch and Kortholt, 2016; West and Cookson, 2016). Moreover, recent studies have further demonstrated a higher level of LRRK2 in immune cells, including circulating B lymphocytes, dendritic cells, and macrophages, suggesting the involvement of LRRK2 in the immune system (Hakimi et al., 2011). More than 40 mutations in LRRK2 have been found, of which eight (N1437H, R1441C/G/H/S, Y1699C, G2019S, and I2020T) cause PD and A419V, R1628P, M1646T and G2385R are considered risk factors for PD (Heckman et al., 2013; Li et al., 2015; Mata et al., 2017; Alessi and Sammler, 2018; De Wit et al., 2018).
Of all the LRRK2 mutations, the best-described one is G2019S, with a penetration rate from 28% at the age of 59 to 74% at the age of 79, which is indistinguishable from that of iPD (Aasly et al., 2005; Healy et al., 2008). LRRK2 mutations typically manifest as late-onset PD, and the LRRK2 carriers have a good response to levodopa (Deng et al., 2005). PD patients with LRRK2 mutations often show postural instability gait difficulty phenotype compared with noncarriers, and progress at a rate similar to iPD in a longitudinal study (Nabli et al., 2015). The prospective longitudinal follow-up study of PD patients with or without the LRRK2 G2019S mutation by Saunders-Pullman et al. (2018) from the cross-sectional study, shows a slower decline in motor UPDRS scores among those PD patients with LRRK2 G2019S.
LRRK2 monogenic PD patients have fewer NMS than iPD. vs. iPD patients, monogenic PD patients carrying LRRK2-G2019S mutation have relatively better performance in attention, executive function, and language domains (Alcalay et al., 2015b). In other studies, it is reported that patients with LRRK2 monogenic PD also perform better than iPD in terms of cognitive ability, smell, sleep, and emotion (Marras et al., 2011, 2016a; Ben Sassi et al., 2012; Saunders-Pullman et al., 2014, 2015). But dementia also has been reported in PD patients carrying LRRK2 mutation (Tomiyama et al., 2006). Lohmann et al. (2009) reported a comparable rate of depression between PD patients carrying G2019S mutation and unaffected G2019S-carriers, and the prevalence of anxiety is similar between patients with iPD and those G2019S-carriers. However, mice expressing human LRRK2 G2019S exhibit mild cognitive impairment earlier than motor dysfunctions (Adeosun et al., 2017). Higher UPDRS motor scores, more frequent urinary problems, and fewer hours of sleep are found in mutation carriers compared to non-carriers (Johansen et al., 2011). The mutation carriers with UPDRS ≥8 are all aged over 50 years and have shorter overall sleeping hours, more frequent urinary and constipation problems, as well as higher mood scores and body mass index (Johansen et al., 2011). Furthermore, RBD, based on questionnaires or polysomnography, appears to be uncommon in LRRK2 mutation carriers (Ehrminger et al., 2015; Saunders-Pullman et al., 2015).
PARK17-VPS35
The PD-related mutations of the VPS35 gene were firstly identified in 2011 (Vilarino-Guell et al., 2011). In 2012, a large multi-center study involving 15,000 individuals worldwide estimated that PARK-VPS35 mutations account for approximately 0.4% of PD (Sharma et al., 2012). PARK-VPS35 monogenic PD has high heritability and low penetrance, and its phenotype is very similar to that of iPD, but the average age of onset is 50 years old (Sharma et al., 2012). In an autosomal dominant PD population study in Japan, VPS35 mutation is also very rare, and the phenotype of motor symptoms in this study is prominent tremor (Ando et al., 2012). VPS35 is a key molecule in the retromer complex within the cell in terms of regulating endosomal trafficking (Cutillo et al., 2020). However new data suggest that VPS35 also regulates mitochondrial dynamics and homeostasis (Cutillo et al., 2020). Also, VPS35 is a critical player in pathways connected to α-synuclein accumulation and clearance (Cutillo et al., 2020).
The NMS profile in VPS35 monogenic PD is similar to that in iPD, including hyposmia, rare mild cognitive impairment, and rare neuropsychiatric features (Struhal et al., 2014). The autonomic manifestations of VPS35 monogenic PD, such as orthostatic hypotension and constipation, have also been reported (Yamaguchi et al., 2005).
PARK2-Parkin
Parkin mutations are the most common cause of autosomal recessive PD, and the median age at onset is 31 years (Bruggemann and Klein, 1993). Parkin is an E3 ubiquitin ligase protein that catalyzes the transfer of ubiquitin to its specific target protein (Narendra et al., 2012). In one meta-analysis of early-onset PD, Kilarski et al. (2012) found a familial mutation frequency of 15.5%, while the mutation rate in the sporadic was 4.3%. At least 60 Parkin mutations and variants have been identified, which may vary with specific mutations and presents a major challenge in determining pathogenicity (Hedrich et al., 2004). Also, deletions and duplications of Parkin are particularly common, which complicates Parkin genotyping (Klein et al., 2000).
The clinical characteristics of Parkin monogenic PD are early-onset and slow progression with a good response to dopaminergic treatment, but are usually complicated by dystonia and prominent freezing of gait, with sleep benefit on most symptoms (Ishikawa and Tsuji, 1996; Grunewald et al., 2013).
Overall, NMS appears to be less severe in patients with Parkin monogenic PD than in iPD. Compared with iPD or PD patients with Parkin heterozygotes, PD patients with Parkin homozygotes and compound heterozygotes may have less olfactory dysfunction (Khan et al., 2004). A follow-up study (Consortium on Risk for Early-Onset PD) examined the cognitive function in early-onset PD with the long-duration disease (>14 years) and found that Parkin carriers performed better on the Mini-Mental State Examination, clinical dementia ratings, attention, memory, and visuospatial performance than noncarriers (Alcalay et al., 2014). Psychosis has been rarely reported in Parkin carriers, and if occurs it is usually before motor symptom onset. Psychotic symptoms are usually related to dopamine replacement therapy (Khan et al., 2003; Kim et al., 2014). However, Parkin carriers may have more serious impulse control disorders, frequent compulsive shopping, overeating, and punding/hobbyism (Morgante et al., 2016). The score of the Questionnaire for Impulsive-Compulsive Disorders in PD–Rating Scale in patients with parkin-PD is higher than nonmutated PD patients (Morgante et al., 2016). In some other studies, the frequency of RBD in Parkin-related PD is no different from iPD. However, Parkin carriers are more likely to have restless leg syndrome than iPD (Kumru et al., 2004; Limousin et al., 2009). Tijero et al. (2015) found that the rate of autonomic symptoms in Parkin-related PD is much lower than iPD, and the heart noradrenergic innervation is relatively preserved.
PARK6-PINK1
Mutations in the PINK1 gene are the second most common cause of autosomal recessive early-onset PD after Parkin (Valente et al., 2004a). According to previous reports, the frequency of PINK1 mutations in sporadic early-onset PD is 4% to 7% (Valente et al., 2004b; Bonifati et al., 2005). PINK1 is a serine/threonine kinase containing key regulatory sites, which activates ubiquitin by phosphorylation and cooperates with the downstream ubiquitin ligase PARKIN, to exert quality control and regulate autophagic degradation of mitochondria and misfolded proteins in all cell types (Valente et al., 2001; Truban et al., 2017). Recent research shows PINK1 deficiency as an early modulator of innate immunity in neurons, which precedes the late stages of neuroinflammation during α-synuclein spreading (Torres-Odio et al., 2017).
PINK1 monogenic PD progresses slowly with a 32-year average age of onset, good levodopa response, and sleep benefit. The most common motor symptom of PINK1 monogenic PD is bradykinesia followed by rigidity (Valente et al., 2004b; Bonifati et al., 2005). The PINK1 monogenic PD patients occasionally have pyramidal signs or hyperreflexia (Kasten et al., 2018).
It has been reported that psychiatric symptoms such as anxiety and depression are more prominent in PINK1-linked PD (Ephraty et al., 2007). According to a recent study, the incidence of cognitive dysfunction is higher in PINK1 mutations (29.4%), and executive function and attention are most affected (Piredda et al., 2020). Regarding the mental symptoms, depression is the most common one (37.5%; Piredda et al., 2020). In a research survey of 20 members of a family (four homozygous, 11 heterozygous, and five non-mutation carriers), Steinlechner et al. (2007) found that 61% mutation carriers and 20% non-mutation carriers have predominantly affective and schizophrenia spectrum disorders. Affective and psychotic symptoms may be part of the phenotypic spectrum or even the sole manifestation of patients with PINK1 mutations (Funayama et al., 2008). Hyposmia appears to be a common NMS in patients with PINK1 monogenic PD (Ferraris et al., 2009). A review of the clinical phenotype of PINK1-related PD cases confirm that autonomic symptoms are not common; among them, the urinary dysfunction consists of 44%, and orthostatic hypotension consists of 22% (Chaudhuri et al., 2006). Ricciardi et al. (2014) conducted a 12-year follow-up study in 5 PINK1 homozygous and 14 heterozygous mutation carriers from two large Italian families. They found all but one patient has sleep impairment, impulse control disorders, anxiety, apathy, and global cognitive impairment (Ricciardi et al., 2014).
PARK7-DJ-1
The mutation in DJ-1 was firstly identified in two close relative families from the Netherlands and Italy (Bonifati et al., 2003), with a very low frequency of mutation. DJ-1 mutation accounts for less 1% of early-onset PD (Sironi et al., 2013). The meta-analysis by Kilarski et al. (2012) found that the overall mutation frequency of DJ-1 was 0.4%, which was slightly higher in familial PD (0.8%) than sporadic PD (0.4%). The median age of disease onset is 27 years (Weissbach et al., 2019).
The clinical manifestations of patients with DJ-1 mutations are similar to those with Parkin or PINK1 mutations. However, the NMS in PD patients carrying DJ-1 mutation is more prominent, including mental disorders and cognitive decline (Kilarski et al., 2012; Kasten et al., 2018). Dystonia is particularly common (16/22; 73%) in DJ-1 mutation carriers, as well as the postural tremor (8/12; 67%), and psychotic signs (8/11; 73%; Weissbach et al., 2019). All but one of the described patients suffer from depression, suggesting that psychotic features may be particularly common among DJ-1 mutations carriers and may be more common than in iPD (Weissbach et al., 2019).
GBA
It is often presented as Gaucher’s disease (GD) with PD in an aggressive phenotype, in which both homozygous and heterozygous GBA mutations seem to be prone to PD (Zhao et al., 2016). GBA mutations become the most important genetic susceptibility factor for PD (Zhao et al., 2016). Multiple studies have established a significantly increased risk of PD associated with GBA mutations (Sidransky et al., 2009; Huang et al., 2011). Studies have shown that PD cases with GBA mutations are on average 5 years younger than iPD (Clark et al., 2007; Sidransky et al., 2009). Penetrance is incomplete and increases with age (Gan-Or et al., 2015a). GBA encodes the lysosomal enzyme glucocerebrosidase (GCase), which cleaves the β-glucosyl linkage of glucosylceramide and glucosylsphingosine (Beavan and Schapira, 2013). Alcalay et al. (2015a) found that lower GCase activity may be associated with a faster course of disease progression.
Since glucosylceramide, the substrate of GCase, is mainly stored in cells of the reticuloendothelial system, macrophages carrying matrix-filled lysosomes (the classic cell marker of GD) accumulate in the spleen and liver when GBA is mutated. Organ enlargement and inflammation occur, and sometimes the nervous system is also involved. According to the clinical progress of GD and the speed of nervous system involvement, GD is described as three types (Grabowski, 2008). Type I GD is classically defined as a non-neurological pathology (the N370 mutation is the most common) with its typical features of hepatosplenomegaly, bone and hematopoietic system abnormalities (Bultron et al., 2010). Type II and type III GD (type L444P mutation is the most common, point mutations or complex allelic mutations can occur) can be distinguished from type I GD by whether there is any neurodegeneration in the central nervous system. While the development of Type II GD is fast, the development of Type III GD is slow (Mazzulli et al., 2011).
The characteristic clinical phenotype of GBA monogenic PD is similar to those of sporadic PD, but the onset is earlier and the course is more severe (Brockmann et al., 2015; Ryan et al., 2019). Furthermore, PD patients with GBA mutations usually have an earlier and more frequent occurrence of NMS (Petrucci et al., 2020). It is worth noting that the NMS in PD patients carrying GBA mutations seems to be more prominent than iPD (Sidransky et al., 2009). Studies have shown that GBA mutations can cause more serious cognitive impairments, characterized by greater declines in working memory, executive function, and visual-spatial ability (Mata et al., 2016). Previous studies suggested that 45% of GBA monogenic patients have visual hallucinations unrelated to dopamine therapy (Neumann et al., 2009). However, a high frequency (78%) of visual hallucinations under dopamine replacement therapy has also been reported in GBA, higher than that of LRRK2 (38%) or iPD (53%) (Wang et al., 2014). Similar results have been found in other studies, with rates of psychosis in GBA patients receiving dopamine replacement therapy ranging between 46% and 53% (Oeda et al., 2015; Yahalom et al., 2019). Finally, a recent meta-analysis revealed that GBA is associated with a 1.83-fold increase in the risk of mental illness (Creese et al., 2018). After onset, GBA has visual hallucinations, often accompanied by cognitive decline or dementia (Neumann et al., 2009; Oeda et al., 2015). The prevalence of depression (33%) in PD patients with GBA mutation, especially in male patients, is also significantly higher than that in iPD (13%) (Swan et al., 2016). The impact of GBA mutation on olfactory in PD is similar to that in iPD (Alcalay et al., 2012), whereas autonomic dysfunction may be more common (Brockmann et al., 2011). Also, GBA mutations are associated with RBD in PD patients (Gan-Or et al., 2015b). Thaler et al. (2017) recently reported a gene-dose effect, in which homozygotes and compound heterozygotes have earlier disease onset and more severe motor and NMS compared with both heterozygotes and iPD.
Conclusion
The exploration of PD has never stopped in recent years. Although gene mutations only exist in a small portion of PD patients, genetic studies have greatly promoted the biggest advances in PD research. In the observational study of PD, the clinical phenotype of different gene mutations has different characteristics. NMS is very common and usually appear earlier than motor symptoms of PD. Different gene mutations are also characterized by various NMS. Such as PD patients with SNCA mutations have a relatively common cognitive decline. LRRK2 mutations in late-onset PD present less NMS than iPD with a similar rate of progression. NMS in patients with PARKIN mutations are not as severe as iPD. The mental symptoms of PINK1 monogenic PD are more frequent, including anxiety and depression. NMS in DJ-1 monogenic PD is more prominent, including depression, anxiety, and other mental illness and cognitive decline. The PD patients carrying GBA mutations usually present with a high frequency of cognitive disorders, such as impaired working memory, executive function, and visual-spatial ability. Also, the incidence of anxiety and depression is high, and autonomic dysfunction may be more severe in GBA monogenic PD (Table 1). The profiling and characteristics of NMS in monogenic PD should be helpful for the early diagnosis and clinical management of the disease.
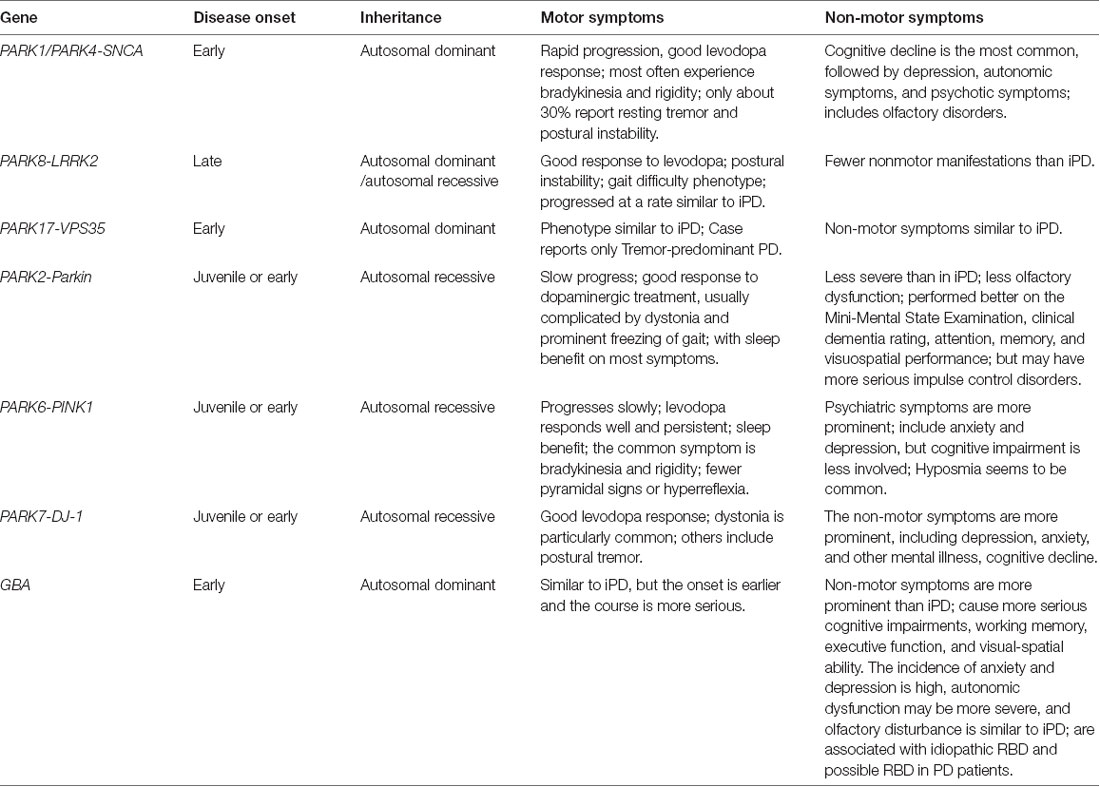
Table 1. Phenotypes of different monogenic Parkinson’s disease with motor and non-motor symptoms.
Author Contributions
XL and WL contributed to the review of the available literature. XL drafted the manuscript. WL revised the text to the final form. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the funding from the National Natural Science Foundation of China (NSFC 81771521) and Guangdong Provincial Key R & D Program 20188030337001.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
My deepest gratitude goes first and foremost to Professor WL for his guidance and help in the topic selection of this article. Second, I would like to express my heartfelt gratitude to Professor Song Li, who was a great help in writing and revising my article.
References
Aasly, J. O., Toft, M., Fernandez-Mata, I., Kachergus, J., Hulihan, M., White, L. R., et al. (2005). Clinical features of LRRK2-associated Parkinson’s disease in central Norway. Ann. Neurol. 57, 762–765. doi: 10.1002/ana.20456
Adeosun, S. O., Hou, X., Zheng, B., Melrose, H. L., Mosley, T., and Wang, J. M. (2017). Human LRRK2 G2019S mutation represses post-synaptic protein PSD95 and causes cognitive impairment in transgenic mice. Neurobiol. Learn. Mem. 142, 182–189. doi: 10.1016/j.nlm.2017.05.001
Alcalay, R. N., Caccappolo, E., Mejia-Santana, H., Tang, M., Rosado, L., Orbe Reilly, M., et al. (2012). Cognitive performance of GBA mutation carriers with early-onset PD: the CORE-PD study. Neurology 78, 1434–1440. doi: 10.1212/WNL.0b013e318253d54b
Alcalay, R. N., Caccappolo, E., Mejia-Santana, H., Tang, M. X., Rosado, L., Orbe Reilly, M., et al. (2014). Cognitive and motor function in long-duration PARKIN-associated Parkinson disease. JAMA Neurol. 71, 62–67. doi: 10.1001/jamaneurol.2013.4498
Alcalay, R. N., Levy, O. A., Waters, C. C., Fahn, S., Ford, B., Kuo, S. H., et al. (2015a). Glucocerebrosidase activity in Parkinson’s disease with and without GBA mutations. Brain 138, 2648–2658. doi: 10.1093/brain/awv179
Alcalay, R. N., Mejia-Santana, H., Mirelman, A., Saunders-Pullman, R., Raymond, D., Palmese, C., et al. (2015b). Neuropsychological performance in LRRK2 G2019S carriers with Parkinson’s disease. Parkinsonism Relat. Disord. 21, 106–110. doi: 10.1016/j.parkreldis.2014.09.033
Alessi, D. R., and Sammler, E. (2018). LRRK2 kinase in Parkinson’s disease. Science 360, 36–37. doi: 10.1126/science.aar5683
Ando, M., Funayama, M., Li, Y., Kashihara, K., Murakami, Y., Ishizu, N., et al. (2012). VPS35 mutation in Japanese patients with typical Parkinson’s disease. Mov. Disord. 27, 1413–1417. doi: 10.1002/mds.25145
Bae, E.-J., Kim, D.-K., Kim, C., Mante, M., Adame, A., Rockenstein, E., et al. (2018). LRRK2 kinase regulates alpha-synuclein propagation via RAB35 phosphorylation. Nat. Commun. 9:3465. doi: 10.1038/s41467-018-05958-z
Beavan, M. S., and Schapira, A. H. (2013). Glucocerebrosidase mutations and the pathogenesis of Parkinson disease. Ann. Med. 45, 511–521. doi: 10.3109/07853890.2013.849003
Ben Sassi, S., Nabli, F., Hentati, E., Nahdi, H., Trabelsi, M., Ben Ayed, H., et al. (2012). Cognitive dysfunction in Tunisian LRRK2 associated Parkinson’s disease. Parkinsonism Relat. Disord. 18, 243–246. doi: 10.1016/j.parkreldis.2011.10.009
Bonifati, V., Rizzu, P., van Baren, M. J., Schaap, O., Breedveld, G. J., Krieger, E., et al. (2003). Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 299, 256–259. doi: 10.1126/science.1077209
Bonifati, V., Rohe, C. F., Breedveld, G. J., Fabrizio, E., De Mari, M., Tassorelli, C., et al. (2005). Early-onset parkinsonism associated with PINK1 mutations: frequency, genotypes, and phenotypes. Neurology 65, 87–95. doi: 10.1212/01.wnl.0000167546.39375.82
Brockmann, K., Srulijes, K., Hauser, A. K., Schulte, C., Csoti, I., Gasser, T., et al. (2011). GBA-associated PD presents with nonmotor characteristics. Neurology 77, 276–280. doi: 10.1212/WNL.0b013e318225ab77
Brockmann, K., Srulijes, K., Pflederer, S., Hauser, A. K., Schulte, C., Maetzler, W., et al. (2015). GBA-associated Parkinson’s disease: reduced survival and more rapid progression in a prospective longitudinal study. Mov. Disord. 30, 407–411. doi: 10.1002/mds.26071
Bruggemann, N., and Klein, C. (1993). “Parkin type of early-onset Parkinson disease,” in GeneReviews((R)), eds M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. H. Bean, K. Stephens and A. Amemiya (Seattle, WA: University of Washington).
Bultron, G., Kacena, K., Pearson, D., Boxer, M., Yang, R., Sathe, S., et al. (2010). The risk of Parkinson’s disease in type 1 Gaucher disease. J. Inherit. Metab. Dis. 33, 167–173. doi: 10.1007/s10545-010-9055-0
Carmona-Abellan, M., Gabilondo, I., Murueta-Goyena, A., Khurana, V., Tijero, B., Luquin, M. R., et al. (2019). Small fiber neuropathy and phosphorylated alpha-synuclein in the skin of E46K-SNCA mutation carriers. Parkinsonism Relat. Disord. 65, 139–145. doi: 10.1016/j.parkreldis.2019.05.038
Chaudhuri, K. R., Martinez-Martin, P., Schapira, A. H., Stocchi, F., Sethi, K., Odin, P., et al. (2006). International multicenter pilot study of the first comprehensive self-completed nonmotor symptoms questionnaire for Parkinson’s disease: the NMSQuest study. Mov. Disord. 21, 916–923. doi: 10.1002/mds.20844
Chen, Y., Gu, X., Ou, R., Zhang, L., Hou, Y., Liu, K., et al. (2020). Evaluating the role of SNCA, LRRK2, and GBA in chinese patients with early-onset Parkinson’s disease. Mov. Disord. doi: 10.1002/mds.28191 [Epub ahead of print].
Clark, L. N., Ross, B. M., Wang, Y., Mejia-Santana, H., Harris, J., Louis, E. D., et al. (2007). Mutations in the glucocerebrosidase gene are associated with early-onset Parkinson disease. Neurology 69, 1270–1277. doi: 10.1212/01.wnl.0000276989.17578.02
Creese, B., Bell, E., Johar, I., Francis, P., Ballard, C., and Aarsland, D. (2018). Glucocerebrosidase mutations and neuropsychiatric phenotypes in Parkinson’s disease and Lewy body dementias: review and meta-analyses. Am. J. Med. Genet. B Neuropsychiatr. Genet. 177, 232–241. doi: 10.1002/ajmg.b.32549
Cutillo, G., Simon, D. K., and Eleuteri, S. (2020). VPS35 and the mitochondria: connecting the dots in Parkinson’s disease pathophysiology. Neurobiol. Dis. 145:105056. doi: 10.1016/j.nbd.2020.105056
Dan, X., Wang, C., Zhang, J., Gu, Z., Zhou, Y., Ma, J., et al. (2016). Association between common genetic risk variants and depression in Parkinson’s disease: a dPD study in Chinese. Parkinsonism Relat. Disord. 33, 122–126. doi: 10.1016/j.parkreldis.2016.09.029
De Wit, T., Baekelandt, V., and Lobbestael, E. (2018). LRRK2 phosphorylation: behind the scenes. Neuroscientist 24, 486–500. doi: 10.1177/1073858418756309
Deng, H., Le, W., Guo, Y., Hunter, C. B., Xie, W., and Jankovic, J. (2005). Genetic and clinical identification of Parkinson’s disease patients with LRRK2 G2019S mutation. Ann. Neurol. 57, 933–934. doi: 10.1002/ana.20510
Deng, H., Wang, P., and Jankovic, J. (2018). The genetics of Parkinson disease. Ageing Res. Rev. 42, 72–85. doi: 10.1016/j.arr.2017.12.007
Dodson, M. W., Zhang, T., Jiang, C., Chen, S., and Guo, M. (2012). Roles of the Drosophila LRRK2 homolog in Rab7-dependent lysosomal positioning. Hum. Mol. Genet. 21, 1350–1363. doi: 10.1093/hmg/ddr573
Dorsey, E. R., Constantinescu, R., Thompson, J. P., Biglan, K. M., Holloway, R. G., Kieburtz, K., et al. (2007). Projected number of people with Parkinson disease in the most populous nations, 2005 through 2030. Neurology 68, 384–386. doi: 10.1212/01.wnl.0000247740.47667.03
Ehrminger, M., Leu-Semenescu, S., Cormier, F., Corvol, J. C., Vidailhet, M., Debellemaniere, E., et al. (2015). Sleep aspects on video-polysomnography in LRRK2 mutation carriers. Mov. Disord. 30, 1839–1843. doi: 10.1002/mds.26412
Ephraty, L., Porat, O., Israeli, D., Cohen, O. S., Tunkel, O., Yael, S., et al. (2007). Neuropsychiatric and cognitive features in autosomal-recessive early parkinsonism due to PINK1 mutations. Mov. Disord. 22, 566–569. doi: 10.1002/mds.21319
Farrer, M., Kachergus, J., Forno, L., Lincoln, S., Wang, D. S., Hulihan, M., et al. (2004). Comparison of kindreds with parkinsonism and alpha-synuclein genomic multiplications. Ann. Neurol. 55, 174–179. doi: 10.1002/ana.10846
Ferraris, A., Ialongo, T., Passali, G. C., Pellecchia, M. T., Brusa, L., Laruffa, M., et al. (2009). Olfactory dysfunction in Parkinsonism caused by PINK1 mutations. Mov. Disord. 24, 2350–2357. doi: 10.1002/mds.22816
Fuchs, J., Nilsson, C., Kachergus, J., Munz, M., Larsson, E. M., Schule, B., et al. (2007). Phenotypic variation in a large Swedish pedigree due to SNCA duplication and triplication. Neurology 68, 916–922. doi: 10.1212/01.wnl.0000254458.17630.c5
Funayama, M., Li, Y., Tsoi, T. H., Lam, C. W., Ohi, T., Yazawa, S., et al. (2008). Familial Parkinsonism with digenic parkin and PINK1 mutations. Mov. Disord. 23, 1461–1465. doi: 10.1002/mds.22143
Gan-Or, Z., Amshalom, I., Kilarski, L. L., Bar-Shira, A., Gana-Weisz, M., Mirelman, A., et al. (2015a). Differential effects of severe vs. mild GBA mutations on Parkinson disease. Neurology 84, 880–887. doi: 10.1212/WNL.0000000000001315
Gan-Or, Z., Mirelman, A., Postuma, R. B., Arnulf, I., Bar-Shira, A., Dauvilliers, Y., et al. (2015b). GBA mutations are associated with rapid eye movement sleep behavior disorder. Ann. Clin. Transl. Neurol. 2, 941–945. doi: 10.1002/acn3.228
Grabowski, G. A. (2008). Phenotype, diagnosis, and treatment of Gaucher’s disease. Lancet 372, 1263–1271. doi: 10.1016/S0140-6736(08)61522-6
Grunewald, A., Kasten, M., Ziegler, A., and Klein, C. (2013). Next-generation phenotyping using the parkin example: time to catch up with genetics. JAMA Neurol. 70, 1186–1191. doi: 10.1001/jamaneurol.2013.488
Hakimi, M., Selvanantham, T., Swinton, E., Padmore, R. F., Tong, Y., Kabbach, G., et al. (2011). Parkinson’s disease-linked LRRK2 is expressed in circulating and tissue immune cells and upregulated following recognition of microbial structures. J. Neural Transm. 118, 795–808. doi: 10.1007/s00702-011-0653-2
Healy, D. G., Falchi, M., O’Sullivan, S. S., Bonifati, V., Durr, A., Bressman, S., et al. (2008). Phenotype, genotype and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: a case-control study. Lancet Neurol. 7, 583–590. doi: 10.1016/S1474-4422(08)70117-0
Heckman, M. G., Soto-Ortolaza, A. I., Aasly, J. O., Abahuni, N., Annesi, G., Bacon, J. A., et al. (2013). Population-specific frequencies for LRRK2 susceptibility variants in the genetic epidemiology of Parkinson’s disease (GEO-PD) consortium. Mov. Disord. 28, 1740–1744. doi: 10.1002/mds.25600
Hedrich, K., Eskelson, C., Wilmot, B., Marder, K., Harris, J., Garrels, J., et al. (2004). Distribution, type and origin of Parkin mutations: review and case studies. Mov. Disord. 19, 1146–1157. doi: 10.1002/mds.20234
Hirsch, E. C., Jenner, P., and Przedborski, S. (2013). Pathogenesis of Parkinson’s disease. Mov. Disord. 28, 24–30. doi: 10.1002/mds.25032
Huang, X., Ng, S. Y.-E., Chia, N.-S., Setiawan, F., Tay, K.-Y., Au, W.-L., et al. (2019). Non-motor symptoms in early Parkinson’s disease with different motor subtypes and their associations with quality of life. Eur. J. Neurol. 26, 400–406. doi: 10.1111/ene.13803
Huang, C.-L., Wu-Chou, Y.-H., Lai, S.-C., Chang, H.-C., Yeh, T.-H., Weng, Y.-H., et al. (2011). Contribution of glucocerebrosidase mutation in a large cohort of sporadic Parkinson’s disease in Taiwan. Eur. J. Neurol. 18, 1227–1232. doi: 10.1111/j.1468-1331.2011.03362.x
Ishikawa, A., and Tsuji, S. (1996). Clinical analysis of 17 patients in 12 Japanese families with autosomal-recessive type juvenile parkinsonism. Neurology 47, 160–166. doi: 10.1212/wnl.47.1.160
Jeong, G. R., Jang, E. H., Bae, J. R., Jun, S., Kang, H. C., Park, C. H., et al. (2018). Dysregulated phosphorylation of Rab GTPases by LRRK2 induces neurodegeneration. Mol. Neurodegener. 13:8. doi: 10.1186/s13024-018-0240-1
Johansen, K. K., White, L. R., Farrer, M. J., and Aasly, J. O. (2011). Subclinical signs in LRRK2 mutation carriers. Parkinsonism Relat. Disord. 17, 528–532. doi: 10.1016/j.parkreldis.2011.04.014
Kalia, L. V., and Lang, A. E. (2015). Parkinson’s disease. Lancet 386, 896–912. doi: 10.1016/S0140-6736(14)61393-3
Kara, E., Kiely, A. P., Proukakis, C., Giffin, N., Love, S., Hehir, J., et al. (2014). A 6.4 Mb duplication of the alpha-synuclein locus causing frontotemporal dementia and Parkinsonism: phenotype-genotype correlations. JAMA Neurol. 71, 1162–1171. doi: 10.1001/jamaneurol.2014.994
Kasten, M., and Klein, C. (2013). The many faces of alpha-synuclein mutations. Mov. Disord. 28, 697–701. doi: 10.1002/mds.25499
Kasten, M., Hartmann, C., Hampf, J., Schaake, S., Westenberger, A., Vollstedt, E. J., et al. (2018). Genotype-phenotype relations for the Parkinson’s disease genes Parkin, PINK1, DJ1: MDSGene systematic review. Mov. Disord. 33, 730–741. doi: 10.1002/mds.27352
Kasten, M., Marras, C., and Klein, C. (2017). Nonmotor signs in genetic forms of Parkinson’s disease. Int. Rev. Neurobiol. 133, 129–178. doi: 10.1016/bs.irn.2017.05.030
Khan, N. L., Graham, E., Critchley, P., Schrag, A. E., Wood, N. W., Lees, A. J., et al. (2003). Parkin disease: a phenotypic study of a large case series. Brain 126, 1279–1292. doi: 10.1093/brain/awg142
Khan, N. L., Katzenschlager, R., Watt, H., Bhatia, K. P., Wood, N. W., Quinn, N., et al. (2004). Olfaction differentiates parkin disease from early-onset parkinsonism and Parkinson disease. Neurology 62, 1224–1226. doi: 10.1212/01.wnl.0000118281.66802.81
Kilarski, L. L., Pearson, J. P., Newsway, V., Majounie, E., Knipe, M. D., Misbahuddin, A., et al. (2012). Systematic review and UK-based study of PARK2 (parkin), PINK1, PARK7 (DJ-1) and LRRK2 in early-onset Parkinson’s disease. Mov. Disord. 27, 1522–1529. doi: 10.1002/mds.25132
Kim, T. J., Kim, T. J., Lee, H., Kim, Y. E., and Jeon, B. S. (2014). A case of Parkin disease (PARK2) with schizophrenia: evidence of regional selectivity. Clin. Neurol. Neurosurg. 126, 35–37. doi: 10.1016/j.clineuro.2014.07.034
Klein, C., Schumacher, K., Jacobs, H., Hagenah, J., Kis, B., Garrels, J., et al. (2000). Association studies of Parkinson’s disease and parkin polymorphisms. Ann. Neurol. 48, 126–127. doi: 10.1002/1531-8249(200007)48:1<126::AID-ANA22>3.0.CO;2-K
Kruger, R., Kuhn, W., Leenders, K. L., Sprengelmeyer, R., Muller, T., Woitalla, D., et al. (2001). Familial parkinsonism with synuclein pathology: clinical and PET studies of A30P mutation carriers. Neurology 56, 1355–1362. doi: 10.1212/wnl.56.10.1355
Kumru, H., Santamaria, J., Tolosa, E., Valldeoriola, F., Munoz, E., Marti, M. J., et al. (2004). Rapid eye movement sleep behavior disorder in parkinsonism with parkin mutations. Ann. Neurol. 56, 599–603. doi: 10.1002/ana.20272
Li, Y., Kang, W., Zhang, L., Zhou, L., Niu, M., and Liu, J. (2017). Hyposmia is associated with RBD for PD patients with variants of SNCA. Front. Aging Neurosci. 9:303. doi: 10.3389/fnagi.2017.00303
Li, K., Tang, B. S., Liu, Z. H., Kang, J. F., Zhang, Y., Shen, L., et al. (2015). LRRK2 A419V variant is a risk factor for Parkinson’s disease in Asian population. Neurobiol. Aging 36, 2908.e11–2908.e15. doi: 10.1016/j.neurobiolaging.2015.07.012
Limousin, N., Konofal, E., Karroum, E., Lohmann, E., Theodorou, I., Durr, A., et al. (2009). Restless legs syndrome, rapid eye movement sleep behavior disorder and hypersomnia in patients with two parkin mutations. Mov. Disord. 24, 1970–1976. doi: 10.1002/mds.22711
Lohmann, E., Leclere, L., De Anna, F., Lesage, S., Dubois, B., Agid, Y., et al. (2009). A clinical, neuropsychological and olfactory evaluation of a large family with LRRK2 mutations. Parkinsonism Relat. Disord. 15, 273–276. doi: 10.1016/j.parkreldis.2008.06.008
MacLeod, D. A., Rhinn, H., Kuwahara, T., Zolin, A., Di Paolo, G., McCabe, B. D., et al. (2013). RAB7L1 interacts with LRRK2 to modify intraneuronal protein sorting and Parkinson’s disease risk. Neuron 77, 425–439. doi: 10.1016/j.neuron.2012.11.033
Marras, C., Alcalay, R. N., Caspell-Garcia, C., Coffey, C., Chan, P., Duda, J. E., et al. (2016a). Motor and nonmotor heterogeneity of LRRK2-related and idiopathic Parkinson’s disease. Mov. Disord. 31, 1192–1202. doi: 10.1002/mds.26614
Marras, C., Lang, A., van de Warrenburg, B. P., Sue, C. M., Tabrizi, S. J., Bertram, L., et al. (2016b). Nomenclature of genetic movement disorders: recommendations of the international Parkinson and movement disorder society task force. Mov. Disord. 31, 436–457. doi: 10.1002/mds.26527
Marras, C., Schüle, B., Munhoz, R. P., Rogaeva, E., Langston, J. W., Kasten, M., et al. (2011). Phenotype in parkinsonian and nonparkinsonian LRRK2 G2019S mutation carriers. Neurology 77, 325–333. doi: 10.1212/WNL.0b013e318227042d
Mata, I. F., Davis, M. Y., Lopez, A. N., Dorschner, M. O., Martinez, E., Yearout, D., et al. (2017). The discovery of LRRK2 p.R1441S, a novel mutation for Parkinson’s disease, adds to the complexity of a mutational hotspot. Am. J. Med. Genet. B Neuropsychiatr. Genet. 174:113. doi: 10.1002/ajmg.b.32510
Mata, I. F., Leverenz, J. B., Weintraub, D., Trojanowski, J. Q., Chen-Plotkin, A., Van Deerlin, V. M., et al. (2016). GBA Variants are associated with a distinct pattern of cognitive deficits in Parkinson’s disease. Mov. Disord. 31, 95–102. doi: 10.1002/mds.26359
Mazzulli, J. R., Xu, Y. H., Sun, Y., Knight, A. L., McLean, P. J., Caldwell, G. A., et al. (2011). Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 146, 37–52. doi: 10.1016/j.cell.2011.06.001
Meissner, W. G., Frasier, M., Gasser, T., Goetz, C. G., Lozano, A., Piccini, P., et al. (2011). Priorities in Parkinson’s disease research. Nat. Rev. Drug Discov. 10, 377–393. doi: 10.1038/nrd3430
Morgante, F., Fasano, A., Ginevrino, M., Petrucci, S., Ricciardi, L., Bove, F., et al. (2016). Impulsive-compulsive behaviors in parkin-associated Parkinson disease. Neurology 87, 1436–1441. doi: 10.1212/WNL.0000000000003177
Nabli, F., Ben Sassi, S., Amouri, R., Duda, J. E., Farrer, M. J., and Hentati, F. (2015). Motor phenotype of LRRK2-associated Parkinson’s disease: a Tunisian longitudinal study. Mov. Disord. 30, 253–258. doi: 10.1002/mds.26097
Narendra, D., Walker, J. E., and Youle, R. (2012). Mitochondrial quality control mediated by PINK1 and Parkin: links to parkinsonism. Cold Spring Harb. Perspect. Biol. 4:a011338. doi: 10.1101/cshperspect.a011338
Neumann, J., Bras, J., Deas, E., O’Sullivan, S. S., Parkkinen, L., Lachmann, R. H., et al. (2009). Glucocerebrosidase mutations in clinical and pathologically proven Parkinson’s disease. Brain 132, 1783–1794. doi: 10.1093/brain/awp044
Oeda, T., Umemura, A., Mori, Y., Tomita, S., Kohsaka, M., Park, K., et al. (2015). Impact of glucocerebrosidase mutations on motor and nonmotor complications in Parkinson’s disease. Neurobiol. Aging 36, 3306–3313. doi: 10.1016/j.neurobiolaging.2015.08.027
Paisán-Ruiz, C., Jain, S., Evans, E. W., Gilks, W. P., Simon, J., van der Brug, M., et al. (2004). Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron 44, 595–600. doi: 10.1016/j.neuron.2004.10.023
Paisán-Ruiz, C., Lewis, P. A., and Singleton, A. B. (2013). LRRK2: cause, risk, and mechanism. J. Parkinsons Dis. 3, 85–103. doi: 10.3233/JPD-130192
Papadimitriou, D., Antonelou, R., Miligkos, M., Maniati, M., Papagiannakis, N., Bostantjopoulou, S., et al. (2016). Motor and nonmotor features of carriers of the p.A53T alpha-synuclein mutation: a longitudinal study. Mov. Disord. 31, 1226–1230. doi: 10.1002/mds.26615
Petrucci, S., Ginevrino, M., Trezzi, I., Monfrini, E., Ricciardi, L., Albanese, A., et al. (2020). GBA-related Parkinson’s disease: dissection of genotype-phenotype correlates in a large italian cohort. Mov. Disord. doi: 10.1002/mds.28195 [Epub ahead of print].
Pfeiffer, R. F. (2016). Non-motor symptoms in Parkinson’s disease. Parkinsonism Relat. Disord. 22, S119–S122. doi: 10.1016/j.parkreldis.2015.09.004
Piredda, R., Desmarais, P., Masellis, M., and Gasca-Salas, C. (2020). Cognitive and psychiatric symptoms in genetically determined Parkinson’s disease: a systematic review. Eur. J. Neurol. 27, 229–234. doi: 10.1111/ene.14115
Ricciardi, L., Petrucci, S., Guidubaldi, A., Ialongo, T., Serra, L., Ferraris, A., et al. (2014). Phenotypic variability of PINK1 expression: 12 Years’ clinical follow-up of two Italian families. Mov. Disord. 29, 1561–1566. doi: 10.1002/mds.25994
Rosenbusch, K. E., and Kortholt, A. (2016). Activation mechanism of LRRK2 and its cellular functions in Parkinson’s disease. Parkinsons Dis. 2016:7351985. doi: 10.1155/2016/7351985
Rotter, A., Lenz, B., Pitsch, R., Richter-Schmidinger, T., Kornhuber, J., and Rhein, C. (2019). Alpha-synuclein RNA expression is increased in major depression. Int. J. Mol. Sci. 20:2029. doi: 10.3390/ijms20082029
Ryan, E., Seehra, G., Sharma, P., and Sidransky, E. (2019). GBA1-associated parkinsonism: new insights and therapeutic opportunities. Curr. Opin. Neurol. 32, 589–596. doi: 10.1097/WCO.0000000000000715
Saunders-Pullman, R., Alcalay, R. N., Mirelman, A., Wang, C., Luciano, M. S., Ortega, R. A., et al. (2015). REM sleep behavior disorder, as assessed by questionnaire, in G2019S LRRK2 mutation PD and carriers. Mov. Disord. 30, 1834–1839. doi: 10.1002/mds.26413
Saunders-Pullman, R., Mirelman, A., Alcalay, R. N., Wang, C., Ortega, R. A., Raymond, D., et al. (2018). Progression in the LRRK2-asssociated Parkinson disease population. JAMA Neurol. 75, 312–319. doi: 10.1001/jamaneurol.2017.4019
Saunders-Pullman, R., Mirelman, A., Wang, C., Alcalay, R. N., San Luciano, M., Ortega, R., et al. (2014). Olfactory identification in LRRK2 G2019S mutation carriers: a relevant marker? Ann. Clin. Transl. Neurol. 1, 670–678. doi: 10.1002/acn3.95
Seppi, K., Ray Chaudhuri, K., Coelho, M., Fox, S. H., Katzenschlager, R., Perez Lloret, S., et al. (2019). Update on treatments for nonmotor symptoms of Parkinson’s disease-an evidence-based medicine review. Mov. Disord. 34, 180–198. doi: 10.1002/mds.27602
Sharma, M., Ioannidis, J. P., Aasly, J. O., Annesi, G., Brice, A., Bertram, L., et al. (2012). A multi-centre clinico-genetic analysis of the VPS35 gene in Parkinson disease indicates reduced penetrance for disease-associated variants. J. Med. Genet. 49, 721–726. doi: 10.1136/jmedgenet-2012-101155
Shin, N., Jeong, H., Kwon, J., Heo, H. Y., Kwon, J. J., Yun, H. J., et al. (2008). LRRK2 regulates synaptic vesicle endocytosis. Exp. Cell Res. 314, 2055–2065. doi: 10.1016/j.yexcr.2008.02.015
Sidransky, E., Nalls, M. A., Aasly, J. O., Aharon-Peretz, J., Annesi, G., Barbosa, E. R., et al. (2009). Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N. Engl. J. Med. 361, 1651–1661. doi: 10.1056/NEJMoa0901281
Singleton, A., Gwinn-Hardy, K., Sharabi, Y., Li, S. T., Holmes, C., Dendi, R., et al. (2004). Association between cardiac denervation and parkinsonism caused by alpha-synuclein gene triplication. Brain 127, 768–772. doi: 10.1093/brain/awh081
Singleton, A. B., Farrer, M., Johnson, J., Singleton, A., Hague, S., Kachergus, J., et al. (2003). alpha-Synuclein locus triplication causes Parkinson’s disease. Science 302:841. doi: 10.1126/science.1090278
Sironi, F., Primignani, P., Ricca, S., Tunesi, S., Zini, M., Tesei, S., et al. (2013). DJ1 analysis in a large cohort of Italian early onset Parkinson disease patients. Neurosci. Lett. 557, 165–170. doi: 10.1016/j.neulet.2013.10.048
Steger, M., Diez, F., Dhekne, H. S., Lis, P., Nirujogi, R. S., Karayel, O., et al. (2017). Systematic proteomic analysis of LRRK2-mediated Rab GTPase phosphorylation establishes a connection to ciliogenesis. eLife 6:e31012. doi: 10.7554/eLife.31012
Steinlechner, S., Stahlberg, J., Volkel, B., Djarmati, A., Hagenah, J., Hiller, A., et al. (2007). Co-occurrence of affective and schizophrenia spectrum disorders with PINK1 mutations. J. Neurol. Neurosurg. Psychiatry 78, 532–535. doi: 10.1136/jnnp.2006.105676
Struhal, W., Presslauer, S., Spielberger, S., Zimprich, A., Auff, E., Bruecke, T., et al. (2014). VPS35 Parkinson’s disease phenotype resembles the sporadic disease. J. Neural Transm. 121, 755–759. doi: 10.1007/s00702-014-1179-1
Swan, M., Doan, N., Ortega, R. A., Barrett, M., Nichols, W., Ozelius, L., et al. (2016). Neuropsychiatric characteristics of GBA-associated Parkinson disease. J. Neurol. Sci. 370, 63–69. doi: 10.1016/j.jns.2016.08.059
Tambasco, N., Nigro, P., Romoli, M., Prontera, P., Simoni, S., and Calabresi, P. (2016). A53T in a parkinsonian family: a clinical update of the SNCA phenotypes. J. Neural Transm. 123, 1301–1307. doi: 10.1007/s00702-016-1578-6
Thaler, A., Gurevich, T., Bar Shira, A., Gana Weisz, M., Ash, E., Shiner, T., et al. (2017). A “dose” effect of mutations in the GBA gene on Parkinson’s disease phenotype. Parkinsonism Relat. Disord. 36, 47–51. doi: 10.1016/j.parkreldis.2016.12.014
Tijero, B., Gabilondo, I., Lezcano, E., Teran-Villagra, N., Llorens, V., Ruiz-Martinez, J., et al. (2015). Autonomic involvement in Parkinsonian carriers of PARK2 gene mutations. Parkinsonism Relat. Disord. 21, 717–722. doi: 10.1016/j.parkreldis.2015.04.012
Tomiyama, H., Li, Y., Funayama, M., Hasegawa, K., Yoshino, H., Kubo, S., et al. (2006). Clinicogenetic study of mutations in LRRK2 exon 41 in Parkinson’s disease patients from 18 countries. Mov. Disord. 21, 1102–1108. doi: 10.1002/mds.20886
Torres-Odio, S., Key, J., Hoepken, H. H., Canet-Pons, J., Valek, L., Roller, B., et al. (2017). Progression of pathology in PINK1-deficient mouse brain from splicing via ubiquitination, ER stress and mitophagy changes to neuroinflammation. J. Neuroinflammation 14:154. doi: 10.1186/s12974-017-0928-0
Trinh, J., Zeldenrust, F. M. J., Huang, J., Kasten, M., Schaake, S., Petkovic, S., et al. (2018). Genotype-phenotype relations for the Parkinson’s disease genes SNCA, LRRK2, VPS35: MDSGene systematic review. Mov. Disord. 33, 1857–1870. doi: 10.1002/mds.27527
Truban, D., Hou, X., Caulfield, T. R., Fiesel, F. C., and Springer, W. (2017). PINK1, parkin and mitochondrial quality control: what can we learn about Parkinson’s disease pathobiology? J. Parkinsons Dis. 7, 13–29. doi: 10.3233/JPD-160989
Valente, E. M., Abou-Sleiman, P. M., Caputo, V., Muqit, M. M., Harvey, K., Gispert, S., et al. (2004a). Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 304, 1158–1160. doi: 10.1126/science.1096284
Valente, E. M., Salvi, S., Ialongo, T., Marongiu, R., Elia, A. E., Caputo, V., et al. (2004b). PINK1 mutations are associated with sporadic early-onset parkinsonism. Ann. Neurol. 56, 336–341. doi: 10.1002/ana.20256
Valente, E. M., Bentivoglio, A. R., Dixon, P. H., Ferraris, A., Ialongo, T., Frontali, M., et al. (2001). Localization of a novel locus for autosomal recessive early-onset parkinsonism, PARK6, on human chromosome 1p35–p36. Am. J. Hum. Genet. 68, 895–900. doi: 10.1086/319522
Vilarino-Guell, C., Wider, C., Ross, O. A., Dachsel, J. C., Kachergus, J. M., Lincoln, S. J., et al. (2011). VPS35 mutations in Parkinson disease. Am. J. Hum. Genet. 89, 162–167. doi: 10.1016/j.ajhg.2011.06.001
Wang, C., Cai, Y., Gu, Z., Ma, J., Zheng, Z., Tang, B. S., et al. (2014). Clinical profiles of Parkinson’s disease associated with common leucine-rich repeat kinase 2 and glucocerebrosidase genetic variants in Chinese individuals. Neurobiol. Aging 35, 725.e1–725.e6. doi: 10.1016/j.neurobiolaging.2013.08.012
Weissbach, A., Wittke, C., Kasten, M., and Klein, C. (2019). ‘Atypical’ Parkinson’s disease - genetic. Int. Rev. Neurobiol. 149, 207–235. doi: 10.1016/bs.irn.2019.10.011
West, A. B., and Cookson, M. R. (2016). Identification of bona-fide LRRK2 kinase substrates. Mov. Disord. 31, 1140–1141. doi: 10.1002/mds.26647
Yahalom, G., Greenbaum, L., Israeli-Korn, S., Fay-Karmon, T., Livneh, V., Ruskey, J. A., et al. (2019). Carriers of both GBA and LRRK2 mutations, compared to carriers of either, in Parkinson’s disease: risk estimates and genotype-phenotype correlations. Parkinsonism Relat. Disord. 62, 179–184. doi: 10.1016/j.parkreldis.2018.12.014
Yamaguchi, K., Cochran, E. J., Murrell, J. R., Polymeropoulos, M. H., Shannon, K. M., Crowther, R. A., et al. (2005). Abundant neuritic inclusions and microvacuolar changes in a case of diffuse Lewy body disease with the A53T mutation in the alpha-synuclein gene. Acta Neuropathol. 110, 298–305. doi: 10.1007/s00401-005-1042-4
Keywords: Parkinson’s disease, monogenic, diagnosis, non-motor symptoms, SNCA
Citation: Liu X and Le W (2020) Profiling Non-motor Symptoms in Monogenic Parkinson’s Disease. Front. Aging Neurosci. 12:591183. doi: 10.3389/fnagi.2020.591183
Received: 04 August 2020; Accepted: 02 October 2020;
Published: 30 October 2020.
Edited by:
Guanghui Wang, Soochow University, ChinaReviewed by:
Jifeng Guo, Central South University, ChinaRoberta Marongiu, Cornell University, United States
Copyright © 2020 Liu and Le. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Weidong Le, wdle_sibs@163.com