 Jun-Jun Liu1*
Jun-Jun Liu1* Richard A. Sniezko2
Richard A. Sniezko2 Arezoo Zamany1Holly Williams1
Arezoo Zamany1Holly Williams1 Kangakola Omendja1Angelia Kegley2Douglas P. Savin2
Kangakola Omendja1Angelia Kegley2Douglas P. Savin2- 1Canadian Forest Service, Natural Resources Canada, Victoria, BC, Canada
- 2USDA Forest Service, Dorena Genetic Resource Center, Cottage Grove, OR, United States
Breeding programs of five-needle pines have documented both major gene resistance (MGR) and quantitative disease resistance (QDR) to Cronartium ribicola (Cri), a non-native, invasive fungal pathogen causing white pine blister rust (WPBR). WPBR is one of the most deadly forest diseases in North America. However, Cri virulent pathotypes have evolved and can successfully infect and kill trees carrying resistance (R) genes, including vcr2 that overcomes MGR conferred by the western white pine (WWP, Pinus monticola) R gene (Cr2). In the absence of a reference genome, the present study generated a vcr2 reference transcriptome, consisting of about 20,000 transcripts with 1,014 being predicted to encode secreted proteins (SPs). Comparative profiling of transcriptomes and secretomes revealed vcr2 was significantly enriched for several gene ontology (GO) terms relating to oxidation-reduction processes and detoxification, suggesting that multiple molecular mechanisms contribute to pathogenicity of the vcr2 pathotype for its overcoming Cr2. RNA-seq-based bulked segregant analysis (BSR-Seq) revealed genome-wide DNA variations, including about 65,617 single nucleotide polymorphism (SNP) loci in 7,749 polymorphic genes shared by vcr2 and avirulent (Avcr2) pathotypes. An examination of the distribution of minor allele frequency (MAF) uncovered a high level of genomic divergence between vcr2 and Avcr2 pathotypes. By integration of extreme-phenotypic genome-wide association (XP-GWAS) analysis and allele frequency directional difference (AFDD) mapping, we identified a set of vcr2-associated SNPs within functional genes, involved in fungal virulence and other molecular functions. These included six SPs that were top candidate effectors with putative activities of reticuline oxidase, proteins with common in several fungal extracellular membrane (CFEM) domain or ferritin-like domain, polysaccharide lyase, rds1p-like stress responsive protein, and two Cri-specific proteins without annotation. Candidate effectors and vcr2-associated genes provide valuable resources for further deciphering molecular mechanisms of virulence and pathogenicity by functional analysis and the subsequent development of diagnostic tools for monitoring the virulence landscape in the WPBR pathosystems.
Introduction
White pine blister rust (WPBR) caused by Cronartium ribicola (Cri) J.C. Fisch is the most severe and damaging disease of five-needle pine species (subgenus Strobus) world-wide. This fungal pathogen depends on pine trees as its primary hosts and Ribes as the principal alternate hosts to complete its life-cycle (Geils et al., 2010). Following its accidental introduction to North America in 1906, Cri had spread to most geographical regions where commercial five-needle pines were originally distributed by the 1950s (Maloy, 2018). All nine native five-needle pine species in the United States, including four native to Canada, are very susceptible to WPBR (Tomback and Achuff, 2010).
Cronartium ribicola has a complex life cycle with five spore stages (aeciospores, urediniospores, teliospores, basidiospores, and spermatia), requiring an alternate hosts (mainly species of Ribes in North America, and Pedicularis in some areas of Asia) to complete its life cycle over at least 2 years. Of the five spore stages of the Cri life cycle, only basidiospores are able to infect pine needles, which occurs in late summer or early fall when they develop on the underside of infected Ribes leaves during cool, wet weather conditions. Basidiospores germinate on the needle surface, and their germ tubes penetrate into needle tissues through stomatal pores. The mycelia grow at infection sites and spread to stems through vascular tissues, and stem cankers develop in the coming spring of next year. As an exotic, invasive fungal pathogen, Cri directly kills western white pine (WWP; Pinus monticola Dougl. ex D. Don) and other native five-needle pines of all ages across North America and predisposes older trees to secondary attack by insects and other fungi.
Due to the high economic and ecological importance of five-needle pines, screening of host resistance to this rust fungus to develop resistant populations is seen as the key to using these species in restoration and reforestation. In some species this work has been ongoing for more than 50 years, and different levels and types of resistance have been found among five-needle pine species (Sniezko et al., 2014, 2020). Resistance (R) loci for major gene resistance (MGR) have been found in several five-needle pine species. Host trees with different resistant genotypes have allowed distinct virulent pathotypes to evolve from predominantly avirulent wild-type races, overcoming MGR in sugar pine and WWP (Kinloch et al., 2004), as well as in Ribes (Tanguay et al., 2013). The Cri pathotype vcr2 was discovered because of its ability to kill WWP trees with the major R gene (Cr2). vcr2 was originally detected in the Champion Mine population on the Umpqua National Forest in Oregon, a presence probably as early as the mid-1950s (Kinloch et al., 2004); and it has now been documented in much of western Oregon and at Happy Camp, CA, United States (Kinloch et al., 2003). Cr2 progeny in field trials in Washington now show infection, suggesting that vcr2 may arise, or migrate from western Oregon. These observations suggest that the Cri virulent pathotype arises relatively quickly under selection where Cr2 trees are present. Planting resistant seedlings is seen as the most effective and environmentally friendly way to manage WPBR. However, increased field planting of resistant trees also promotes selection pressure on Cri and accelerates the spread of vcr races and other genotypes with enhanced virulence. More information is required about compatible and incompatible white pine-blister rust (WP-BR) interactions to understand the durability and stability of MGR and quantitative disease resistance in different species (Sniezko et al., 2020).
Understanding the mechanisms underlying variations in pathogenicity is necessary for disease management and utilization of tools for genetic resistance. However, the pathogen side of molecular tree-microbe interactions is poorly understood in WPBR pathosystems. Previously, investigation of Cri genetic diversity revealed the global movement, local host selection, and genetic shifts in local populations (Richardson et al., 2008; Brar et al., 2015). Despite this progress, the absence of a Cri reference genome sequence means that little molecular information is available for the Cronartium genus. Recently, the genome of Cronartium quercuum f.sp. fusiforme (Cqf) was sequenced (Pendleton et al., 2014), allowing comparative genomics studies to identify avirulence genes and secreted effectors in this rust genus. Transcriptome profiling of Cri avirulent pathotypes described genes and putative effectors that were differentially expressed among different stages of its life cycle (Liu et al., 2015). An effector secreted by Cri avirulent pathotypes was functionally characterized (Ma et al., 2019). Such genomics and transcriptomics studies are invaluable in the discovery of gene functions and metabolic pathways in this rust group. However, genetics and molecular understanding of the genomic basis of Cri virulence is still lacking at this point (Kinloch et al., 2004).
In this paper, we report on the profiling of Cri vcr2 transcriptomes and secretomes. To understand the molecular mechanisms contributing to pathogenic virulence, pathotype-specific and conserved transcriptomic programs were revealed by a comparative transcriptomic approach. Candidate effectors and vcr2-associated genes were further identified by RNA-seq-based bulked segregant analysis (BSR-Seq). These genomic resources provide a strong foundation for future efforts to isolate the vcr2 and Avcr2 genes for investigations of their biological properties and development of new diagnostic tests for monitoring virulence across Cri’s landscape.
Materials and Methods
Fungal and Plant Materials
Cronartium ribicola (Cri) vcr2 aeciospores were collected from infected trees of wind-pollinated progeny of two Cr2/Cr2 parent trees, 20046-031 and 023225 on March 12, 2014. Their seedlings were planted as part of a field trial (RV7) in 2000 at Travis Tyrrell seed orchard, Oregon, United States (Sniezko et al., 2020), where vcr2 was documented (Kinloch et al. 2004). All Cri aeciospore samples were collected separately from different trees as biological repeats. Infected stems showing disease symptoms of discolored bark tissues were collected from 20-month-old seedlings ~14 months post Cri infection as described previously (Liu et al., 2014). In brief, seeds were sown in June 2010 after 4 months stratification. Seedlings were grown in a greenhouse and inoculated with Cri basidiospores in September 2010 using infected leaves of Ribes spp. (the alternate host of C. ribicola). The Cri sources in these inoculations were collected from eight field locations in eastern Oregon as a heterogenous mixture. Phenotypic traits were assessed at periodic intervals in 2011 when infection symptoms were evident on needles and stems. Based on phenotypes of needle disease spot types (i.e., all HR-like; all susceptible; mixed; and un-identified disease spots) and stem symptoms (i.e., cankers present or absent), each seedling was determined as a resistant (Cr2/−) or susceptible (cr2/cr2) genotype, and their genotypes were further confirmed by SNP genotyping using Cr2-linked DNA markers (Liu et al., 2020). Cankered WWP (Cr2/−) stems were harvested for the vcr2 pathotype with three biological repeats, each pooled with at least 15 seedlings. Uninfected healthy seedlings were used as negative controls. The samples were frozen using liquid nitrogen and stored at −80°C before RNA extraction.
RNA-Seq Analysis and de novo Assembly and Post Filtering of the Transcriptomes
Total RNAs were extracted from aeciospores and cankered WWP stems following a protocol described previously (Liu, 2012). After genomic DNA digestion with DNase, RNA was re-purified using an RNeasy plant mini kit (Qiagen). Following mRNA separation using an RNA-seq sample preparation kit (Illumina, San Diego, CA, United States), cDNA libraries were constructed with sample-specific 6-bp bar-coding tags. Paired-ends (PE) were sequenced on an Illumina HiSeq 2500 instrument (Illumina), which yielded 23–56 million 100-bp PE reads per library at the National Research Council of Canada (Saskatoon, Canada). Raw reads of the Illumina RNA-seq 100-bp PE sequences from nine samples were deposited in the NCBI SRA under accession number SRR11101722–SRR11101730.
Trimmomatic with default settings was used to remove adapter sequences and low-quality bases (phred score <20) from the raw RNA-seq reads (Bolger et al., 2014). The clean reads were de novo assembled into transcripts for the aeciospores and cankered stems separately using Trinity (version: trinityrnaseq_r2013-02-25) with default k-mer length of 25 (Haas et al., 2013). A workflow as outlined by De Fine Licht et al. (2017) was used to remove host transcripts and reduce sequence redundancy. Putative WWP transcripts were first removed from two assemblies by BLASTn search (Altschul et al., 1990) against healthy WWP stem transcriptome (Liu et al., 2014) with a cut-off at e-value < e-6. The highest expressed isoform was then selected for each component using “pick_isoform_trinity_RSEM.py” (Yang and Smith, 2013), and overlapping sequences were assembled into contigs using CAP3 Sequence Assembly Program with setting -o 200 -p 99 (Huang and Madan, 1999). After combining contigs and singlets, CD-Hit-EST (Fu et al., 2012) was run to combine transcripts at 97% identity of the nucleotide sequences. Finally, each sequence was assigned to a putative taxonomical origin with BLASTx searches (e-value < 1e-6) against the UNIPROT database (ver. 2015_1) using Metagenome Analyzer (MEGAN, ver 5.7.1; Huson et al., 2011). Transcripts were assigned to four categories: “fungi”, “no hits” (sequences for which BLASTx did not find any hits), “not assigned” (sequences had BLASTx hits with bitscores below the threshold, thus no taxonomy was assigned to them), and “other” for those sequences that were assigned a taxonomy but beyond any of the above three categories. Sequences in the category of “not assigned” were also excluded where BLASTx search against the proteome of C. quercuum f. sp. fusiforme (Pendleton et al., 2014; downloaded at https://mycocosm.jgi.doe.gov/Croqu1/Croqu1.home.html) had E values >e-6. The sequences were further filtered by total length >200-bp with a minimum open reading frame (ORF) long 50 codons as predicted by TransDecoder. The putative protein sequences were combined by a run of CD-Hit at 97% of amino acid sequence identity. The resulting transcript dataset, including all sequences in the categories “fungi” and “no hits” and a part of “not assigned” sequences, was considered as a vcr2 reference transcriptome for further analysis. This transcriptome shotgun assembly project has been deposited at DDBJ/EMBL/GenBank under the accession GIKE00000000 in Bioproject PRJNA261951.
Ortholog cluster analysis was performed to compare the reference transcriptomes between vcr2 and Avcr2 (Liu et al., 2015) using OrthoVenn2 with the default settings of E-values ≤1e-5 and an inflation value of 1.5 (Xu et al., 2019). Gene names and gene ontology (GO) terms were assigned to the Cri genes based on their homologies to the available databases (NCBI-nr, PIR, KEGG, and GO) using BLAST2GO (Götz et al., 2008). GO-term enrichment was analyzed by Fisher’s exact test with correction for multiple testing (p values < 0.01) as implemented using BLAST2GO.
Prediction of Secreted Proteins
The vcr2 secretome was determined in complete ORFs based on bioinformatic prediction. The N-terminal signal peptide was predicted by combining SignalP v5.0 (Petersen et al., 2011) and Phobius (Kall et al., 2004). Transmembrane (TM) domains were predicted by allowing one TM domain in the first 60 amino acids using TMHMM1; and proteins with endoplasmic reticulum (ER) targeting sequence (Prosite: PS00014) were removed by using ScanProsite (de Castro et al., 2006). CD-HIT was used to cluster secreted proteins from vcr2 and Avcr2 (Liu et al., 2015) into clusters at a threshold of 98% amino acid sequence identity. Localization of effectors to apoplast was predicted by ApoplastP based on depletion in glutamic acid, acidic amino acids and charged amino acids and enrichment in small amino acids (Sperschneider et al., 2018). Carbohydrate-active enzymes (CAZy) in the secretome were annotated using the dbCAN2 meta server (Zhang et al., 2018). Pfam domain predictions were performed using HMMSCAN at the European Bioinformatics Institute (EMBL-EBI).2
Global Gene Expression Analysis
For comparative transcriptomic study of vcr2 with Avcr2, RNA-seq 100-bp PE reads from Avcr2-related samples were downloaded from the NCBI SRA. They included cankered stems of susceptible (cr2/cr2) WWP seedlings infected by the Avcr2 pathotype (SRR3273235–SRR3273237), Avcr2 aeciospores (SRR1583540, SRR1583545, and SRR1583552), avcr2 urediniospores (SRR1583557–SRR1583559), as well as the shoot-tip samples of healthy WWP (Cr2/−) seedlings (SRR1574690–1574692) as negative controls.
RNA-Seq reads were mapped back to the vcr2 transcriptome using CLC with parameters as: no masking, mismatch cost = 2, insertion/deletion cost = 3, length fraction = 0.9, similarity fraction = 0.9, auto-detect paired distances = yes, global alignment = yes, and non-specific match handling = ignore. As only paired reads were counted, transcript expression values were normalized by fragments per kilobase of exon per million fragments mapped (FPKM, Mortazavi et al., 2008) using CLC Genomics Workbench 5.5 (CLC bio, QIAgen, Aarhus, Denmark). Genes were counted as expressed in a specific pathotype using a cut-off of an average FPKM ≥ 1 in all sample types. Differentially expressed genes (DEGs) were identified by Baggerley’s test with weighted proportions fold change > |2| and Bonferroni corrected p < 0.05.
Identification of DNA Polymorphisms and Estimation of Minor Allele Frequency
Using the vcr2 reference transcriptome de novo assembled in this study as a reference, the variant detection tool embedded in CLC Genomics Workbench was used to analyze DNA variants in vcr2 and Avcr2 pathotypes as two pooled samples. Clean RNA-seq reads were pooled from six and 12 samples for the vcr2 and Avcr2 pathotypes separately. The two sets of pooled reads were mapped to the reference using CLC with parameters set the same as in the above gene expression study. DNA variants were called by the quality-based variant detection method using CLC with parameters set as: neighborhood radius = 5, maximum gap and mismatch count = 2, minimum neighborhood quality = 15, minimum central quality = 20, ignore non-specific matches = yes, ignore broken pairs = no, minimum coverage = 4, minimum variant frequency (%) = 1.0, and maximum expected alleles = 2. DNA variants were further filtered by a minimum contig depth of 10 reads covering the polymorphic site. These genes with low expression levels were not included in further analysis of genetic diversity. The vcr2 and Avcr2 pools were compared to identify variants specific to either pool as well as those common to both pools. Minor allele frequency (MAF) was estimated in the two data sets based on pathotypes. The distribution of MAFs was estimated according to the proportion of SNPs with MAF values that fall within the following six ranges: < 0.05, ≥0.05 to < 0.1, ≥ 0.1 to < 0.2, ≥ 0.2 to < 0.3, ≥ 0.3 to < 0.4 and ≥ 0.4 to ≤ 0.5. Chi-squared tests were used to compare frequency differences between the two pathotypes.
Detection of vcr2-Associaed SNPs
Bulked segregant analysis RNA-seq (BSR-Seq, Michelmore et al., 1991; Liu et al., 2012; Trick et al., 2012) was used to detect vcr2-associated SNPs. RNA-seq reads were bulked from individual samples from at least 90 C ribicola infected WWP trees based on vcr2 and Avcr2 pathotypes and mapped to the reference transcriptome as described above to calculate sequencing depth of the SNPs, counts of reference and alternative alleles at each SNP locus, as well as allele frequency for each SNP across the transcriptome throughout the whole genome.
DNA variants common to both vcr2 and Avcr2 pools were used to detect vcr2-associated SNPs, which are genetically informative due to the elimination of false positive variants, a process which is challenging in NGS data processing (Ribeiro et al., 2015). Extreme-phenotype genome-wide association study (XP-GWAS) and allele frequency directional difference (AFDD) mapping were integrated to identify vcr2-associated SNPs.
Extreme-phenotype genome-wide association study was generally performed using a generalized linear mode as described by Yang et al. (2015). To attenuate over-dispersion of the X2 test statistic from RNA-seq data with large variation of gene expression levels, counts of reference and alternative alleles were first normalized based on allele frequency for each SNP loci prior to the XP-GWAS run, where normalized count = log10 (count/10) x the allele frequency in percentage point (i.e., between 1 and 100). Phenotypic pool numbers were set as 1 = vcr1, 3 = Avcr2, and 2 = random that was generated by randomly pooling RNA-seq reads equally from both vcr2 and Avcr2. The likelihood ratio test statistic was computed for testing the null model of no association between success probability and phenotypic pool number; and then corrected by dividing by λ, an inflation factor proposed for consideration of cryptic within-group relatedness (Devlin and Roeder, 1999). The inflation factor (λ) was estimated using the R add-on package “gap”.3 To identify significant variants, the values of p derived from the likelihood ratio test statistic were corrected by the FDR method at the 5% level (Yang et al., 2015).
Allele frequency directional difference mapping was performed using two pooled samples as described previously (Fekih et al., 2013; Takagi et al., 2013). Avcr2 was presumed to be a dominant trait while virulent races, expected to be homozygous (vcr2/vcr2), were relatively rare across Cri landscape. Based on this hypothesis, genotypes were proposed for the bulked samples of virulence (ll) and avirulence (lm), where the allele “l” is assumed to be linked to vcr2. Under this scenario, the expected frequencies of the vcr2-linked alleles were close to 100% in the vcr2 pool and close to 50% in the Avcr2 pool. Therefore, SNP loci with allele frequency >0.90 as homozygote (ll) in the vcr2 pool and allele frequency in the range of 0.4~0.6 as heterozygote (lm) in the Avcr2 pool were selected for association testing.
Although considered less likely, the opposite hypothesis was also considered where the vcr2 pathotype has a heterozygous genotype (lm) and the Avcr2 pathotype has a homozygous genotype (mm). Under this assumption, the vcr2-linked allele (l) would have frequencies close to 50% in the vcr2 pool and its frequency would be close to 0 in the Avcr2 pool. SNP loci were then selected for virulence association tests with allele frequency in the range of 0.4~0.6 as heterozygote (lm) in the vcr2 pool and allele frequency < 0.1 as the homozygote (mm) in the Avcr2 pool. Under both assumptions, the AFDD threshold was set with the minimum of 30% points with the goal being to search for variants with AFDD of 50% points as a practical measurement (Fekih et al., 2013; Dougherty et al., 2018). The AFDD values were plotted against values of p from XP-GWAS for identification of significant association.
Results
De novo Assembly of the Cri vcr2 Reference Transcriptome by RNA-Seq Analysis
A total of 102 and 161 million 100-bp RNA-seq PE reads were generated from RNA prepared from vcr2 aeciospores and cankered WWP stems with Cr2/− genotypes, respectively, (Supplementary Table S1). Because infected host tissues contained a mixture of mRNAs expressed by both C. ribicola mycelia and WWP cells, transcriptomes were de-novo assembled for the spore samples and cankered WWP stem samples separately. The Cri samples collected in the present study came from greenhouses and the field, because culturing rust fungi in vitro is very difficult. In addition to Cri and WWP, a wide range of other organisms might contribute to RNA-seq reads of these environmental samples. After removal of host WWP transcripts, MEGAN analysis assigned transcripts and RNA-seq reads into the four categories: fungi, no hits, not assigned, and other. Although fungal transcripts accounted for only 54.2% of total de novo assembled transcriptomes, 93.3% of total mapped RNA-seq reads were assigned to them (Supplementary Figure S1). Detailed analysis of the WWP host transcriptome and biodiversity of the WPBR field samples will be described in a separate report. Following removal of transcripts potentially expressed by other taxa from the primary assemblies by bioinformatic processing as outlined in the methods, a final set of 24K unique transcripts was generated, representing the Cri vcr2 reference transcriptome with a total length of ~22 Mb (GenBank TSA accession GIKE00000000) for further analysis. Searching open reading frames (ORFs) at a cut-off of 150-bp identified 6,859 transcripts with putatively complete ORFs, while the remainder had incomplete ORFs, with a total ORF length of ~15 Mb (Supplementary Table S2).
This vcr2 reference transcriptome was compared to that of the Cri Avcr2 pathotype (Liu et al., 2015) using OrthoVenn2 (Supplementary Table S3). Both pathotypes had a similar number of clusters (8,498 vs. 8,063), but singleton clusters (proteins that do not cluster with others) were about two times more prevalent in vcr2 than in Avcr2 (15,793 vs. 6,324). The Cri vcr2 and Avcr2 pathotypes shared 6,944 clusters between them while 435 and 1,119 unique clusters were detected in vcr2 and Avcr2, respectively (Supplementary Figure S2). Both the unique clusters and the unique singletons might represent novel pathotype-specific proteins, or they may reflect differences in filtering of the transcriptome assemblies. OrthoVenn2 analysis did reveal that 435 vcr2-unique clusters were enriched with three GO terms of biological processes (cellulose catabolic process, hydrogen peroxide catabolic process, and mitochondrial electron transport, cytochrome c to oxygen) while 1,119 Avcr2-unique clusters were enriched with only one biological process termed as translation (Supplementary Table S4).
Prediction of the Cri Secretome
Fungal pathogens secrete effector proteins into the plant apoplast or cytoplasm to trigger molecular plant-microbe interactions. The full-length proteins encoded by the 6,859 complete ORFs were used to mine secreted proteins (SPs) in the Cri vcr2 reference transcriptome. Presence of secretory signal peptides at the N termini of proteins and absence of transmembrane domains were used to identify candidate effectors. A combination of four in-silico prediction programs (SignalP-v5.0, Phobius, TMHMM, and ScanProsite) revealed 519 candidate genes expressed as secreted proteins in the Cri vcr2 reference transcriptome (Figure 1), which accounted for 7.6% of unigenes with complete ORFs. After clustering with a set of 734 proteins identified previously as the Cri avcr secretome (Liu et al., 2015) and by using CD-Hit with a threshold of 98% identity between amino acid sequences, a total of 1,014 SPs were identified in transcriptomes of both vcr2 and Avcr2 pathotypes (Figure 1; Supplementary Table S5).
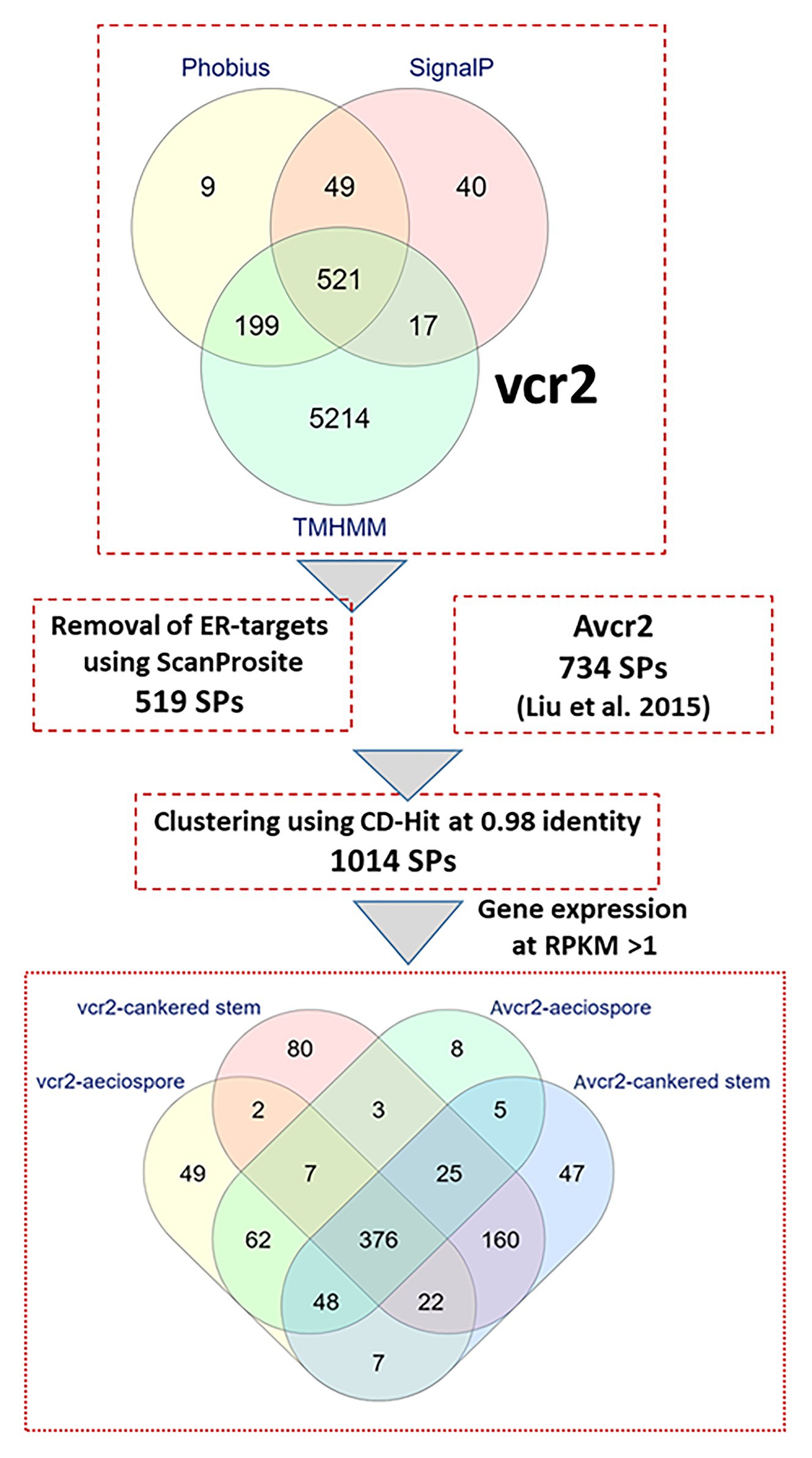
Figure 1. In-silico secretome prediction and identification of pathotype-specific effectors using a combination of computational programs. Putative full-length proteins were used to predict presence of signal peptides by using SignalP-v5.0 and Phobius. Proteins with transmembrane (TM) domains were removed by using TMHMM-v2.0 and Phobius. Proteins targeted to endoplasmic reticulum (ER) were removed by using ScanProsite. Cri secreted proteins from vcr2 and Avcr2 pathotypes were clustered by using CD-Hit. Within the Cri secretome, pathotype-specific effectors and those exclusively expressed in cankered host tissues were determined by mapping RNA-seq reads with a threshold of FPKM >1. The Venn diagram at bottom showing number of unique and shared proteins identified in Cri vcr2 and Avcr2 secretomes.
B2G based annotation showed 687 SPs had significant hits (Blastx E values < e-6) in the GeneBank nr database. Of all SPs, 109 and 321 SPs were further classified within CAZy and apoplast groups, respectively, (Supplementary Table S6). In addition, HMMScan revealed Pfam domains in 360 Cri SPs (Supplementary Table S6). The secreted CAZyes included 63 glycoside hydrolases (GHs), 3 glycosyltransferases (GTs), 4 polysaccharide lyases (PLs), 20 carbohydrate esterases (CEs), and 19 proteins with redox-active auxiliary activities (AAs). Protease/peptidase-like proteins comprised 29 annotated SPs in the Cri secretome, while others included multiple members of gene families for multi-copper oxidase laccase-like proteins, endoglucanases, nucleases, peroxidases, L-ascorbate oxidases, thioredoxins, Cu/Zn superoxide dismutase (SOD), pathogenesis-related proteins (PR-1-like and thaumatin-like proteins), catalases, and expansin proteins, Egh16-like virulence factors, common in several fungal extracellular membrane proteins (CFEM)-domain proteins, heat shock proteins (HSPs) and their chaperones with DnaJ domain (Supplementary Table S6). Hypothetical proteins with unknown function comprised 53.6% of the total SPs, but the majority had top Blast hits related to species of other rust fungi belonging to the genus Melampsora and Puccinia (Supplementary Table S6).
Expression Profiling of the Transcriptomes Revealed Pathotype-Specific Pathways
Expression profiling was performed by mapping RNA-seq reads to the Cri vcr2 reference transcriptome, including the Cri secretome. About 72~73% of total RNA-seq reads from aeciospores were mapped, while mapped reads from cankered WWP stems only accounted for 0.08~8% of the total RNA-seq reads (Supplementary Table S1). Abundance of fungal RNA-seq reads was much lower in vcr2-infected stems of the WWP resistant trees with Cr2/− genotype than in Avcr2-infected stems of the WWP susceptible trees with cr2/cr2 genotype (0.5 ± 0.6% vs. 7.6 ± 0.9%, t-test p = 1e-3). Transcripts expressed in pathotypes were filtered by a cut-off of FPKM = 1 on average, per sample type, resulting in detection of 18,014 and 12,771 genes expressed in vcr2 and Avcr2 pathotypes, respectively, (Supplementary Table S1). A core set of 7,092 genes was expressed in both vcr2 and Avcr2 pathotypes for both aeciospores and infected WWP stems (Figure 2A).
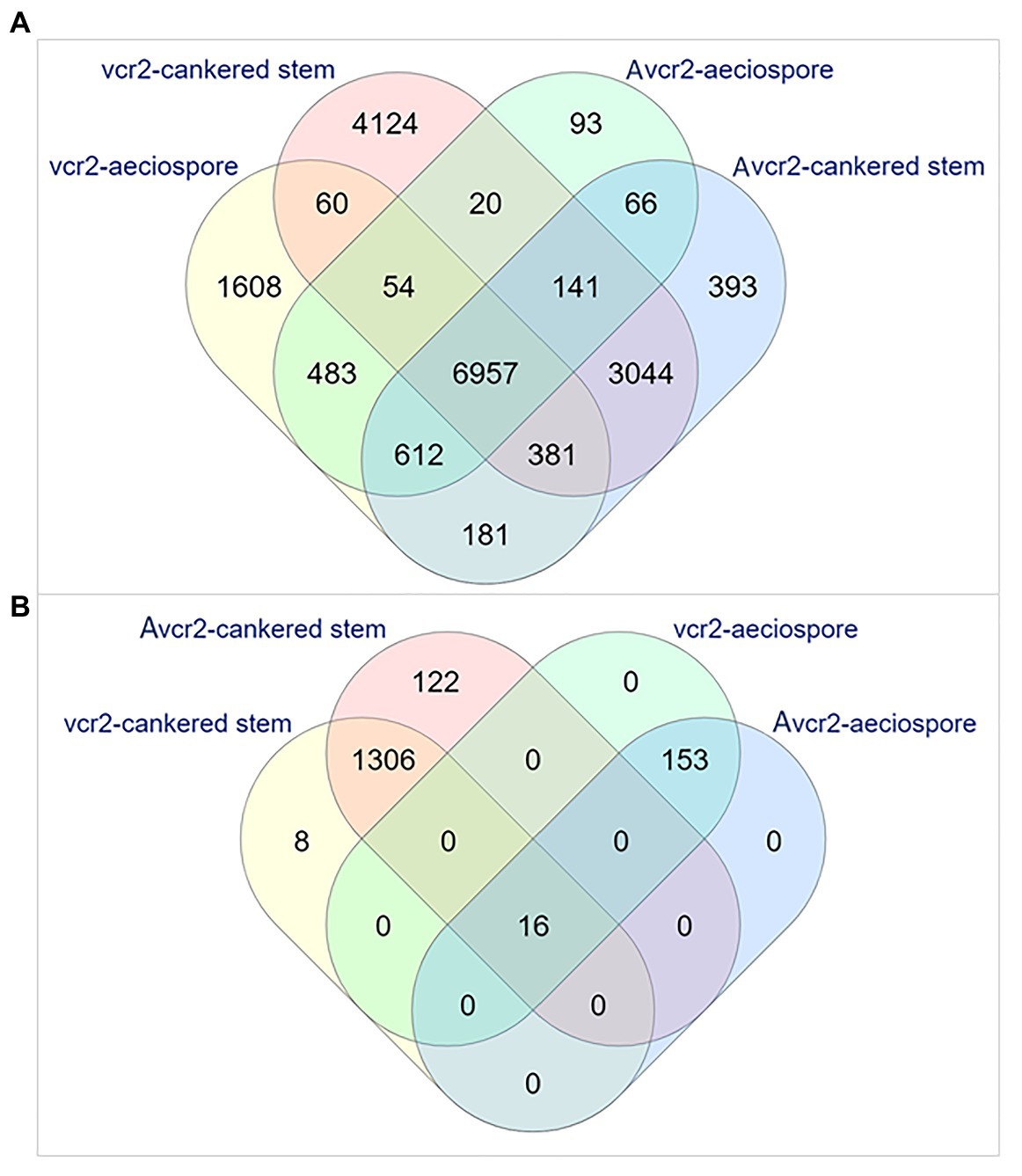
Figure 2. Venn diagrams representing numbers of the expressed unigenes. The expressed unigenes were counted in aeciospore and cankered stem with FPKM > 1. (A) Overlap of total genes expressed in vcr2 and Avcr2 pathotypes. (B) Distribution of differentially expressed genes (DEGs) by comparison of vcr2 with Avcr2.
Following B2G-based gene annotation, a comparison of the top 20 GO-terms at level-3 shows both pathotypes shared five main biological processes (biosynthetic process, cellular metabolic process, organic substance metabolic process, primary metabolic process, and nitrogen compound metabolic process), each accounting for >10% of the total annotated transcripts. Consistently with results of orthologous clustering analysis by OrthoVenn2, X2 test revealed that the vcr2 transcriptome had significant enrichment of oxidation-reduction process (GO: 0055114, p < 1e-5), response to chemical (GO: 0042221, p < 1e-5), oxidoreductase activity (GO: 0016491, p < 1e-3), lipid binding (GO: 0008289, p < 1e-5), and proteasome complex (GO: 0000502, p < 1e-5) while the Avcr2 transcriptome had enrichment of cell cycle (GO: 0007049, p < 1e-5), enzyme regulator activity (GO: 0030234, p < 1e-5), membrane-enclosed lumen (GO: 0031974, p < 3e-2), and Sm-like protein family complex (GO: 0120114, p < 1e-5; Figure 3).
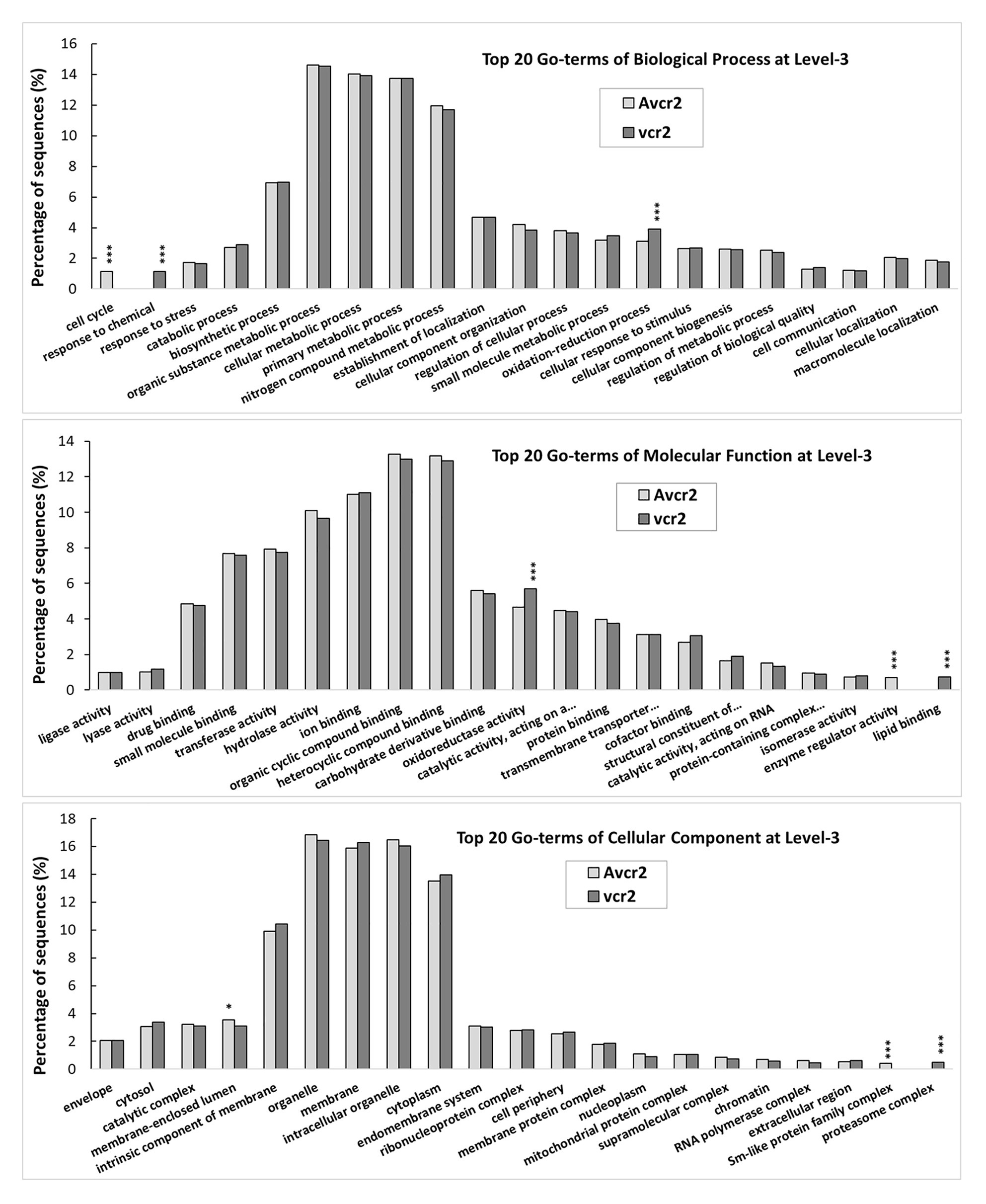
Figure 3. GO-term comparison between vcr2 and Avcr2 transcriptomes. The vcr2 and Avcr2 transcriptomes consist of 18,596 and 13,139 transcripts with the mean FPKM >1. A comparison of the top 20 GO-terms at level-3 shows the vcr2 transcriptome with a significant enrichment of oxidation-reduction process (GO:0055114), response to chemical (GO:0042221), oxidoreductase activity (GO:0016491), lipid binding (GO:0008289), and proteasome complex (GO:0000502) while the Avcr2 transcriptome with enrichment of cell cycle (GO:0007049), enzyme regulator activity (GO:0030234), membrane-enclosed lumen (GO:0031974), Sm-like protein family complex (GO:0120114). One and three stars indicate chi-square test p < 0.05 and 0.001, respectively.
Examination of pathotype-specific transcripts detected 1,610 and 4,178 genes exclusively expressed in Avcr2 aeciospores and cankered stems of WWP seedlings with genotypes Cr2/–, respectively. Relatively fewer genes were detected with Avcr2-specific expression patterns with 99 and 437 genes in aeciospores and cankered WWP stems, respectively, (Figure 2A). Similar to the transcriptome results shown above, Fisher’s exact tests showed vcr2-specifically expressed genes significantly enriched with seven GO-terms of molecular functions and 16 GO-terms of biological processes (FDR-adjusted p < 1e-3; Supplementary Table S7). The most enriched biological processes included oxidation-reduction process (GO: 0055114), carbohydrate metabolic process (GO: 0005975), detoxification (GO: 0098754), cellular response to toxic substance (GO: 0097237), involving 568, 230, 74, and 73 genes, respectively. Consistently, the most enriched molecular functions included oxidoreductase activity (GO: 0016491), cofactor binding (GO: 0048037), and coenzyme binding (GO: 0050662). In contrast, there were no enriched GO term detected in Avcr2-specific transcripts.
In addition to the exclusive presence or absence of transcripts, a total of 1,605 DEGs (Bonferroni adjusted p < 0.05 and fold changes ≥ 2) were detected between vcr2 and Avcr2 pathotypes, including 169 in aeciospores and 1,452 in cankered stems (Figure 2B). Of these, 8 and 122 DEGs were not detectable (FPKM = 0) in cankered stems infected by Avcr2 and vcr2, respectively (Figure 2B). The majority of aeciospore DEGs (160 of 169) were upregulated in vcr2 compared to Avcr2, while the majority of cankered stem DEGs (1,361 of 1,452) were upregulated in Avcr2 compared to vcr2. GO enrichment revealed seven biological processes enriched in DEGs in aeciospore (FDR-adjusted p < 1e-3), including cellular process, macromolecule modification, regulation of cellular component organization, cellular component organization or biogenesis, cellular protein modification process, regulation of organelle organization, and organelle organization. DEGs in cankered stems were enriched with four other biological processes (FDR-adjusted 0.01 < p < 0.05), nucleic acid metabolic process, macromolecule modification, regulation of nucleic acid-templated transcription, and regulation of cellular process (Supplementary Table S7).
Expression Profiling of the Cri secretome Identified vcr2 Candidates
Most SPs belonged to gene families with multiple members with differential expression among family members. Among the top 60 genes expressed at the highest levels (FPKM > 1,000) in infected pine stems, 40 of them were predicted to be SPs and annotated with functions such as GH5, GH12, GH17, GH18, GH27, PL3, HSP12, subtilisin protease, and thaumatin-like protein (Supplementary Table S8). A total of 901 SPs were found with average FPKM >1; and 376 of those were expressed in both pathotypes in aeciospores and mycelium growing inside the pine stems (Figure 1). These conserved genes likely represent a core group of effector candidates enriched for effectors that may be essential for successful fungal infection into five-needle pines and Ribes.
Furthermore, 131 and 60 SPs were specifically expressed in either the vcr2 or Avcr2 pathotype, respectively, at a cut-off of FPKM ≥ 1 (Figure 1; Supplementary Table S6). Of these, 36 and 16 SPs were not detectable in the other pathotype, i.e., with FFPKM = 0 in any of the Avcr2 and vcr2 samples. Specifically expressed SPs in vcr2 cankered stems were annotated as ATP-NAD kinase, cutinase 1, endo-beta-1,6-glucanase, exonuclease, GT24, CE8, isoleucyl-tRNA synthetase, ligase, N-acylphosphatidylethanolamine-specific phospholipase D (NAPEPLD), nucleotidyltransferase, oxidoreductase, PR-1-like protein, protein disulfide isomerase (PDI), protein disulfide oxidoreductase (PDOR), subtilisin-like protein, UDP-N-acetylglucosamine-dolichyl-phosphate N-acetylglucosaminephosphotransferase, and other hypothetical proteins. In contrast, few specifically expressed SPs in Avcr2 cankered stems were annotated; this list included homologs to ATP-NAD kinase, carboxylesterase, PL1, GH5, GH8, and pupal cuticle 27-like protein. Comparisons between expression profiles and functional annotations revealed that the SPs specifically expressed in vcr2 cankered stems most likely have an effect on the virulence of vcr2 pathotype for overcoming Cr2-conferred resistance.
In addition, 131 of the 1,605 DEGs identified above, were predicted to be SP-coding DEGs, of which 93% (122/131) were candidate effector genes upregulated in Avcr2 cankered stem compared with vcr2. B2G analysis revealed that 77% of the DEGs (101/131) had significant homology hits to CE4, CBM13 and 18, GH of various families, PL3, proteases/peptidase, peroxidase, thaumatin-like protein, aromatic compound dioxygenases, and others. Apparently, the majority of these differentially expressed SPs are conserved and play indispensable roles during tree infection in compatible Cr2-Avcr2 interactions. In contrast, only four SP-coding DEGs were upregulated in vcr2 cankered stem, annotated as cutinase, GH10, GH61 domain protein, and hypothetical protein.
Genomic Variations and Association With Cri Virulence
To compare genotypes between the two pathotypes, small genomic variations, including single nucleotide variant/polymorphism (SNV/SNP), multi-nucleotide variant (MNV), and insertion/deletion (InDel), were examined based on mapping reads to the vcr2 reference transcriptome. A total of 696,284 and 609,783 DNA variants were identified for vcr2 and Avcr2, respectively. The distribution of MAF showed vcr2 had lower proportions of variants with MAF ≥ 0.3, but higher proportion of indel and ns-SNPs than Avcr2 (X2 test, p < 1e-3; Supplementary Figure S3), suggesting greater genetic diversity of Avcr2 populations obtained from sequencing data. Analysis of DNA variants, showed that both pathotypes shared 65,617 SNPs (Figure 4A), distributed within 7,749 polymorphic transcripts, averaging 4.4 SNPs/kb.
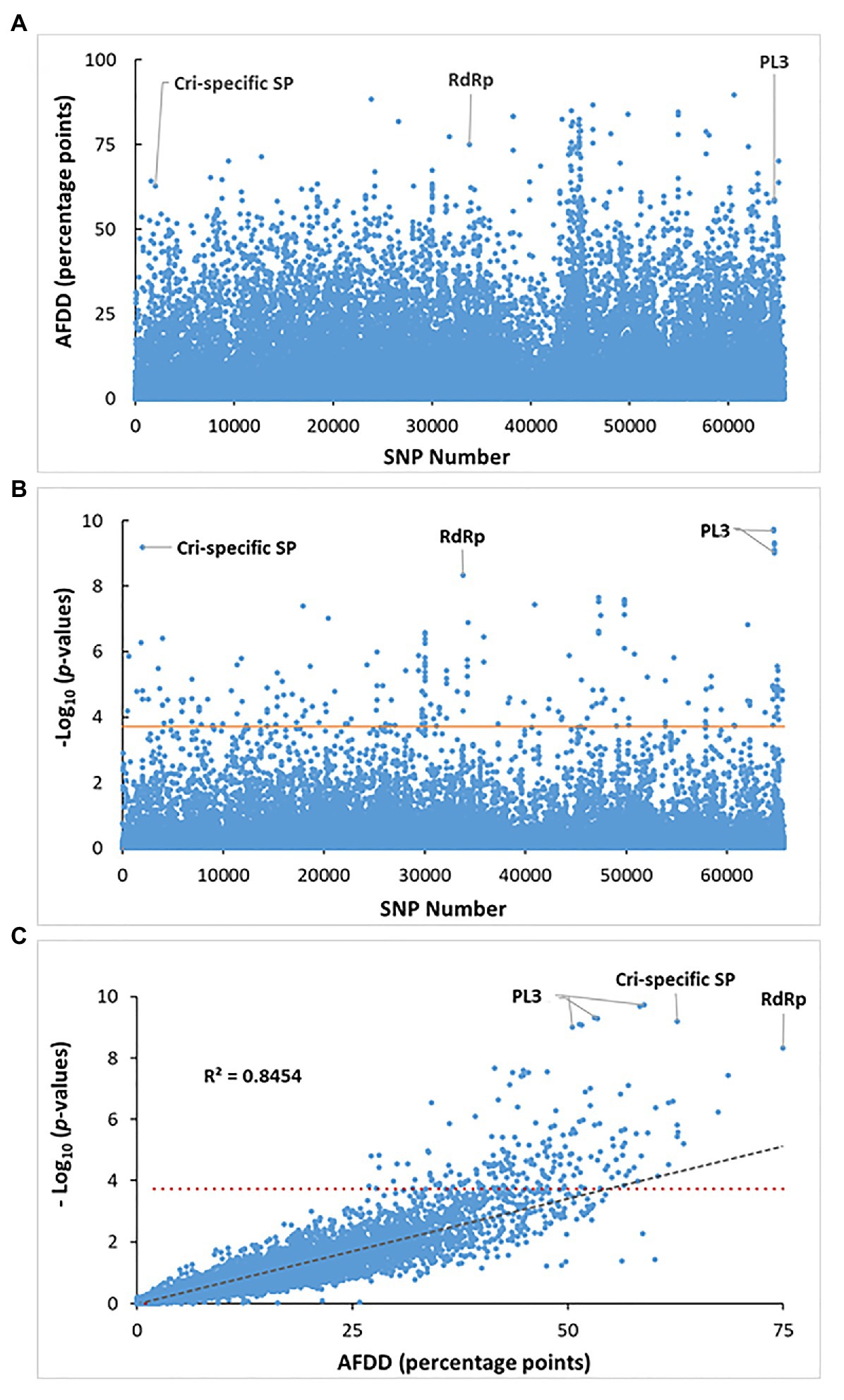
Figure 4. Identification of SNP loci for significant association with the virulent phenotype. (A) Distribution of allele frequency directional difference (AFDD) across all tested 65,617 SNPs shared between the vcr2 and wild Avcr2 pools. About 3% of total SNPs showed AFDD >30. (B) Manhattan plot of association p-values across all tested 65,617 SNPs analyzed by XP-GWAS. Three genes are labeled to show their SNPs with association at the most significant levels. (C) Scatter plotting AFDD values against p values analyzed by XP-GWAS across all tested SNPs. The horizontal red lines in (B) and (C) indicates the FDR-corrected threshold of p = 2e-04 for SNP loci with significant association. The gray dashed line in (C) shows that FADD values are highly correlated with the p-values analyzed by XP-GWAS for the corresponding SNP loci.
The allele frequencies for the 65,617 SNPs shared by both vcr2 and Avcr2 pools were calculated (Supplementary Figure S4) and used to normalize reference and alternative allele counts for XP-GWAS analysis. Of the 65,617 shared SNPs, 2011 SNPs (3% of the total) showed FADD > 30 (Figure 4A). The statistical analysis by XP-GWAS identified 204 SNP loci with significant vcr2-association after FDR multiple testing correction at a threshold of p = 2e-04 (Figure 4B). Of the 204 significantly associated SNPs, 198 of them (97% of the total) had FADD > 30 percentage points (Figures 4B,C). These associated SNPs were distributed in 119 genes (Supplementary Table S9). As shown in Figure 4C, by plotting the XP-GWAS calculated values of p against FADD values across all 65,617 SNPs, we demonstrated that the results were highly correlated with each other (R2 = 0.8454). Top associated SNPs were detected in three genes denoted as contig401 (1.82E-10 ≤ p ≥ 9.74E-10), Spr127863_c1_seq3 (p = 6.24E-10), and F_Contig7773 (p = 4.79E-9); which encoded PL3, Cri-specific SP (without any BLAST hit in the NCBI-nr database), and RNA-dependent RNA polymerase (RdRp), respectively (Figure 4; Supplementary Tables S6, S9, S10).
The 198 associated SNPs were filtered with FADD > 40 and MAF < 0.1, resulting in 87 SNPs within 52 genes (Supplementary Table S10), including 63 SNPs in 41 genes under the hypothesis of dominant avirulence and 24 SNPs in 12 genes under the hypothesis of dominant virulence. Their annotations and BLAST top-hit homologs from corresponding species are shown in Supplementary Table S10. The putative RdRp-encoding gene (F_Contig7773) was the sole one that was detected with significantly associated SNPs under both hypothesis. Of these 52 top-associated genes, four genes (SB-contig463, F_Contig7868, SB-contig401, and F_OR-Aec15165_c0_seq1) were highly polymorphic, with eight, six, and five, and five significant SNPs, respectively.
Of the 41 vcr2-associated genes with presumed dominant avirulence, five genes (Spr127863_c1_seq3, F_OR-Aec12603_c0_seq1, NA_HC37594_c0_seq1, SB-Spr-contig11, and SB-contig463) encoded SPs as predicted above. Notably, the most polymorphic gene SB-contig463 encoded a SP with putative reticuline oxidase activity, with the highest level of similarity (BLAST hit E-value of 4.8e-168) to a homolog from Amborella trichopoda (GenBank accession no. ERN08152.1). Furthermore, six of eight associated SNPs were nsSNPs (418cac/gac=His/Asp, 422atc/gtc=Ile/Val, 1156aaa/caa=Lys/Gln, 1190aac/agc=Asn/Ser, 1195tat/gat=Tyr/Asp, and 1240ccc/tcc=Pro/Ser), and may functionally alter Cri reticuline oxidase activity. Interestingly, SB-contig463 was de novo induced in cankered stems infected by vcr2, with 23-fold higher expression than in cankered stems infected by Avcr2 (Supplementary Table S5). F_OR-Aec12603_c0_seq1 encoded an rds1p-like stress responsive protein predicted as a SP, and the BLAST top hit revealed its closest homolog (BLAST E value = 3.2e-168) in Melampsora larici-populina (GenBank accession no. XP_007404719.1). One of three associated SNPs was nsSNP (714ttc/ctc=Phe/Leu) in F_OR-Aec12603_c0_seq1. This gene was mainly expressed in aeciospores with very low level or no expression in the cankered stems of WWP trees. SB-Spr-contig11 was also detected with one nsSNP (481gct/act=Ala/Thr). This gene encoded a predicted SP and was annotated as a fungal-specific CFEM domain protein, with the highest similarity to a putative SP (GenBank accession no. EGG12953.1) from M. larici-populina but at a low homology level (BLAST E-value of 1.4e-44). The other two SP-coding genes, Spr127863_c1_seq3_m.45366 and NA_HC37594_c0_seq1, were Cri-specific without any BLAST hit when searching against NCBI nr database (Supplementary Table S10).
Of 12 vcr2-associated genes with presumed dominant virulence, SB-contig401 encoded a SP that was annotated as PL3 protein. Four of its five associated SNPs were ns-SNPs (56gat/ggt=Asp/Gly, 67tct/gct=Ser/Ala, 76tac/gac=Tyr/Asp, and 82gtt/att=Val/Ile). This Cri PL3 protein was highly divergent from its homologs in other fungi, with the BLAST top-hit to a Mixia osmundae PL3 (GenBank accession no. XP_014566663.1) at low similarity level (BLAST E-value of 2.8e-32). Its transcript was highly induced in cankered stems infected by Avcr2 with 7-fold higher expression than that infected by vcr2 (Supplementary Tables S6, S9, S10).
Among other top associated genes, both F_OR-Aec15165_c0_seq1 and F_Contig7868 were highly polymorphic, and they encoded a putative alpha-2 subunit of Gpa2-Guanine nucleotide-binding protein (Gpa2) and a PH domain containing protein, respectively. Both annotated functions were involved in signaling pathways. The Cri Gpa2 was a fungal conserved protein, with the highest homology (BLAST E-value of 5.4e-156) to a Puccinia triticina protein (GenBank accession no. OAV91176.1). Within F_OR-Aec15165_c0_seq1, three of its five-associated SNPs were nsSNPs (652tga/tgc=Stop/Cys, 691ctc/cac=Leu/His, and 1133tac/cac=Tyr/His). Only one nsSNP (1004gaa/gga=Glu/Gly) was detected in five SNPs of F_Contig7868, but its change from acidic to neutral amino acid suggests a functional variation. These putative functional variations of signaling components imply that both genes may be functional candidates contributing to vcr2-related virulence.
Among the top vcr2-associated genes (Supplementary Table S10), other annotated functions included RdRp (F_Contig7773), arginyl-tRNA synthetase (RARS, F_Contig1146), ubiquitin system component Cue (F_Contig1284), deoxyhypusine synthase (F_Contig4649), CAMK/CAMKL/KIN4 protein kinase (F_Contig7285), RraA-like protein (F_Contig811), Eisosome component PIL1-domain-containing protein (F_Contig9075), glucose-repressible alcohol dehydrogenase transcriptional effector (F_OR-Aec12969_c0_seq1), DUF21 and cystathionine beta synthase (CBS) domain containing protein (F_OR-Aec7227_c0_seq1), and splicing factor U2AF 65 kDa subunit (F_Contig4947), and others (Supplementary Table S10).
Discussion
An Integrative Genomic Approach for Investigating Cri Virulence
Genetics of pathogen infectivity and host resistance are of fundamental importance to pathogen persistence and patterns of disease incidence and prevalence. The present study integrated research approaches of global profiling of transcriptomes and secretomes and BSR-Seq-based association studies to elucidate molecular mechanisms and candidate effectors involved in Cri vcr2 virulence.
Our results demonstrated that BSR-Seq was very effective for identifying vcr2-candidates and associated genes by comparing DNA variants between vcr2 and Avcr2. Because next-generation sequencing (NGS) can detect a large number of DNA variants at an affordable price, genome-wide association study (GWAS) has been developed and widely applied to identify phenotype-associated genes for functional verification. Using GWAS, candidate effectors and other pathogenicity-related genes were identified for their potential interaction with R genes in a few crop pathosystems (Bruce et al., 2014; Plissonneau et al., 2017; Wu et al., 2017). Exploration of allele frequency through NGS-based BSA is a powerful tool for determining genetic architecture causing phenotypic changes through associated DNA markers in targeted genomic regions (Schneeberger, 2014). A key step is exploration of AFDD by comparing allele frequencies between bulked samples. AFDD mapping is widely applied to genetic mapping of genetic loci underlying both qualitative and quantitative traits (Fekih et al., 2013; Takagi et al., 2013).
Of various GWAS approaches available, we used a BSR-Seq approach to identify vcr2-associated genes through integration of XP-GWAS analysis and FADD mapping. By combining RNA-seq with BSA, BSR-Seq has been demonstrated as an efficient approach for fine genetic mapping of agronomic traits in crops (Trick et al., 2012; Li et al., 2013; Ramirez-Gonzalez et al., 2015; Liu et al., 2016b) and their wild relatives (Edae and Rouse, 2019). Due to enrichment of genomic variants of the expressed genes, BSR-Seq facilitates identification of the genes causing or contributing to phenotypic traits of interest. Our BSR-Seq identified over 600K DNA variants in either vcr2 and Avcr2 bulked samples. Of the total variants, 65K SNPs common to vcr2 and Avcr2 were selected to detect vcr2-linked DNA markers. Shared markers are genetically informative because elimination of false positive variants is a challenging issue in NGS data processing (Huang et al., 2015; Ribeiro et al., 2015).
Our results indicated that without availability of a species reference sequence, BSR-Seq using a de-novo assembled transcriptome is an advantageous option to identify genome-wide SNPs. High consistency between XP-GWAS run and FADD mapping allowed us to refine candidate vcr2 effectors in the Cri secretome. The effectors that are shared or distinct between Cri Avcr2 and vcr2 pathotypes will be important in understanding host resistance and WP-BR interactions. However, RNA-Seq-based SNP discovery and related BSR-Seq-based association studies have limitations, in that they probably miss SNPs with low coverage due to low transcript expression, or with locations on the non-transcribed strand (O’Brien et al., 2015). Inter-individual variation of gene expression and allele-specific gene expression also influence allele frequency estimation accuracy (Konczal et al., 2014). As Cri samples were diploid or heterokaryotic, allele specific gene expression (ASGE) could conceivably impact allele frequency estimates. Therefore, the candidate vcr2 effectors need functional verification in a future study.
Transcriptomic Landscapes Underlying vcr2 Virulence
Comparative transcriptomics through enrichment analysis of GO terms revealed oxidation-reduction process, cellular response to toxic substance, and cellular oxidant detoxification as the main biological processes that are enhanced in vcr2 pathotypes relative to Avcr2 pathotypes, and thus may be key to vcr2 overcoming Cr2-mediated MGR in WWP seedlings. Pathogenic infection is accompanied by the formation of the reactive oxygen species (ROS) in both plants and pathogens. For successful infection, fungal pathogens must activate a series of intracellular biological processes to counteract negative effects resulting from the imbalance of redox homeostasis. A large set of Cri SPs were commonly expressed in both vcr2 and Avcr2, and their putative enzymatic activities included multi-copper oxidase laccase, peroxidases, L-ascorbate oxidases, SOD, and catalase, suggesting that they may act coordinately as ROS producers and scavengers for the maintenance of intracellular redox balance through a wide range of antioxidant defense mechanisms. Worth noting, several vcr2 exclusively expressed SPs were annotated as oxidoreductase, PDOR, PDI, and other redox-associated enzymes. PDO and PDI are key players for enzymatic disulfide bond reduction, oxidation and isomerization, contributing to essential steps in the protein folding pathway and to the stability of the native state of the proteins (Ladenstein and Ren, 2006). PDI-1 assists a set of glycoproteins in folding and disulfide bonds formation during effector secretion into the apoplast of maize plants during Ustilago maydis infection and its deletion led to a strong reduction in virulence (Marín-Menguian et al., 2019).
In addition to ROS stresses, Cri infection also induces host cells to produce and accumulate a large array of related anti-microbial peptides and PR proteins (Liu et al., 2013), as well as defensive chemicals (Bullington et al., 2018). Therefore, pathogens have adapted a wide spectrum of gene functions to fight against plant defense responses, constituting a diverse array of pathogenicity-related components for fungi to infect a broad range of hosts (Nakajima and Akutsu, 2014). Cri SPs belonging to several functional classes, such as GHs, GTs, subtilisin-like proteases, cutinase, endo-beta-1,6-glucanase, lipase, PR-1-like, and other enzymes involved in cell wall degradation and the synthesis of secondary metabolites with potential phytotoxic effects, were shown to be expressed in a vcr2-specific pattern, suggesting contributions to the successful infection of WWP Cr2-seedlings. Many fungal SPs with activities as degradative enzymes of plant cell wall components play functional roles in pathogenicity and virulence, evidenced by their triggering of host cell-death and other plant immunity responses (Ma et al., 2015; Zhu et al., 2017a; Gui et al., 2018). Cutinases from several fungal pathogens were shown to have activity as necrotrophic effectors (Liu et al., 2016a; Wang et al., 2017; Gui et al., 2018; Lu et al., 2018). A Fusarium oxysporum PR-1-like protein (Fpr1) is required for full virulence of F. oxysporum on a mammalian host (Prados-Rosales et al., 2012). Candida albicans PR1-like proteins displayed sterol binding activity with a link to virulence, which may affect fungal recognition by host cells through immune evasion by masking pathogen-associated molecular patterns (PAMPs; Bantel et al., 2018). Fungal subtilisin-like proteases disrupt the physiological integrity of the hosts during penetration and colonization, and are proposed as virulence factors in pathogenic fungi (Huang et al., 2004; Bryant et al., 2009). Our profiling of Cri transcriptomes and secretomes revealed both shared and pathotype-specific transcriptional programs. Annotated molecular functions of candidate genes imply their involvement in Cri pathogenesis. SP genes identified by transcript presence/absence provide vcr2-candidates with a high priority for further functional dissection of the virulence mechanism in the WPBR pathosystems.
Cri Effector Candidates and vcr2-Associated Genes
vcr2-associated genes were determined in either a homozygous or heterozygous status based on two separate assumptions of dominancy for either avirulence or virulence. These two assumptions are based on gene-for-gene and matching allele models, respectively, the main models representing the basic genetic mechanisms underlying plant-microbe molecular interactions (Flor, 1951; Lambrechts et al., 2006; Thrall et al., 2016). Gene-for-gene interaction assume that vcr2 overcomes MGR through escaping recognition by the WWP Cr2 gene. The WWP Cr2 protein is probably an immune receptor encoded by a NBS-LRR gene that specifically interacts with the corresponding Avcr2 effector expressed by Cri (Liu et al., 2017). Interaction of an avirulent (Avr) effector and its specific R protein resulting in incompatibility is known as effector-triggered immunity (Jones et al., 2016). Therefore, evolution of a rust virulent race (such as vcr2) is thought to involve a mutation (including deletion) of an Avr gene for a loss of function. Absence of the Avr effector leads to the pathogen being unrecognizable by the corresponding host R protein, resulting in successful infection of previously resistant hosts (Dodds and Rathjen, 2010).
Annotation of significantly associated SNPs revealed six SPs as effector candidates: reticuline oxidase, rds1p-like stress response protein, fungal-specific CFEM domain protein, PL3, and two Cri-specific SPs (Spr127863_c1_seq3 and NA_HC37594_c0_seq1) without any annotated function.
Reticuline oxidase belongs to the super-family of the berberine bridge enzymes (BBEs) found in bacteria, fungi, and plants. Six of eight associated SNPs in the Cri BBE homolog were ns-SNPs (the highest FADD = 51 with FDR-corrected p = 0.003). In addition, in planta expression of the Cri BBE homolog was 23-fold higher in vcr2 than in Avcr2. Nonsynonymous mutations of Avr genes have resulted in an acquired virulence for several R genes (Mesarich et al., 2014; Chen et al., 2017; Salcedo et al., 2017). BBEs are flavoenzymes that catalyze carbohydrate oxidation, either for the biosynthesis of berberine type alkaloids, or for H2O2 generation (Daniel et al., 2017). Several fungal BBEs showed activity for oxidation of oligosaccharides degraded from chitin and xylan. As oligosaccharides are fungal elicitors, their deactivation would lead to downregulation of the plant immune response (Daniel et al., 2017). Among fungal BBEs with oxidation activity, a Phytophthora infestans BBE-like enzyme (PITG_02930) proved to be required for the invasion of host plants (Raffaele et al., 2010). On the other hand, a few fungal BBEs catalyze complex reactions for biosynthesis of alkaloids that are involved in anti-biotic mechanisms (Daniel et al., 2017).
Fungal-specific CFEM domain proteins contain eight cysteines. They were highly divergent across different species and have been proposed to play roles in fungal pathogenesis (Kulkarni et al., 2003). A recent study reported contribution of a Botrytis cinerea CFEM domain protein (BcCFEM1) to virulence, conidial production, and stress tolerance (Zhu et al., 2017b). The Cri rds1p-like protein contained a ferritin-like domain, belonging to the Ferritin-like superfamily, whose role in defense against toxic oxygen species are well documented (Andrews, 2010). The Cri rds1p-like gene was highly expressed in aeciospores and urediniospores, but at a low to no expression levels in cankered pine stems, suggesting that it may play a role during interactions of Cri with its alternative host plants.
PL3 enzymes hydrolyze long-chain acyl-triglycerides into di‐ and monoglycerides, glycerol, and free fatty acids, and their activities appear to be important for the pathogenicity of several fungi (Gaillardin, 2010). A Cri PL3 homolog was identified with two ns-SNPs under the assumption of virulence dominancy. This gene was also among DEG, with an expression level 7-fold higher in Avcr2 than in vcr2, suggesting its potential role as effector in avirulence. Several fungal PLs have been implicated as having effector roles in plant pathogens (Ben-Daniel et al., 2011; Yang et al., 2018). Additionally, almost all effectors are highly expressed in planta to play important roles in pathogenicity and virulence (Mesarich et al., 2014). In planta upregulation of effector expression may help fungal mycelia to extract nutrition, in addition to the more obvious role of physically facilitating invasion of the host tissue. Consistently, both Cri BBE and PL3 homologs are highly expressed in cankered stems relative to their expression during the aeciospore stage.
In addition to SPs, annotations of other top vcr2-associated genes implied their involvement in virulence, including PH domain containing protein, Gpa2, RdRp, arginyl-tRNA synthetase, deoxyhypusine synthase, eisosome component PIL1-domain-containing protein, and DUF21 and CBS domain containing protein. PH domain proteins play a role in intracellular signaling or as constituents of the cytoskeleton (Lemmon and Ferguson, 2000). The PH domain has been found in different proteins, such as regulators of small G-proteins and G protein receptor kinases (Fort and Blangy, 2017; Komolov and Benovic, 2018). A Cryptococcus neoformans PH domain containing protein (Cin1) was found to have pleiotropic functions in morphogenesis, endocytosis, exocytosis, and fungal virulence (Wang and Shen, 2011). Gpa2 is another modulator with roles in various transmembrane signaling systems, including response to environmental nutrients (Harashima et al., 2006), regulation of fungal morphogenesis and hypha formation (Miwa et al., 2004), and heat resistance (Kraakman et al., 1999).
Mutations of aminoacyl-tRNA synthetase (ArgRS) genes have been implicated in a spectrum of inherited, single-gene (Mendelian) human disorders (Oprescu et al., 2017). One of Cri ARS (Ile-tRNA synthetase gene) was specially expressed in vcr2-infected cankered stems. Arginyl-tRNA synthetase is well known because its homozygous variant causes a rare Mendelian recessive disorder called Pelizaeus-Merzbacher disease in humans (Nafisinia et al., 2017). Overexpression of plant proteins containing CBS and DUF21 domains conferred higher tolerance against various abiotic stresses such as salinity, heavy metals, oxidative stress, and low nitrogen conditions (Singh et al., 2012; Hao et al., 2016). Deoxyhypusine synthase catalyzes the biosynthesis of hypusine, a modification of a specific lysine residue in the precursor of eukaryotic translation initiation factor 5A (eIF-5). A yeast deoxyhypusine synthase mutant (dys1-1) exhibited defects associated with protein synthesis, translation machinery, and cell wall integrity; and a complete growth dependency on an osmotic stabilizer (1 M sorbitol; Galvao et al., 2013). Fungal proteins Pil1 and Lsp1 are key components for formation of eisosomes. At least another 17 proteins including two PH domain containing proteins, were detected in the eisosomes. Eisosome mutants display abnormal spatial regulation of cell wall synthesis, higher sensitivity to a variety of oxidants, and a greatly reduced virulence (Foderaro et al., 2017). RdRps are key components of RNA viruses. Several RNA mycoviruses are well documented for the harmful effect on their host fungi by causing mitigation of fungal virulence or hypovirulence (Hillman et al., 2018). A previous study showed a Cri totivirus (CrTV4) with significant association with vcr2 virulence (Liu et al., 2019), supporting a proposed hypothesis of cytoplasmic inheritance of Cri virulence (Kinloch et al., 2004). All of the vcr2-associated functions might target genomic regions which are potentially responsible for Cri virulence adaptation. Additional annotations of the top vcr2-associated genes were CAMK/CAMKL/KIN4 protein kinase, RraA-like protein, glucose-repressible alcohol dehydrogenase transcriptional effector, and splicing factor U2AF 65 kDa subunit. However, networking of these functions with pathogenic virulence awaits future studies.
Currently identification of Cri virulence pathotypes is time-consuming, totally dependent on a race’s ability to overcome resistance of MGR trees with well-known genotypes by natural infection or controlled inoculation. Through this approach, we collected Cri samples of the vcr2 pathotype from both western and eastern Oregon, several areas of the latter region were previously surveyed but neither vcr2 nor Cr2 was found (Kinloch et al. 2003, 2004). Our inoculation utilized very high spore loads (>6,000 spores/cm2) on MGR seedlings, allowing visualization and sampling of vcr2 races even though they were presumably at low frequency in a heterogeneous mixture from eastern Oregon. The identification of effector candidates and vcr2-associated genes would allow future efforts to isolate the virulent effectors and determine their functions for development of diagnostic tools for monitoring Cri virulence across North America. The SNP assay will be developed from vcr2-associated genes, which could be useful in the future for monitoring for presence of vcr2, or determining if vcr2 is present in infected stems of Cr2/– genotypes rather than potential other explanations. Discovery of a large number of genes as well as related biological processes contributing to Cri pathogenicity provides a valuable insight into the comprehensive understanding of molecular WP-BR interactions.
Data Availability Statement
All sequencing data generated in this study are available from the SRA-Archive (http://www.ncbi.nlm.nih.gov/sra) under the study accession SRP031625. Raw reads of the Illumina RNA-seq 100-bp PE sequences from nine samples were deposited in the NCBI SRA under accession number SRR11101722‐ SRR11101730. A Cronartium ribicola vcr2 reference transcriptome was deposited at DDBJ/EMBL/GenBank under TSA accession GIKE00000000. All other supporting data are included in the Supplementary Material.
Author Contributions
J-JL and RS conceived the research and supervised the project. RS, AK, and DS provided the genetic materials, conducted the disease ratings, and analyzed phenotypic data. J-JL, AZ, HW, and KO conducted biochemical experiments and analyzed NGS data. J-JL wrote the original draft with contributions from all authors. All authors contributed to the article and approved the submitted version.
Funding
This project was supported in part by fund of the CFS Pest Risk Management Project awarded to J-JL. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank E. Ayala, B. Rancourt, and G. Zhang at PFC-CFS for the kind help with sample processing and computing programming and technicians at DGRC who assisted with sample collections.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.602812/full#supplementary-material
Supplementary Table S1 | Statistics of RNA-seq reads and gene expression analysis of reads mapped back to the vcr2 reference transcriptome.
Supplementary Table S2 | Statistics of the vcr2 reference transcriptome generated in the present study.
Supplementary Table S3 | Output of OrthoVenn2 ortholog cluster analysis of Cronartium ribicola vcr2 and Avcr2 pathotypes.
Supplementary Table S4 | GO-term enrichment analysis of pathotype-unique clusters detected by OrthoVenn2.
Supplementary Table S5 | Amino acid sequences of 1014 secreted proteins predicted in Cronartium ribicola transcriptomes.
Supplementary Table S6 | Annotation and expression levels of 1014 proteins identified in the Cronartium ribicola secretome.
Supplementary Table S7 | Enriched GO-terms of pathotype-specifically expressed genes and differentially expressed genes as detected by RNA-seq Analysis.
Supplementary Table S8 | Cronartium ribicola unigenes with the most abundant transcripts (FPKM > 1,000) expressed in cankered stems of western white pine with the Cr2/− genotype.
Supplementary Table S9 | Putative protein sequences of 119 associated genes.
Supplementary Table S10 | Annotation of 87 top vcr2-associated SNPs in 52 polymorphic genes.
Supplementary Figure S1 | Distributions of transcripts and RNA-seq reads for the transcriptomes de-novo assembled from Cronartium ribicola vcr2 samples. Western white pine transcripts from vcr2-infected host tissues were pre-removed by BLASTn search against transcriptomes of healthy host tissues. MEGA analysis assigned all transcripts into four categories (fungi, no hits, not assigned, and other). (A) Distribution of transcripts across four categories; (B) Distribution of the mapped RNA-seq reads across four categories.
Supplementary Figure S2 | Ortholog clusters analysis between Cronartium ribicola vcr2 and Avcr2 pathotypes using OrthoVenn2 (Xu et al., 2019).
Supplementary Figure S3 | Comparisons of minor allele frequency (MAF) distributions between vcr2 and Avcr2. (A) Distribution of minor allele frequency (MAF) of all DNA variants. A total of 696K and 609K DNA variants with site coverage >10 were analyzed in vcr2 and Avcr2, respectively. (B) MAFs of 65,617 SNPs shared between vcr2 and Avcr2. (C) Three types of DNA variants (SNV/SNP, MNV, and Indel). (D) Two types of SNPs (synonymous-SNP and nonsynonymous-SNP) detected in the coding regions. Three stars indicate significant difference between vcr2 and Avcr2 (X2 test at p < 0.001).
Supplementary Figure S4 | Distribution of allele frequency (AF) of 65,617 SNPs shared by vcr2 and Avcr2.
Footnotes
References
Altschul, S., Gish, W., and Miller, W. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Andrews, S. C. (2010). The ferritin-like superfamily: evolution of the biological iron storeman from a rubrerythrin-like ancestor. Biochim. Biophys. Acta 1800, 691–705. doi: 10.1016/j.bbagen.2010.05.010
Bantel, Y., Darwiche, R., Rupp, S., Schneiter, R., and Sohn, K. (2018). Localization and functional characterization of the pathogenesis-related proteins Rbe1p and Rbt4p in Candida albicans. PLoS One 13:e0201932. doi: 10.1371/journal.pone.0201932
Ben-Daniel, B. -H., Bar-Zvi, D., and Lahkim, L. T. (2011). Pectate lyase affects pathogenicity in natural isolates of Colletotrichum coccodes and in pelA gene-disrupted and gene-overexpressing mutant lines. Mol. Plant Pathol. 13, 187–197. doi: 10.1111/j.1364-3703.2011.00740.x
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Brar, S., Tsui, C. K., Dhillon, B., Bergeron, M. J., Joly, D. L., Zambino, P. J., et al. (2015). Colonization, history, host distribution, anthropogenic influence and landscape features shape populations of white pine blister rust, an invasive alien tree pathogen. PLoS One 10:e0127916. doi: 10.1371/journal.pone.0127916
Bruce, M., Neugebauer, K. A., Joly, D. L., Migeon, P., Cuomo, C. A., Wang, S., et al. (2014). Using transcription of six Puccinia triticina races to identify the effective secretome during infection of wheat. Front. Plant Sci. 4:520. doi: 10.3389/fpls.2013.00520
Bryant, M. K., Schardl, C. L., Hesse, U., and Scott, B. (2009). Evolution of a subtilisin-like protease gene family in the grass endophytic fungus Epichloë festucae. BMC Evol. Biol. 9:168. doi: 10.1186/1471-2148-9-168
Bullington, L. S., Lekberg, Y., Sniezko, R., and Larkin, B. (2018). The influence of genetics, defensive chemistry and the fungal microbiome on disease outcome in whitebark pine trees. Mol. Plant Pathol. 19, 1847–1858. doi: 10.1111/mpp.12663
Chen, J., Upadhyaya, N. M., Ortiz, D., Sperschneider, J., Li, F., Bouton, C., et al. (2017). Loss of AvrSr50 by somatic exchange in stem rust leads to virulence for Sr50 resistance in wheat. Science 358, 1607–1610. doi: 10.1126/science.aao4810
Daniel, B., Konrad, B., Toplak, M., Lahham, M., Messenlehner, J., Winkler, A., et al. (2017). The family of berberine bridge enzyme-like enzymes: a treasure-trove of oxidative reactions. Arch. Biochem. Biophys. 632, 88–103. doi: 10.1016/j.abb.2017.06.023
de Castro, E., Sigrist, C. J. A., Gattiker, A., Bulliard, V., Langendijk-Genevaux, P. S., Gasteiger, E., et al. (2006). Scan-Prosite: detection of PROSITE signature matches and ProRule-associated functional and structural residues in proteins. Nucleic Acids Res. 34, W362–W365. doi: 10.1093/nar/gkl124
De Fine Licht, H. H., Jensen, A. B., and Eilenberg, J. (2017). Comparative transcriptomics reveal host-specific nucleotide variation in entomophthoralean fungi. Mol. Ecol. 26, 2092–2110. doi: 10.1111/mec.13863
Devlin, B., and Roeder, K. (1999). Genomic control for association studies. Biometrics 55, 997–1004. doi: 10.1111/j.0006-341X.1999.00997.x
Dodds, P. N., and Rathjen, J. P. (2010). Plant immunity: towards an integrated view of plant-pathogen interactions. Nat. Rev. Genet. 11, 539–548. doi: 10.1038/nrg2812
Dougherty, L., Singh, R., Brown, S., Dardick, C., and Xu, K. (2018). Exploring DNA variant segregation types in pooled genome sequencing enables effective mapping of weeping trait in Malus. J. Exp. Bot. 69, 1499–1516. doi: 10.1093/jxb/erx490
Edae, E. A., and Rouse, M. N. (2019). Bulked segregant analysis RNA-seq (BSR-Seq) validated a stem resistance locus in Aegilops umbellulata, a wild relative of wheat. PLoS One 14:e0215492. doi: 10.1371/journal.pone.0215492
Fekih, R., Takagi, H., Tamiru, M., Abe, A., Natsume, S., Yaegashi, H., et al. (2013). MutMap: genetic mapping and mutant identification without crossing in rice. PLoS One 8:e68529. doi: 10.1371/journal.pone.0068529
Flor, H. H. (1951). Genes for resistance to rust in victory flax. Agron. J. 43, 527–531. doi: 10.2134/agronj1951.00021962004300110001x
Foderaro, J. E., Douglas, L. M., and Konopka, J. B. (2017). MCC/eisosomes regulate cell wall synthesis and stress responses in fungi. J. Fungi 3:61. doi: 10.3390/jof3040061
Fort, P., and Blangy, A. (2017). The evolutionary landscape of Dbl-like RhoGEF families: adapting eukaryotic cells to environmental signals. Genome Biol. Evol. 9, 1471–1486. doi: 10.1093/gbe/evx100
Fu, L., Niu, B., Zhu, Z., Wu, S., and Li, W. (2012). CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152. doi: 10.1093/bioinformatics/bts565
Gaillardin, C. (2010). “Lipases as pathogenicity factors of fungi” in Handbook of hydrocarbon and lipid microbiology. ed. K. N. Timmis (Heidelberg, Berlin: Springer), 2010.
Galvao, F. C., Rossi, D., Silveira, W. d. S., Valentini, S. R., and Zanelli, C. F. (2013). The deoxyhypusine synthase mutant dys1-1 reveals the association of eIF5A and Asc1 with cell wall integrity. PLoS One 8:e60140. doi: 10.1371/journal.pone.0060140
Geils, B. W., Hummer, K. E., and Hunt, R. S. (2010). White pines, Ribes, and blister rust: a review and synthesis. For. Pathol. 40, 147–185. doi: 10.1111/j.1439-0329.2010.00654.x
Götz, S., García-Gómez, J. M., Terol, J., Williams, T. D., Nagaraj, S. H., Nueda, M. J., et al. (2008). High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 36, 3420–3435. doi: 10.1093/nar/gkn176
Gui, Y. J., Zhang, W. Q., Zhang, D. D., Zhou, L., Short, D. P. G., Wang, J., et al. (2018). A Verticillium dahliae extracellular cutinase modulates plant immune responses. Mol. Plant-Microbe Interact. 31, 260–273. doi: 10.1094/MPMI-06-17-0136-R
Haas, B., Papanicolaou, A., Yassour, M., Grabherr, M., Blood, P. D., Bowden, J., et al. (2013). De novo transcript sequence reconstruction from RNA-seq using the trinity platform for reference generation and analysis. Nat. Protoc. 8, 1494–1512. doi: 10.1038/nprot.2013.084
Hao, Q., Shang, W., Zhang, C., Chen, H., Chen, L., Yuan, S., et al. (2016). Identification and comparative analysis of CBS domain-containing proteins in soybean (Glycine max) and the primary function of GmCBS21 in enhanced tolerance to low nitrogen stress. Int. J. Mol. Sci. 17:620. doi: 10.3390/ijms17050620
Harashima, T., Anderson, S., Yates, J. R., and Heitman, J. (2006). The kelch proteins Gpb1 and Gpb2 inhibit Ras activity via association with the yeast RasGAP neurofibromin homologs Ira1 and Ira2. J. Mol. Cell. 22, 819–830. doi: 10.1016/j.molcel.2006.05.011
Hillman, B. I., Annisa, A., and Suzuki, N. (2018). Viruses of plant-interacting fungi. Adv. Virus Res. 100, 99–116. doi: 10.1016/bs.aivir.2017.10.003
Huang, H. W., Comparative Sequencing Program, N. I. S. C., Mullikin, J. C., and Hansen, N. F. (2015). Evaluation of variant detection software for pooled next-generation sequence data. BMC Bioinformatics 16:235. doi: 10.1186/s12859-015-0624-y
Huang, X., and Madan, A. (1999). CAP3: a DNA sequence assembly program. Genome Res. 9, 868–877. doi: 10.1101/gr.9.9.868
Huang, X. W., Zhao, N. H., and Zhang, K. Q. (2004). Extracellular enzymes serving as virulence factors in nematophagous fungi involved in infection of the host. Res. Microbiol. 155, 811–816. doi: 10.1016/j.resmic.2004.07.003
Huson, D., Mitra, S., Ruscheweyh, H. -J., Weber, N., and Schuster, S. C. (2011). Integrative analysis of environmental sequences using MEGAN4. Genome Res. 21, 1552–1560. doi: 10.1101/gr.120618.111
Jones, J. D. G., Vance, R. E., and Dangl, J. L. (2016). Intracellular innate immune surveillance devices in plants and animals. Science 354:aaf6395. doi: 10.1126/science.aaf6395
Kall, L., Krogh, A., and Sonnhammer, E. L. L. (2004). A combined transmembrane topology and signal peptide prediction method. J. Mol. Biol. 338, 1027–1036. doi: 10.1016/j.jmb.2004.03.016
Kinloch, B. B. J., Sniezko, R. A., and Dupper, G. E. (2003). Origin and distribution of Cr2, a gene for resistance to white pine blister rust in natural populations of western white pine. Phytopathology 93, 691–694. doi: 10.1094/PHYTO.2003.93.6.691
Kinloch, B. B. J., Sniezko, R. A., and Dupper, G. E. (2004). Virulence gene distribution and dynamics of the white pine blister rust pathogen in western North America. Phytopathology 94, 751–758. doi: 10.1094/PHYTO.2004.94.7.751
Komolov, K. E., and Benovic, J. L. (2018). G protein-coupled receptor kinases: past, present and future. Cell. Signal. 41, 17–24. doi: 10.1016/j.cellsig.2017.07.004
Konczal, M., Koteja, P., Stuglik, M. T., Radwan, J., and Babik, W. (2014). Accuracy of allele frequency estimation using pooled RNA-Seq. Mol. Ecol. Resour. 14, 381–392. doi: 10.1111/1755-0998.12186
Kraakman, L., Lemaire, K., Ma, P., Teunissen, A. W., Donaton, M. C., Van Dijck, P., et al. (1999). A Saccharomyces cerevisiae G-protein coupled receptor, Gpr1, is specifically required for glucose activation of the cAMP pathway during the transition to growth on glucose. Mol. Microbiol. 32, 1002–1012. doi: 10.1046/j.1365-2958.1999.01413.x
Kulkarni, R. D., Kelkar, H. S., and Dean, R. A. (2003). An eight-cysteine-containing CFEM domain unique to a group of fungal membrane proteins. Trends Biochem. Sci. 28, 118–121. doi: 10.1016/S0968-0004(03)00025-2
Ladenstein, R., and Ren, B. (2006). Protein disulfides and protein disulfide oxidoreductases in hyperthermophiles. FEBS J. 273, 4170–4185. doi: 10.1111/j.1742-4658.2006.05421.x
Lambrechts, L., Fellous, S., and Koella, J. C. (2006). Coevolutionary interactions between host and parasite genotypes. Trends Parasitol. 22, 12–16. doi: 10.1016/j.pt.2005.11.008
Lemmon, M. A., and Ferguson, K. M. (2000). Signal-dependent membrane targeting by pleckstrin homology (PH) domains. Biochem. J. 350, 1–18. doi: 10.1042/bj3500001
Li, L., Li, D., Liu, S. Z., Ma, X. L., Dietrich, C. R., Hu, H. C., et al. (2013). The maize glossy13 gene, cloned via BSR-Seq and Seq-walking encodes a putative ABC transporter required for the normal accumulation of epicuticular waxes. PLoS One 8:e82333. doi: 10.1371/journal.pone.0085815
Liu, J. -J. (2012). Ectopic expression of a truncated Pinus radiata AGAMOUS homolog (PrAG1) causes alteration of inflorescence architecture and male sterility in Nicotiana tabacum. Mol. Breed. 30, 453–467. doi: 10.1007/s11032-011-9635-1
Liu, T., Hou, J., Wang, Y., Jin, Y., Borth, W., Zhao, F., et al. (2016a). Genome-wide identification, classification and expression analysis in fungal-plant interactions of cutinase gene family and functional analysis of a putative ClCUT7 in Curvularia lunata. Mol. Gen. Genomics 291, 1105–1115. doi: 10.1007/s00438-016-1168-1
Liu, J. -J., Sniezko, R. A., Sturrock, R. N., and Chen, H. (2014). Western white pine SNP discovery and high-throughput genotyping for breeding and conservation applications. BMC Plant Biol. 14:1586. doi: 10.1186/s12870-014-0380-6
Liu, J. -J., Sniezko, R. A., Zamany, A., Williams, H., Wang, N., Kegley, A., et al. (2017). Saturated genic SNP mapping identified functional candidates and selection tools for the Pinus monticola Cr2 locus controlling resistance to white pine blister rust. Plant Biotechnol. J. 15, 1149–1162. doi: 10.1111/pbi.12705
Liu, J. -J., Sturrock, R. N., and Benton, R. (2013). Transcriptome analysis of Pinus monticola primary needles by RNA-seq provides novel insight into host resistance to Cronartium ribicola. BMC Genomics 14:884. doi: 10.1186/1471-2164-14-884
Liu, J. -J., Sturrock, R. N., Sniezko, R. A., Williams, H., Benton, R., and Zamany, A. (2015). De novo sequencing of Cronartium ribicola transcriptome for understanding molecular white pine blister rust interactions. BMC Genomics 16:678. doi: 10.1186/s12864-015-1861-1
Liu, J. -J., Williams, H., Zamany, A., Li, X. -R., Gellner, S., and Sniezko, R. A. (2020). Development and application of marker-assisted selection (MAS) tools for breeding of western white pine (Pinus monticola Douglas ex D. Don) resistance to blister rust (Cronartium ribicola J.C. Fisch.) in British Columbia. Can. J. Plant Pathol. 42, 250–259. doi: 10.1080/07060661.2019.1638454
Liu, J. -J., Xiang, Y., Sniezko, R. A., Schoettle, A. W., Williams, H., and Arezoo, Z. (2019). Characterization of Cronartium ribicola dsRNAs reveals novel taxa of the family Totiviridae and viral association with fungal virulence. BMC Virol. J. 16:118. doi: 10.1186/s12985-019-1226-5
Liu, S. Z., Yeh, C. T., Tang, H. M., Nettleton, D., and Schnable, P. S. (2012). Gene mapping via bulked segregant RNA-Seq (BSR-Seq). PLoS One 7:e36406. doi: 10.1371/journal.pone.0053178
Liu, C., Zhou, Q., Dong, L., Wang, H., Liu, F., Weng, J., et al. (2016b). Genetic architecture of the maize kernel row number revealed by combining QTL mapping using a high-density genetic map and bulked segregant RNA sequencing. BMC Genomics 17:915. doi: 10.1186/s12864-016-3240-y
Lu, L., Rong, W., Massart, S., and Zhang, Z. (2018). Genome-wide identification and expression analysis of cutinase gene family in Rhizoctonia cerealis and functional study of an active cutinase RcCUT1 in the fungal–wheat interaction. Front. Microbiol. 9:1813. doi: 10.3389/fmicb.2018.01813
Ma, Z., Liu, J. -J., and Zamany, A. (2019). Identification and functional analysis of an effector secreted by Cronartium ribicola. Phytopathology 109, 942–951. doi: 10.1094/PHYTO-11-18-0427-R
Ma, Z., Song, T., Zhu, L., Ye, W., Wang, Y., Shao, Y., et al. (2015). A Phytophthora sojae glycoside hydrolase 12 protein is a major virulence factor during soybean infection and is recognized as a PAMP. Plant Cell 27, 2057–2072. doi: 10.1105/tpc.15.00390
Maloy, O. C. (2018). White pine blister rust: symptoms and signs. Plant Health Instructor. 2003:2018. doi: 10.1094/PHI-I-2003-0908-01.Updated
Marín-Menguiano, M., Moreno-Sánchez, I., Barrales, R. R., Fernández-Álvarez, A., and Ibeas, J. I. (2019). N-glycosylation of the protein disulfide isomerase Pdi1 ensures full Ustilago maydis virulence. PLoS Pathog. 15:e1007687. doi: 10.1371/journal.ppat.1007687
Mesarich, C. H., Griffiths, S. A., van der Burgt, A., Ökmen, B., Beenen, H. G., Etalo, D. W., et al. (2014). Transcriptome sequencing uncovers the Avr5 avirulence gene of the tomato leaf mold pathogen Cladosporium fulvum. Mol. Plant-Microbe Interact. 27, 846–857. doi: 10.1094/MPMI-02-14-0050-R
Michelmore, R. W., Paran, I., and Kesseli, R. V. (1991). Identification of markers linked to disease-resistance genes by bulked segregant analysis: a rapid method to detect markers in specific genomic regions by using segregating populations. Proc. Natl. Acad. Sci. 88, 9828–9832.
Miwa, T., Takagi, Y., Shinozaki, M., Yun, C. W., Schell, W. A., Perfect, J. R., et al. (2004). Gpr1, a putative G-protein-coupled receptor, regulates morphogenesis and hypha formation in the pathogenic fungus Candida albicans. Eukaryot. Cell 3, 919–931. doi: 10.1128/EC.3.4.919-931.2004
Mortazavi, A., Williams, B. A., Schaeffer, L., and Wold, B. (2008). Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5, 621–628. doi: 10.1038/nmeth.1226
Nafisinia, M., Sobreira, N., Riley, L., Gold, W., Uhlenberg, B., Weiß, C., et al. (2017). Mutations in RARS cause a hypomyelination disorder akin to Pelizaeus-Merzbacher disease. Eur. J. Hum. Genet. 25, 1134–1141. doi: 10.1038/ejhg.2017.119
Nakajima, M., and Akutsu, K. (2014). Virulence factors of Botrytis cinerea. J. Gen. Plant Pathol. 80, 15–23. doi: 10.1007/s10327-013-0492-0
O’Brien, T. D., Jia, P., Xia, J., Saxena, U., Jin, H., Vuong, H., et al. (2015). Inconsistency and features of single nucleotide variants detected in whole exome sequencing versus transcriptome sequencing: a case study in lung cancer. Methods 83, 118–127. doi: 10.1016/j.ymeth.2015.04.016
Oprescu, S. N., Griffin, L. B., Beg, A. A., and Antonellis, A. (2017). Predicting the pathogenicity of aminoacyl-tRNA synthetase mutations. Methods 113, 139–151. doi: 10.1016/j.ymeth.2016.11.013
Pendleton, A. L., Smith, K. E., Feau, N., Martin, F. M., Grigoriev, I. V., Hamelin, R., et al. (2014). Duplications and losses in gene families of rust pathogens highlight putative effectors. Front. Plant Sci. 5:299. doi: 10.3389/fpls.2014.00299
Petersen, T. N., Brunak, S., von Heijne, G., and Nielsen, H. (2011). SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods 8, 785–786. doi: 10.1038/nmeth.1701
Plissonneau, C., Benevenuto, J., Mohd-Assaad, N., Fouché, S., Hartmann, F. E., and Croll, D. (2017). Using population and comparative genomics to understand the genetic basis of effector-driven fungal pathogen evolution. Front. Plant Sci. 8:119. doi: 10.3389/fpls.2017.00119
Prados-Rosales, R. C., Roldán-Rodríguez, R., Serena, C., López-Berges, M. S., Guarro, J., Martínez-del-Pozo, Á., et al. (2012). A PR-1-like protein of Fusarium oxysporum functions in virulence on mammalian hosts. J. Biol. Chem. 287, 21970–21979. doi: 10.1074/jbc.M112.364034
Raffaele, S., Win, J., Cano, L. M., and Kamoun, S. (2010). Analyses of genome architecture and gene expression reveal novel candidate virulence factors in the secretome of Phytophthora infestans. BMC Genomics 11:637. doi: 10.1186/1471-2164-11-637
Ramirez-Gonzalez, R. H., Segovia, V., Bird, N., Fenwick, P., Holdgate, S., Berry, S., et al. (2015). RNA-Seq bulked segregant analysis enables the identification of high-resolution genetic markers for breeding in hexaploid wheat. Plant Biotechnol. 13, 613–624. doi: 10.1111/pbi.12281
Ribeiro, A., Golicz, A., Hackett, C. A., Milne, I., Stephen, G., Marshall, D., et al. (2015). An investigation of causes of false positive single nucleotide polymorphisms using simulated reads from a small eukaryote genome. BMC Bioinformatics 16:382. doi: 10.1186/s12859-015-0801-z
Richardson, B. A., Klopfenstein, N. B., Zambino, P. J., McDonald, G. I., Geils, B. W., and Carris, L. M. (2008). Influence of host resistance on the genetic structure of the white pine blister rust fungus in the western United States. Phytopathology 98, 413–420. doi: 10.1094/PHYTO-98-4-0413
Salcedo, A., Rutter, W., Wang, S., Akhunova, A., Bolus, S., Chao, S., et al. (2017). Variation in the AvrSr35 gene determines Sr35 resistance against wheat stem rust race Ug99. Science 358, 1604–1606. doi: 10.1126/science.aao7294
Schneeberger, K. (2014). Using next-generation sequencing to isolate mutant genes from forward genetic screens. Nat. Rev. Genet. 15, 662–676. doi: 10.1038/nrg3745
Singh, A. K., Kumar, R., Pareek, A., Sopory, S. K., and Singla-Pareek, S. L. (2012). Overexpression of rice CBS domain containing protein improves salinity, oxidative, and heavy metal tolerance in transgenic tobacco. Mol. Biotechnol. 52, 205–216. doi: 10.1007/s12033-011-9487-2
Sniezko, R. A., Johnson, J. S., and Savin, D. P. (2020). Assessing the durability, stability, and usability of genetic resistance to a non-native fungal pathogen in two pine species. Plants People Planet 2, 57–68. doi: 10.1002/ppp3.49
Sniezko, R. A., Smith, J., Liu, J. -J., and Hamelin, R. (2014). Genetic resistance to fusiform rust in southern pines and white pine blister rust in white pines – a contrasting tale of two rust pathosystems ‐ current status and future prospects. Forests 5, 2050–2083. doi: 10.3390/f5092050
Sperschneider, J., Dodds, P. N., Singh, K. B., and Taylor, J. M. (2018). ApoplastP: prediction of effectors and plant proteins in the apoplast using machine learning. New Phytol. 217, 1764–1778. doi: 10.1111/nph.14946
Takagi, H., Abe, A., Yoshida, K., Kosugi, S., Natsume, S., Mitsuoka, C., et al. (2013). QTL-seq: rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations. Plant J. 74, 174–183. doi: 10.1111/tpj.12105
Tanguay, P., Bergeron, M. J., Tsui, C., Brar, S., Lamarche, J., and Hamelin, R. C. (2013). The origin of a new race of Cronartium ribicola, virulent on previously immune blackcurrant cultivars, and rapidly spreading in eastern North America. Phytopathology 103:143.
Thrall, P. H., Barrett, L. G., Dodds, P. N., and Burdon, J. J. (2016). Epidemiological and evolutionary outcomes in gene-for-gene and matching allele models. Front. Plant Sci. 6:1084. doi: 10.3389/fpls.2015.01084
Tomback, D. F., and Achuff, P. (2010). Blister rust and western forest biodiversity: ecology, values, and outlook for white pines. For. Pathol. 40, 186–225. doi: 10.1111/j.1439-0329.2010.00655.x
Trick, M., Adamski, N. M., Mugford, S. G., Jiang, C. C., Febrer, M., and Uauy, C. (2012). Combining SNP discovery from next-generation sequencing data with bulked segregant analysis (BSA) to fine-map genes in polyploid wheat. BMC Plant Biol. 12:14. doi: 10.1186/1471-2229-12-14
Wang, Y., Chen, J., Li, D. W., Zheng, L., and Huang, J. (2017). CglCUT1 gene required for cutinase activity and pathogenicity of Colletotrichum gloeosporioides causing anthracnose of Camellia oleifera. Eur. J. Plant Pathol. 147, 103–114. doi: 10.1007/s10658-016-0983-x
Wang, P., and Shen, G. (2011). The endocytic adaptor proteins of pathogenic fungi: charting new and familiar pathways. Med. Mycol. 49, 449–457. doi: 10.3109/13693786.2011.553246
Wu, J. Q., Sakthikumar, S., Dong, C., Zhang, P., Cuomo, C. A., and Park, R. F. (2017). Comparative genomics integrated with association analysis identifies candidate effector genes corresponding to Lr20 in phenotype-paired Puccinia triticina isolates from Australia. Front. Plant Sci. 8:148. doi: 10.3389/fpls.2017.00148
Xu, L., Dong, Z., Fang, L., Luo, Y., Wei, Z., Guo, H., et al. (2019). OrthoVenn2: a web server for genome wide comparison and annotation of orthologous clusters across multiple species. Nucleic Acids Res. 47, W52–W58. doi: 10.1093/nar/gkz333
Yang, J., Jiang, H., Yeh, C. T., Yu, J., Jeddeloh, J. A., Nettleton, D., et al. (2015). Extreme-phenotype genome-wide association study (XP-GWAS): a method for identifying trait-associated variants by sequencing pools of individuals selected from a diversity panel. Plant J. 84, 587–596. doi: 10.1111/tpj.13029
Yang, Y., and Smith, S. A. (2013). Optimizing de novo assembly of short-read RNA-seq data for phylogenomics. BMC Genomics 14:328. doi: 10.1186/1471-2164-14-328
Yang, Y., Zhang, Y., Li, B., Yang, X., Dong, Y., and Qiu, D. (2018). A Verticillium dahliae pectate lyase induces plant immune responses and contributes to virulence. Front. Plant Sci. 9:1271. doi: 10.3389/fpls.2018.01271
Zhang, H., Yohe, T., Huang, L., Entwistle, S., Wu, P., Yang, Z., et al. (2018). dbCAN2: a meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 46, W95–W101. doi: 10.1093/nar/gky418
Zhu, W., Ronen, M., Gur, Y., Minz-Dub, A., Masrati, G., Ben-Tal, N., et al. (2017a). BcXYG1, a secreted xyloglucanase from Botrytis cinerea, triggers both cell death and plant immune responses. Plant Physiol. 175, 438–456. doi: 10.1104/pp.17.00375
Keywords: bulked segregant analysis-based RNA-seq (BSR-Seq), candidate effectors, comparative transcriptomics, major gene resistance, white pine-blister rust (WP-BR) interaction
Citation: Liu J-J, Sniezko RA, Zamany A, Williams H, Omendja K, Kegley A and Savin DP (2021) Comparative Transcriptomics and RNA-Seq-Based Bulked Segregant Analysis Reveals Genomic Basis Underlying Cronartium ribicola vcr2 Virulence. Front. Microbiol. 12:602812. doi: 10.3389/fmicb.2021.602812
Edited by:
Chia-Lin Chung, National Taiwan University, TaiwanReviewed by:
Dawei Xin, Northeast Agricultural University, ChinaJohn G. Gibbons, University of Massachusetts Amherst, United States
Copyright © 2021 Liu, Sniezko, Zamany, Williams, Omendja, Kegley and Savin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jun-Jun Liu, jun-jun.liu@canada.ca