Primary Hyperoxaluria Type 1 Disease Manifestations and Healthcare Utilization: A Multi-Country, Online, Chart Review Study
 Xiangling Wang1
Xiangling Wang1  David Danese2
David Danese2  Thomas Brown2 Jessica Baldwin2
Thomas Brown2 Jessica Baldwin2  Gautam Sajeev3
Gautam Sajeev3  Erin E. Cook3 Yao Wang3 Chunyi Xu3 Hongbo Yang3 Michael L. Moritz4*
Erin E. Cook3 Yao Wang3 Chunyi Xu3 Hongbo Yang3 Michael L. Moritz4*- 1Center for Personalized Genetic Healthcare, Department of Nephrology and Hypertension, Department of Molecular Medicine, Cleveland Clinic, Cleveland, OH, United States
- 2Alnylam Pharmaceuticals, Inc., Cambridge, MA, United States
- 3Analysis Group, Inc., Boston, MA, United States
- 4Division of Nephrology, Department of Pediatrics, University of Pittsburgh School of Medicine, Pittsburgh, PA, United States
Background: Primary hyperoxaluria type 1 (PH1) is a rare genetic disease that can result in irreversible damage to the kidneys and, eventually, extrarenal organs. While kidney failure is a known consequence of PH1, few studies to date have characterized clinical consequences of PH1 prior to kidney failure, and data on healthcare resource use outcomes across different stages of disease severity in PH1 are also limited. To help fill this knowledge gap, this study characterized the clinical and healthcare resource use (HRU) burden in patients with PH1 with varying stages of kidney disease.
Methods: Nephrologists in the United States, Canada, United Kingdom, France, Germany, and Italy abstracted chart data from patients with PH1 under their care via an online questionnaire. Eligible patients had confirmed PH1 and ≥2 office visits from 2016 to 2019.
Results: A total of 120 patients were analyzed (median age at diagnosis, 17.4 years old, median age at index 19.5 years old, median eGFR at index 45 ml/min/1.73 m2; median follow-up 1.7 years). During follow-up, the most common PH1 manifestations were kidney stones and urinary tract infections (UTIs, both 56.8%), and the most common symptoms were fatigue/weakness (71.7%) and pain (64.6%). With regard to HRU during follow-up, 37.4% required lithotripsy, 31.3% required ureteroscopy, and 9.6% required nephrolithotomy. PH1-related hospitalizations and emergency/urgent care visits were noted for 84.0 and 81.6% of patients, respectively.
Conclusions: The current study demonstrated that patients with PH1 across various stages of kidney disease exhibited a substantial clinical burden, including kidney stones, UTIs, fatigue/weakness, and pain, and required frequent HRU, including kidney stone procedures, hospitalizations, and emergency visits. These findings highlight the significant morbidity and HRU burden in patients with PH1.
Introduction
Primary hyperoxaluria type 1 (PH1) is a rare, autosomal recessive genetic disease resulting from mutations in the AGXT gene. PH1 causes irreparable damage to the kidneys and other vital organs, with potentially life-threatening consequences. The estimated prevalence of PH1 is 1–3 per 1,000,000 in Europe and North America, with greater prevalence in populations with higher levels of consanguinity (e.g., Middle Eastern and North African populations) or where founder mutations are present (e.g., Canary Islander populations) (1–6). While progression to kidney failure has been well-characterized in PH1 natural history studies, a better understanding of the disease burden prior to kidney failure and the healthcare resource use (HRU) outcomes experienced across all stages of PH1 will be important for development of efficient treatments.
In patients with PH1, AGXT gene mutations lead to deficiency in the alanine-glyoxylate aminotransferase (AGT) enzyme, which catalyzes the conversion of glyoxylate to glycine in the liver (7). As a result, glyoxylate accumulates and, rather than being primarily metabolized to glycine, is converted to oxalate, which is then excreted by the kidneys (7). In the kidneys, the excess oxalate combines with calcium to form insoluble calcium oxalate (CaOx) crystals, which can result in kidney stones, nephrocalcinosis and progressive kidney damage, ultimately leading to kidney failure (4, 7, 8). The eventual loss of kidney function and the inability of kidneys to fully clear oxalate from blood can result in systemic oxalosis, in which oxalate accumulates in plasma and leads to extrarenal CaOx crystal formation, with resultant damage to organs such as bone, heart, skin, and eyes (9, 10). In addition to carrying a significant clinical burden, PH1 also has important humanistic impacts. A recent survey found that manifestations of PH1 and disease management measures often create challenges for patients in terms of pain and other physical trauma while also disrupting the lives of patients and their caregivers (11). The same survey found that a majority of patients and caregivers experienced ongoing emotional stress due to uncertainty over when a painful stone event, complication from oxalosis, or kidney failure might occur.
Common supportive care measures, including hyperhydration and crystallization inhibitors, are effective to increase oxalate solubility and prevent CaOx crystallization but do not target the underlying pathophysiology of PH1 (4, 12, 13). Pyridoxine may stabilize certain pathogenic AGT variants to enhance AGT enzyme activity, with some degree of efficacy in~30% of patients (9). Later-stage management consists of intensive dialysis as a bridge to transplant or when no other viable treatment option exists (13–15). Liver transplantation, by addressing the metabolic defect in PH1, can normalize oxalate levels and, if done preemptively, prevent progression to kidney failure; however, it carries substantial morbidity and mortality risk and must be paired with kidney transplantation to restore lost kidney function in patients with kidney failure. To address the limitations of current options, multiple novel PH1 therapies are in clinical testing, including RNA interference (RNAi) therapeutics that suppress expression of proteins involved in oxalate production and an enteric oxalate-degrading bacterial preparation (16–18).
To date, literature on the natural history of PH1 has typically focused on the outcomes of kidney failure and mortality. Data on other outcomes, including clinical outcomes occurring prior to kidney failure and HRU outcomes in PH1, are less common. To complement the existing evidence base, a multi-country, retrospective, online chart review study was conducted to characterize the clinical and HRU burden of PH1 at various stages of kidney disease, with a focus on disease burden prior to kidney failure.
Materials and Methods
Study Overview
A retrospective chart review study of patients with PH1 under nephrologists' care in the United States (US), United Kingdom (UK), France, Germany, Italy, and Canada was conducted. Participating nephrologists were asked to abstract chart data for randomly selected patients who met the study eligibility criteria and enter this data into a standardized online questionnaire prepared by the investigators. The online questionnaire was pilot tested for clarity and usability by 2 US-based nephrologists with experience managing PH1. The form was revised based on their input and translated into French, German, and Italian. All patient data collected were anonymous and non-identifiable. Physicians completed data entry between October 2019 and January 2020. The study was approved by the New England Independent Review Board in October 2019.
Data Source and Physician Eligibility
Adult and pediatric nephrologists were recruited from the Schlesinger Group healthcare and partner panels for this study. These online panels include over 125,000 physicians whose credentials, including demographics and education, are verified by Schlesinger Group. Panel members are preregistered to be contacted for opportunities to participate in surveys and chart review studies. An invitation link for the current study was sent to adult and pediatric nephrologists in the panel. In addition, following the initial invitation, panel participants were contacted periodically with follow-up reminders to encourage participation and maximize response rates. Respondents to the survey link were asked to first complete screening questions to determine their eligibility to participate in this chart review study (see Supplementary Figure 1, Part I: Physician Practice Characteristics). To allow collection of data characterizing a contemporary cohort of patients with PH1, physicians were required to meet the following criteria to be eligible to participate: (1) he/she was a practicing adult or pediatric nephrologist; (2) he/she was the healthcare provider primarily responsible for managing ≥1 patient with PH1 during the past 3 years; and (3) he/she had ≥1 patient with PH1 who met the patient selection criteria summarized below. Physicians were excluded if they did not complete the eligibility questions, did not meet these study eligibility criteria, or if they met the criteria but did not provide data for any eligible patients. As compensation for their time and effort, physicians received an honorarium of $95–155 (USD) for each patient chart provided.
Patient Selection
Participating physicians reviewed the patient selection criteria to determine the eligibility of their patients. To be eligible, patients had to have: a PH1 diagnosis confirmed by genetic test or liver biopsy-based AGT assay; ≥2 office visits to the participating physician in the last 3 years; and available medical charts with information on signs, symptoms, disease manifestations, and PH1-related HRU since their first visit to the participating physician in the last 3 years. If a physician indicated that they had more than one eligible PH1 patient under their care, they were asked to randomly select patients for inclusion in this study, one at a time, up to a maximum of 5 patients. To facilitate random selection of each patient, the online questionnaire interface displayed a random letter of the alphabet, and physicians were instructed to select the patient whose surname started with the letter closest to that randomly generated letter.
Study Period
The index date was defined as the date of patients' first office visit in the past 3 years prior to chart abstraction. Restriction of data collection to the past 3 years was done to yield an approximate snapshot of the current burden of prevalent PH1 cases. The baseline period was defined as the time prior to the index date and the follow-up period defined as the time from the index date to the most recent office visit prior to chart abstraction.
Study Measures
All data were collected via the standardized online questionnaire, which included questions about the participating physician and chart data for each patient (Supplementary Figure 1). Data collected on participating physicians included specialty, years in practice, practice setting, and number of patients with PH1 seen in the last 3 years. Data collected on patients included demographics at index, diagnostic information, and clinical, HRU, and disease management history prior to index and during the follow-up period. Unless otherwise noted in the questionnaire, clinical and HRU outcomes were defined pragmatically, as recorded by physicians in patient charts based on their interpretation of the available data (rather than by an external “gold standard” definition based on laboratory criteria, imaging, and/or any other criteria specified in the online questionnaire). After starting data entry, physicians could pause and return to the questionnaire as needed, with no time limit for completion. Data were included for all eligible patients for whom the physician reached the end of the questionnaire and confirmed completion via the online platform (with no required minimum number of items to complete).
Statistical Analyses
Descriptive analyses of physician and patient data were performed. For each variable, these analyses included physicians/patients with available data on that variable (with the number of physicians/patients with available data for each analysis reported as ‘N’ in the Results). Categorical variables were summarized with counts and percentages and continuous variables summarized with means, standard deviations (SDs), medians, and interquartile ranges (IQRs). Rates of acute clinical events and HRU during follow-up were calculated as the total number of events per person-year (PY).
Results
Physician Characteristics
The study invitation was sent to approximately 9,350 physicians and accepted by 935 (Figure 1). Of these, 609 did not meet the eligibility criteria and 269 did not complete the questionnaire. The remaining 57 physicians met the eligibility criteria and provided information on 120 patients with PH1. Approximately one-third were based in the US (19 physicians/37 patients), with smaller proportions in France (10/19), Italy (8/15), the UK (8/18), Canada (7/16), and Germany (5/15). All participating physicians were nephrologists, including 17.5% who were pediatric nephrologists. These physicians had been practicing in their field for a mean of 16.8 years (SD: 7.4), had a median of 6 patients (IQR: 3, 23) with PH1 under their care with ≥2 office visits in the last 3 years, and provided data on a mean of 2.11 eligible patients under their care. Similar percentages of participating physicians were practicing at academic (53%) and non-academic institutions (47%).
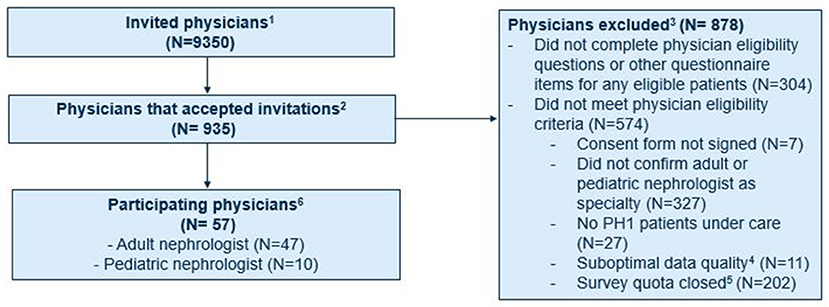
Figure 1. Recruitment of physicians participating in chart review study. 1Adult and pediatric nephrologists who were part of the Schlesinger Group healthcare and partner panels who were notified of the opportunity to participate in this study. 2Physicians who accepted the invitation and were asked to complete screening questions to determine their eligibility to participate in the study. 3Physicians who did not complete the eligibility criteria, did not provide data for any eligible patients, and did not meet the study eligibility criteria.4Included providers who selected responses that were illogical (e.g., reported number of patients under their care was larger than possible based on responses to initial screening questions) and were thus excluded from the study. 5Includes providers who accepted invitation after quotas for their respective country and/or specialty (as implemented during physician recruitment to ensure representation of patients from different countries and age groups) had been met. 6Physicians who met the study eligibility criteria and provided information on 120 eligible patients with PH1.
Patient Characteristics at PH1 Diagnosis
Median patient age at diagnosis (N = 82) was 17.4 years (IQR: 11.1, 24.9). The majority of patients were white (78.2%) and male (64.4%). Of those with available eGFR data at diagnosis (N = 86), 15.1% had eGFR ≥ 90 ml/min/1.73 m2, 33.7% had eGFR 60–89, 41.9% had eGFR 30–59, 7.0% had eGFR 15–29, and 2.3% had eGFR < 15. Most patients (82.0%) presented with signs and/or symptoms of PH1, with a median age of 13.0 years at symptom onset (N = 111). The most common signs and symptoms at diagnosis (N = 111) were kidney stones (51.4%), fatigue/weakness (44.1%), and pain (44.1%). Mean and median times from symptom onset to diagnosis were 5.7 (SD: 8.3) and 2.8 years (IQR: 0.9, 6.7), respectively (N = 82).
Patient, Disease, and Treatment Characteristics at Index
Median patient age at index (N = 99) was 19.5 years, with 58.6% of patients ≥18 years old (Table 1). Median duration of post-index follow-up was 1.7 years. Of those with genetic data available (N = 87), 9.2% had two p.Gly170Arg pathogenic variant AGXT alleles, 21.8% had one p.Gly170Arg AGXT allele, and the remainder had two pathogenic variant AGXT alleles not encoding the p.Gly170Arg amino acid substitution. The remaining patients included in the cohort had their PH1 diagnosed via biopsy-based assessment of hepatic AGT enzyme activity. Among patients with available data (N = 81), median eGFR at index was 45 ml/min/1.73 m2 (IQR: 35, 75); 37.1% of patients had eGFR ≥ 60 ml/min/1.73 m2 at index. With regard to PH1 management history (N = 111), hyperhydration (70.3%), and crystallization inhibitor use (60.4%) were reported in most patients; 40.5% of patients had a history of pyridoxine use.

Table 1. Baseline patient demographics and clinical characteristics.
Disease Manifestations During Follow-Up
During the follow-up period, the most common signs and symptoms (N = 113–114) of PH1 were fatigue/weakness (71.7%), pain (64.6%), and hematuria (defined per data collection form as “blood in urine”: 57.9%, Table 2). Common clinical events (N = 118) included kidney stone events and urinary tract infections (UTIs; each reported ≥ 1 time in 56.8%). In the overall population, kidney stone events occurred at a rate of 0.8 per PY and UTIs at a rate of 1.0 per PY (Supplementary Figure 2).

Table 2. Prevalence of clinical signs, symptoms, and other PH1 manifestations during follow-up.
With regard to kidney function, 32 patients (27.1%) had an episode of acute decline (N = 118); among the 27 of these 32 patients for whom more detailed information was provided, common causes of acute decline included dehydration, infection, and obstructive stone events (Supplementary Figure 3). Of the 27 patients in question, 18 (66.7%) experienced a permanent loss of kidney function (defined as failure to recover to the level of kidney function seen immediately prior to the episode) in association with an episode of acute decline. About one fifth of patients (20.4%) in the overall cohort (n/N = 23/113) ever had kidney failure (captured as “end-stage renal disease” in the data collection form), which was diagnosed at a median age of 25.3 years (N = 16, Table 3), and approximately one-fifth (21.4%, n/N = 24/112) of patients required dialysis (before or during follow-up), with a median age of 24.6 years at the time of initiation (N = 16). In addition, transplant (before or during follow-up) was performed for 15.9% of patients (n/N = 18/113), of whom 61.1% had combined liver-kidney transplant.

Table 3. Late-stage outcomes of PH1.
Aside from renal manifestations, systemic manifestations of PH1 (n/N = 36/104) were reported in 34.6% of patients during the follow-up period—most often anemia, arrhythmias, and bone pain (Supplementary Figure 4).
HRU During Follow-Up
During the follow-up period, 84.0% of patients required hospitalization (N = 94) for reasons related to PH1 and 81.6% required ≥1 emergency visit (N = 87, Table 4) related to PH1. PH1-related hospitalizations and emergency visits each occurred at a rate of 1.1 per PY, and median length of stay was 5.0 days (IQR: 3.0, 7.5).
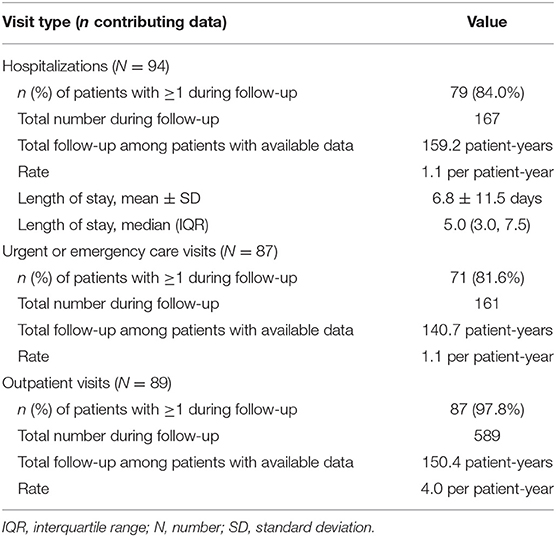
Table 4. PH1-related hospitalizations, urgent/emergency care visits, and outpatient visits during follow-up.
With regard to specific medical procedures (N = 115) related to PH1, approximately one-third of patients had ≥1 lithotripsy and approximately one-third had ≥1 ureteroscopy to manage kidney stones during follow-up (Table 5), while 9.6% had ≥1 percutaneous nephrolithotomy (PCNL). Patients with ≥1 lithotripsy required these procedures at a rate of 1.2 per PY; corresponding rates for patients with ≥1 ureteroscopy and ≥1 PCNL were 1.0 and 0.6 procedures per PY, respectively (Table 5). In addition, 4.3% of patients underwent gastrostomy tube (G-tube) placement and 6.1% underwent nasogastric tube (NG-tube) placement during follow-up.
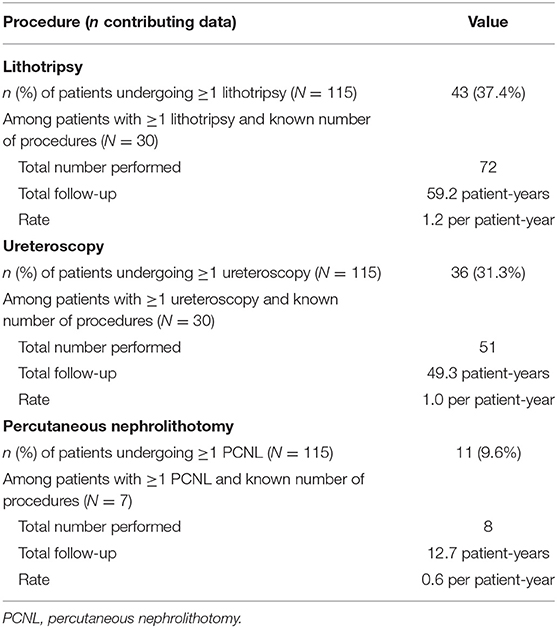
Table 5. PH1-related medical procedures during follow-up.
Discussion
This multi-country, retrospective online chart review study characterizes a relatively large cohort of patients with PH1 in North America and Europe, particularly relevant given the rarity of PH1. Outside of large cohort studies involving the two leading PH registries (5, 19) and an additional study led by a French PH1 reference center (20), most other published reports on PH1 have involved <100 patients (6, 21–23). Prior published reports have provided limited data on clinical outcomes of PH1 before progression to kidney failure and mortality, and data on HRU in PH1 are scarce (6, 22, 24, 25). The current study aimed to better characterize the clinical and HRU burden of PH1 in patients at different stages of disease, most without kidney failure. In this study, many patients experienced adverse clinical events during the follow-up period, and over 80% had ≥1 hospitalization and/or emergency visit related to PH1. These findings highlight the significant clinical and HRU burden of PH1 even prior to kidney failure.
The clinical burden in this study was notable in terms of kidney stone events and UTIs, as in other published PH1 cohorts (6, 24, 26). In addition, pain, fatigue/weakness, and hematuria (defined as “blood in urine” for data collection purposes) were reported as common PH1 signs and symptoms in the current cohort. This clinical burden was accompanied by substantial HRU. Overall, 84% of patients required ≥1 hospitalization and 82% required ≥1 emergency visit related to PH1 during the follow-up period, with each occurring at a rate of approximately 1 per PY. To provide context, national data from the US, Europe, and Canada suggest hospitalization rates of 0.08–0.25 per PY and emergency visit rates of 0.08–0.48 per PY in the general population, despite its older age relative to the PH1 population (27–29).
Stone interventions were also common in the current study population, and patients who required lithotripsy, ureteroscopy, and PCNL to manage stones underwent such procedures on an annual to biannual basis, on average. In this study, clinical and HRU burden were present despite the typically moderate degree of kidney impairment and the finding that most patients were receiving supportive care for PH1, and despite the heterogeneous nature of the disease.
The current study helps further elucidate the clinical burden of PH1, particularly in terms of clinically evident stone events and other symptoms prior to development of kidney failure, and is among the first to collect detailed HRU data in PH1. Moreover, this study collected data from all (rather than only newly diagnosed) PH1 patients over a recent 3 year period. As a result, findings are more likely to reflect the burden of PH1 in the current, prevalent population.
For context, it is important to note that the current study cohort differed from previously reported PH1 cohorts in terms of age and degree of kidney disease at diagnosis. Patients in this study had a median age at diagnosis of 17.4 years, somewhat older than in other PH1 cohorts (7–17 years) (6, 30, 31). As most participating physicians were adult nephrologists, patients closer to the documented smaller, later peak in the bimodal distribution of age at PH1 diagnosis (23) may have been overrepresented in the current study. Additionally, age at diagnosis was unknown in this study for 32% of patients. Pediatric onset may have been more common in these patients, as dates of diagnosis may not have been accessible to physicians caring for adults who were diagnosed as children. Nonetheless, the median age at diagnosis in this study may make the findings less generalizable to patients diagnosed in infancy or early childhood.
With regard to kidney function, patients in the current study had varying levels of impairment at diagnosis, but overall, there were fewer patients with eGFR <15 mL/min/1.73m2 at diagnosis than in other published cohorts (6, 7, 24). The current study may have captured a milder population, given the comparatively high proportion of adult nephrologists involved and given that pediatric-onset PH1 tends to progress more rapidly. The findings of this study may thus be more generalizable to patients with PH1 diagnosed prior to kidney failure. As a result, they provide evidence of a substantial burden of PH1 even in patients with less severe kidney impairment than previously published cohorts.
Other methodological considerations should also be taken into account in interpreting these results. Due to differences among physicians in terms of chart data capture, some data elements were not consistently available. Despite an initial feasibility assessment of data availability, some data elements were less available than expected (e.g., eGFR at index, age at diagnosis). In addition, although pilot tests were conducted and the online questionnaire revised accordingly, the questionnaire may have been challenging to complete due to its comprehensive and detailed nature, which may have impacted data quality and completeness, particularly for clinical and HRU event counts. Also of note is that data on urine oxalate, a key indicator of PH1 disease activity, were not considered sufficiently complete for reporting and interpretation (unavailable within 31 days of index for 61% of patients). Analysis was further complicated by inter-laboratory variability in terms of urine oxalate assays and methods for calculating and reporting oxalate assay results. Aside from these considerations, the retrospective nature of the study itself introduces potential issues such as selection bias and non-random missingness of data.
An additional methodological consideration is that comprehensive data validation via review of source medical charts was not possible, given the panel-based design. To minimize the impact of this limitation, steps were taken to prospectively reduce the potential for invalid data entry—specifically, by adopting feedback from pilot testing (as previously noted) with nephrologists to optimize survey clarity and usability, and by incorporating automated logic checks throughout the survey to help ensure accurate data entry and flag potentially erroneous entries for review and revision as needed. In addition, post-survey queries on certain key data fields were made to selected participants whose responses for those fields were identified as being potentially invalid and required further clarification. Even in a scenario that would allow validation of source medical charts, it remains a limitation of chart review studies in general that the accuracy of chart documentation fundamentally limits data quality, as events documented in patient charts cannot be “re-observed” prospectively for the purposes of the study.
These considerations notwithstanding, the current work suggests several areas for additional investigation. For instance, further research in a population with younger age at presentation and diagnosis would be valuable to provide a more complete picture of the clinical and HRU burden of PH1, given the frequency with which PH1 presents in childhood. Selected findings from the current cohort also suggest previously underappreciated elements of clinical burden in PH1 that may warrant further exploration. For example, prior anecdotal reports and small case series have noted that progressive kidney damage in PH1 may be accelerated by acute declines in kidney function (e.g., due to obstruction of renal outflow by CaOx stones) (21, 24, 32). In the current study, acute kidney decline was reported in 27% of patients and was often triggered by common events that would be relatively benign in a non-PH1 setting (e.g., dehydration). Similarly, anemia and arrhythmia were reported in an appreciable portion of patients who experienced systemic manifestations of PH1 during follow-up. Both have been documented in PH1 (33–35) and are also known complications of CKD more broadly (36, 37); however, their prevalence in PH1 is not well characterized. While findings from the current study regarding acute declines in kidney function and systemic manifestations of PH1 are preliminary in nature, more in-depth investigation into these events could further elucidate their contribution to the disease burden of PH1.
Additional economic analyses also represent a valuable direction for future research in PH1. The ability to translate clinical and HRU data from the current study to PH1-related cost estimates is limited, given the multinational nature of the patient cohort (and the associated variation across countries in terms of healthcare systems and resource use costs) and the chart-based study design, as study designs based on healthcare reimbursement claims data are best-suited to generate quantitative cost estimates (38). Country-specific claims studies of this type may therefore be valuable for more precisely characterizing hospitalization-related and total costs specifically attributable to PH1.
In summary, the current study found that patients with PH1 experienced substantial clinical and HRU burden. Patients commonly experienced stone events and UTIs as well as symptoms such as pain, fatigue and weakness. This clinical burden was accompanied by an appreciable burden in terms of HRU. Most patients required hospitalizations and emergency visits for their PH1 (each annually, on average), and a number also required stone management interventions, which are known to carry risk of clinical complications and impair quality of life. The clinical and HRU burden of PH1 were evident despite the finding that most patients were being managed with standard supportive care measures and the moderate level of kidney impairment at index. In addition, this burden is superimposed on a background of progressive kidney decline that is well-documented in PH1. These findings highlight an ever-present risk of ongoing morbidity and significant HRU, which accompany the well-recognized, ongoing risk of progressive kidney decline in PH1, and underscore the urgency of effective PH1 treatment throughout the disease course.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by the New England Independent Review Board. Written informed consent from the participants' legal guardian/next of kin was not required to participate in this study in accordance with the national legislation and the institutional requirements.
Author Contributions
All authors participated in the study design, analysis and interpretation, manuscript composition and editing.
Funding
This study was funded by Alnylam Pharmaceuticals, Inc.
Conflict of Interest
DD and TB are employees of Alnylam. JB is a former employee of Alnylam; she is currently an employee of Vertex Pharmaceuticals. MM and XW are consultants of Alnylam. GS, EC, YW, CX, and HY are employees of Analysis Group Inc., which received funding for this research from Alnylam.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmed.2021.703305/full#supplementary-material
References
1. Kamoun A, Lakhoua R. End-stage renal disease of the Tunisian child: epidemiology, etiologies, and outcome. Pediatr Nephrol. (1995) 10:479–82. doi: 10.1007/s004670050143
2. Lorenzo V, Alvarez A, Torres A, Torregrosa V, Hernández D, Salido E. Presentation and role of transplantation in adult patients with type 1 primary hyperoxaluria and the I244T AGXT mutation: single-center experience. Kidney Int. (2006) 70:1115–9. doi: 10.1038/sj.ki.5001758
3. Cochat P, Deloraine A, Rotily M, Olive F, Liponski I, Deries N. Epidemiology of primary hyperoxaluria type 1. Nephrol Dial Transplant. (1995) 10:3–7. doi: 10.1093/ndt/10.supp8.3
4. Cochat P, Rumsby G. Primary hyperoxaluria. N Engl J Med. (2013) 369:649–58. doi: 10.1056/NEJMra1301564
5. Hopp K, Cogal AG, Bergstralh EJ, Seide BM, Olson JB, Meek AM, et al. Phenotype-genotype correlations and estimated carrier frequencies of primary hyperoxaluria. J Am Soc Nephrol. (2015) 26:2559–70. doi: 10.1681/ASN.2014070698
6. van Woerden CS, Groothoff JW, Wanders RJA, Davin JC, Wijburg FA. Primary hyperoxaluria type 1 in The Netherlands: prevalence and outcome. Nephrol Dial Transplant. (2003) 18:273–9. doi: 10.1093/ndt/18.2.273
7. Danpure CJ, Jennings PR. Peroxisomal alanine:glyoxylate aminotransferase deficiency in primary hyperoxaluria type I. FEBS Lett. (1986) 201:20–34. doi: 10.1016/0014-5793(86)80563-4
8. McGregor TL, Hunt KA, Nioi P, Mason D, Ticau S, Pelosi M, et al. Deep phenotyping of a healthy human HAOI knockout informs therapeutic development for primary hyperoxaluria type I. BioRxiv. (2019) doi: 10.1101/524256
9. Cochat P, Hulton S-A, Acquaviva C, Danpure CJ, Daudon M, De Marchi M, et al. Primary hyperoxaluria type 1: indications for screening and guidance for diagnosis and treatment. Nephrol Dial Transplant. (2012) 27:1729–36. doi: 10.1093/ndt/gfs078
10. Perinpam M, Enders FT, Mara KC, Vaughan LE, Mehta RA, Voskoboev N, et al. Plasma oxalate in relation to eGFR in patients with primary hyperoxaluria, enteric hyperoxaluria and urinary stone disease. Clin Biochem. (2017) 50:1014–9. doi: 10.1016/j.clinbiochem.2017.07.017
11. Lawrence JE, Wattenberg DJ. Primary hyperoxaluria: the patient and caregiver perspective. Clin J Am Soc Nephrol. (2020) 15:909–11. doi: 10.2215/cjn.13831119
12. Leumann E, Hoppe B, Neuhaus T. Management of primary hyperoxaluria: efficacy of oral citrate administration. Pediatr Nephrol. (1993) 7:207–11. doi: 10.1007/BF00864405
13. Milliner DS, Harris PC, Cogal AG, Lieske JC. Primary Hyperoxaluria Type 1 (GeneReviews®). Available online at: https://www.ncbi.nlm.nih.gov/books/NBK1283/ (accessed April 3, 2020).
14. Hoppe B, Graf D, Offner G, Latta K, Byrd DJ, Michalk D, et al. Oxalate elimination via hemodialysis or peritoneal dialysis in children with chronic renal failure. Pediatr Nephrol. (1996) 10:488–92. doi: 10.1007/s004670050145
15. Marangella M, Petrarulo M, Cosseddu D, Vitale C, Linari F. Oxalate balance studies in patients on hemodialysis for type I primary hyperoxaluria. Am J Kidney Dis. (1992) 19:546–53. doi: 10.1016/S0272-6386(12)80833-X
16. Dindo M, Conter C, Oppici E, Ceccarelli V, Marinucci L, Cellini B. Molecular basis of primary hyperoxaluria: clues to innovative treatments. Urolithiasis. (2019) 47:67–78. doi: 10.1007/s00240-018-1089-z
17. Wang X, Bhutani G, Vaughan LE, Enders FT, Haskic Z, Milliner D, et al. Urinary monocyte chemoattractant protein 1 associated with calcium oxalate crystallization in patients with primary hyperoxaluria. BMC Nephrol. (2020) 21:133. doi: 10.1186/s12882-020-01783-z
18. Garrelfs SF, Frishberg Y, Hulton SA, Koren MJ, O'Riordan WD, Cochat P, et al. Lumasiran, an RNAi therapeutic for primary hyperoxaluria type 1. N Engl J Med. (2021) 384:1216–26. doi: 10.1056/NEJMoa2021712
19. Mandrile G, van Woerden CS, Berchialla P, Beck BB, Acquaviva Bourdain C, Hulton S-A, et al. Data from a large European study indicate that the outcome of primary hyperoxaluria type 1 correlates with the AGXT mutation type. Kidney Int. (2014) 86:1197–204. doi: 10.1038/ki.2014.222
20. Harambat J, Fargue S, Acquaviva C, Gagnadoux M-F, Janssen F, Liutkus A, et al.: Genotype-phenotype correlation in primary hyperoxaluria type 1: the p.Gly170Arg AGXT mutation is associated with a better outcome. Kidney Int. (2010) 77:443–9. doi: 10.1038/ki.2009.435
21. Hasan A, Maynard S, Santoriello D, Schairer H: Primary hyperoxaluria type 1 with thrombophilia in pregnancy: a case report. Case Rep Nephrol Dial. (2018) 8:223–9. doi: 10.1159/000493091
22. Hoppe B, Langman CB. A United States survey on diagnosis, treatment, and outcome of primary hyperoxaluria. Pediatr Nephrol. (2003) 18:986–91. doi: 10.1007/s00467-003-1234-x
23. Shapiro R, Weismann I, Mandel H, Eisenstein B, Ben-Ari Z, Bar-Nathan N, et al. Primary hyperoxaluria type 1: improved outcome with timely liver transplantation: a single-center report of 36 children. Transplantation. (2001) 72:428–32. doi: 10.1097/00007890-200108150-00012
24. Fargue S, Harambat J, Gagnadoux M-F, Tsimaratos M, Janssen F, Llanas B, et al. Effect of conservative treatment on the renal outcome of children with primary hyperoxaluria type 1. Kidney Int. (2009) 76:767–3. doi: 10.1038/ki.2009.237
25. Hoppe B, Latta K, von Schnakenburg C, Kemper MJ. Primary hyperoxaluria—the German experience. Am J Nephrol. (2005) 25:276–81. doi: 10.1159/000086358
26. Jamieson NV. A 20-year experience of combined liver/kidney transplantation for primary hyperoxaluria (PH1): the European PH1 transplant registry experience 1984–2004. Am J Nephrol. (2005) 25:282–9. doi: 10.1159/000086359
27. Berchet C. Emergency care services: trends, drivers and interventions to manage the demand. In: OECD Health Working Papers 83. OECD Publishing, Paris (2015).
28. Organisation for Economic Cooperation and Development (OECD). Hospital Discharge Rates. Available online at: https://www.oecd-ilibrary.org/content/data/5880c955-en (accessed August 12, 2020).
30. Lieske JC, Monico CG, Holmes WS, Bergstralh EJ, Slezak JM, Rohlinger AL, et al. International registry for primary hyperoxaluria. Am J Nephrol. (2005) 25:290–6. doi: 10.1159/000086360
31. Takayama T, Nagata M, Ichiyama A, Ozono S. Primary hyperoxaluria type 1 in Japan. Am J Nephrol. (2005) 25:297–302. doi: 10.1159/000086361
32. El-Reshaid K, Al-Bader D, Madda JP. Primary hyperoxaluria in an adult male: a rare cause of end-stage kidney disease yet potentially fatal if misdiagnosed. Saudi J Kidney Dis Transpl. (2016) 27:606–9. doi: 10.4103/1319-2442.182440
33. Karadag S, Gursu M, Aydin Z, Uzun S, Dogan O, Ozturk S, et al. Primary hyperoxaluria in an adult presenting with end-stage renal failure together with hypercalcemia and hypothyroidism. Hemodial Int. (2011) 15:573–6. doi: 10.1111/j.1542-4758.2011.00573.x
34. Mookadam F, Smith T, Jiamsripong P, Moustafa SE, Monico CG, Lieske JC, et al. Cardiac abnormalities in primary hyperoxaluria. Circ J. (2010) 74:2403–9. doi: 10.1253/circj.cj-10-0107
35. Mykytiv V, Campoy Garcia F. Anemia in patient with primary hyperoxaluria and bone marrow involvement by oxalate crystals. Hematol Oncol Stem Cell Ther. (2018) 11:118–21. doi: 10.1016/j.hemonc.2017.07.007
36. Bonato FO, Lemos MM, Cassiolato JL, Canziani ME. Prevalence of ventricular arrhythmia and its associated factors in nondialyzed chronic kidney disease patients. PLoS ONE. (2013) 8:e66036. doi: 10.1371/journal.pone.0066036
37. Stauffer ME, Fan T. Prevalence of anemia in chronic kidney disease in the United States. PLoS ONE. (2014) 9:e84943. doi: 10.1371/journal.pone.0084943
38. HCUPnet. HCUPnet, Healthcare Cost and Utilization Project. Available online at: https://hcupnet.ahrq.gov/ (accessed January 15, 2001).
Keywords: primary hyperoxaluria type 1, oxaluria, chart review, healthcare resource utilization, kidney stones, chronic kidney disease, urinary tract infections
Citation: Wang X, Danese D, Brown T, Baldwin J, Sajeev G, Cook EE, Wang Y, Xu C, Yang H and Moritz ML (2021) Primary Hyperoxaluria Type 1 Disease Manifestations and Healthcare Utilization: A Multi-Country, Online, Chart Review Study. Front. Med. 8:703305. doi: 10.3389/fmed.2021.703305
Received: 30 April 2021; Accepted: 26 August 2021;
Published: 20 September 2021.
Edited by:
Asha Moudgil, Children's National Hospital, United StatesReviewed by:
Gaurav Gupta, Virginia Commonwealth University, United StatesNicola Brunetti-Pierri, Telethon Institute of Genetics and Medicine (TIGEM), Italy
Copyright © 2021 Wang, Danese, Brown, Baldwin, Sajeev, Cook, Wang, Xu, Yang and Moritz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael L. Moritz, moritzml@upmc.edu