 Mengqi Xiang
Mengqi Xiang Haijiao Jing1
Haijiao Jing1 Jialan Shi
Jialan Shi- 1Department of Hematology, First Affiliated Hospital of Harbin Medical University, Harbin Medical University, Harbin, China
- 2Department of Research, Veterans Affairs Boston Healthcare System, Harvard Medical School, Boston, MA, United States
- 3Department of Medical Oncology, Dana-Farber Cancer Institute, Harvard Medical School, Boston, MA, United States
Lung injury may persist during the recovery period of COVID-19 as shown through imaging, six-minute walk, and lung function tests. The pathophysiological mechanisms leading to long COVID have not been adequately explained. Our aim is to investigate the basis of pulmonary susceptibility during sequelae and the possibility that prothrombotic states may influence long-term pulmonary symptoms of COVID-19. The patient’s lungs remain vulnerable during the recovery stage due to persistent shedding of the virus, the inflammatory environment, the prothrombotic state, and injury and subsequent repair of the blood-air barrier. The transformation of inflammation to proliferation and fibrosis, hypoxia-involved vascular remodeling, vascular endothelial cell damage, phosphatidylserine-involved hypercoagulability, and continuous changes in serological markers all contribute to post-discharge lung injury. Considering the important role of microthrombus and arteriovenous thrombus in the process of pulmonary functional lesions to organic lesions, we further study the possibility that prothrombotic states, including pulmonary vascular endothelial cell activation and hypercoagulability, may affect long-term pulmonary symptoms in long COVID. Early use of combined anticoagulant and antiplatelet therapy is a promising approach to reduce the incidence of pulmonary sequelae. Essentially, early treatment can block the occurrence of thrombotic events. Because impeded pulmonary circulation causes large pressure imbalances over the alveolar membrane leading to the infiltration of plasma into the alveolar cavity, inhibition of thrombotic events can prevent pulmonary hypertension, formation of lung hyaline membranes, and lung consolidation.
Introduction
While the majority of patients with coronavirus disease 2019 (COVID-19) will develop only mild, self-limited illness, up to 20% will progress to a more serious form, including severe pneumonia, acute respiratory distress syndrome (ARDS), and pulmonary fibrosis (1–8). The potential risk of pulmonary impairment and parenchymal fibrosis in long COVID is of particular concern (9–13), and studies of multiple treatment options for COVID-19 do not consider their effects on subsequent risk and progression of long-term COVID-19 symptoms (14). Multiple mechanisms of lung injury in COVID-19 patients have been tentatively described, but the long-term pathogenicity of SARS-CoV-2 in discharged patients remains unclear. It has been reported that the consequences of severe COVID-19 are similar to those of severe acute respiratory syndrome (SARS) and Middle East respiratory syndrome (MERS) in terms of clinical sequelae, respiratory function, mental illness, and health-related quality of life (15–17). After infection, virus-induced immunopathological events are believed to be responsible for the pulmonary manifestations of SARS and MERS. Specifically, the virus replicates rapidly, infects type II alveolar epithelial cells and vascular endothelial cells, and increases the production of proinflammatory cytokines and chemokines. These, in turn, recruit fibroblasts and induce their differentiation into myofibroblasts, resulting in impaired O2 and CO2 exchange (18). In addition, viral antagonism and delayed interferon responses further aggravate inflammation (19, 20).
When SARS-CoV-2 replicates in large numbers, immune cells and inflammatory mediators respond strongly, forming cytokine storms and damaging alveolar structures. The virus invades vascular endotheliocytes from the blood-air barrier. As the disease progresses, endothelial dysfunction leads to more rigid and therefore vulnerable pulmonary vessels. Vascular endothelium expresses more protease activated receptor 1, tissue factor (TF), P-selectin and phosphatidylserine (PS) on the membrane surfaces, releasing microparticles, von Willebrand Factor (vWF), and factor VIII (21). These alterations, together with soluble thrombomodulin (sTM) and increased surface chemokines, causes platelet overactivation and thrombosis (22). With the enhanced permeability of the alveolar membrane, the pulmonary edema causes further hypoxemia and deterioration (23, 24). Pulmonary (micro)thrombus is key to severe hypoxemia, multiple organ dysfunction, and prolonged COVID-19 syndrome (25–33). Microthrombi can block microvessels in the alveolar capillaries, making it difficult for red blood cells to pass through. Slow blood flow and local congestion lead to elevated pulmonary capillary pressure and then to pulmonary hypertension (4). The pressure difference between the two sides of the blood-air barrier increases, while severe inflammation causes diffuse alveolar damage, resulting in the inability of the alveolar membrane to maintain normal permeability (34). Various components in the blood, including macromolecules (mainly albumin and globulin), enter the alveolar cavity. This fluid in the alveolar cavity then induces aggravated dyspnea (35–37). The alveolar liquid evaporates under airflow action, leaving behind plasma proteins and necrotic alveolar epithelial debris to form a transparent membrane, leading to lung consolidation (3–6, 38, 39). Although hypoxemia results from a combination of many mechanisms, the amplifying effects of hypoxia promote the exacerbation of cytokine storms, endothelial injury and thrombosis (23, 24). In long COVID, patients often show substandard six-minute walk test (6mWT), abnormal chest imaging findings (such as bilateral interstitial infiltration, ground-glass opacity (GGO), and fibrosis), and lung diffusing capacity for carbon monoxide (DLCO) < 80%, all indicating persistent lung damage (11–13, 40). In a follow-up study of 113 COVID-19 patients with ARDS, 55% reported dyspnea eight months after diagnosis. Adjusted for age, more than 50% of patients who undertook a 6mWT reached less than 80% of the theoretical distance. Abnormal chest radiographs were reported in 49% of cases, with bilateral interstitial infiltration predominating (87.5%). Chest computerized tomography (CT) scans showing GGO (55%) and fibrosis (19%) were common. Additionally, DLCO was less than 80% in 77.8% of patients (41).
The frequently reported pulmonary arterial, venous, and capillary thrombotic events at autopsy suggest that the transformation of stable vascular endothelial cells to the prothrombotic state is not negligible (8, 25, 27–29, 35, 38–47). We therefore hypothesize that the effects of thrombosis may persist long after the patient has met the criteria for discharge (27). In the recovery stage, it is worth considering whether the prothrombotic status is neglected in patients without thrombotic complications and whether procoagulant factors (such as PS exposure) return to normal in patients with thrombosis. We will investigate pulmonary susceptibility in long COVID and the possibility that prothrombotic states may influence long-term pulmonary symptoms.
Continuous Shedding of the Virus
Tarhini et al. reported cases of severely immunocompromised COVID-19 patients shedding infectious virus up to four months following symptom onset. In one instance, they reported a single persistent infection with high load culture-positive virus and positive reverse transcription polymerase chain reaction (RT-PCR) on day 103 (27 days after readmission). This study also included a discharged patient who developed post-COVID pneumonia with active virus replication in the lower respiratory tract and finally developed a double infection after second admission (48). The immune system can be suppressed or even depleted by fighting off the increasing viral load (23, 49). Therefore, even if patients have no compromised immune system before SARS-CoV-2 infection, they may show similar symptoms during the disease. In these cases, the virus may retain the ability to transmit over time, as evidenced by positive viral cultures. A variety of scenarios (such as asymptomatic carrier, symptom resolution, or secondary infection) allow for prolonged infectious virus emission (12, 50). In a cross-sectional study, 10 of 60 discharged COVID-19 patients tested positive for SARS-CoV-2 by RT-PCR 4-24 days after discharge. Since all discharged patients were instructed to stay at home and local cases were rare, the researchers assumed that the positive result was persistent virus shedding rather than reinfection (51). Viral persistence is associated with more extensive tissue invasion and worse recovery outcomes. Another study found that the average shedding time of the virus was 19 days in asymptomatic patients, ranging from 6-45 days. While circulating antibodies to other coronaviruses (such as SARS-CoV or MERS-CoV) have been shown to last for at least a year, antibodies to SARS-CoV-2 wane relatively quickly. In both the asymptomatic and symptomatic groups, IgG levels fell by more than 70% in more than 90% of cases during the early recovery period (8 weeks after discharge) (52). In a cohort study, the positive serum rate and median titer of neutralizing antibodies were significantly lower in the convalescent follow-up than during acute infection (12). A report assessed 30,082 patients with mild-to-moderate COVID-19 indicated that antibody titers remained stable at three months but declined slightly at the 5-month time point (49). There is evidence that the antigenicity of SARS-CoV-2 spike protein changes. Spike of amino acid substitutions and deletions impact neutralizing antibodies and the variants are resistant to antibody-mediated immunity elicited by vaccines (53). Therefore, the risk of reinfection should be monitored, especially in patients with prolonged viral shedding (54). SARS-CoV-2 is likely to persist in certain tissues to drive chronic symptoms. In a follow-up trial, approximately 4-6 months after diagnosis, positive SARS-CoV-2 ribonucleic acid (RNA) was detected in olfactory mucosa samples from four patients with negative nasopharyngeal swabs for SARS-CoV-2 RNA (55).
Pulmonary Vulnerability
Early Stage
SARS-CoV-2 enters local alveolar type II cells along the airway, replicates, and damages the targeted cells. Inflammatory mediators are produced when sentinel cells (pulmonary macrophages in the lung interstitium) surrounding the injured tissue recognize the damaged target cells. These mediators trigger neutrophils and monocytes in the blood circulation to migrate to the injured site under the action of chemokines. Various immune cells are mobilized and activated wherever the virus goes, inducing the release of inflammatory factors such as monocyte chemoattractant protein-1 (MCP-1), granulocyte-macrophage colony-stimulating factor (GM-CSF), interleukin-1β (IL-1β), tumor necrosis factor-α (TNF-α), interleukin 6 (IL-6), interferon-γ (IFN-γ), etc (56). Leukocytes, while necessary to phagocytose inflammatory substances, can also release lysosomal enzymes, reactive oxygen species, and free radicals into the extracellular stroma, damaging normal alveolar type I and II cells. Another function of leukocytes is to recognize membrane expression of PS, an ‘eat me’ signal for macrophages lest more extensive pro-coagulant surfaces appear (57, 58). Ideally, SARS-CoV-2 is gradually cleared by three defensive walls consisting of airway secretions, ciliary oscillations, and innate and acquired immune cells. Even if local ciliated goblet cells, mucous goblet cells, and alveolar epithelial cells are damaged, mild conditions will not evolve into persistent symptoms, and virus numbers may quickly start to decrease. The damaged target cells in the airway can be repaired by the proliferation and differentiation of basal cells. Still, alveolar cells are difficult to regenerate, which is often the precursor to long-term symptoms (59). The virus has the opportunity to invade vascular endothelial cells by crossing the local air-blood barrier (60, 61) (Figure 1A). However, the extent of the damage to the local vascular endothelial cells is difficult to predict in early stage. Nevertheless, these impaired vascular endothelial cells often serve as the initial site of thrombosis. At this time, activated vascular endothelial cells are often overlooked, but they are the vulnerable basis of long COVID after discharge (31, 33, 39, 43). Although vascular endothelial damage is difficult to distinguish clinically, it can be used as a differentiating point in pathology. This is important, because the chain reaction caused by damaged endothelial cells will affect the vascular and blood system (45).
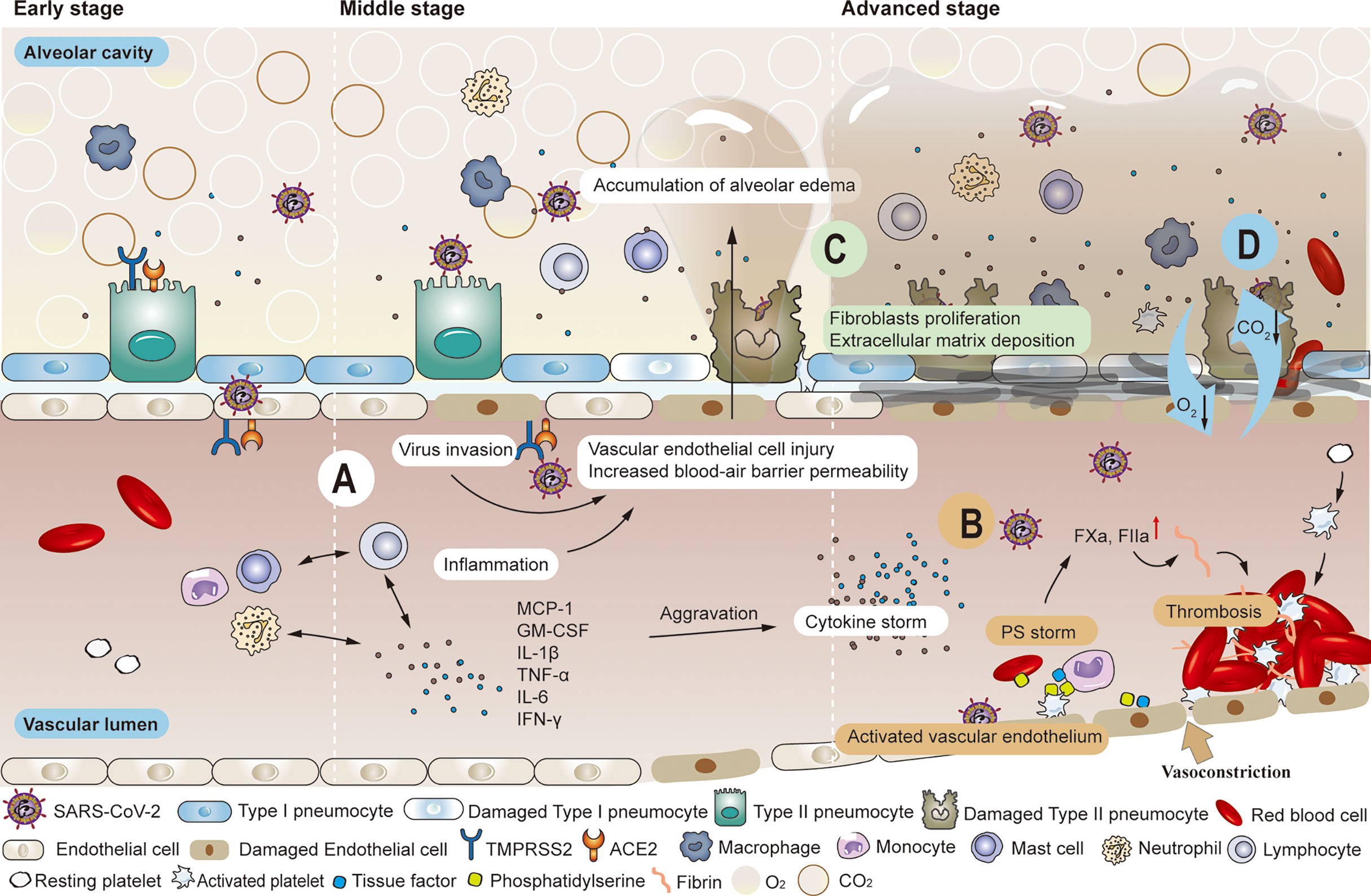
Figure 1 Common pathological changes in the lungs of patients with COVID-19. (A) SARS-COV-2 enters local alveolar type II cells along the airway, replicates, and damages the targeted cells. Various immune cells are mobilized and activated wherever the virus goes, inducing the release of inflammatory factors such as MCP-1, GM-CSF, IL-1β, TNF-α, IL-6, IFN-γ, etc. The inflammatory response produced in killing the virus also leads to the injury of alveolar type I and type II cells. When high viral load accompanies the inflammatory response, the air-blood barrier is destroyed on the alveolar side. The virus has the opportunity to invade vascular endothelial cells by crossing the local air-blood barrier, and strong cytokine responses can also spread to vascular endothelial cells. As the permeability of the blood-air barrier increases, blood components enter the alveolar cavity, forming pulmonary edema. (B) Significantly enhanced thrombin and elevated levels of endothelial cell biomarkers (vWF: Ag, vWFpp, FVIII, and sTM) were observed in the convalescence period. Impaired pulmonary vascular endothelium can cause uncontrolled activation of coagulation cascades, further leading to vascular thrombosis or fatal pulmonary fibrosis. (C) The initial response to the destruction of the alveolar epithelial-endothelial barrier is edematous infiltration in the alveoli and interstitial portion, followed by proliferation as the alveolar barrier is rebuilt by removing exudate. Extracellular matrix deposition occurs. Fibroblasts migrate and transform into muscle cells. (D) The damaged blood-air barrier, impaired pulmonary blood perfusion, reduced effective volume of alveolar cavities and the appearance of fibrosis all lead to the obstruction of the exchange of oxygen and carbon dioxide.
Middle Stage
Particular attention should be paid to patients with intermediate disease who have not developed to a severe stage. The increasing viral loads infect alveolar capillary endothelial cells at the blood-air barrier through damaged alveolar epithelial cells and alveolar interstitium (21). The enhanced defense system will inevitably cause tissue damage while killing the virus. However, the overall impact of the defense system is more positive than negative. The injured vascular endothelial cells initiate the coagulation cascade system, activating coagulation factor X and promoting thrombin production. Then, this catalyzes the conversion of fibrinogen to fibrin and form pulmonary microthrombi (21, 31, 33, 39, 43–47, 61–65). As weak parts of the alveolar membrane are destroyed, blood components such as water molecules, plasma proteins, and platelets enter the alveolar cavity, forming pulmonary edema (6) (Figure 1A). Many histopathological findings showed that the most frequently reported morphological feature of COVID-19 disease is diffuse alveolar damage, characterized by a variable degree of edema in the exudate phase (42, 66, 67). As long as reduction of alveolar volume is compensated and thrombosis can be prevented or dissolved, the trend towards severe complications can be blocked. However, even if the criteria for discharge are met after effective treatment, the influence of impaired alveolar ventilation and pulmonary microcirculation in the course of COVID continues, and the pulmonary tissues are vulnerable to further damage (11–13, 40, 41). In another study, no significant differences in forced expiratory volume in 1s (FEV1), forced vital capacity (FVC), or their rates were observed nearly one month after discharge, regardless of the severity of COVID-19. DLCO values decreased significantly with increasing severity of clinical symptoms (total 47.2%, mild 30.4%, moderate 42.4%, severe pneumonia 84.2%) (68). Huang et al. observed that 30 days after discharge, DLCO values of patients were notably different (< 80%), 42.5% in non-critical patients versus 75.6% in severe patients (69).
Advanced Stage
When the disease progresses to the severe/critical stage, the primary clinical task is to prevent and treat multiple organ failures and prolong life regardless of the risk of subsequent sequelae. With the exponential increase in SARS-CoV-2 particles, a large number of immune cells activate and release cytokines while gathering around and infiltrating into the lung tissue, thus initiating relevant transduction pathways and a cascade of inflammatory reactions. This creates a vicious cycle that eventually leads to a cytokine storm (70–77). Severe cases can also be accompanied by lymphocytic depletion, leading to suppression or even failure of the immune system (2, 78). Direct and rapid cytotoxic effects of plasma from critically ill patients on umbilical cord blood tubule cells were found in vitro (79). Researchers extracted plasma from healthy donors, non-intensive care unit (non-ICU) patients with COVID-19, intensive care unit (ICU) patients with COVID-19, and convalescent patients with COVID-19. Results showed that plasma from both COVID-19 patients and convalesced patients significantly reduced human pulmonary microvascular endothelial cells activity compared to healthy plasma, but plasma from ICU patients induced the greatest cytotoxicity. Blood vessel involvement through endotheliitis is also one of the distinguishing features of COVID-19. Microthrombi within alveolar capillaries, precapillary arteries, and postcapillary venules were frequently reported (66). Studies have shown that alveolar capillary microthrombi were 9 times more common in patients with COVID-19 than in patients with influenza, and the amount of new pulmonary vessel growth were 2.7 times higher than in patients with influenza (80). Damaged vascular endothelium contributes to a pre-thrombotic state, further activating the clotting pathway, accelerating (micro) thrombogenesis, and reducing alveolar blood flow. Impaired pulmonary vascular endothelium can cause uncontrolled activation of coagulation cascades, further leading to vascular thrombosis or fatal pulmonary symptoms of fibrosis (21) (Figure 1B). Many studies have suggested that severe and critical COVID-19 is associated with an increased incidence of diffuse thrombosis or pulmonary blood vessel thrombosis (2, 81–83). In severe cases, hypoxic capillary constriction and pulmonary microthrombus, thrombosis, and/or embolism cause slow blood flow, local blocked microvasculature, elevated pulmonary capillary pressure, and overall pulmonary hypertension (84). Changes in the vasculature, coupled with extensive inflammation of lung tissue, enhance the permeability of the air-blood barrier, resulting in vascular leakage with plasma and blood cells entering the alveolar cavity. Pulmonary hyaline membrane formation, acute respiratory distress, and pulmonary fibrosis exacerbate dyspnea (1–8) Impaired pulmonary vascular endothelium can cause uncontrolled activation of coagulation cascades, further leading to vascular thrombosis or fatal pulmonary symptoms of fibrosis (21) (Figure 1C). Therefore, besides damaged alveolar structure, reduced effective volume of the alveolar cavity, and difficulty in gas dispersion (85), (Figure 1D) insufficient alveolar blood flow caused by thrombus also needs timely improvement. However, severe or critical illness can present challenges. The incidence of bleeding events and sequelae is relatively high and monitoring is essential to maintain the patient’s health status (86–92). The importance of thrombus and embolic events in severe and critically ill patients is widely recognized (93–95). But the occurrence of hypoxemia even with good lung compliance in the early stage also indicates that early abnormal pulmonary blood perfusion may also exist (26, 96, 97). Autopsy results showed microthrombi in the lung but no destruction of surrounding alveolar structures (98), further suggesting that pulmonary blood perfusion is of great importance in the formation of microthrombi. Since this circumvention of traditional ARDS formation has been found, attention should also be paid to lung damage caused by microthrombi in patients recovering from COVID-19.
Lung Injury in Long COVID
The most commonly reported lingering symptoms of COVID-19 at discharge are fatigue, muscle weakness, sleep disturbances, abnormal lung dispersion, anxiety, and depression (12). Although fatigue and weakness are the most common effects in long COVID, some survivors also report persistent severe symptoms and organ dysfunction (88). A meta-analysis of 16 cohort studies showed that discharged patients could develop residual symptoms in multiple organs, including cardiopulmonary (chest pain, dyspnea, cough, sore throat, and palpitations), nerve (dysmnesia, cognitive disorder, headache, dysgeusia, and dysosmia), gastrointestinal tract (diarrhea, vomiting, abdominal pain, and anorexia), eyes (conjunctivitis), skin (urticaria), musculoskeletal system (myalgia, and arthralgia), etc. (86) In addition to the psychological impact, there is overwhelming evidence that the lung is the most severely affected organ in COVID-19 patients, both in the progressive and convalescent stages (99, 100). In a 6-month follow-up study involving 1733 discharged patients, those requiring high flow nasal catheter (HFNC), non-invasive ventilation (NIV), or intermittent mandatory ventilation (IMV) had an odds ratio (OR) of 4.60 (after multivariable adjustment) for diffusion disorders compared with those requiring no supplemental oxygen. 36% of patients in the severest group had dyspnea with a modified Medical Research Council (mMRC) score > 1 (severe dyspnea) at six months. 50% of patients who completed high-resolution computed tomography chest scans across different severity scales had at least one CT anomaly, with GGO being the most common, followed by irregular lines (12). Revisiting the survivors after 12 months showed a slight increase in the rate of dyspnea. There was no improvement in pulmonary diffusion impairment. And the incidence of pulmonary diffusion impairment was 23% in the no oxygen group, 31% in the oxygen-required group, and 54% in the group with HFNC, NIV, or IMV. The proportion of CT abnormalities decreased significantly over time. But 76% of patients in the severe group still had GGO, and the proportion of patients with thickened interlobular septa increased significantly (11). Wu et al. tested lung function in 83 survivors of severe COVID-19 pneumonia. Although the 6mWT and dyspnea score showed significant improvement at 12 months, 33% had DLCO < 80%, and 24% had GGO radiological abnormalities (13). While most studies have focused on the long-term effects of COVID-19 on hospitalized patients, little is known about the statistics of long COVID in patients with mild or asymptomatic disease. SARS-CoV-2 infection can have subtle effects on the body, even if the patient does not require hospitalization. A study of 8,983 non-hospitalized patients two weeks after a positive test showed that these individuals had a slightly increased risk of initial diagnosis of dyspnea (1.2% vs. 0.7%) and venous thromboembolism (0.2% vs. 0.1%) compared with matched SARS-CoV-2-negative individuals. However, similar results were not found for increased risk of serious complications (such as ischemic stroke, encephalitis, psychosis, or multisystem inflammatory syndrome in children), as previously seen in severe COVID-19 hospitalizations. Positive patients were more likely to initiate bronchodilator therapy, particularly short-acting beta2 agonists (17% vs. 13%), which may be associated with dyspnea (40).
In one meta-analysis of 894 subjects from seven studies, lung function tests showed that low diffusion ability was the most common abnormality, followed by reduced lung volume, while airflow obstruction was relatively uncommon (86). Damage and repair of the blood-air barrier play an essential role in long COVID. Alveolar epithelium is a single layer of epithelial cells in which a subpopulation of alveolar type II cells undergoes self-repair after injury and act as precursors of type I cells. Alveolar type II epithelia are the dominant target cells for SARS-CoV-2. Therefore, impaired type II cells can significantly impede epithelial repair mechanisms, resulting in incomplete repair, scarring, and fibrosis (59). The initial response to the destruction of the alveolar epithelial-endothelial barrier is edematous infiltration in the alveoli and interstitial portion. This is followed by proliferation as the alveolar barrier is rebuilt by removing exudate. However, in some patients, it progresses to excessive fibrosis rather than dissipating inflammation. During the recovery of influenza and SARS, evidence of parenchymal fibrous bands and tractive bronchiectasis has been observed (101, 102). Studies have also found that elevated growth factor receptor B1 mediates extracellular interstitial protein deposition, chemotactic fibroblast migration, and the transformation of fibroblasts into myocytes (4). It has also been suggested that respiratory virus infection may induce significant fibroblast activation during convalescence (12). It is unclear whether COVID-19-associated ARDS causes irreversible pulmonary fibrosis. Studies have observed dramatic increases in the number of lung fibroblasts and collagen deposits in cases of fatal COVID-19 disease (1–8). However, whether long COVID fibrosis will stabilize and subside in subsequent years remains uncertain (5).
When injured host cells release damage-related molecular patterns, the pro-inflammatory molecules and activated immune cells lead to endothelial cell damage, hypoxia, and dysfunction in pulmonary vessels (21). Hypoxia is a driver of vascular remodeling, inducing the activation of endothelial, mesenchymal, and immune cells and promotes thrombotic fibrosis and epithelial-mesenchymal transformation in COVID-19 patients. Similar vascular remodeling can occur in pulmonary hypertension and chronic obstructive pulmonary disease, but the degree of remodeling is greater in patients with COVID-19. In COVID-19, the endothelial cells transform into smooth muscle cells. Proliferation, migration, and hypertrophy of vascular smooth muscle cells were observed at the cellular level (7). In addition, overexpression and high levels of pro-angiogenic factors (such as vascular endothelial growth factor (VEGF), hypoxia-inducible factor 1α (HIF-1α), IL-6, tumor necrosis factor receptor superfamilies 1a and 12, and angiotensin-converting enzyme 2(ACE2)) have been found in both living and dead COVID-19 patients (103). In one study, compared with healthy volunteers, patients with COVID-19 had more pulmonary vessels with a 5-30mm2 cross-sectional area and fewer small vessels (0-< 5mm2). However, there was no difference in overall lung blood volume, suggesting blood redistribution between blood vessels of different sizes (43). Histological evaluation of early COVID-19 showed an abnormal increase in the number of pulmonary blood vessels, accompanied by hyperemia, dilation, and distortion. CD4+ T lymphocytes infiltrated the edema wall and thickened post-capillary venules (44). Compared with the normal vascular endothelium, these new blood vessels, together with the damaged vascular endothelium, are still activated in the convalescence period. They are therefore unable to fully fulfill their role in maintaining the normal blood-air barrier in an anti-coagulant state. As a result, patients can still suffer from respiratory insufficiency and other pulmonary symptoms following COVID-19.
New Point: PS and Thrombosis
Pulmonary vascular endothelial cells prevent thrombosis by binding to TF pathway inhibitors (TFPIs) and blocking the action of the FVIIa-TF complex (104). The presence of various endothelial injury biomarkers, including extracellular vesicles, confirms the persistence of vascular damage in convalescent COVID-19. Significantly elevated thrombin and endothelial cell biomarkers (vWF antigen (vWF: Ag), vWF propeptide (vWFpp), FVIII, and sTM) were observed in the convalescence period. At this point, most patients have normalized acute phase markers, including C-reactive protein, neutrophil counts, white blood cell counts, IL-6, and sCD25 levels (105). Persistent endothelial lesions were also observed during recovery in non-hospitalized patients (31). Impaired pulmonary vascular endothelium can cause uncontrolled activation of coagulation cascades, further leading to vascular thrombosis or fatal pulmonary symptoms of fibrosis (21). Evasio et al. monitored serological markers in 75 patients who had been discharged from the hospital for two months after COVID-19. They found high concentrations of D-dimer, and this persistent change raised the long-term risk of thromboembolic disease in convalescence patients (106).
Ongoing monitoring of COVID-19 patients after discharge from the hospital is necessary to understand the breadth and severity of long-term effects. However, COVID-19 has not existed long enough to complete large-scale cohort studies to examine its long-term impacts on infected patients in detail. Although a critical factor in the development of disease, thrombus-related indicators have rarely been comprehensively studied (106). Thrombosis is a pathological outcome of the local microenvironment. The research on thrombosis should not be limited to its subsequent influence on tissues or organs but should also include the mechanisms involved in thrombosis formation, such as the close connection with vascular endothelium, damage to blood cells, formation of the procoagulant state, and the existence of microthrombi (83, 107–110). Fibrin clumps formed in hypercoagulable conditions are difficult to detect, in contrast to thrombus formation (111). But during the transition period between disease progression and recovery period, it is difficult to judge the extent of the risk of locally developing arteriovenous thrombosis. It is also difficult to confirm whether there has been improvement of the local endothelial injury and return to an anti-coagulant state (21). However, these processes are undeniably common in COVID-19. Therefore, in long COVID, the influence of thrombosis should be assessed from the onset of the prothrombotic state. The degree of early injury and the progression from functional to organic lesions are associated with dyspnea that affects long-term quality of life.
Elevated endothelial stress products are present in the circulating blood of COVID-19 patients. Although endothelial alterations are not specific, thrombogenesis caused by endothelial alterations in COVID-19 results in fibrin deposition in small blood vessels in the lungs and other organs. An early step in the thrombogenesis process is the expression of the pro-coagulant phospholipid PS. In normal conditions, PS is confined to the inner layer of cell membranes by the actions of floppase and flippase. When intracellular Ca2+ increases, the ATP-dependent translocation enzyme is blocked, and scramblase is activated, resulting in a random distribution of PS to both sides of the membrane (112). Once exposed on the outer membrane, PS mediates TF decryption and activation, initiating the coagulation cascade (113). PS also provides an active catalytic surface for the formation of the TF-FVIIa, factor X-enzyme (FIXa-FVIIIa-Ca2+-PL), and prothrombinase (FXa-FVa-Ca2+-PL) complexes, leading to the conversion of fibrinogen to fibrin. Pulmonary microthrombi can further develop into pulmonary arteriovenous thrombosis and decrease alveolar blood flow (Figure 2B). In addition to providing a negatively charged surface to initiate and maintain clotting functions, PS also acts as a signal to be engulfed by macrophages, avoiding the activation of inflammation and autoimmunity (58). There are two modes of recognition between macrophages and PS-expressing cells. One is direct recognition by the phagocytic receptors: brain-specific angiogenesis inhibitor 1 (BAI1), T cell immunoglobulin mucin 4 (TIM-4), and Stabilin 2. The other is indirect recognition. The bridging molecules milk fat globule epidermal growth factor 8 (MFG-E8, also known as lactadherin) and growth arrest-specific 6 (GAS6) bind to PS (114, 115), which is recognized by membrane proteins MER and αVβ3. These two recognition patterns are not mutually exclusive and may co-occur (57) (Figure 2A). Biomarkers of platelet activation are associated with thrombosis and mortality risk in COVID-19 (116, 117). Several studies have pointed to microvesicles (EVs) and platelet-derived microvesicles (pEVs) as potential biomarkers in COVID-19. Elevated levels of circulating pEVs have been observed in patients with SARS-CoV-2 infection and significantly elevated levels of pEVs in patients with severe disease (118–122). One study using flow cytometry of patient samples found that the frequency of PS+ cells in the blood of all COVID-19 patients within a week of diagnosis was considerably higher than that of peripheral blood mononuclear cells (PBMC) from healthy or recovered donors. The number of PS+ PBMC is strongly correlated with the severity of disease and can better predict the need for respiratory support (123). Corresponding autoantibodies to the PS/prothrombin complex have also been found in COVID-19 patients (124). EVs, approximately 100 to 1000 nm in diameter, are produced by budding and shedding of the plasma membrane of various blood cells. Since EVs are unable to maintain membrane asymmetry, they are characterized by PS externalization and can affect the regulation of coagulation and inflammation (125–127). In both sepsis and COVID-19, upregulation of PS exposure can occur on cell surfaces (including endothelial cells, platelets, red blood cells, neutrophils, and lymphocytes) and extracellular particles (128–130). Karina et al. reported significantly increased depolarization of mitochondrial inner transmembrane potential and cytosolic Ca2+ and PS externalization in ICU patients compared with healthy controls and non-ICU patients with COVID-19 (131). Since the localization of PS on subcellular organelles was first reported in 2008, we have been focusing on PS-induced hypercoagulability and thrombotic events. The presence of PS+ blood cells, endothelial cells, and particles has been found in experimental studies of acute promyelocytic leukemia, nephrotic syndrome, sepsis, inflammatory bowel disease, acute stroke, and triple-negative breast cancer, suggesting that PS-induced procoagulant activity may be common in various diseases (132–136).
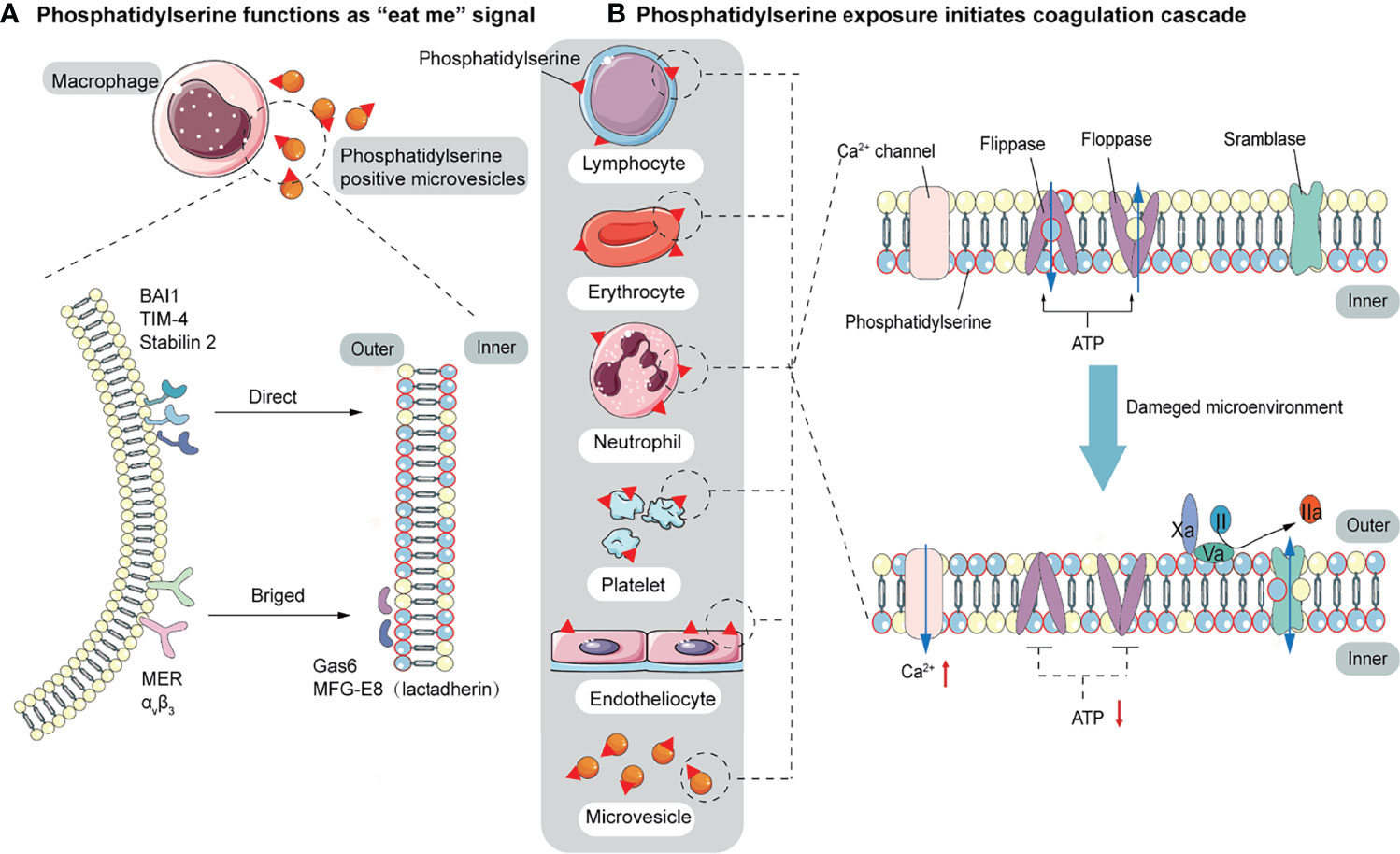
Figure 2 The function of PS. (A) PS acts as a signal to be engulfed by macrophages with two recognition modes. One is directly recognized by phagocytic receptors BAI1, TIM-4, and Stabilin 2. The other is indirect recognition. The bridging molecules MFG-E8 (also known as lactadherin) and GAS6 bind to PS, which is recognized by membrane proteins MER and αVβ3. These two recognition patterns are not mutually exclusive and may co-occur. (B) PS provides a negatively charged surface to initiate and maintain coagulation. In normal conditions, PS is sequestered in the inner layer of cell membranes by the action of floppase and flippase. When intracellular Ca2+ increases, the ATP-dependent translocation enzyme is blocked, and the scramblase is activated, resulting in a random distribution of PS to both sides of the membrane. PS, initially located in the cell’s inner membrane, is exposed to the outer side. On the outer membrane, PS provides active clotting catalytic surfaces for forming TF-FVIIa complex, factor X-enzyme complex (FIXa-FVIIIa-Ca2+-PL), and prothrombinase complex (FXa-FVa-Ca2+-PL).
Inhibiting the Prothrombotic State
In terms of thrombosis, thrombogenesis should be blocked from the beginning, and the procoagulant state should be alleviated to reduce the incidence of sequelae. D-dimer is commonly used as a marker for thrombosis, and a higher D-dimer level is independently associated with a higher risk of death. In some studies, changes in D-dimer level are used to distinguish the severity of COVID-19 in the middle and late stages (137–140). Because existing anti-thrombotic interventions appear to have limited effects in the severe and critical stage, it is crucial to take measures at moderate or even mild stage to improve patient outcomes and reduce the occurrence of sequelae. In late stage COVID-19 patients, the maximum solubility assessed by rotary thromboelastometer analysis was significantly less in patients with thrombus compared with patients without thrombotic events (141–143). Meanwhile, the increased levels of tissue plasminogen activator and plasminogen activator inhibitor-1 in patients’ blood circulation further suggest impaired fibrinolysis (144). It is important to relieve thrombus formation tendency or remove (micro) thrombi early. Therefore, the focus should be on early antithrombotic therapy, including anticoagulation, anti-platelet activation, and thrombolytic therapy as appropriate.
As a means to prevent thrombosis and relieve hypercoagulability, anticoagulant therapy has been studied primarily in the acute stage of COVID-19. Currently, all guidelines agree that low-molecular-weight heparin thromboprophylaxis should be used in all hospitalized patients with COVID-19, recognizing that hypercoagulability can contribute to more severe disease progression (145–149). The commonly used anticoagulant drugs are low molecular weight heparin (LMWH) (such as enoxaparin) and direct oral anticoagulants (such as Rivarxaban and dabigatran). Heparin can also play a part in controlling leukocyte migration and complement activation (150). Treatment with high-dose prophylactic anticoagulation was associated with a significantly reduced risk of pulmonary embolism (hazard ratio, 0.72, 95%CI, 0.53-0.98) in a study of patients admitted to ICU 14 days after COVID-19 diagnosis (151). A New York study, using the Cox proportional risk model to assess the effect of therapeutic-dose anticoagulation on in-hospital mortality, found that patients treated with anticoagulant had a 22.5% in-hospital mortality and a median survival of 21 days, compared with 22.8% who did not receive anticoagulant and a median survival of 14 days. Systemic therapeutic dose anticoagulation may be associated with improved outcomes in hospitalized patients with COVID-19 (152). One study found that for non-critical patients with COVID-19, therapeutic anticoagulation improved hospital discharge survival without organ support (153). Theoretically, inhibition of hypercoagulability can block the occurrence of microthrombotic events, especially with early treatment. As the site of initial infection, lungs are the most susceptible organ where the effects of inflammation and thrombosis appear early. With early anticoagulation, the pulmonary circulation remains unblocked so that inflammatory substances formed in the lungs that enter systemic circulation can be removed by the immune system. Limiting the level of inflammation in the lungs can reduce the damage to the alveoli and prevent the generation of cytokine storms and PS storms. Effective control of inflammation and improvement of hypoxia reduces damage to endothelial cells and prevents a large number of endothelial cells from transitioning to a defensive state.
Some studies have failed to show prolonged survival time or improved survival rate as a result of anticoagulant therapy. In these studies, most samples are patients with severe or critical disease. A large number of clots have formed, resulting in the depletion of clotting factors resulting in a low fibrinolytic state. Under these conditions, anticoagulants do not protect the patient (138, 154). After the necessary thrombolysis or thrombectomy to narrow and remove the clot, the alveolar perfusion blood flow is improved, and function is gradually restored. However, the inflammatory storm and PS storm, which were originally confined to the lung, can also quickly enter the systemic circulation, accelerate injury to the extrapulmonary organs, and even lead to death. Another possible explanation is the dosage of anticoagulant therapy. Some studies have used therapeutic doses of LMWH for thromboprophylaxis in critically ill patients with thrombotic risk factors (146). The incidence of bleeding events with therapeutic dose anticoagulants was 3.0%, and 1.7% with the prophylactic dose. Among all bleeding events, the gastrointestinal tract was the most common (50.7%), followed by mucosa (19.4%), bronchia (14.9%), and intracalvarium (6%), but fatal bleeding events were rare (155–157). This suggests that therapeutic dose anticoagulants improve overall survival. Meanwhile, some studies have shown that therapeutic doses do not increase the risk of bleeding (158). However, some studies have avoided an increased dose of thromboprophylaxis due to a slight increase in bleeding events (159, 160). The difficulty of using anti-thrombotic therapy during the period of severe and critical illness highlights the importance of timely comprehensive antithrombotic therapy before the late stage. From this perspective, treatment to prevent mild or moderate disease from progressing to a severe or critical condition will significantly improve the overall prognosis. Temporary appropriate comprehensive treatment can effectively block the trend of severe disease development and reduce the occurrence of sequelae in the long run (161). In a prospective study in France, analysis using propensity score matching confirmed that pre-hospital anticoagulant treatment was associated with a better outcome, with a risk of 0.43(95% CI, 0.29-0.63) for admission to intensive care (162). The difference in therapeutic anticoagulant efficacy between moderate and critically ill patients may be attributed to severe inflammatory responses, when thrombotic complications in critically ill patients are too pronounced to recover. In non-ICU patients, therapeutic anticoagulant therapy may still help maintain an appropriate balance (163). As for the risk of bleeding events, because the vascular endothelial cells are relatively undamaged and the coagulation factors are not yet depleted, the risk of antithrombotic bleeding is lower than the risks associated with respiratory distress syndrome, multiple organ failure, and/or sequelae. Christopher T Rentsch and colleagues found that patients who received prophylactic anticoagulation within 24 hours of admission had a 27% reduction in 30-day mortality (hazard ratio, 0.73; 95% confidence interval, 0.66 to 0.81) compared with those who did not. Receiving prophylactic anticoagulant therapy was not associated with an increased risk of bleeding requiring transfusion (hazard ratio, 0.87,0.71 to 1.05) (164). Unobstructed blood flow ensures adequate blood perfusion to the alveoli, reducing hypoxemia incidence. Unimpeded pulmonary circulation slows or prevents the leakage of plasma into the alveolar cavity, which is aggravated when there is a large pressure difference between the two sides of the alveolar membrane. Thus improved circulation prevents pulmonary hypertension, lung hyaline membrane formation, and lung consolidation, thereby reducing the risk of death and sequelae. Current guidelines have no routine precautions for discharged patients. Some recommend anticoagulant prophylaxis (LMWH or direct oral anticoagulants) in high-risk patients with a low risk of bleeding (165).
The application of antiplatelet drugs mainly includes aspirin (cyclooxyganese inhibitor), clopidogrel (P2Y12 inhibitor), or dipyridamole (adenosine deaminase and phosphodiesterase) (166, 167). It has been reported that clopidogrel may interact with antiviral drugs and should be used with caution. Dipyridamole may be considered for antiplatelet therapy in the presence of renal insufficiency to reduce bleeding due to drug build-up. Aspirin is the most commonly used antiplatelet drug with anti-inflammatory, antipyretic, analgesic and antiplatelet functions. In infectious diseases, aspirin is associated with a reduction in thrombotic inflammation, clinical complications, and in-hospital mortality (168). In a retrospective cohort study of COVID-19, aspirin had some benefits in reducing the risk of mechanical ventilation, ICU admission, and in-hospital mortality (169). Similar results were found in a small observational cohort study of adults with COVID-19 when aspirin was taken at least seven days before or within 24 hours of hospitalization compared with no aspirin (170). In another recent observational study, 730 patients who received antiplatelet therapy had lower mortality and shorter mechanical ventilation duration during hospitalization than 6986 patients who did not receive antiplatelet treatment (171). However there are clinical trials showing that aspirin is not associated with a reduced risk of ARDS (172). Aspirin, as an irreversible platelet inhibitor, is cheap and available, and trials are investigating its effect on the risk of thrombosis.
PS: Novel Therapeutic Targets
PS, an initial factor of the coagulation cascade, could potentially be used as a new therapeutic target. Lactadherin combined with PS is a more targeted “eat me” signal that could prevent or reduce hypercoagulability (173). PS+ EVs and cells provide a platform for the anchoring of coagulation factors. Annexin V and lactadherin interrupt coagulation cascades by selectively binding PS (174). Lactadherin is structurally homologous to FVIII and FV and effectively blocks the availability of PS for coagulation reactions (175). The levels of PS+ cells and pEVs in COVID-19 patients were higher than those in healthy controls and are positively correlated with the severity of the disease. Due to the high likelihood of diffuse microthrombi and arteriovenous thrombus in critically ill patients and the procoagulant role of PS, we think that Annexin V or lactadherin could reduce the incidence of thrombosis in COVID-19 patients (118, 123). In the presence of a large amount of PS, the inhibition of upstream FXIa, FXIIa, and FXa generation can block IIa generation and avoid the formation of thrombus. While the anticoagulant effect of lactadherin has been confirmed in vitro, it’s anticoagulant effect in vivo requires additional study (176). As an under-recognized hemostatic regulator, lactadherin is a potential therapeutic agent in preventing COVID-19 thrombosis (177).
Discussion
Due to the high prevalence of respiratory failure and the need for mechanical ventilation in COVID-19, a significant number of patients will be at risk of long-term complications following severe lung disease. Currently, the world has limited knowledge of long-term lung disease in survivors, and long COVID is still a public health concern. There may be variation in the phenotypes of long COVID, and different post-infection states exist in survivors of COVID-19 ARDS. Many people continue to experience respiratory symptoms for months after an acute infection, especially those with underlying asthma. Other subgroups of patients appear to worsen within three to four weeks of initial infection and brief recovery. Some patients start with a very mild course of illness (do not require medical care or hospitalization) but go on to develop infectious symptoms and ARDS after a few weeks. Thrombus is a crucial factor in the progression of COVID-19 to severe disease, and its importance in long COVID has not been fully assessed. Early treatment of microthrombi can reduce not only mortality but also reduce the incidence of sequelae. PS expression is common when cells are damaged or undergoing apoptosis. And epidemic diseases often have a large number of cell damage and death, forming PS storms and promoting thrombosis. While vaccines are essential in preventing severe disease, effective treatment of COVID-19 remains essential given the rise of virus variants and the waning effectiveness of vaccines.
Author Contributions
MX prepared figures and wrote the manuscript. HJ and CW provided helpful comments and acquired data. JS came up with the project, designed the study, contributed to successive drafts, and reviewed this manuscript. VN gave the revision advice and polished this review. All authors read and approved the final manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank all participants for their contribution to our study and the reviewers for the suggestions provided.
References
1. Rello J, Storti E, Belliato M, Serrano R. Clinical Phenotypes of SARS-CoV-2: Implications for Clinicians and Researchers. Eur Respir J (2020) 55(5):2001028. doi: 10.1183/13993003.01028-2020
2. Attaway AH, Scheraga RG, Bhimraj A, Biehl M, Hatipoğlu U. Severe COVID-19 Pneumonia: Pathogenesis and Clinical Management. BMJ (2021) 372:n436. doi: 10.1136/bmj.n436
3. Oronsky B, Larson C, Hammond TC, Oronsky A, Kesari S, Lybeck M, et al. A Review of Persistent Post-COVID Syndrome (PPCS). Clin Rev Allergy Immunol (2021) 20:1–9. doi: 10.1007/s12016-021-08848-3
4. Cueto-Robledo G, Porres-Aguilar M, Puebla-Aldama D, Barragán-Martínez MDP, Jurado-Hernández MY, García-César M, et al. Severe Pulmonary Hypertension: An Important Sequel After Severe Post-Acute COVID-19 Pneumonia. Curr Probl Cardiol (2021) 30:101004. doi: 10.1016/j.cpcardiol.2021.101004
5. Michalski JE, Kurche JS, Schwartz DA. From ARDS to Pulmonary Fibrosis: The Next Phase of the COVID-19 Pandemic? Transl Res (2022) 241:13–24. doi: 10.1016/j.trsl.2021.09.001
6. Archer SL, Sharp WW, Weir EK. Differentiating COVID-19 Pneumonia From Acute Respiratory Distress Syndrome and High Altitude Pulmonary Edema: Therapeutic Implications. Circulation (2020) 142(2):101–4. doi: 10.1161/CIRCULATIONAHA.120.047915
7. John AE, Joseph C, Jenkins G, Tatler AL. COVID-19 and Pulmonary Fibrosis: A Potential Role for Lung Epithelial Cells and Fibroblasts. Immunol Rev (2021) 302(1):228–40. doi: 10.1111/imr.12977
8. Fox SE, Akmatbekov A, Harbert JL, Li G, Quincy Brown J, Vander Heide RS. Pulmonary and Cardiac Pathology in African American Patients With COVID-19: An Autopsy Series From New Orleans. Lancet Respir Med (2020) 8(7):681–6. doi: 10.1016/S2213-2600(20)30243-5
9. Blanco JR, Cobos-Ceballos MJ, Navarro F, Sanjoaquin I, Arnaiz de Las Revillas F, Bernal E, et al. Pulmonary Long-Term Consequences of COVID-19 Infections After Hospital Discharge. Clin Microbiol Infect (2021) 27(6):892–6. doi: 10.1016/j.cmi.2021.02.019
10. Higgins V, Sohaei D, Diamandis EP, Prassas I. COVID-19: From an Acute to Chronic Disease? Potential Long-Term Health Consequences. Crit Rev Clin Lab Sci (2021) 58(5):297–310. doi: 10.1080/10408363.2020.1860895
11. Huang L, Yao Q, Gu X, Wang Q, Ren L, Wang Y, et al. 1-Year Outcomes in Hospital Survivors With COVID-19: A Longitudinal Cohort Study. Lancet (2021) 398(10302):747–58. doi: 10.1016/S0140-6736(21)01755-4
12. Huang C, Huang L, Wang Y, Li X, Ren L, Gu X, et al. 6-Month Consequences of COVID-19 in Patients Discharged From Hospital: A Cohort Study. Lancet (2021) 397(10270):220–32. doi: 10.1016/S0140-6736(20)32656-8
13. Olliaro PL. An Integrated Understanding of Long-Term Sequelae After Acute COVID-19. Lancet Respir Med (2021) 9(7):679–80. doi: 10.1016/S2213-2600(21)00206-X
14. Adeloye D, Elneima O, Daines L, Poinasamy K, Quint JK, Walker S, et al. The Long-Term Sequelae of COVID-19: An International Consensus on Research Priorities for Patients With Pre-Existing and New-Onset Airways Disease. Lancet Respir Med (2021) 9(12):1467–78. doi: 10.1016/S2213-2600(21)00286-1
15. Naeije R, Caravita S. Phenotyping Long COVID. Eur Respir J (2021) 58(2):2101763. doi: 10.1183/13993003.01763-2021
16. Zhang YY, Li BR, Ning BT. The Comparative Immunological Characteristics of SARS-CoV, MERS-CoV, and SARS-CoV-2 Coronavirus Infections. Front Immunol (2020) 11:2033. doi: 10.3389/fimmu.2020.02033
17. Peng Y, Tao H, Satyanarayanan SK, Jin K, Su H. A Comprehensive Summary of the Knowledge on COVID-19 Treatment. Aging Dis (2021) 12(1):155–91. doi: 10.14336/AD.2020.1124
18. Chen W, Lan Y, Yuan X, Deng X, Li Y, Cai X, et al. Detectable 2019-Ncov Viral RNA in Blood Is a Strong Indicator for the Further Clinical Severity. Emerg Microbes Infect (2020) 9(1):469–73. doi: 10.1080/22221751.2020.1732837
19. Pinney SP, Giustino G, Halperin JL, Mechanick JI, Neibart E, Olin JW, et al. Coronavirus Historical Perspective, Disease Mechanisms, and Clinical Outcomes: JACC Focus Seminar. J Am Coll Cardiol (2020) 76(17):1999–2010. doi: 10.1016/j.jacc.2020.08.058
20. Sa Ribero M, Jouvenet N, Dreux M, Nisole S. Interplay Between SARS-CoV-2 and the Type I Interferon Response. PloS Pathog (2020) 16(7):e1008737. doi: 10.1371/journal.ppat.1008737
21. Maiuolo J, Mollace R, Gliozzi M, Musolino V, Carresi C, Paone S, et al. The Contribution of Endothelial Dysfunction in Systemic Injury Subsequent to SARS-CoV-2 Infection. Int J Mol Sci (2020) 21(23):9309. doi: 10.3390/ijms21239309
22. Godoy LC, Goligher EC, Lawler PR, Slutsky AS, Zarychanski R. Anticipating and Managing Coagulopathy and Thrombotic Manifestations of Severe COVID-19. CMAJ (2020) 192(40):E1156–61. doi: 10.1503/cmaj.201240
23. McElvaney OJ, McEvoy NL, McElvaney OF, Carroll TP, Murphy MP, Dunlea DM, et al. Characterization of the Inflammatory Response to Severe COVID-19 Illness. Am J Respir Crit Care Med (2020) 202(6):812–21. doi: 10.1164/rccm.202005-1583OC
24. Noubouossie DF, Reeves BN, Strahl BD, Key NS. Neutrophils: Back in the Thrombosis Spotlight. Blood (2019) 133(20):2186–97. doi: 10.1182/blood-2018-10-862243
25. Mackman N, Antoniak S, Wolberg AS, Kasthuri R, Key NS. Coagulation Abnormalities and Thrombosis in Patients Infected With SARS-CoV-2 and Other Pandemic Viruses. Arterioscler Thromb Vasc Biol (2020) 40(9):2033–44. doi: 10.1161/ATVBAHA.120.314514
26. Bhatia P, Mohammed S. Severe Hypoxemia in Early COVID-19 Pneumonia. Am J Respir Crit Care Med (2020) 202(4):621–2. doi: 10.1164/rccm.202004-1313LE
27. Piazza G, Morrow DA. Diagnosis, Management, and Pathophysiology of Arterial and Venous Thrombosis in COVID-19. JAMA (2020) 324(24):2548–9. doi: 10.1001/jama.2020.23422
28. Price LC, McCabe C, Garfield B, Wort SJ. Thrombosis and COVID-19 Pneumonia: The Clot Thickens! Eur Respir J (2020) 56(1):2001608. doi: 10.1183/13993003.01608-2020
29. Silva Andrade B, Siqueira S, de Assis Soares WR, de Souza Rangel F, Santos NO, Dos Santos Freitas A, et al. Long-COVID and Post-COVID Health Complications: An Up-To-Date Review on Clinical Conditions and Their Possible Molecular Mechanisms. Viruses (2021) 13(4):700. doi: 10.3390/v13040700
30. Pretorius E, Vlok M, Venter C, Bezuidenhout JA, Laubscher GJ, Steenkamp J, et al. Persistent Clotting Protein Pathology in Long COVID/Post-Acute Sequelae of COVID-19 (PASC) Is Accompanied by Increased Levels of Antiplasmin. Cardiovasc Diabetol (2021) 20(1):172. doi: 10.1186/s12933-021-01359-7
31. Fogarty H, Townsend L, Morrin H, Ahmad A, Comerford C, Karampini E, et al. Persistent Endotheliopathy in the Pathogenesis of Long COVID Syndrome. J Thromb Haemost (2021) 19(10):2546–53. doi: 10.1111/jth.15490
32. Khismatullin RR, Ponomareva AA, Nagaswami C, Ivaeva RA, Montone KT, Weisel JW, et al. Pathology of Lung-Specific Thrombosis and Inflammation in COVID-19. J Thromb Haemost (2021) 19(12):3062–72. doi: 10.1111/jth.15532
33. Korompoki E, Gavriatopoulou M, Fotiou D, Ntanasis-Stathopoulos I, Dimopoulos MA, Terpos E. Late-Onset Hematological Complications Post COVID-19: An Emerging Medical Problem for the Hematologist. Am J Hematol (2022) 97(1):119–28. doi: 10.1002/ajh.26384
34. Oudkerk M, Büller HR, Kuijpers D, van Es N, Oudkerk SF, McLoud T, et al. Diagnosis, Prevention, and Treatment of Thromboembolic Complications in COVID-19: Report of the National Institute for Public Health of the Netherlands. Radiology (2020) 297(1):E216–22. doi: 10.1148/radiol.2020201629
35. Cheung PC, Eisch AR, Maleque N, Polly DM, Auld SC, Druey KM. Fatal Exacerbations of Systemic Capillary Leak Syndrome Complicating Coronavirus Disease. Emerg Infect Dis (2021) 27(10):2529–34. doi: 10.3201/eid2710.211155
36. Lacout C, Rogez J, Orvain C, Nicot C, Rony L, Julien H, et al. A New Diagnosis of Systemic Capillary Leak Syndrome on a Patient With COVID-19. Rheumatology (Oxford) (2021) 60(1):e19–20. doi: 10.1093/rheumatology/keaa606
37. Veluswamy P, Wacker M, Stavridis D, Reichel T, Schmidt H, Scherner M, et al. The SARS-CoV-2/Receptor Axis in Heart and Blood Vessels: A Crisp Update on COVID-19 Disease With Cardiovascular Complications. Viruses (2021) 13(7):1346. doi: 10.3390/v13071346
38. Gattinoni L, Chiumello D, Caironi P, Busana M, Romitti F, Brazzi L, et al. COVID-19 Pneumonia: Different Respiratory Treatments for Different Phenotypes? Intensive Care Med (2020) 46(6):1099–102. doi: 10.1007/s00134-020-06033-2
39. D’Agnillo F, Walters KA, Xiao Y, Sheng ZM, Scherler K, Park J, et al. Lung Epithelial and Endothelial Damage, Loss of Tissue Repair, Inhibition of Fibrinolysis, and Cellular Senescence in Fatal COVID-19. Sci Transl Med (2021) 13(620):eabj7790. doi: 10.1126/scitranslmed.abj7790
40. Lund LC, Hallas J, Nielsen H, Koch A, Mogensen SH, Brun NC, et al. Post-Acute Effects of SARS-CoV-2 Infection in Individuals Not Requiring Hospital Admission: A Danish Population-Based Cohort Study. Lancet Infect Dis (2021) 21(10):1373–82. doi: 10.1016/S1473-3099(21)00211-5
41. Aranda J, Oriol I, Martín M, Feria L, Vázquez N, Rhyman N, et al. Long-Term Impact of COVID-19 Associated Acute Respiratory Distress Syndrome. J Infect (2021) 83(5):581–8. doi: 10.1016/j.jinf.2021.08.018
42. Milross L, Majo J, Cooper N, Kaye PM, Bayraktar O, Filby A, et al. Post-Mortem Lung Tissue: The Fossil Record of the Pathophysiology and Immunopathology of Severe COVID-19. Lancet Respir Med (2022) 10(1):95–106. doi: 10.1016/S2213-2600(21)00408-2
43. Jadaun PK, Chatterjee S. COVID-19 and Dys-Regulation of Pulmonary Endothelium: Implications for Vascular Remodeling. Cytokine Growth Factor Rev (2021) 63:69–77. doi: 10.1016/j.cytogfr.2021.10.003
44. Chilosi M, Poletti V, Ravaglia C, Rossi G, Dubini A, Piciucchi S, et al. The Pathogenic Role of Epithelial and Endothelial Cells in Early-Phase COVID-19 Pneumonia: Victims and Partners in Crime. Mod Pathol (2021) 34(8):1444–55. doi: 10.1038/s41379-021-00808-8
45. Teuwen LA, Geldhof V, Pasut A, Carmeliet P. COVID-19: The Vasculature Unleashed. Nat Rev Immunol (2020) 20(7):389–91. doi: 10.1038/s41577-020-0343-0
46. Varga Z, Flammer AJ, Steiger P, Haberecker M, Andermatt R, Zinkernagel AS, et al. Endothelial Cell Infection and Endotheliitis in COVID-19. Lancet (2020) 395(10234):1417–8. doi: 10.1016/S0140-6736(20)30937-5
47. Pons S, Fodil S, Azoulay E, Zafrani L. The Vascular Endothelium: The Cornerstone of Organ Dysfunction in Severe SARS-CoV-2 Infection. Crit Care (2020) 24(1):353. doi: 10.1186/s13054-020-03062-7
48. Tarhini H, Recoing A, Bridier-Nahmias A, Rahi M, Lambert C, Martres P, et al. Long-Term Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Infectiousness Among Three Immunocompromised Patients: From Prolonged Viral Shedding to SARS-CoV-2 Superinfection. J Infect Dis (2021) 223(9):1522–7. doi: 10.1093/infdis/jiab075
49. Vardhana SA, Wolchok JD. The Many Faces of the Anti-COVID Immune Response. J Exp Med (2020) 217(6):e20200678. doi: 10.1084/jem.20200678
50. Proal AD, VanElzakker MB. Long COVID or Post-Acute Sequelae of COVID-19 (PASC): An Overview of Biological Factors That May Contribute to Persistent Symptoms. Front Microbiol (2021) 12:698169. doi: 10.3389/fmicb.2021.698169
51. Wu J, Liu X, Liu J, Liao H, Long S, Zhou N, et al. Coronavirus Disease 2019 Test Results After Clinical Recovery and Hospital Discharge Among Patients in China. JAMA Netw Open (2020) 3(5):e209759. doi: 10.1001/jamanetworkopen.2020.9759
52. Long QX, Tang XJ, Shi QL, Li Q, Deng HJ, Yuan J, et al. Clinical and Immunological Assessment of Asymptomatic SARS-CoV-2 Infections. Nat Med (2020) 26(8):1200–4. doi: 10.1038/s41591-020-0965-6
53. Harvey WT, Carabelli AM, Jackson B, Gupta RK, Thomson EC, Harrison EM, et al. SARS-CoV-2 Variants, Spike Mutations and Immune Escape. Nat Rev Microbiol (2021) 19(7):409–24. doi: 10.1038/s41579-021-00573-0
54. Wajnberg A, Amanat F, Firpo A, Altman DR, Bailey MJ, Mansour M, et al. Robust Neutralizing Antibodies to SARS-CoV-2 Infection Persist for Months. Science (2020) 370(6521):1227–30. doi: 10.1126/science.abd7728
55. De Melo GD, Lazarini F, Levallois S, Hautefort C, Michel V, Larrous F, et al. COVID-19-Associated Olfactory Dysfunction Reveals SARS-CoV-2 Neuroinvasion and Persistence in the Olfactory System. bioRxiv (2020) 2020:2020.11.18.388819. doi: 10.1101/2020.11.18.388819
56. Hu B, Guo H, Zhou P, Shi ZL. Characteristics of SARS-CoV-2 and COVID-19 [Published Correction Appears in Nat Rev Microbiol. 2022 Feb 23]. Nat Rev Microbiol (2021) 19(3):141–54. doi: 10.1038/s41579-020-00459-7
57. Ravichandran KS. Beginnings of a Good Apoptotic Meal: The Find-Me and Eat-Me Signaling Pathways. Immunity (2011) 35(4):445–55. doi: 10.1016/j.immuni.2011.09.004
58. Bagalkot V, Deiuliis JA, Rajagopalan S, Maiseyeu A. “Eat Me” Imaging and Therapy. Adv Drug Deliv Rev (2016) 99(Pt A):2–11. doi: 10.1016/j.addr.2016.01.009
59. Bridges JP, Vladar EK, Huang H, Mason RJ. Respiratory Epithelial Cell Responses to SARS-CoV-2 in COVID-19. Thorax (2022) 77(2):203–9. doi: 10.1136/thoraxjnl-2021-217561
60. Trougakos IP, Stamatelopoulos K, Terpos E, Tsitsilonis OE, Aivalioti E, Paraskevis D, et al. Insights to SARS-CoV-2 Life Cycle, Pathophysiology, and Rationalized Treatments That Target COVID-19 Clinical Complications. J BioMed Sci (2021) 28(1):9. doi: 10.1186/s12929-020-00703-5
61. Wazny V, Siau A, Wu KX, Cheung C. Vascular Underpinning of COVID-19. Open Biol (2020) 10(8):200208. doi: 10.1098/rsob.200208
62. Prasad M, Leon M, Lerman LO, Lerman A. Viral Endothelial Dysfunction: A Unifying Mechanism for COVID-19. Mayo Clin Proc (2021) 96(12):3099–108. doi: 10.1016/j.mayocp.2021.06.027
63. Higashikuni Y, Liu W, Obana T, Sata M. Pathogenic Basis of Thromboinflammation and Endothelial Injury in COVID-19: Current Findings and Therapeutic Implications. Int J Mol Sci (2021) 22(21):12081. doi: 10.3390/ijms222112081
64. Ma Z, Yang KY, Huang Y, Lui KO. Endothelial Contribution to COVID-19: An Update on Mechanisms and Therapeutic Implications. J Mol Cell Cardiol (2021) 164:69–82. doi: 10.1016/j.yjmcc.2021.11.010
65. Kander T. Coagulation Disorder in COVID-19. Lancet Haematol (2020) 7(9):e630–2. doi: 10.1016/S2352-3026(20)30218-0
66. Zarrilli G, Angerilli V, Businello G, Sbaraglia M, Traverso G, Fortarezza F, et al. The Immunopathological and Histological Landscape of COVID-19-Mediated Lung Injury. Int J Mol Sci (2021) 22(2):974. doi: 10.3390/ijms22020974
67. Deinhardt-Emmer S, Wittschieber D, Sanft J, Kleemann S, Elschner S, Haupt KF, et al. Early Postmortem Mapping Of SARS-Cov-2 RNA in Patients With COVID-19 and the Correlation With Tissue Damage. Elife (2021) 10:e60361. doi: 10.7554/eLife.60361
68. Mo X, Jian W, Su Z, Chen M, Peng H, Peng P, et al. Abnormal Pulmonary Function in COVID-19 Patients at Time of Hospital Discharge. Eur Respir J (2020) 55(6):2001217. doi: 10.1183/13993003.01217-2020
69. Huang Y, Tan C, Wu J, Chen M, Wang Z, Luo L, et al. Impact of Coronavirus Disease 2019 on Pulmonary Function in Early Convalescence Phase. Respir Res (2020) 21(1):163. doi: 10.1186/s12931-020-01429-6
70. Fajgenbaum DC, June CH. Cytokine Storm. N Engl J Med (2020) 383(23):2255–73. doi: 10.1056/NEJMra2026131
71. Leisman DE, Ronner L, Pinotti R, Taylor MD, Sinha P, Calfee CS, et al. Cytokine Elevation in Severe and Critical COVID-19: A Rapid Systematic Review, Meta-Analysis, and Comparison With Other Inflammatory Syndromes. Lancet Respir Med (2020) 8(12):1233–44. doi: 10.1016/S2213-2600(20)30404-5
72. Gao YM, Xu G, Wang B, Liu BC. Cytokine Storm Syndrome in Coronavirus Disease 2019: A Narrative Review. J Intern Med (2021) 289(2):147–61. doi: 10.1111/joim.13144
73. Kang S, Tanaka T, Inoue H, Ono C, Hashimoto S, Kioi Y, et al. IL-6 Trans-Signaling Induces Plasminogen Activator Inhibitor-1 From Vascular Endothelial Cells in Cytokine Release Syndrome. Proc Natl Acad Sci USA (2020) 17(36):22351–6. doi: 10.1073/pnas.2010229117
74. Kaur S, Bansal R, Kollimuttathuillam S, Gowda AM, Singh B, Mehta D, et al. The Looming Storm: Blood and Cytokines in COVID-19. Blood Rev (2021) 46:100743. doi: 10.1016/j.blre.2020.100743
75. Caricchio R, Gallucci M, Dass C, Zhang X, Gallucci S, Fleece D, et al. Preliminary Predictive Criteria for COVID-19 Cytokine Storm. Ann Rheum Dis (2021) 80(1):88–95. doi: 10.1136/annrheumdis-2020-218323
76. Chen R, Lan Z, Ye J, Pang L, Liu Y, Wu W, et al. Cytokine Storm: The Primary Determinant for the Pathophysiological Evolution of COVID-19 Deterioration. Front Immunol (2021) 12:589095. doi: 10.3389/fimmu.2021.589095
77. Mangalmurti N, Hunter CA. Cytokine Storms: Understanding COVID-19. Immunity (2020) 53(1):19–25. doi: 10.1016/j.immuni.2020.06.017
78. Shah A, Frost JN, Aaron L, Donovan K, Drakesmith H, Collaborators. Systemic Hypoferremia and Severity of Hypoxemic Respiratory Failure in COVID-19. Crit Care (2020) 24(1):320. doi: 10.1186/s13054-020-03051-w
79. Rauch A, Dupont A, Goutay J, Caplan M, Staessens S, Moussa M, et al. Endotheliopathy Is Induced by Plasma From Critically Ill Patients and Associated With Organ Failure in Severe COVID-19. Circulation (2020) 142(19):1881–4. doi: 10.1161/CIRCULATIONAHA.120.050907
80. Ackermann M, Verleden SE, Kuehnel M, Haverich A, Welte T, Laenger F, et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in COVID-19. N Engl J Med (2020) 383(2):120–8. doi: 10.1056/NEJMoa2015432
81. Tan BK, Mainbourg S, Friggeri A, Bertoletti L, Douplat M, Dargaud Y, et al. Arterial and Venous Thromboembolism in COVID-19: A Study-Level Meta-Analysis. Thorax (2021) 76(10):970–9. doi: 10.1136/thoraxjnl-2020-215383
82. Bellmunt-Montoya S, Riera C, Gil D, Rodríguez M, García-Reyes M, Martínez-Carnovale L, et al. COVID-19 Infection in Critically Ill Patients Carries a High Risk of Venous Thrombo-Embolism. Eur J Vasc Endovasc Surg (2021) 61(4):628–34. doi: 10.1016/j.ejvs.2020.12.015
83. McFadyen JD, Stevens H, Peter K. The Emerging Threat of (Micro)Thrombosis in COVID-19 and Its Therapeutic Implications. Circ Res (2020) 127(4):571–87. doi: 10.1161/CIRCRESAHA.120.317447
84. Voicu S, Ketfi C, Stépanian A, Chousterman BG, Mohamedi N, Siguret V, et al. Pathophysiological Processes Underlying the High Prevalence of Deep Vein Thrombosis in Critically Ill COVID-19 Patients. Front Physiol (2021) 11:608788. doi: 10.3389/fphys.2020.608788
85. Dhawan RT, Gopalan D, Howard L, Vicente A, Park M, Manalan K, et al. Beyond the Clot: Perfusion Imaging of the Pulmonary Vasculature After COVID-19. Lancet Respir Med (2021) 9(1):107–16. doi: 10.1016/S2213-2600(20)30407-0
86. Long Q, Li J, Hu X, Bai Y, Zheng Y, Gao Z. Follow-Ups on Persistent Symptoms and Pulmonary Function Among Post-Acute COVID-19 Patients: A Systematic Review and Meta-Analysis. Front Med (Lausanne) (2021) 8:702635. doi: 10.3389/fmed.2021.702635
87. Sivan M, Rayner C, Delaney B. Fresh Evidence of the Scale and Scope of Long COVID. BMJ (2021) 373:n853. doi: 10.1136/bmj.n853
88. Al-Aly Z, Xie Y, Bowe B. High-Dimensional Characterization of Post-Acute Sequelae of COVID-19. Nature (2021) 594(7862):259–64. doi: 10.1038/s41586-021-03553-9
89. Yomogida K, Zhu S, Rubino F, Figueroa W, Balanji N, Holman E. Post-Acute Sequelae of SARS-CoV-2 Infection Among Adults Aged ≥18 Years - Long Beach, California, April 1-December 10, 2020. MMWR Morb Mortal Wkly Rep (2021) 70(37):1274–7. doi: 10.15585/mmwr.mm7037a2
90. Daugherty SE, Guo Y, Heath K, Dasmariñas MC, Jubilo KG, Samranvedhya J, et al. Risk of Clinical Sequelae After the Acute Phase of SARS-CoV-2 Infection: Retrospective Cohort Study. BMJ (2021) 373:n1098. doi: 10.1136/bmj.n1098
91. Anaya JM, Rojas M, Salinas ML, Rodríguez Y, Roa G, Lozano M, et al. Post-COVID Syndrome. A Case Series and Comprehensive Review. Autoimmun Rev (2021) 20(11):102947. doi: 10.1016/j.autrev.2021.102947
92. Michelen M, Manoharan L, Elkheir N, Cheng V, Dagens A, Hastie C, et al. Characterising Long COVID: A Living Systematic Review. BMJ Glob Health (2021) 6(9):e005427. doi: 10.1136/bmjgh-2021-005427
93. O’Donnell JS, Peyvandi F, Martin-Loeches I. Pulmonary Immuno-Thrombosis in COVID-19 ARDS Pathogenesis. Intensive Care Med (2021) 47(8):899–902. doi: 10.1007/s00134-021-06419-w
94. Fang XZ, Wang YX, Xu JQ, He YJ, Peng ZK, Shang Y. Immunothrombosis in Acute Respiratory Dysfunction of COVID-19. Front Immunol (2021) 12:651545. doi: 10.3389/fimmu.2021.651545
95. Lim MS, Mcrae S. COVID-19 and Immunothrombosis: Pathophysiology and Therapeutic Implications. Crit Rev Oncol Hematol (2021) 168:103529. doi: 10.1016/j.critrevonc.2021.103529
96. Gattinoni L, Coppola S, Cressoni M, Busana M, Rossi S, Chiumello D. COVID-19 Does Not Lead to a “Typical” Acute Respiratory Distress Syndrome. Am J Respir Crit Care Med (2020) 201(10):1299–300. doi: 10.1164/rccm.202003-0817LE
97. Poor HD. Pulmonary Thrombosis and Thromboembolism in COVID-19. Chest (2021) 160(4):1471–80. doi: 10.1016/j.chest.2021.06.016
98. Magro C, Mulvey JJ, Berlin D, Nuovo G, Salvatore S, Harp J, et al. Complement Associated Microvascular Injury and Thrombosis in the Pathogenesis of Severe COVID-19 Infection: A Report of Five Cases. Transl Res (2020) 220:1–13. doi: 10.1016/j.trsl.2020.04.007
99. Synowiec A, Szczepański A, Barreto-Duran E, Lie LK, Pyrc K. Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2): A Systemic Infection. Clin Microbiol Rev (2021) 34(2):e00133-20. doi: 10.1128/CMR.00133-20
100. Writing Committee for the COMEBAC Study Group, Morin L, Savale L, Pham T, Colle R, Figueiredo S, et al. Four-Month Clinical Status of a Cohort of Patients After Hospitalization for COVID-19. JAMA (2021) 325(15):1525–34. doi: 10.1001/jama.2021.3331
101. Antonio GE, Wong KT, Hui DS, Wu A, Lee N, Yuen EH, et al. Thin-Section CT in Patients With Severe Acute Respiratory Syndrome Following Hospital Discharge: Preliminary Experience. Radiology (2003) 228(3):810–5. doi: 10.1148/radiol.2283030726
102. Das KM, Lee EY, Singh R, Enani MA, Al Dossari K, Van Gorkom K, et al. Follow-Up Chest Radiographic Findings in Patients With MERS-CoV After Recovery. Indian J Radiol Imaging (2017) 27(3):342–9. doi: 10.4103/ijri.IJRI_469_16
103. Norooznezhad AH, Mansouri K. Endothelial Cell Dysfunction, Coagulation, and Angiogenesis in Coronavirus Disease 2019 (COVID-19). Microvasc Res (2021) 137:104188. doi: 10.1016/j.mvr.2021.104188
104. Libby P, Lüscher T. COVID-19 Is, in the End, an Endothelial Disease. Eur Heart J (2020) 41(32):3038–44. doi: 10.1093/eurheartj/ehaa623
105. Goshua G, Pine AB, Meizlish ML, Chang CH, Zhang H, Bahel P, et al. Endotheliopathy in COVID-19-Associated Coagulopathy: Evidence From a Single-Centre, Cross-Sectional Study. Lancet Haematol (2020) 7(8):e575–82. doi: 10.1016/S2352-3026(20)30216-7
106. Pasini E, Corsetti G, Romano C, Scarabelli TM, Chen-Scarabelli C, Saravolatz L, et al. Serum Metabolic Profile in Patients With Long-Covid (PASC) Syndrome: Clinical Implications. Front Med (Lausanne) (2021) 8:714426. doi: 10.3389/fmed.2021.714426
107. de Candia P, Prattichizzo F, Garavelli S, Matarese G. T Cells: Warriors of SARS-CoV-2 Infection. Trends Immunol (2021) 42(1):18–30. doi: 10.1016/j.it.2020.11.002
108. Jarjour NN, Masopust D, Jameson SC. T Cell Memory: Understanding COVID-19. Immunity (2021) 54(1):14–8. doi: 10.1016/j.immuni.2020.12.009
109. Chen W, Pan JY. Anatomical and Pathological Observation and Analysis of SARS and COVID-19: Microthrombosis Is the Main Cause of Death. Biol Proced Online (2021) 23(1):4. doi: 10.1186/s12575-021-00142-y
110. Loo J, Spittle DA, Newnham M. COVID-19, Immunothrombosis and Venous Thromboembolism: Biological Mechanisms. Thorax (2021) 76(4):412–20. doi: 10.1136/thoraxjnl-2020-216243
111. Grobler C, Maphumulo SC, Grobbelaar LM, Bredenkamp JC, Laubscher GJ, Lourens PJ, et al. COVID-19: The Rollercoaster of Fibrin(Ogen), D-Dimer, Von Willebrand Factor, P-Selectin and Their Interactions With Endothelial Cells, Platelets and Erythrocytes. Int J Mol Sci (2020) 21(14):5168. doi: 10.3390/ijms21145168
112. Bevers EM, Williamson PL. Getting to the Outer Leaflet: Physiology of Phosphatidylserine Exposure at the Plasma Membrane. Physiol Rev (2016) 96(2):605–45. doi: 10.1152/physrev.00020.2015
113. Langer F, Ruf W. Synergies of Phosphatidylserine and Protein Disulfide Isomerase in Tissue Factor Activation. Thromb Haemost (2014) 111(4):590–7. doi: 10.1160/TH13-09-0802
114. Hanayama R, Tanaka M, Miwa K, Shinohara A, Iwamatsu A, Nagata S. Identification of a Factor That Links Apoptotic Cells to Phagocytes. Nature (2002) 417(6885):182–7. doi: 10.1038/417182a
115. Fricker M, Neher JJ, Zhao JW, Théry C, Tolkovsky AM, Brown GC. MFG-E8 Mediates Primary Phagocytosis of Viable Neurons During Neuroinflammation. J Neurosci (2012) 32(8):2657–66. doi: 10.1523/JNEUROSCI.4837-11.2012
116. Barrett TJ, Lee AH, Xia Y, Lin LH, Black M, Cotzia P, et al. Platelet and Vascular Biomarkers Associate With Thrombosis and Death in Coronavirus Disease. Circ Res (2020) 127(7):945–7. doi: 10.1161/CIRCRESAHA.120.317803
117. Barrett TJ, Lee A, Xia Y, Lin L, Black M, Cotzia P, et al. Abstract 15888: Platelet Activation Is Associated With All-Cause Mortality in Patients With COVID-19. Circulation (2020) 142(Suppl_3):A15888–A. doi: 10.1161/circ.142.suppl_3.15888
118. Puhm F, Flamand L, Boilard E. Platelet Extracellular Vesicles in COVID-19: Potential Markers and Makers. J Leukoc Biol (2022) 111(1):63–74. doi: 10.1002/JLB.3MIR0221-100R
119. Balbi C, Burrello J, Bolis S, Lazzarini E, Biemmi V, Pianezzi E, et al. Circulating Extracellular Vesicles Are Endowed With Enhanced Procoagulant Activity in SARS-CoV-2 Infection. EBioMedicine (2021) 67:103369. doi: 10.1016/j.ebiom.2021.103369
120. Georgescu A, Simionescu M. Extracellular Vesicles: Versatile Nanomediators, Potential Biomarkers and Therapeutic Agents in Atherosclerosis and COVID-19-Related Thrombosis. Int J Mol Sci (2021) 22(11):5967. doi: 10.3390/ijms22115967
121. Rosell A, Havervall S, von Meijenfeldt F, Hisada Y, Aguilera K, Grover SP, et al. Patients With COVID-19 Have Elevated Levels of Circulating Extracellular Vesicle Tissue Factor Activity That Is Associated With Severity and Mortality-Brief Report. Arterioscler Thromb Vasc Biol (2021) 41(2):878–82. doi: 10.1161/ATVBAHA.120.315547
122. Zaid Y, Puhm F, Allaeys I, Naya A, Oudghiri M, Khalki L, et al. Platelets Can Associate With SARS-CoV-2 RNA and Are Hyperactivated in COVID-19. Circ Res (2020) 127(11):1404–18. doi: 10.1161/CIRCRESAHA.120.317703
123. Rausch L, Lutz K, Schifferer M, Winheim E, Gruber R, Oesterhaus EF, et al. Binding of Phosphatidylserine-Positive Microparticles by Pbmcs Classifies Disease Severity in COVID-19 Patients. J Extracell Vesicles (2021) 10(14):e12173. doi: 10.1002/jev2.12173
124. Zhang Y, Xiao M, Zhang S, Xia P, Cao W, Jiang W, et al. Coagulopathy and Antiphospholipid Antibodies in Patients With COVID-19. N Engl J Med (2020) 382(17):e38. doi: 10.1056/NEJMc2007575
125. Semeraro F, Ammollo CT, Esmon NL, Esmon CT. Histones Induce Phosphatidylserine Exposure and a Procoagulant Phenotype in Human Red Blood Cells. J Thromb Haemost (2014) 12(10):1697–702. doi: 10.1111/jth.12677
126. Yeung T, Gilbert GE, Shi J, Silvius J, Kapus A, Grinstein S. Membrane Phosphatidylserine Regulates Surface Charge and Protein Localization. Science (2008) 319(5860):210–3. doi: 10.1126/science.1152066
127. Krishnamachary B, Cook C, Kumar A, Spikes L, Chalise P, Dhillon NK. Extracellular Vesicle-Mediated Endothelial Apoptosis and EV-Associated Proteins Correlate With COVID-19 Disease Severity. J Extracell Vesicles (2021) 10(9):e12117. doi: 10.1002/jev2.12117
128. Argañaraz GA, Palmeira JDF, Argañaraz ER. Phosphatidylserine Inside Out: A Possible Underlying Mechanism in the Inflammation and Coagulation Abnormalities of COVID-19. Cell Commun Signal (2020) 18(1):190. doi: 10.1186/s12964-020-00687-7
129. Ma R, Xie R, Yu C, Si Y, Wu X, Zhao L, et al. Phosphatidylserine-Mediated Platelet Clearance by Endothelium Decreases Platelet Aggregates and Procoagulant Activity in Sepsis. Sci Rep (2017) 7(1):4978. doi: 10.1038/s41598-017-04773-8
130. Gao C, Xie R, Yu C, Wang Q, Shi F, Yao C, et al. Procoagulant Activity of Erythrocytes and Platelets Through Phosphatidylserine Exposure and Microparticles Release in Patients With Nephrotic Syndrome. Thromb Haemost (2012) 107(4):681–9. doi: 10.1160/TH11-09-0673
131. Althaus K, Marini I, Zlamal J, Pelzl L, Singh A, Häberle H, et al. Antibody-Induced Procoagulant Platelets in Severe COVID-19 Infection. Blood (2021) 137(8):1061–71. doi: 10.1182/blood.2020008762
132. Fu Y, Zhou J, Li H, Cao F, Su Y, Fan S, et al. Daunorubicin Induces Procoagulant Activity of Cultured Endothelial Cells Through Phosphatidylserine Exposure and Microparticles Release. Thromb Haemost (2010) 104(6):1235–41. doi: 10.1160/TH10-02-0102
133. Zhou J, Shi J, Hou J, Cao F, Zhang Y, Rasmussen JT, et al. Phosphatidylserine Exposure and Procoagulant Activity in Acute Promyelocytic Leukemia. J Thromb Haemost (2010) 8(4):773–82. doi: 10.1111/j.1538-7836.2010.03763.x
134. Cao M, Li T, He Z, Wang L, Yang X, Kou Y, et al. Promyelocytic Extracellular Chromatin Exacerbates Coagulation and Fibrinolysis in Acute Promyelocytic Leukemia. Blood (2017) 129(13):1855–64. doi: 10.1182/blood-2016-09-739334
135. He Z, Si Y, Jiang T, Ma R, Zhang Y, Cao M, et al. Phosphotidylserine Exposure and Neutrophil Extracellular Traps Enhance Procoagulant Activity in Patients With Inflammatory Bowel Disease. Thromb Haemost (2016) 115(4):738–51. doi: 10.1160/TH15-09-0710
136. Xie R, Gao C, Li W, Zhu J, Novakovic V, Wang J, et al. Phagocytosis by Macrophages and Endothelial Cells Inhibits Procoagulant and Fibrinolytic Activity of Acute Promyelocytic Leukemia Cells. Blood (2012) 119(10):2325–34. doi: 10.1182/blood-2011-06-362186
137. Viecca M, Radovanovic D, Forleo GB, Santus P. Enhanced Platelet Inhibition Treatment Improves Hypoxemia in Patients With Severe COVID-19 and Hypercoagulability. A Case Control, Proof of Concept Study. Pharmacol Res (2020) 158:104950. doi: 10.1016/j.phrs.2020.104950
138. Flaczyk A, Rosovsky RP, Reed CT, Bankhead-Kendall BK, Bittner EA, Chang MG. Comparison of Published Guidelines for Management of Coagulopathy and Thrombosis in Critically Ill Patients With COVID 19: Implications for Clinical Practice and Future Investigations. Crit Care (2020) 24(1):559. doi: 10.1186/s13054-020-03273-y
139. Billett HH, Reyes-Gil M, Szymanski J, Ikemura K, Stahl LR, Lo Y, et al. Anticoagulation in COVID-19: Effect of Enoxaparin, Heparin, and Apixaban on Mortality. Thromb Haemost (2020) 120(12):1691–9. doi: 10.1055/s-0040-1720978
140. Short SAP, Gupta S, Brenner SK, Hayek SS, Srivastava A, Shaefi S, et al. D-Dimer and Death in Critically Ill Patients With Coronavirus Disease 2019. Crit Care Med (2021) 49(5):e500–11. doi: 10.1097/CCM.0000000000004917
141. Kruse JM, Magomedov A, Kurreck A, Münch FH, Koerner R, Kamhieh-Milz J, et al. Thromboembolic Complications in Critically Ill COVID-19 Patients Are Associated With Impaired Fibrinolysis. Crit Care (2020) 24(1):676. doi: 10.1186/s13054-020-03401-8
142. Meizoso JP, Moore HB, Moore EE. Fibrinolysis Shutdown in COVID-19: Clinical Manifestations, Molecular Mechanisms, and Therapeutic Implications. J Am Coll Surg (2021) 232(6):995–1003. doi: 10.1016/j.jamcollsurg.2021.02.019
143. Bachler M, Bösch J, Stürzel DP, Hell T, Giebl A, Ströhle M, et al. Impaired Fibrinolysis in Critically Ill COVID-19 Patients. Br J Anaesth (2021) 126(3):590–8. doi: 10.1016/j.bja.2020.12.010
144. Nougier C, Benoit R, Simon M, Desmurs-Clavel H, Marcotte G, Argaud L, et al. Hypofibrinolytic State and High Thrombin Generation May Play a Major Role in SARS-CoV2 Associated Thrombosis. J Thromb Haemost (2020) 18(9):2215–9. doi: 10.1111/jth.15016
145. Thachil J, Tang N, Gando S, Falanga A, Cattaneo M, Levi M, et al. ISTH Interim Guidance on Recognition and Management of Coagulopathy in COVID-19. J Thromb Haemost (2020) 18(5):1023–6. doi: 10.1111/jth.14810
146. Moores LK, Tritschler T, Brosnahan S, Carrier M, Collen JF, Doerschug K, et al. Prevention, Diagnosis, and Treatment of VTE in Patients With Coronavirus Disease 2019: CHEST Guideline and Expert Panel Report. Chest (2020) 158(3):1143–63. doi: 10.1016/j.chest.2020.05.559
147. Cuker A, Tseng EK, Nieuwlaat R, Angchaisuksiri P, Blair C, Dane K, et al. American Society of Hematology 2021 Guidelines on the Use of Anticoagulation for Thromboprophylaxis in Patients With COVID-19. Blood Adv (2021) 5(3):872–88. doi: 10.1182/bloodadvances.2020003763
148. Spyropoulos AC, Levy JH, Ageno W, Connors JM, Hunt BJ, Iba T, et al. Scientific and Standardization Committee Communication: Clinical Guidance on the Diagnosis, Prevention, and Treatment of Venous Thromboembolism in Hospitalized Patients With COVID-19. J Thromb Haemost (2020) 18(8):1859–65. doi: 10.1111/jth.14929
149. Gerotziafas GT, Catalano M, Colgan MP, Pecsvarady Z, Wautrecht JC, Fazeli B, et al. Guidance for the Management of Patients With Vascular Disease or Cardiovascular Risk Factors and COVID-19: Position Paper From VAS-European Independent Foundation in Angiology/Vascular Medicine. Thromb Haemost (2020) 120(12):1597–628. doi: 10.1055/s-0040-1715798
150. Buijsers B, Yanginlar C, Maciej-Hulme ML, de Mast Q, van der Vlag J. Beneficial Non-Anticoagulant Mechanisms Underlying Heparin Treatment of COVID-19 Patients. EBioMedicine (2020) 59:102969. doi: 10.1016/j.ebiom.2020.102969
151. Tacquard C, Mansour A, Godon A, Godet J, Poissy J, Garrigue D, et al. Impact of High-Dose Prophylactic Anticoagulation in Critically Ill Patients With COVID-19 Pneumonia. Chest (2021) 159(6):2417–27. doi: 10.1016/j.chest.2021.01.017
152. Paranjpe I, Fuster V, Lala A, Russak AJ, Glicksberg BS, Levin MA, et al. Association of Treatment Dose Anticoagulation With In-Hospital Survival Among Hospitalized Patients With COVID-19. J Am Coll Cardiol (2020) 76(1):122–4. doi: 10.1016/j.jacc.2020.05.001
153. Donato AA. In Non-Critically Ill Patients With COVID-19, Therapeutic Anticoagulation Improved Survival to Discharge Without Organ Support. Ann Intern Med (2021) 174(12):JC134. doi: 10.7326/ACPJ202112210-134
154. Jonmarker S, Hollenberg J, Dahlberg M, Stackelberg O, Litorell J, Everhov ÅH, et al. Dosing of Thromboprophylaxis and Mortality in Critically Ill COVID-19 Patients. Crit Care (2020) 24(1):653. doi: 10.1186/s13054-020-03375-7
155. Al-Samkari H, Karp Leaf RS, Dzik WH, Carlson JCT, Fogerty AE, Waheed A, et al. COVID-19 and Coagulation: Bleeding and Thrombotic Manifestations of SARS-CoV-2 Infection. Blood (2020) 136(4):489–500. doi: 10.1182/blood.2020006520
156. Nadkarni GN, Lala A, Bagiella E, Chang HL, Moreno PR, Pujadas E, et al. Anticoagulation, Bleeding, Mortality, and Pathology in Hospitalized Patients With COVID-19. J Am Coll Cardiol (2020) 76(16):1815–26. doi: 10.1016/j.jacc.2020.08.041
157. Connors JM, Levy JH. COVID-19 and Its Implications for Thrombosis and Anticoagulation. Blood (2020) 135(23):2033–40. doi: 10.1182/blood.2020006000
158. Pavoni V, Gianesello L, Pazzi M, Stera C, Meconi T, Frigieri FC. Venous Thromboembolism and Bleeding in Critically Ill COVID-19 Patients Treated With Higher Than Standard Low Molecular Weight Heparin Doses and Aspirin: A Call to Action. Thromb Res (2020) 196:313–7. doi: 10.1016/j.thromres.2020.09.013
159. Lopes RD, de Barros E Silva PGM, Furtado RHM, Macedo AVS, Bronhara B, Damiani LP, et al. Therapeutic Versus Prophylactic Anticoagulation for Patients Admitted to Hospital With COVID-19 and Elevated D-Dimer Concentration (ACTION): An Open-Label, Multicentre, Randomised, Controlled Trial. Lancet (2021) 397(10291):2253–63. doi: 10.1016/S0140-6736(21)01203-4
160. Kolanko E, Senderek T, Prokop-Staszecka A, Kruk A, Broniatowska E, Konieczyńska M, et al. Thromboprophylaxis in Hospitalized COVID-19 Patients: The Efficacy and Safety of the Approved Hospital Protocol. Pol Arch Intern Med (2021) 131(10):16102. doi: 10.20452/pamw.16102
161. Kabir AA. Anticoagulation Is the Answer in Treating Noncritical COVID-19 Patients. Open Med (Wars) (2021) 16(1):1486–92. doi: 10.1515/med-2021-0354
162. Chocron R, Galand V, Cellier J, Gendron N, Pommier T, Bory O, et al. Critical COVID-19 France Investigators. Anticoagulation Before Hospitalization Is a Potential Protective Factor for COVID-19: Insight From a French Multicenter Cohort Study. J Am Heart Assoc (2021) 10(8):e018624. doi: 10.1161/JAHA.120.018624
163. ATTACC Investigators, ACTIV-4a Investigators, REMAP-CAP Investigators, Lawler PR, Goligher EC, Berger JS, et al. Therapeutic Anticoagulation With Heparin in Noncritically Ill Patients With COVID-19. N Engl J Med (2021) 385(9):790–802. doi: 10.1056/NEJMoa2105911
164. Rentsch CT, Beckman JA, Tomlinson L, Gellad WF, Alcorn C, Kidwai-Khan F, et al. Early Initiation of Prophylactic Anticoagulation for Prevention of Coronavirus Disease 2019 Mortality in Patients Admitted to Hospital in the United States: Cohort Study. BMJ (2021) 372:n311. doi: 10.1136/bmj.n311
165. Cuker A, Tseng EK, Nieuwlaat R, Angchaisuksiri P, Blair C, Dane K, et al. American Society of Hematology Living Guidelines on the Use of Anticoagulation for Thromboprophylaxis in Patients With COVID-19: July 2021 Update on Postdischarge Thromboprophylaxis. Blood Adv (2022) 6(2):664–71. doi: 10.1182/bloodadvances.2021005945
166. Kanthi Y, Knight JS, Zuo Y, Pinsky DJ. New (Re)Purpose for an Old Drug: Purinergic Modulation May Extinguish the COVID-19 Thromboinflammatory Firestorm. JCI Insight (2020) 5(14):e140971. doi: 10.1172/jci.insight.140971
167. Sriram K, Insel PA. Inflammation and Thrombosis in COVID-19 Pathophysiology: Proteinase-Activated and Purinergic Receptors As Drivers and Candidate Therapeutic Targets. Physiol Rev (2021) 101(2):545–67. doi: 10.1152/physrev.00035.2020
168. Bianconi V, Violi F, Fallarino F, Pignatelli P, Sahebkar A, Pirro M. Is Acetylsalicylic Acid a Safe and Potentially Useful Choice for Adult Patients With COVID-19? Drugs (2020) 80(14):1383–96. doi: 10.1007/s40265-020-01365-1
169. Meizlish ML, Goshua G, Liu Y, Fine R, Amin K, Chang E, et al. Intermediate-Dose Anticoagulation, Aspirin, and In-Hospital Mortality in COVID-19: A Propensity Score-Matched Analysis. Am J Hematol (2021) 96(4):471–9. doi: 10.1002/ajh.26102
170. Chow JH, Khanna AK, Kethireddy S, Yamane D, Levine A, Jackson AM, et al. Aspirin Use Is Associated With Decreased Mechanical Ventilation, Intensive Care Unit Admission, and In-Hospital Mortality in Hospitalized Patients With Coronavirus Disease 2019. Anesth Analg (2021) 132(4):930–41. doi: 10.1213/ANE.0000000000005292
171. Santoro F, Nuñez-Gil IJ, Vitale E, Viana-Llamas MC, Reche-Martinez B, Romero-Pareja R, et al. Antiplatelet Therapy and Outcome in COVID-19: The Health Outcome Predictive Evaluation Registry. Heart (2022) 108(2):130–6. doi: 10.1136/heartjnl-2021-319552
172. Kor DJ, Carter RE, Park PK, Festic E, Banner-Goodspeed VM, Hinds R, et al. Effect of Aspirin on Development of ARDS in At-Risk Patients Presenting to the Emergency Department: The LIPS-A Randomized Clinical Trial. JAMA (2016) 315(22):2406–14. doi: 10.1001/jama.2016.6330
173. Hou J, Fu Y, Zhou J, Li W, Xie R, Cao F, et al. Lactadherin Functions As A Probe for Phosphatidylserine Exposure and As An Anticoagulant in the Study of Stored Platelets. Vox Sang (2011) 100(2):187–95. doi: 10.1111/j.1423-0410.2010.01375.x
174. Dasgupta SK, Guchhait P, Thiagarajan P. Lactadherin Binding and Phosphatidylserine Expression on Cell Surface-Comparison With Annexin A5. Transl Res (2006) 148(1):19–25. doi: 10.1016/j.lab.2006.03.006
175. Shi J, Gilbert GE. Lactadherin Inhibits Enzyme Complexes of Blood Coagulation by Competing for Phospholipid-Binding Sites. Blood (2003) 101(7):2628–36. doi: 10.1182/blood-2002-07-1951
176. Dasgupta SK, Abdel-Monem H, Niravath P, Le A, Bellera RV, Langlois K, et al. Lactadherin and Clearance of Platelet-Derived Microvesicles. Blood (2009) 113(6):1332–9. doi: 10.1182/blood-2008-07-167148
Keywords: COVID-19, long COVID, thrombosis, phosphatidylserine, therapy, anticoagulation
Citation: Xiang M, Jing H, Wang C, Novakovic VA and Shi J (2022) Persistent Lung Injury and Prothrombotic State in Long COVID. Front. Immunol. 13:862522. doi: 10.3389/fimmu.2022.862522
Received: 26 January 2022; Accepted: 17 March 2022;
Published: 07 April 2022.
Edited by:
Tessa Barrett, NYU Grossman School of Medicine, United StatesReviewed by:
Marcin Sowa, NYU Grossman School of Medicine, United StatesZhimin Tao, Jiangsu University, China
Copyright © 2022 Xiang, Jing, Wang, Novakovic and Shi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jialan Shi, Jialan_shi@dfci.harvard.edu; shi73661@gmail.com